


Abstract
Some of the most efficacious antiviral therapeutics are ribonucleos(t)ide analogs. The presence of a 3′-to-5′ proofreading exoribonuclease (ExoN) in coronaviruses diminishes the potency of many ribonucleotide analogs. The ability to interfere with ExoN activity will create new possibilities for control of SARS-CoV-2 infection. ExoN is formed by a 1:1 complex of nsp14 and nsp10 proteins. We have purified and characterized ExoN using a robust, quantitative system that reveals determinants of specificity and efficiency of hydrolysis. Double-stranded RNA is preferred over single-stranded RNA. Nucleotide excision is distributive, with only one or two nucleotides hydrolyzed in a single binding event. The composition of the terminal basepair modulates excision. A stalled SARS-CoV-2 replicase in complex with either correctly or incorrectly terminated products prevents excision, suggesting that a mispaired end is insufficient to displace the replicase. Finally, we have discovered several modifications to the 3′-RNA terminus that interfere with or block ExoN-catalyzed excision. While a 3′-OH facilitates hydrolysis of a nucleotide with a normal ribose configuration, this substituent is not required for a nucleotide with a planar ribose configuration such as that present in the antiviral nucleotide produced by viperin. Design of ExoN-resistant, antiviral ribonucleotides should be feasible.
INTRODUCTION
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, has led to a global pandemic and caused immeasurable consequences to humankind even more substantial than the incidence of disease and death. While the development of safe and effective vaccines has diminished overall morbidity and mortality, transmission of SARS-CoV-2 continues. Current therapeutics against SARS-CoV-2 infection include polymerase inhibitors: remdesivir and molnupiravir (1,2); and a protease inhibitor: nirmatrelvir, which is boosted with ritonavir (3). All of these therapeutic agents have complications that are tolerable in the midst of the pandemic. However, safer and more effective antiviral agents are needed against multiple targets to support the use of drug cocktails to maximize therapeutic efficacy and minimize the possibility of resistance.
Ribonucleos(t)ide analogs are among the most effective antiviral therapeutics for treatment of RNA virus infections (4–7). This class of compounds is generally administered as the ribonucleoside or ribonucleoside monophosphate prodrug. Cellular kinases then produce the active metabolite, a triphosphorylated ribonucleotide (rNTP) (7). Utilization of the rNTP analog by viral polymerases leads to one or more consequences: chain termination, lethal mutagenesis, backtracking, pausing, and/or recombination, all of which exhibit an antiviral effect (5,6,8–10).
The coronavirus genome is on the order of 30 000 nt, among the largest RNA genome known (11). Because the fidelity of viral RNA polymerases is usually lower than 1 error per 10 000 nt, how coronaviruses resist lethal mutagenesis has been a longstanding question for those studying these viruses (12–14). Efforts initiated in response to emergence of the first SARS-CoV led to the discovery of a 3′-to-5′ proofreading exoribonuclease, termed ExoN (15). ExoN is a member of the DEDDh/DEEDh subfamily in the DEED family of exonucleases (13,14,16). These enzymes use a two-metal-ion mechanism for catalysis (13,14,17,18). ExoN is composed of a 1:1 complex of non-structural proteins 14 (60 kDa) and 10 (15 kDa). Nsp14 harbors two catalytic domains: an N-terminal exoribonuclease and a C-terminal methyltransferase (13,14). Nsp10 serves as an accessory factor, stabilizing the active conformation of the exoribonuclease active site and thereby stimulating its activity (19–22).
Consistent with a role of ExoN in proofreading and therefore genome stability, previous studies have shown that genetic inactivation is most often lethal (15,23,24). For those coronaviruses, which are viable in the absence of active ExoN, the genomes contain a higher mutational load. Also, these viruses exhibit enhanced sensitivity to some antiviral ribonucleotides (25). Collectively, these observations suggest that inhibitors of ExoN will exhibit antiviral activity and may synergize with ribonucleotide-based inhibitors of the viral polymerase (26). Moreover, an understanding of the determinants of the substrate nucleotide that promote or interfere with excision may guide the design of ExoN-resistant ribonucleotide analogs.
Proofreading DNA exonucleases are generally a subunit of the DNA polymerase holoenzyme and exhibit a preference for mispaired ends (27). This circumstance appears to reflect the preferential partitioning of the mispaired end to the active site of the exonuclease instead of the active site of the polymerase (27,28). How proofreading by ExoN is initiated is not known. Given the dimensions of ExoN and the location of the polymerase active site within the SARS-CoV-2 replication-transcription complex (RTC), an active-site-switching mechanism is unlikely (29,30). It is easy to imagine how an end that cannot be extended by the RTC could become a substrate for repair by ExoN after the RTC dissociates. The ability for nucleotide analogs like remdesivir and molnupiravir to display efficacy in the presence of active ExoN may reflect the inability of these analogs to perturb elongation upon incorporation, with the antiviral activity manifested at the level of the analog-substituted template (31–33).
We have expressed and purified ExoN, and established a robust, quantitative system to study the determinants of the scissile phosphodiester bond and terminal ribonucleotide driving efficient excision by ExoN. ExoN prefers a primed-template-like dsRNA substrate over a ssRNA substrate and cannot access the 3′-terminus at a nick. ExoN only cleaves one or two nucleotides in a single binding event, as expected for a proofreading enzyme (27,28). Cleavage can be completely inhibited by the presence of the phosphorothioate Rp isomer at the scissile phosphodiester bond. A mispaired end does not appear to be highly favored by ExoN relative to a paired end in the absence or presence of a replicating RTC. However, stalling the RTC at the 3′-end of nascent RNA blocks excision regardless of the nature of the basepair. The inability of a mispaired end to promote dissociation of the RTC suggests a role for additional factors in this process. Finally, we identify modifications to the 3′-terminal nucleotide that interfere with excision by ExoN. The most unexpected finding was that the ribose conformation determines whether the 3′-OH is required for efficient turnover. The antiviral nucleotide, ddhCTP, produced by the antiviral protein, viperin, is readily excised by ExoN. This molecule lacks a 3′-OH but also lacks sugar pucker because of the presence of a double bond forcing the ribose into a planar conformation. This observation is consistent with repair of ddhC-terminated RNA as a major driver for acquisition of ExoN and evolution of its substrate specificity.
MATERIALS AND METHODS
Materials
DNA oligonucleotides and dsDNA fragments, GBlocks, were from Integrated DNA Technologies. RNA oligonucleotides were either from Horizon Discovery Ltd. (Dharmacon) or Integrated DNA Technologies. Restriction enzymes and T4 PNK (3′-phosphatase minus) were from New England Biolabs. IN-FUSION HD enzyme was from TakaraBio. Phusion DNA polymerase and T4 polynucleotide kinase were from ThermoFisher. pBirACm plasmid DNA was from Avidity. Streptactin XT 4F High-Capacity resin was from IBA Life Sciences. [γ-32P]ATP (6000 Ci/mmol) and [α-32P]ATP (3000 Ci/mmol) were from Perkin Elmer. Nucleoside 5′-triphosphates (ultrapure solutions) were from Cytiva. pET16b-RtcA-NTerm-His expression plasmid was provided by Stewart Shuman (Sloan Kettering) (34). Cytidine 5′-O-(1-thiotriphosphate) and 3′-deoxycytidine 5′-triphosphate were from TriLink. Adenosine 5′-O-(1-thiotriphosphate) (Sp isomer) was from BioLog. Remdesivir-terminated RNA was synthesized by both Dharmacon and the Harki lab as previously described (35). Remdesivir was provided to Dharmacon by Gilead Sciences, and this was chemically converted to the phosphoramidite to be synthetically incorporated into RNA. ddhCTP was synthesized by the Harki lab as previously described (36); 2′-C-methylcytidine 5′-triphosphate was provided by Gilead Sciences. All other reagents were of the highest grade available from MilliporeSigma, VWR, or Fisher Scientific.
Construction of modified pSUMO vectors containing AviTag
The pSUMO system allows for production of SUMO fusion proteins containing an amino-terminal affinity tag fused to SUMO that can be purified by affinity chromatography and subsequently processed by the SUMO protease, Ulp1 (37). After cleavage this will produce an authentic untagged protein target of interest. The pSUMO vector (LifeSensors) (37) was modified such that the coding sequence for the six-histidine tag was replaced with a DNA sequence coding for an AviTag codon optimized for bacterial expression (38,39). The AviTag is a short 15 amino acid sequence (GLNDIFEAQKIEWHE) that can be specifically biotinylated on the lysine residue by the biotin ligase, BirA (38,39). The construct contains two tags separated by a short linker to increase the affinity to the chromatography resin during purification. The biotinylated AviTag allows affinity purification of the fusion protein to be isolated using streptavidin or Strep-Tactin resins. The DNA sequences (GBlocks encoding the tags) were cloned into pSUMO using XbaI and SalI by IN-FUSION. The final constructs (pAviTag_SUMO) were confirmed by sanger sequencing performed by Genewiz.
Codon-optimized sequence for AviTag
5′ATGGGACTAAATGATATATTTGAAGCTCAAAAGATCGAGTGGCACGAGGGTGGTGGCAGCGGTGGCGGCTCCGGCGGTAGCGGCCTGAACGACATCTTCGAGGCGCAGAAAATTGAATGGCATGAAGGTGGCTCTAGCGGTGGT3′
(amino acid sequence: MGLNDIFEAQKIEWHEGGGSGGGSGGSGLNDIFEAQKIEWHEGGSSGG)
Construction of SARS-CoV-2 nsp10 and nsp14 bacterial expression plasmids
The SARS-CoV-2 nsp10 and nsp14 genes were codon optimized for expression in E. coli and obtained from Genescript. The amino acid sequences for nsp10 and nsp14 were derived from SARS-CoV-2 isolate 2019-nCoV/USA-WA1/2020 (GenBank MN985325.1). The genes were amplified by PCR using Phusion DNA polymerase. The nsp10 gene was amplified using the synthetic nsp10 gene as template and DNA oligonucleotides (5′-GAACAGATTGGAGGTGCCGGGAATGCTACGGAA-3′ and 5′-CCGCAAGCTTGTCGACTTATCATTGAAGCATAGGTTCACGCAA-3′). The nsp14 gene was amplified using the synthetic nsp14 gene as template and DNA oligonucleotides (5′-GAACAGATTGGAGGTGCGGAGAACGTTACAGGT and 5′-CCGCAAGCTTGTCGACTTATCATTGAAGGCCAGTAAACGTATTCCA-3′). The PCR products were gel purified and cloned into either the pAviTag-pSUMO bacterial expression plasmids using BsaI and SalI. The final constructs were confirmed by sanger sequencing performed by Genewiz. To construct a catalytically inactive nsp14, the WT nsp14 expression plasmid was modified such that D90 and E92 were both changed to alanine. This was performed using Quickchange mutagenesis using the WT nsp14 expression plasmid as template and DNA oligonucleotides (5′-GCGTGGATTGGTTTTGCTGTTGCGGGTTGCCACGCGACCCGT-3′ and 5′-ACGGGTCGCGTGGCAACCCGCAACAGCAAAACCAATCCACGC-3′). The final construct was confirmed by sanger sequencing performed by Genewiz.
Codon optimized sequence for SARS-CoV-2 nsp10
5′GCCGGGAATGCTACGGAAGTTCCAGCTAACTCGACCGTTCTTAGCTTTTGTGCTTTTGCAGTCGATGCAGCGAAAGCGTATAAGGACTATCTGGCGTCAGGGGGACAACCCATTACTAACTGTGTCAAGATGCTGTGTACCCATACCGGCACGGGTCAAGCGATTACTGTTACACCAGAAGCTAACATGGACCAGGAATCTTTTGGTGGTGCCAGTTGCTGCTTGTACTGCCGCTGTCATATCGATCACCCCAATCCAAAAGGTTTCTGCGATCTGAAGGGAAAATACGTGCAAATCCCCACCACTTGTGCTAATGACCCGGTCGGATTTACGCTGAAGAACACCGTTTGTACTGTTTGCGGGATGTGGAAAGGGTATGGGTGTTCTTGCGACCAGTTGCGTGAACCTATGCTTCAA3′.
Codon optimized sequence for SARS-CoV-2 nsp14
5′GCGGAGAACGTTACAGGTTTATTTAAGGATTGCTCTAAAGTAATTACCGGCCTGCACCCAACGCAGGCACCAACTCATCTTAGCGTGGATACAAAATTTAAGACAGAAGGACTGTGTGTGGACATTCCTGGCATCCCAAAGGACATGACATACCGCCGTTTGATCTCCATGATGGGGTTCAAAATGAACTACCAGGTAAACGGATACCCTAATATGTTCATTACACGTGAGGAGGCGATTCGTCATGTCCGCGCCTGGATCGGATTCGACGTAGAAGGTTGCCACGCCACCCGTGAGGCTGTGGGGACGAACTTACCCCTTCAGCTTGGCTTCTCAACTGGGGTAAACTTGGTGGCCGTCCCGACAGGGTATGTTGACACTCCTAATAACACTGATTTCTCGCGTGTATCTGCAAAGCCACCACCAGGGGACCAGTTCAAACACCTGATCCCCCTGATGTATAAGGGTCTTCCTTGGAATGTGGTCCGTATTAAAATCGTCCAGATGCTGTCAGACACCCTTAAGAATCTGTCAGATCGTGTGGTATTTGTATTGTGGGCGCACGGATTCGAGTTAACAAGCATGAAATATTTTGTGAAAATTGGCCCCGAACGCACATGCTGCTTATGCGATCGTCGCGCTACTTGCTTTAGTACTGCTTCAGACACTTATGCCTGCTGGCACCACTCTATTGGATTTGACTACGTGTATAACCCATTCATGATTGATGTCCAGCAGTGGGGCTTCACCGGGAACTTGCAGTCCAACCATGACCTTTATTGTCAGGTTCACGGAAATGCCCACGTGGCAAGCTGCGACGCGATTATGACACGCTGTCTGGCGGTACATGAGTGCTTTGTAAAGCGTGTCGATTGGACCATCGAGTATCCAATCATTGGAGACGAACTTAAGATCAATGCCGCATGCCGTAAAGTTCAACACATGGTAGTAAAGGCCGCCCTTCTTGCGGATAAGTTTCCGGTTCTGCATGACATTGGCAACCCTAAGGCGATTAAGTGTGTCCCGCAGGCGGATGTCGAATGGAAATTCTATGACGCGCAACCCTGCTCGGATAAAGCATATAAAATCGAAGAGCTGTTTTATTCATACGCTACGCATTCCGACAAGTTTACAGATGGCGTTTGTCTTTTTTGGAATTGTAACGTTGATCGCTACCCGGCGAACTCAATCGTTTGCCGCTTTGACACACGTGTGCTGTCTAACTTGAACTTGCCTGGTTGCGATGGAGGCTCGTTGTATGTTAATAAACATGCGTTTCATACCCCCGCCTTCGACAAGTCCGCTTTCGTAAACCTGAAGCAGTTGCCATTTTTCTACTATAGCGACTCACCGTGCGAGTCCCACGGTAAGCAAGTAGTGTCTGACATTGATTATGTACCTTTAAAAAGTGCTACCTGCATCACCCGTTGCAACTTGGGCGGAGCGGTTTGCCGCCACCATGCGAACGAATATCGCTTATACCTTGATGCCTATAATATGATGATTAGCGCGGGATTTAGCCTTTGGGTTTATAAACAGTTCGATACTTATAACCTGTGGAATACGTTTACTCGCCTTCAA3′.
Expression and purification of SARS-CoV-2 nsp10
Escherichia coli BL21(DE3)pBirACm competent cells were transformed with the pAviTag_SUMO-SARS-CoV-2-nsp10 plasmid for protein expression. BL21(DE3)pBirACm cells containing the pAviTag_SUMO-SARS-CoV-2-nsp10 plasmid were grown in 100 mL of media (NZCYM) supplemented with kanamycin (K25, 25 μg/ml) and chloramphenicol (C20, 20 μg/ml) at 37°C until an OD600 of 1.0 was reached. This culture was then used to inoculate 4 l of K25,C20 media to an OD600 = 0.1. Biotin (25 mM in 500 mM Bicine pH 8.0) was added to the media to a final concentration of 50 μM. The cells were grown at 37°C to an OD600 of 0.8 to 1.0, cooled to 25°C and then IPTG (500 μM) was added to induce protein expression. Cultures were then grown for an additional 4 h at 25°C. Cells were harvested by centrifugation (6000 × g, 10 min) and the cell pellet was washed once in 200 ml of TE buffer (10 mM Tris, 1 mM EDTA), centrifuged again, and the cell paste weighed. The cells were then frozen and stored at –80°C until used. Frozen cell pellets were thawed on ice and suspended in lysis buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 2 mM TCEP, 20% glycerol, 1.4 μg/ml leupeptin, 1.0 μg/ml pepstatin A and two Roche EDTA-free protease tablet per 5 g cell pellet), with 5 ml of lysis buffer per 1 gram of cells. The cell suspension was lysed by passing through a French press (SLM-AMINCO) at 15 000 psi. After lysis, phenylmethylsulfonylfluoride (PMSF) and NP-40 were added to a final concentration of 1 mM and 0.1% (v/v), respectively. While stirring the lysate, polyethylenimine (PEI) was slowly added to a final concentration of 0.25% (v/v) to precipitate nucleic acids from cell extracts. The lysate was stirred for an additional 30 min at 4°C after the last addition of PEI, and then centrifuged at 75 000 x g for 30 min at 4°C. The PEI supernatant was then loaded onto a Strep-Tactin XT 4F HC resin (IBA Life Sciences) at a flow rate of 1 ml/min (approximately 1 mL bed volume per 100 mg total protein) equilibrated with buffer A (25 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM TCEP, 20% glycerol). After loading, the column was washed with twenty column volumes of buffer A. The resin was then suspended in two column volumes of buffer A and Ulp1 (5 μg per 1 ml bed volume) was added with the resin overnight at 4°C to cleave the SUMO-nsp10 fusion protein. The column was then washed with 5 column volumes of buffer A and fractions were collected and assayed for purity by SDS-PAGE. The protein concentration was determined by measuring the absorbance at 280 nm by using a Nanodrop spectrophotometer and using a calculated molar extinction coefficient of 13 700 M−1 cm−1. Purified, concentrated protein was aliquoted and frozen at –80°C until use. Typical nsp10 yields were 1 mg/1 g of E. coli cells.
Expression and purification of SARS-CoV-2 nsp14
Escherichia coli BL21(DE3)pBirACm competent cells were transformed with the pAviTag_SUMO-SARS-CoV-2-nsp14 plasmid for protein expression. BL21(DE3)pBirACm cells containing the pAviTag_SUMO-SARS-CoV-2-nsp14 plasmid were grown in 100 ml of media (NZCYM) supplemented with kanamycin (K25, 25 μg/ml) and chloramphenicol (C20, 20 μg/ml) at 37°C until an OD600 of 1.0 was reached. This culture was then used to inoculate 4 l of K25,C20 media to an OD600 = 0.1. Biotin (25 mM in 500 mM Bicine pH 8.0) was added to the media to a final concentration of 50 μM. The cells were grown at 37°C to an OD600 of 0.8 to 1.0, cooled to 25°C and then IPTG (500 μM) was added to induce protein expression. Cultures were then grown for an additional 4 h at 25°C. Cells were harvested by centrifugation (6000 × g, 10 min) and the cell pellet was washed once in 200 mL of TE buffer (10 mM Tris, 1 mM EDTA), centrifuged again, and the cell paste weighed. The cells were then frozen and stored at -80°C until used. Frozen cell pellets were thawed on ice and suspended in lysis buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 2 mM TCEP, 20% glycerol, 1.4 μg/ml leupeptin, 1.0 μg/ml pepstatin A and two Roche EDTA-free protease tablet per 5 g cell pellet), with 5 ml of lysis buffer per 1 g of cells. The cell suspension was lysed by passing through a French press (SLM-AMINCO) at 15 000 psi. After lysis, phenylmethylsulfonylfluoride (PMSF) and NP-40 were added to a final concentration of 1 mM and 0.1% (v/v), respectively. While stirring the lysate, polyethylenimine (PEI) was slowly added to a final concentration of 0.25% (v/v) to precipitate nucleic acids from cell extracts. The lysate was stirred for an additional 30 min at 4°C after the last addition of PEI, and then centrifuged at 75 000 × g for 30 min at 4°C. The PEI supernatant was then loaded onto a Strep-Tactin XT 4F HC resin (IBA Life Sciences) at a flow rate of 1 ml/min (∼1 ml bed volume per 100 mg total protein) equilibrated with buffer A (25 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM TCEP, 20% glycerol). After loading, the column was washed with twenty column volumes of buffer A. The resin was then suspended in two column volumes of buffer A and Ulp1 (5 μg per 1 ml bed volume) was added with the resin overnight at 4°C to cleave the SUMO-nsp14 fusion protein. The column was then washed with 5 column volumes of buffer A and fractions were collected and assayed for purity by SDS-PAGE. The protein concentration was determined by measuring the absorbance at 280 nm by using a Nanodrop spectrophotometer and using a calculated molar extinction coefficient of 93 625 M−1 cm−1. Purified, concentrated protein was aliquoted and frozen at –80°C until use. Typical nsp14 yields were 0.2 mg/1 g of E. coli cells. The catalytically inactive nsp14 D90A E92A was expressed and purified using the exact same conditions as WT nsp14.
SARS CoV-2 nsp7, nsp8 and nsp12 recombinant proteins
Expression and purification of SARS CoV-2 nsp7, nsp8 and nsp12 are described in detail in (8).
Expression and purification of RtcA
Escherichia coli Rosetta(DE3) competent cells were transformed with the pET16b-RtcA-NTerm-His plasmid for protein expression. E. coli Rosetta(DE3) cells containing the pET16b-RtcA-NTerm-His plasmid were grown in 100 ml of A300,C60-supplemented ZYP-5052 auto-induction media at 37°C (40,41). The cells were grown at 37°C to an OD600 of 0.8 to 1.0, cooled to 15°C and then grown for 36–44 h. After ∼40 h at 15°C the OD600 reached ∼10– 15. Cells were harvested by centrifugation (6000 × g, 10 min) and the cell pellet was washed once in 200 ml of TE buffer (10 mM Tris, 1 mM EDTA), centrifuged again, and the cell paste weighed. The cells were then frozen and stored at -80°C until used. Frozen cell pellets were thawed on ice and suspended in lysis buffer (25 mM HEPES pH 7.5, 500 mM NaCl, 2 mM TCEP, 20% glycerol, 5 mM imidazole, 1.4 μg/ml leupeptin, 1.0 μg/ml pepstatin A and two Roche EDTA-free protease tablet per 5 g cell pellet), with 5 ml of lysis buffer per 1 g of cells. The cell suspension was lysed by passing through a French press (SLM-AMINCO) at 15 000 psi. After lysis, phenylmethylsulfonylfluoride (PMSF) and NP-40 were added to a final concentration of 1 mM and 0.1% (v/v), respectively. The lysate was centrifuged at 75 000 × g for 30 min at 4°C. The lysate was then loaded onto Qiagen Ni-Spin columns (a total of 5 mL per one Ni spin column). After loading, the column was washed with two 0.5 ml volumes of buffer B (25 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM TCEP, 20% glycerol) with 5 mM imidazole, then washed with two 0.5 ml volumes of buffer B with 50 mM imidazole and then eluted in four 0.1 ml volumes of buffer B with 500 mM imidazole. Elution fractions were collected and assayed for purity by SDS-PAGE. The protein concentration was determined by measuring the absorbance at 280 nm by using a Nanodrop spectrophotometer and using a calculated molar extinction coefficient of 11 585 M−1 cm−1. Purified, concentrated protein was aliquoted and frozen at –80°C until use. RtcA yields were 0.5 mg/2 g of E. coli cells.
5′-32P-labeling of RNA substrates
RNA oligonucleotides were end-labeled by using [γ-32P]ATP and T4 polynucleotide kinase. Reaction mixtures, with a typical volume of 50 μl, contained 0.5 μM [γ-32P]ATP, 10 μM RNA oligonucleotide, 1 × kinase buffer, and 0.4 unit/μl T4 polynucleotide kinase. Reaction mixtures were incubated at 37°C for 60 min and then held at 65°C for 5 min to heat inactivate T4 PNK. For RNAs containing a 3′phosphate T4 PNK (minus 3′phosphatase) was used using the same reaction conditions.
Cyclization reactions to produce 2′,3′-cyclic phosphate containing RNAs
Reactions contained 25 mM HEPES pH 7.5, 10 mM MgCl2, 10 mM TCEP, 50 mM NaCl, 100 μM ATP, 1 μM 32P-labeled RNA (3′-phosphate termini) and 5 μM RtcA. Reactions were performed at 37°C for 30 min. Reactions were quenched by addition of EDTA to a final concentration of 10 mM and placed at 65°C to heat inactivate RtcA enzyme.
Annealing of dsRNA substrates
dsRNA substrates were produced by annealing 10 μM RNA oligonucleotides in T10E1 [10 mM Tris pH 8.0 and 1 mM EDTA] and 50 mM NaCl in a Progene Thermocycler (Techne). Annealing reaction mixtures were heated to 90°C for 1 min and slowly cooled (5°C/min) to 10°C. Specific scaffolds are described in the figure legends.
Native PAGE
5′-P32-labeled ssRNA and dsRNA substrates were mixed with an equal volume of loading buffer (10% glycerol, 0.025% bromophenol blue and 0.025% xylene cyanol) and loaded onto a 10% or 20% acrylamide, 0.5% bisacrylamide native polyacrylamide gel containing 1× TBE (89 mM Tris base, 89 mM boric acid and 2 mM EDTA). Electrophoresis was performed in 1× TBE at 15 mA. Gels were visualized by using a Phosphorimager.
SARS-CoV-2 nsp10/nsp14-catalyzed exoribonuclease assays
Reactions contained 25 mM HEPES pH 7.5, 2 mM MgCl2, 1 mM TCEP, 10 mM KCl, and 50 mM NaCl and were performed at 30°C. Reactions were quenched by addition of EDTA to a final concentration of 25 mM. Specific concentrations of RNA substrate, ExoN, along with any deviations from the above, are indicated in the appropriate figure legend. Typical concentrations for RNA substrate and enzyme were between 0.01 to 2 μM. Enzymes were diluted immediately prior to use in 25 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM TCEP, and 20% glycerol. SARS-CoV-2 nsp10 was pre-mixed with SARS-CoV-2 nsp14 on ice in 25 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM TCEP, and 20% glycerol for 5 min prior to initiating the reaction with ExoN. The volume of enzyme added to any reaction was always less than or equal to one-tenth the total volume. The ExoN concentration refers to the nsp14 concentration and the ratio of nsp14 to nsp10 is also indicated in cases where nsp10 was in excess of nsp14. For example, 0.1 μM ExoN (1:5) refers to final concentrations of nsp14 of 0.1 μM and nsp10 at 0.5 μM. Products were resolved from substrates by denaturing PAGE.
Incorporation of modified nucleoside triphosphates into dsRNA substrates prior to challenge for removal by ExoN
Reactions contained 25 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM TCEP and 50 mM NaCl and were performed at 30°C. Human mitochondrial RNA polymerase, POLRMT, was used to incorporate modified nucleoside triphosphates. POLRMT was expressed and purified as described previously (42). 1 μM POLRMT was mixed with 1 μM 32P-labeled dsRNA nucleic acid scaffold (primed-template) in the presence of 10 μM ATP, 10 μM UTP and either 10 μM CTP or 100 μM of the modified nucleoside 5′-triphosphate. The RNA primer was P9 (5′-CCGGGCGGC-3′) and RNA template was T21 (Table 1). This primer-template pair allows ATP to be incorporated at the n + 1 position, UTP at n + 2 and the modified nucleoside 5′-triphosphate to be incorporated at the n + 3 position. Reactions were allowed to proceed for 30 min to allow complete extension to n + 3 at which point 0.1 μM ExoN (1:5) was added to the reaction. Reactions were quenched at various times by addition of 25 mM EDTA. Products were resolved from substrates by denaturing PAGE.
Table 1.
RNA oligonucleotides. RNA substrates used in this study. The name, length, type of modification (if any) and sequence are indicated
| # | Name | Length (nt) | Modification | Sequence |
|---|---|---|---|---|
| 1 | P10A | 10 | - | 5′-CCGGGCGGCA-3′ |
| 2 | P10C | 10 | - | 5′-CCGGGCGGCC-3′ |
| 3 | P10G | 10 | - | 5′-CCGGGCGGCG-3′ |
| 4 | P10U | 10 | - | 5′-CCGGGCGGCU-3′ |
| 5 | P10R | 10 | remdesivir | 5′-CCGGGCGGCR-3′ |
| 6 | P10A-2′d | 10 | 2′-deoxy | 5′-CCGGGCGGCA/2′d/-3′ |
| 7 | P10A-3′d | 10 | 3′-deoxy | 5′-CCGGGCGGCA/3′d/-3′ |
| 8 | P10A-3′P | 10 | 3′-phosphate | 5′-CCGGGCGGCA/3′P/-3′ |
| 9 | PS110A | 10 | phosphorothioate | 5′-CCGGGCGGC/s/A-3′ |
| 10 | PS210A | 10 | phosphorothioate | 5′-CCGGGCGG/s/CA-3′ |
| 11 | PS310A | 10 | phosphorothioate | 5′-CCGGGCG/s/GCA-3′ |
| 12 | PS2,310A | 10 | phosphorothioate | 5′-CCGGGCG/s/G/s/CA-3′ |
| 13 | P10U-2′F | 10 | 2′-fluoro | 5′-CCGGGCGGCU/2′F/-3′ |
| 14 | P10U-2′NH2 | 10 | 2′-amino | 5′-CCGGGCGGCU/2′NH2/-3′ |
| 15 | P10A-2′OMe | 10 | 2′-O-methyl | 5′-CCGGGCGGCA/2′OMe/-3′ |
| 16 | P15 | 15 | - | 5′-AAGAAAGGAGGGAGG-3′ |
| 17 | P25 | 25 | 5′-GGAAAGGGAAAGGGAAGGAGGAAGA-3′ | |
| 18 | P40 | 40 | - | 5′-GGAAAGGGAAAGGGAAGGAGGAAGAAAGAAAGGAGGGAGG-3′ |
| 19 | P50 | 50 | - | 5′-GGAAAGGGAAAGGGAAGGAGGAAGAAAGAAAGGAGGGAGGCCGGGCGGCA-3′ |
| 20 | T21 | 21 | - | 5′-CCCCCGAUGCCGCCCGGCCCC-3′ |
| 21 | T57 | 57 | - | 5′-CCCCCGAUGCCGCCCGGCCUCCCUCCUUUCUUUCUUCCUCCUUCCCUUUCCCUUUCC-3′ |
| 22 | P9 | 9 | - | 5′-CCGGGCGGC-3′ |
| 23 | P8 | 8 | - | 5′-CCGGGCGG-3′ |
| 24 | P7 | 7 | - | 5′-CCGGGCG-3′ |
| 25 | P6 | 6 | - | 5′-CCGGGC-3′ |
| 26 | P5 | 5 | - | 5′-CCGGG-3′ |
| 27 | P4 | 4 | - | 5′-CCGG-3′ |
| 28 | P3 | 3 | - | 5′-CCG-3′ |
| 29 | P2 | 2 | - | 5′-CC-3′ |
Incorporation of correct and incorrect nucleotides by SARS-CoV-2 replicase into dsRNA substrates prior to challenge for removal by ExoN
Reactions contained 25 mM HEPES pH 7.5, 2 mM MgCl2, 1 mM TCEP and 50 mM NaCl and were performed at 30°C. Reactions contained 0.1 μM nsp12, 0.3 μM nsp7, 0.3 μM nsp8, 0.1 μM ExoN (1:5), 0.1 μM dsRNA primed/template (P9-P40:T57), 1 μM ATP, 0.1 μCi/μl [α-32P]ATP, 1 μM UTP, and either 10 μM CTP or 100 μM of the modified nucleoside 5′-triphosphate. Initially, nsp12/7/8 was incubated with dsRNA substrate and ATP in the absence or presence of UTP for 60 min to form n + 1 or n + 2 (with UTP) product, at which point either CTP, 3′-dCTP, ddhCTP, 2′-C-Me-CTP was added to promote further extension. After 2 min, ExoN was added, and the reaction was quenched at various times by the addition of EDTA to 25 mM.
Denaturing PAGE analysis of Exonuclease-catalyzed reaction products
An equal volume of loading buffer (85% formamide, 0.025% bromophenol blue and 0.025% xylene cyanol) was added to quenched reaction mixtures and heated to 90°C for 5 min prior to loading 5 μl on a denaturing either 15% or 23% polyacrylamide gel containing 1× TBE (89 mM Tris base, 89 mM boric acid, and 2 mM EDTA) and 7 M urea. For reactions that contained dsRNA substrates an excess (50-fold) of unlabeled RNA oligonucleotide (trap strand) that is the exact same sequence to the 32P-labeled RNA oligonucleotide in the reaction was present in the loading buffer to ensure complete separation and release of the 32P-lableled RNA oligonucleotide prior to gel electrophoresis (43). This procedure allows efficient strand separation of 32P-labeled RNA oligos that are in the presence of their RNA complements (43). Electrophoresis was performed in 1× TBE at 90 W. Gels were visualized by using a PhosphorImager (GE) and quantified by using ImageQuant TL software (GE).
Data analysis
All gels shown are representative, single experiments that have been performed at least three to four individual times to define the concentration or time range shown with similar results. In all cases, values for parameters measured during individual trials were within the limits of the error reported for the final experiments. Data were fit by either linear or nonlinear regression using the program GraphPad Prism v7.03 (GraphPad Software Inc.).
RESULTS
Expression and purification of SARS-CoV-2 ExoN: nsp10 and nsp14
Many proteins and enzymes encoded by SARS-CoV-2 contain zinc ions coordinated by side chains of histidine and/or cysteine residues, including the nsp10 and nsp14 subunits of the 3′-to-5′ exoribonuclease complex (ExoN) (11,13,14,19). For the nsp12 gene-encoded RdRp, the natural ligand has been proposed to be a four-iron, four-sulfur cluster (44). To avoid the potential for displacement of metal ions during protein purification that were incorporated during protein expression, we used the AviTag-SUMO system, which avoids metal-affinity chromatography (Supplementary Figure S1A) (38,39). The AviTag is a 15 amino acid sequence (GLNDIFEAQKIEWHE) that is biotinylated on the lysine residue by biotin ligase, BirA, co-expressed with the fusion protein (38,39). The biotinylated AviTag-SUMO fusion protein binds to streptavidin or streptactin resins (Supplementary Figure S1B) (38,39). The protein of interest is released from the resin by cleavage at the carboxyl terminus of SUMO by the Ulp1 protease (Supplementary Figure S1C–F) (39). Purified proteins used in this study are shown in Figure 1A. We also purified a catalytically inactive derivative of nsp14, referred to as MUT (Supplementary Figure S1F).
Figure 1.

ExoN prefers primed-template-like dsRNA substrates. (A) Purified SARS-CoV-2 nsp14 (60 kDa) and nsp10 (15 kDa) proteins used in this study. Proteins (1 μg) were resolved on a 15% polyacrylamide gel containing SDS and stained with Coomassie. Broad-range molecular weight markers (Mr) and corresponding molecular weights are indicated. (B) Schematic of ssRNA and dsRNA substrates used to measure exoribonuclease activity. RNA sequences are provided in Table 1 and/or Supplementary Figure S2. RNAs were labeled on the 5′-end with 32P, also indicated by an asterisk (*). RNAs are designated as primers (P) or templates (T). The numbers indicate the length of the RNA. When a base is designated, this reflects the 3′-terminal nucleotide. (C) ssRNA and dsRNA substrates evaluated by Native PAGE. (D) Evaluation of ExoN-catalyzed hydrolysis of RNA. Reaction products were resolved by denaturing PAGE and visualized by phosphorimaging. Reactions contained 0.1 μM ExoN (1:1 nsp14:nsp10) and 0.1 μM of the indicated RNA, were incubated at 30°C for the indicated time, then quenched by addition of EDTA. In both cases, primed-template-like dsRNA substrates were cleaved more efficiently than ssRNA substrates. (E) Exoribonuclease activity is dependent on the nsp14 active site. Residues of nsp14 required for catalysis were changed as follows: D90A, E92A; this derivative is referred to as MUT. Reactions contained 0.1 μM ExoN (1:1 nsp14(WT or MUT):nsp10) and 1 μM RNA and were run as described above. MUT did not exhibit any detectable exoribonuclease activity.
ExoN Prefers a dsRNA substrate containing a recessed 3′-end, a ‘primed-template’
We used ssRNA and dsRNA substrates of various lengths in the assay (Figure 1B, C, Table 1, Supplementary Figure S2). We chose these substrates because they can also be used as primers (P) and templates (T) for the replication-transcription complex (RTC). Native PAGE confirmed these substrates to be either single or double stranded (Figure 1C). We use denaturing polyacrylamide gel electrophoresis followed by phosphorimaging to monitor the hydrolysis of the 32P-labeled RNA strand (indicated by an asterisk) in the absence (ssRNA) or presence of an annealed RNA strand (dsRNA). The concentration of ExoN and nsp14:nsp10 stoichiometry used was selected to reflect conditions used by others (19,20,23,29,45–52), to facilitate a comparison of the results obtained here to those. Our studies demonstrated that ssRNA was not a good substrate by ExoN (*P10A and *T57 in Figure 1D and Supplementary Figure S3A–C). The 3′-end of a blunt-ended duplex was also a poor substrate for ExoN (P50A:*T57 in Figure 1D and Supplementary Figure S3D). dsRNA with a recessed 3′-end was the most active substrate (*P10A:T21 in Figure 1D and Supplementary Figure S3F). Comparable results were obtained with P50A and T57 RNAs (Supplementary Figure S3). Mutagenesis of the nsp14 ligands required for Mg2+-dependent catalysis (D90A, E92A) inactivated ExoN (compare MUT to WT in Figure 1D) (13–16). Neither nsp10 nor nsp14 alone exhibited exonuclease activity on ssRNA or dsRNA substrates (Supplementary Figure S3B,C). Therefore, none of the activity measured here derived from a contaminating ribonuclease.
Products of the nsp15 endonuclease are not substrates for ExoN
Coronaviruses also encode an uridylate-specific, ssRNA or dsRNA endonuclease, referred to as NendoU and is encoded by the nsp15-coding sequence of the SARS-CoV-2 genome (53,54). While the activity on dsRNA has been suggested to clear dsRNA that would otherwise activate intrinsic antiviral defenses (53,55,56), the possibility existed that this enzyme could contribute to a post-transcriptional, mismatch-repair mechanism. For example, nsp15 might also cleave the phosphodiester backbone at or near a mismatch. If ExoN could cleave at a nick or at a 2′–3′ cyclic-phosphate or 3′-phosphate terminus produced by nsp15, then such a repair mechanism might exist.
To test this possibility, we assembled substrates in which the P50A RNA was fragmented into three segments: P-1, P-2 and P-3 in the 5′-to-3′ direction, respectively (Figure 2A, B and Supplementary Figure S2A). We evaluated three conditions: I, where P-3 was labeled; II, where P-3 was omitted and P-2 was labeled; and III, where P-2 was labeled (Figure 2A). The labeled, 3′-terminal P-3 primer was cleaved efficiently (I in Figure 2B). Similarly, P-2 was cleaved efficiently when present as the 3′-terminal RNA (II in Figure 2B). However, P-2 was not cleaved when embedded between P-1 and P-3 (III in Figure 2B), suggesting that ExoN is incapable of initiating hydrolysis from the 3′-OH at a nick.
Figure 2.

Products of nsp15-catalyzed endonucleolytic cleavage are not substrates for ExoN. (A) Schematic of dsRNA substrates used. Both substrates I and II contain 32P-labeled RNAs (*P-3 and *P-2) annealed to the template such that both RNAs have an exposed 3′-end. Substrate III contains a 32P-labeled RNA (*P-2) annealed to the template at a position where an additional RNA (P-3) is annealed downstream and blocks access to the 3′-end of the labeled RNA. Substrate III permits assessment of ExoN cleavage at a nick, as produced by nsp15 endonucleolytic cleavage. (B) RNA substrates evaluated by Native PAGE. Shown are the RNAs (*P-2 and *P-3) alone and when annealed to form the dsRNA substrates I, II or III. (C) ExoN does not initiate hydrolysis at a nick. The 10-nt and 15-nt RNAs are indicated. Reactions contained 0.5 μM ExoN (1:1) and 0.1 μM RNA and were quenched at the indicated times. The 32P-labeled RNAs (*P-3 and *P-2) in substrates I and II that have an exposed 3′-end were efficiently cleaved by ExoN. The 32P-labeled RNA (*P-2) in substrate III was not cleaved. (D–F) ExoN does not hydrolyze termini containing a 3′-phosphate or 2′,3′-cyclic phosphate. The structures of these modifications are shown in panel D. Schematic of dsRNA substrates containing 3′-phosphate and 2′,3′-cyclic phosphate modifications used are shown in panel E. *P10A:T21 dsRNA were used as substrates. Reactions contained 0.5 μM ExoN (1:1) and 0.1 μM RNA, were incubated for the indicated time, then quenched. Products are shown in panel F. The unmodified RNA is completely degraded; however, the 3′-phosphate and 2′,3′-cyclic modifications block excision by ExoN. Note, the 3′-phosphate and 2′,3′-cyclic modifications alter the apparent mobility of the RNA as it is more negatively charged and runs faster on the gel; the mobilities of each full-length RNA are indicated by n.
Cleavage by nsp15 leaves a 3′-end with a 2′-3′-cyclic phosphate (cycP) that can be hydrolyzed to a 3′-phoshphate (3′-OPO3) and a 5′-end with a hydroxyl (Figure 2D). Using approaches developed by the Shuman laboratory (Supplementary Figure S4) (34), we prepared RNAs to test as substrates for ExoN that contained either a cycP or a 3′-OPO3 at the recessed end of the dsRNA (Figure 2D, E). Neither of these RNAs served as substrates (Figure 2F); note that the presence of a 3′-phosphate and cycP group facilitates faster migration in the denaturing PAGE gel than the unmodified RNA with a 3′-OH (Figure 2F). So, even if the RNA downstream of the nick produced by nsp15 cleavage were removed, this product would be incapable of being degraded by ExoN without production of a 3′-hydroxyl.
Nucleotide hydrolysis by ExoN occurs in a distributive manner, as expected for a proofreading exonuclease.
Studies of nucleotide hydrolysis by ExoN that either employ conditions of ExoN in excess of substrate or evaluate product formation after long incubation times (30 min to hours) can give the appearance that ExoN is processive (18,23,29,45–48,50–52). In this case, processive means hydrolysis of multiples of ten nucleotides per binding event and is in contrast to a distributive enzyme, which would hydrolyze one or two nucleotides per binding event. Proofreading exonucleases associated with DNA polymerases have evolved to function in a distributive manner. This circumstance eliminates the mismatch without the need for re-synthesis of stretches of nucleic acid that were correctly basepaired, as would be the case if the exonuclease acted processively (27,28).
The ability to split the hydrolyzed strand into multiple components (Figure 2), offered the opportunity to create a substrate with sufficient dsRNA to form a processive, elongation complex using the SARS-CoV-2 RTC (30,57,58) but at the same time permit hydrolysis by ExoN to be monitored with single-nucleotide resolution. The substrate used is shown in Figure 3A. We evaluated utilization of this substrate by ExoN under two conditions. We refer to the first condition as substrate excess, which should reveal the length(s) of product formed by ExoN in a single binding event (left panel of Figure 3B). We refer to the second condition as enzyme excess (right panel of Figure 3B). Under these conditions and upon dissociation, the product of one enzyme in the reaction immediately serves as a substrate for the same or a second enzyme in a reiterative manner until the product dissociates into two single-stranded molecules. This latter condition is the most prevalent in the literature (23,45–48,51,59).
Figure 3.

Monitoring ExoN-catalyzed RNA hydrolysis at single-nucleotide resolution. (A) Schematic of dsRNA substrate used to monitor ExoN activity at single-nucleotide resolution. (B) Reactions contained 0.05 μM ExoN (1:1) and 1 μM RNA or 0.1 μM ExoN (1:1) and 0.05 μM RNA, were incubated for the indicated time, then quenched. Products were resolved using denaturing 20 or 23% polyacrylamide gels. The RNA cleavage products, n – 1 to n – 8, are indicated. (C, D) Quantitative analysis of kinetics of substrate RNA utilization and/or product RNA formation. Formation of individual products is shown on the right. Panel C reports on the experiment in which RNA was in excess of enzyme; panel D reports on the experiment in which enzyme was in excess of RNA.
Under both conditions, the earliest products were primarily n – 1 and n – 2 in length (Figure 3B). Quantitative analysis of both conditions showed consumption of >80% of the substrate during each reaction (left panels of Figure 3C and D). More than 50% of the substrate was consumed by the first time point when enzyme was present in excess (left panel of Figure 3D), thus precluding an unambiguous assessment of precursor-product relationships (right panel of Figure 3D). However, under the condition of substrate-excess, the kinetics suggested production of n – 1 and n – 2 at the same rate, with n – 2 accumulating over the time course (right panel of Figure 3C). Therefore, products shorter than n – 3 likely arise from utilization of the n – 1 and/or n – 2 products.
Existence of an equilibrium between nsp14 and nsp10 proteins
Structural studies of ExoN demonstrate unambiguously that the nsp14:nsp10 stoichiometry is 1:1 (19,22,29,46,47,49). However, there has been an assumption that the affinity of nsp14 for nsp10 is sufficiently high that complex never dissociates once formed. This interpretation is based on the fact that most published studies emphasize the nsp14:nsp10 stoichiometry rather than the concentration of each component (20,23,29,45–52,59). Because most published studies have used conditions of enzyme excess, any dependence of the reaction rate on nsp10 concentration would be masked without evaluating product formation on the msec-sec timescale (Supplementary Figure S5).
We evaluated the reaction kinetics, processivity, and substrate specificity of nsp14 as a function of nsp10 concentration (Figure 4). The reaction rate increased as a function of nsp10 concentration, with apparent saturation at a concentration of 2 μM, and nsp14:nsp10 ratio of 1:20 (Figure 4). The primed-template-like dsRNA substrate was still favored over the ssRNA substrate (compare the 10-nt band in Figure 4A to that in Figure 4B). Finally, the distributive nature of the enzyme remained the same, with n – 1 and n – 2 products accumulating at early times for both substrates (Figure 4).
Figure 4.

nsp10 stimulates hydrolysis without affecting processivity or substrate specificity. (A) Effect of nsp10 concentration on the kinetics of nsp14-catalyzed dsRNA hydrolysis. Reactions contained fixed nsp14 (0.1 μM) and varied nsp10 (0–2 μM) concentrations. In all cases, nsp14 was pre-mixed with nsp10 on ice 5 min prior to adding to the reaction. Hydrolysis of dsRNA substrate (*P10A-P40:T57) was monitored over a 30 min time course as indicated. Product analysis is shown. (B) Effect of nsp10 concentration on the kinetics of nsp14-catalyzed hydrolysis of ssRNA. Reactions were as in panel A except a ssRNA substrate (*P10A) was used. Product analysis is shown. Products shorter than n – 4 are not observed when the 10-nt ssRNA is used. (C, D) Analysis of the kinetics of product formation for both dsRNA (panel C) and ssRNA (panel D) at varying concentrations of nsp10, error bars indicate mean ± SD (n = 3).
An understanding of the kinetics and thermodynamics of the binding reaction between nsp14 and nsp10 leading formation of the nsp14-nsp10 complex is warranted. Such studies will require approaches to assess complex formation directly, rather than indirectly by monitoring exoribonuclease activity.
The presence of the phosphorothioate Rp diastereomer at the scissile phosphodiester bond blocks hydrolysis by ExoN
Polymerases prefer use of the Sp diastereomer when a phosphorothioate is placed at the alpha position of a nucleoside triphosphate (60–64). Incorporation yields the Rp diastereomer at the resulting phosphorothioate bond (Figure 5A), and the presence of this diastereomer can block hydrolysis by some exonucleases (65–69). We substituted the ultimate (PS110A in Figure 5B), penultimate (PS210A in Figure 5B), and antepenultimate (PS310A in Figure 5B) phosphodiester bond with a phosphorothioate. Both diastereomers were present at a 50:50 ratio.
Figure 5.
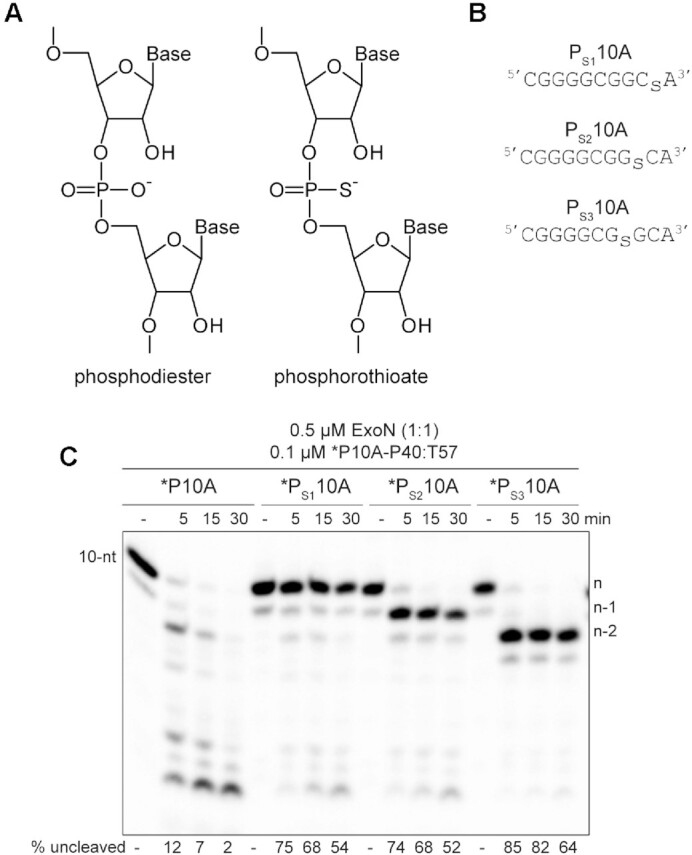
RNA hydrolysis by ExoN is inhibited by a phosphorothioate bond. (A) Structure of phosphorothioate. One of the non-bridging oxygens of the phosphodiester bond is replaced with a sulfur atom, creating Rp and Sp diastereomers. A 50:50 ratio of diastereomers were used in these experiments. (B) Schematic of phosphorothioate-substituted ssRNAs used. (C) Effect of phosphorothioate ExoN-catalyzed hydrolysis of dsRNA. Reactions contained 0.5 μM ExoN (1:1) and 0.1 μM of the indicated RNA, were incubated at the indicated times, then quenched. Product analysis is shown. Fraction of RNA remaining is indicated (% uncleaved). The unmodified RNA is completely degraded; however, ExoN is unable to hydrolyze beyond the phosphorothioate substitution, for half the RNA molecules at least. This observation is consistent with only a single diastereomer being inhibitory.
We assembled each phosphorothioate-substituted RNA into the *P10A-P40:T57 substrate to determine the impact of the phosphorothioate substitution on hydrolysis by ExoN compared to the control RNA (Figure 5C). Under the conditions of enzyme excess, essentially all of the control substrate was consumed (*P10A in Figure 5C). However, only 50% or so of the substrate was consumed when a phosphorothioate was present in the RNA (*PS110A, *PS210A and *PS310A in Figure 5B), suggesting that one diastereomer inhibits hydrolysis. Importantly, the size of the terminal product in the reaction was consistent with the position of the phosphorothioate: n, for *PS110A; n – 1 for *PS210A; and n – 2, for *PS310A (Figure 5B).
To determine if the phosphorothioate Rp diastereomer was the inhibitory species, we used polymerase incorporation of nucleoside-5′-O-(1-thiotriphosphates) to produce RNA with only the Rp diastereomer at the scissile bond (Figure 6A). For these experiments, we used the cryptic RdRp activity present in the mitochondrial DNA-dependent RNA polymerase (DdRp), POLRMT. For these experiments, we used the DdRp to produce an RNA product with the terminal scissile bond containing the phosphorothioate Rp diastereomer (Figure 6B). After ∼50% of the primer was extended to product, we added ExoN and monitored the fate of the product RNA by denaturing PAGE and phosphorimaging (Figure 6B). Extension of only half of the product provides an internal control for the presence of ExoN, because the primer lacks a phosphorothioate bond. We used two different substrates. For both, the presence of the phosphorothioate Rp isomer completely blocked hydrolysis by ExoN (right panels in Figure 6C, D), relative to both the unextended primer (internal control) and the comparable RNA product lacking the phosphorothioate (left panels in Figure 6C, D). Quantitation is provided in Figure 6E and F. Addition of the phosphorothioate to the α-position of an antiviral nucleotide analog should eliminate the possibility for excision by ExoN.
Figure 6.

ExoN is inhibited by the Rp diastereomer at the phosphorothioate-substituted scissile bond. (A) The Sp diastereomer of a nucleoside-5′-O-thiotriphosphate is preferentially incorporated by RNA polymerases. Nucleophilic attack at the alpha phosphorous atom leads to inversion of configuration and creation of Rp diastereomer at the phosphorothioate bond. (B) Schematic of assay. Primer extension is initiated by adding an RNA polymerase in the presence of either UTP and CTPαS or ATPαS. CTPαS is a mixture of both the Rp and Sp diastereomers. ATPαS was obtained as the pure Sp diastereomer. Incorporation yields extended products. The last nucleotide to be incorporated is either CMP(αS) or AMP(αS). Once 50–75% of the primers were extended to the end, ExoN was added to the reaction. The reaction was monitored over time for hydrolysis. Unextended primer in reactions served as a useful control to demonstrate the presence of active ExoN in the reaction. P9:T21 was used as dsRNA substrate for these reactions. (C, D) Analysis of reaction products by denaturing PAGE. Incorporation of CMP and AMP results in removal of the incorporated nucleotide by ExoN. Incorporation of CMPαS and AMPαS (Rp diastereomers) results in a terminated primer that cannot be cleaved by ExoN. (E, F) Kinetics of excision of CMP, CMPαS (Rp diastereomer) and AMP and AMPαS (Rp diastereomer) by ExoN. Data were fit to a single exponential. Rates are provided in Table 2.
Composition of the terminal basepair modulates the kinetics of hydrolysis by ExoN
For proofreading exonucleases of DNA polymerases, it is not that the exonuclease exhibits a preference for a mispaired end relative to a properly paired end (27,28). Rather, a mispaired end is unstable in the polymerase active site, providing the opportunity for the mispaired end to partition to the exonuclease active site (27,28). To assess the impact of the terminal basepair on the kinetics of excision by ExoN, we designed two additional RNA substrates for ExoN. *P10G creates a terminal G:U mispair; *P10R creates a terminal remdesivir:U pair, which is equivalent to an A:U pair at the level of the hydrogen bonding potential between the bases (Figure 7A). We evaluated the kinetics of hydrolysis of the various RNA substrates by ExoN under conditions in which the RNA substrate was present in excess of enzyme. Reaction products were resolved by denaturing PAGE and visualized by phosphorimaging (Figure 7B). The observed rates of consumption of the RNA substrate and formation of all products were the same (Figure 7C). Any difference that would be observed, however, would be related to hydrolysis of 3′-terminal nucleotide to produce the n-1 product and any subsequent excision reactions occurring prior to dissociation of ExoN. We performed a comprehensive analysis of the kinetics of formation of each product for each substrate (Figure 7D). We observed a clear difference in the magnitude of the n – 1 product that accumulated for each substrate (Figure 7D). Such an observation would suggest that the rate of the step governing hydrolysis and/or rate of dissociation of the excised nucleotide product differ between the substrates. Interestingly, excision of remdesivir was substantially slower than the natural nucleotides as the n – 1 product accumulated to the greatest extent for the P10R substrate (Figure 7D).
Figure 7.

Terminal basepair-dependent differences in the kinetics of hydrolysis by ExoN. (A) Schematic of dsRNA substrates used to evaluate the influence of the terminal basepair on kinetics of excision by ExoN. The termini have a A:U, G:U or R:U basepair, where R represents remdesivir. (B) Effect of terminal basepair on hydrolysis by ExoN. Reactions contained 0.05 μM ExoN (1:5) and 1 μM of the indicated dsRNA substrate, were incubated at 30°C for the indicated times, then quenched. Product analysis is shown. (C) Analysis of the kinetics of substrate utilization and product formation. Here, depletion of substrate and total product formation are monitored, showing no difference in the kinetics, error bars indicate mean ± SD (n = 3). (D) Analysis of the kinetics of formation of each individual product. Cleavage products (n – 1 to n – 8) were plotted as a function of time for each RNA substrate, error bars indicate mean ± SD (n = 3). The kinetics of formation and utilization of the n – 1 product vary between substrates, suggesting that some, but not all steps of the mechanism are agnostic to the sequence and/or complementarity of the terminal basepair. Statistical analysis (unpaired t-test) comparing the n – 1 products from P10A to P10R at 5 min is indicated, P = 0.0038.
Some modifications to the 3′-terminal ribose antagonize ExoN-catalyzed excision
To further our understanding of the structure–activity relationships of the 3′-terminal nucleotide, we prepared substrates in which the 2′- or 3′-hydroxyl was changed (Figure 8A). Our standard substrate RNA is P10A. Unfortunately, not all modifications were available as an adenosine phosphoramidite. Therefore, some modifications were analyzed in the context of P10U (Figure 8A and Supplementary Figure S7). We monitored the kinetics of ExoN cleavage of modified RNAs. Products were resolved by denaturing PAGE and visualized by phosphorimaging (Supplementary Figures S6 and S7). We plotted the concentration of total RNA products as a function of time for the two conditions used: excess substrate (Figure 8B) or excess enzyme (Figure 8C). The corresponding observed rates are presented in Table 3. The presence of a 3′-phosphate blocked hydrolysis (Figure 2E). The presence of a 2′-fluoro had no effect. All other substitutions reduced the efficiency of cleavage, with the most effective being 3′-H, 2′-NH2 and 2′-H (Figure 8B, C and Table 3). Together, these data suggest that it may be possible to identify substituents on the 3′-terminal ribose that would not impact utilization by the RTC but would inhibit excision by ExoN.
Figure 8.

Structure-activity relationships for the ribose of the 3′-terminal nucleotide. (A) Modifications to the ribose of the terminal nucleotide studied here in the context of P10A: 3′-deoxy (-H), 3′-phosphate (-O-PO3), 2′-deoxy (-H) and 2′-O-methyl (-O-CH3); or P10U: 2′-amino (-NH2) and 2′-fluoro (-F). (B, C) Kinetics of cleavage of modified dsRNAs. In panel B, substrate was present in excess of enzyme. In panel C, enzyme was present in excess of substrate. The data were fit to a line (panel B) or to a single exponential (panel C). Reactions contained 0.1 μM ExoN (1:20) and either 1 or 0.05 μM dsRNA substrate and were quenched at the indicated times. The order in which the modified RNAs were cleaved is as follows: unmodified = 2′-F > 2′-O-Me > 2′-deoxy > 2′-amino > 3′-deoxy > 3′-phosphate. The kinetics of cleavage using the 3′-phosphate RNA is not shown, because cleavage was not detected. The observed rate of cleavage of modified RNAs and fold difference from the unmodified RNA are shown in Table 3.
Table 3.
Effect of modifications to the 3′-end on the kinetics of excision by ExoN. Data were fit either to a line (S > E) or to a single exponential (E > S) to determine observed rate of cleavage
| Rate (min−1) | Rate (min−1) | |
|---|---|---|
| Modification | S > E | E > S |
| Unmodified | 0.87 ± 01 | 2.2 ± 0.2 |
| 2′-fluoro | 0.85 ± 0.3 | 1.7 ± 0.1 |
| 2′-O-methyl | 0.64 ± 0.02 | 0.46 ± 0.04 |
| 2′-deoxy | 0.41 ± 0.01 | 0.33 ± 0.02 |
| 2′-amino | 0.33 ± 0.01 | 0.15 ± 0.02 |
| 3′-deoxy | 0.035 ± 0.006 | 0.056 ± 0.008 |
| 3′-phosphate | No cleavage | No cleavage |
ExoN efficiently excises the chain-terminating antiviral ribonucleotide produced by viperin, ddhC
The literature is rife with examples of the inability of obligate and non-obligate chain terminators to interfere with SARS-CoV-2 multiplication in cell culture (8,13,70–72), even the antiviral ribonucleotide, ddhCTP, produced by the antiviral protein, viperin (73). However, a 3′-dAMP terminated RNA is not a good substrate for ExoN (Figure 8), and others have made similar observations for other 3′-dNMPs (29,74).
Given this apparent contradiction between the cell-based and biochemical experiments, we asked how ExoN would deal with the presence of the viperin product, ddhCTP, and the non-obligate chain terminator, 2′-C-Me-CTP (structures shown in Figure 9A), when present at the 3′-end of RNA. Because synthetic RNA containing these analogs was not available, we made these RNAs biosynthetically by taking advantage of the cryptic RdRp activity present in the mitochondrial DNA-dependent RNA polymerase (DdRp, aka POLRMT, Figure 9B). CTP and 3′-dCTP were used as controls to permit comparison to data obtained using synthetic RNA. After production of the CMP analog-terminated RNA, ExoN was added, and the reaction proceeded for the indicated time before quenching (Figure 9B). Reaction products were resolved by denaturing PAGE and visualized by phosphorimaging (Figure 9C) and quantified (Figure 9D). The presence of the 2′-C-Me substituent had little to no effect on ExoN activity (2′-C-Me-CTP in Figure 9C, D). In spite of the absence of a 3′-OH on ddhCTP, ddhCMP was excised from RNA almost as efficiently as CTP (ddhCTP in Figure 9C, D). Together, the results presented in Figures 8 and 9 reveal complexity to the interaction between the 3′-terminal nucleotide and the ExoN active site that is not explained by simple docking experiments. A more in-depth characterization of the substrate specificity of ExoN is warranted.
Figure 9.

Viperin product (ddhCMP)-terminated RNA is a good substrate for ExoN but 3′-dCMP-terminated RNA is not. (A) Structure of nucleotide analogs used. (B) Schematic of the assay. Primer extension is initiated by adding an RNA polymerase in the presence of UTP and a CTP analog. Incorporation produces n + 1 and n + 2 products. The last nucleotide to be incorporated is the CTP analog. Once 50–75% of the primers were extended to n + 2, ExoN was added to the reaction. The reaction was monitored over time for hydrolysis. Unextended primer in reactions served as a useful control to demonstrate the presence of active ExoN in the reaction. (C) Analysis of reaction products by denaturing PAGE. Only the 3′-dCMP-terminated RNA exhibited a delay in excision. (D) Kinetics of excision of CMP analogs. The quantitation revealed that only 3′-dCMP-terminated RNA exhibited a significant delay in the rate of excision relative to the CMP control, even though ddhCMP also lacks a 3′-OH. Data were fit to a single exponential. Rates are provided in Table 4.
SARS-CoV-2 RTC prevents access of ExoN to the 3′-end nascent RNA
We have been able to assemble elongation complexes comprised of SARS-CoV-2 RTC and our split-primer:template substrate used for ExoN (Figure 10A). Studies using similarly designed substrates with shorter duplexes fail to assemble a stable complex (data not shown). The ability to split the primer provides the advantage of being able to monitor extension at single-nucleotide resolution.
Figure 10.

Stalled SARS-CoV-2 replication-transcription complexes block access of ExoN to the RNA terminus. (A) Schematic of assay. Primer extension was initiated by adding the SARS-CoV-2 replicase-transcription complex (nsp7/nsp8/nsp12) in the presence of ATP and UTP or ATP, UTP and CTP or a CTP analog. After an incubation time of 60 min, we added ExoN, incubated for the indicated time, and then quenched the reaction. (B) Effect of ExoN on stalled elongation complexes. Incorporation yields n + 1, n + 2 and n + 3 products. When ATP, UTP and CTP are present, the n + 3 product has a properly basepaired 3′-end. The n + 4 product is formed by misincorporation and has a mispaired 3′-end. When only ATP and UTP (low concentration, 1 μM UTP) are present, both the n + 3 and n + 4 products are formed by misincorporation and both products have mispaired 3′-ends. At high concentrations of UTP (100 μM), most of the products observed are at n + 3 from misincorporation of U opposite G. ExoN was added to elongation complexes for the indicated time (5–60 min). Reaction products were visualized by phosphorimaging after denaturing PAGE. No change in the level of properly paired (n + 3) or mispaired (n + 4) elongation products was apparent for any of the reactions performed, consistent with the replicase remaining bound to both products and obstructing access by ExoN. (C) Quantitation of reaction products shown in panel B. The data were fit to a line. Rates are provided in Table 4.
Assembly and incubation of a complex with a correctly basepaired 3′-end for 60 min (Figure 10A; 0 min Figure 10B) remains stable to challenge with ExoN for at least an additional 60 min (Figure 10A; CTP in Figure 10B, C). Judicious selection of substrate nucleotides and/or analogs permitted us to exploit this system to create elongation complexes with different 3′-ends. This experimental design permitted us to determine if the nature of the 3′-terminal basepair under investigation influenced the stability of assembled SARS-CoV-2 RTC and/or accessibility of ExoN. Neither a mispaired end (UTP in Figure 10B, C) nor an end containing a chain terminating analog (2′-C-Me-CTP, 3′-dCTP, ddhCTP in Figure 10B, C) changed the sensitivity to ExoN, so by inference did not change stability with the SARS-CoV-2 RTC either. Extension of shorter duplexes by the SARS-CoV-2 RTC that failed to form a stable complex were excised by ExoN (data not shown).
Together, these data show that the SARS-CoV-2 RTC creates a physical block to cleavage and that the composition of the terminal basepair does not trigger a response by the SARS-CoV-2 RTC that facilitates access by ExoN.
DISCUSSION
Coronaviruses encode a 3′-5′ exoribonuclease (11,13–15). A complex of the nsp10 and nsp14 proteins at a stoichiometry of 1:1 forms the active enzyme (19,22,29,47,49). The nsp14 subunit harbors the catalytic residues; however, this subunit fails to achieve a stable conformation competent for RNA binding and catalysis in the absence of nsp10 (22). Therefore, we have referred to the active complex as ExoN instead of the nsp14 subunit alone. The biological function of ExoN is proofreading, correction of errors introduced by the viral polymerase (11–14). ExoN also contributes to excision of antiviral nucleotides, as the sensitivity of the virus to certain antiviral nucleotides increases in the absence of exoribonuclease activity (25). This latter activity complicates the use of some conventional antiviral nucleotides for the treatment of coronavirus infection. The goal of this study was to discover strategies to interfere with excision of nucleotides by ExoN and thereby inform the development of nucleotide analogs with greater efficacy against coronaviruses.
The catalytic cycle of ExoN includes binding of the RNA substrate to the enzyme followed by hydrolysis (Figure 11A). There is now a consensus that the RNA substrate preferred by ExoN is one that would also be preferred by the replication-transcription complex (RTC), a primed-template (Figure 1) (20,29,49). The number of nucleotides hydrolyzed per binding event was not known before this study. This gap existed for two reasons. First, the design of the RNA substrates used in these studies made difficult – if not impossible – the monitoring of product formation with single-nucleotide resolution (23,29,45,47–51,59). Second, ExoN was present in substantial excess over the RNA substrate, thus requiring monitoring of hydrolysis on the millisecond timescale to observe a product one nucleotide shorter than the substrate, which was never done (23,29,45,47–51,59). The inability of ExoN to hydrolyze nucleotides at a nick permitted us to distribute the overall requirement for dsRNA over multiple segments, which enabled product analysis at single-nucleotide resolution (Figure 2A, B). While performing the experiments under conditions of enzyme excess may have some utility when evaluating inhibitors (45), only conditions of substrate excess can provide information on the number of nucleotides hydrolyzed per binding event when quenching reactions manually (Figure 11A). ExoN excised only one or two nucleotides per binding event (Figure 3). The distributive nature of ExoN makes sense, because a processive enzyme would then require the RTC to resynthesize RNA that lacked any damage.
Figure 11.

Hypothetical model for ExoN-catalyzed excision. (A) Excision uncoupled to RNA synthesis. The active exoribonuclease, referred to here as ExoN, is a complex of two subunits: nsp10 (regulatory) and nsp14 (catalytic). The concentration of ExoN therefore depends on the value of the dissociation constant for this binding reaction, as well as the concentrations of nsp10 and nsp14. Most studies to date have ignored this equilibrium. Under steady-state conditions in which the RNA substrate (RS) is present in excess of ExoN, RS binds, catalysis occurs, releasing one or two nucleotides, followed by dissociation of the RNA product (RP). Under these conditions, the rate-limiting step is likely release of RP. Modifications to the terminal nucleotide that reduce the rate of hydrolysis to values substantially lower than the rate of RP dissociation should interfere with excision. RP consumption depends on its ability to compete with RS, for example when more than 20% of the substrate is consumed or when ExoN is present in excess of substrate and more than a single turnover occurs. The possibility also exists for product dissociation to be driven by dissociation of the nsp10 and nsp14 subunits. (B) Excision coupled to RNA synthesis. Introduction of mispaired nucleotides or chain terminators by the polymerase complex does not lead to dissociation, blocking access of the 3′-end for repair by ExoN. The polymerase complex must therefore be actively dislodged from the terminus to access the 3′-end. Once ExoN binds, only one or two nucleotides will be removed.
For a nucleotide analog to resist excision, the rate constant for excision (Step 2 in Figure 11A) needs to be slower than the rate constant for dissociation of ExoN from the 3′-terminus (Step 4 in Figure 11A). RNA product dissociation is likely the rate-limiting step for this reaction under steady-state conditions (Figure 11A). Both the 2′- and 3′-hydroxyls contribute to the efficiency of excision, as modifications to either appear to reduce the rate of catalysis to levels at or below the rate constant for dissociation (Figure 8, Table 3). Our assumption is that when the rate of product formation is the same under conditions of excess substrate and excess enzyme, then the rate measured is catalysis (Table 3). The 3′-position was most important. Loss of the 3′-OH made catalysis rate limiting (Figure 8, Table 3); addition of a 3′-phosphate eliminated turnover altogether (Table 3). The SARS-CoV-2 RTC has no problem utilizing nucleotide substrates lacking a 3′-OH (8,75). The issue with most analogs is the inability to compete with natural nucleotide pools (8,75). Remdesivir does not suffer this problem (8,75). Perhaps elimination of its 3′-OH will bolster its anti-coronavirus activity.
Table 4.
Kinetics of excision of chain terminating cytidine analogs by ExoN. Data were fit to a single exponential to determine the observed rate of cleavage and half-life (t1/2)
| −SARS replicasea | +SARS replicaseb | |||
|---|---|---|---|---|
| NTP | Rate (min−1) | t 1/2 (min) | Rate (min−1) | t 1/2 (min) |
| CTP | 0.23 ± 0.01 | 3 | 0.0027 ± 0.0004 | 260 |
| UTP | - | - | 0.0046 ± 0.0002 | 150 |
| 3′-dCTP | 0.014 ± 0.002 | 50 | 0.0032 ± 0.0004 | 220 |
| ddhCTP | 0.089 ± 0.01 | 8 | 0.0018 ± 0.0002 | 390 |
| 2′-C-Me-CTP | 0.15 ± 0.02 | 5 | 0.0028 ± 0.0001 | 250 |
The ability of 3′-dCMP to antagonize ExoN-mediated hydrolysis so effectively was not expected, although several other groups have now reported this observation (29,74). Our previous studies showed that the antiviral nucleoside, ddhC, does not interfere with SARS-CoV-2 multiplication in cells, even though the RTC utilized the nucleotide, ddhCTP, quite readily (8). ddhC lacks a 3′-OH, but ExoN readily excised ddhCMP from the 3′-end of RNA (Figure 9). We conclude that the requirement for a 3′-OH is not absolute but is context dependent. The context is likely associated with the ribose conformation. The ribose component of normal nucleotides are not planar and possess ‘sugar pucker’ with the 3′-endo conformation being favored for ribonucleosides. The ribose of ddhC on the other hand is forced into a planar conformation because of the double bond between carbons 3′ and 4′ (Figure 9). Structural studies will be required to provide an explanation for the context-dependent requirement for the 3′-OH.
In retrospect, it is not surprising that coronaviruses have evolved to evade the inhibitory activity of ddhCMP-terminated RNA. ddhCTP is induced by infection (73,76). Indeed, the presence of ddhCTP in cells presents a strong selective pressure for acquisition of an exoribonuclease, especially given the efficiency with which the RTC utilizes this antiviral nucleotide (8). The presence of an exoribonuclease may have also contributed to the polymerase retaining the ability to utilize ddhCTP. The evolutionarily related enteroviral polymerases have lost the ability to utilize ddhCTP (73). Some coronaviruses deal with loss of the exoribonuclease better than others(23), perhaps the efficiency with which the polymerases utilize ddhCTP has something to do with this. It would be interesting to determine the requirement of the exoribonuclease in cells lacking viperin, the enzyme that produces ddhCTP.
The most efficient strategy to interfere with nucleotide excision is to introduce a non-hydrolyzable scissile bond. Phosphorothioate (P-S) substitutions at the scissile phosphodiester bond inhibit excision by DNA exonucleases, usually requiring a specific stereoisomer (SP or RP) (65–69,77). Here we show that the presence of a phosphorothioate in the RP configuration of the scissile phosphodiester bond inhibits hydrolysis by ExoN (Figures 5 and 6 and Table 2). The ability to block cleavage of the terminal nucleotide rules out the existence of ExoN-associated endonuclease activity as suggest recently (45). Introduction of the P-S-substituted bond at ultimate, penultimate, and/or antepenultimate position will create substrates to facilitate elucidation of the details of ExoN-catalyzed hydrolysis by limiting the extent to which a product can be consumed. Current strategies to synthesize P-S-substituted oligonucleotides yield diastereomeric mixtures at phosphorous, but stereoisomer-specific solutions are on the horizon (77).
Table 2.
Kinetics of excision of the Rp diastereomer of CMPαS and AMPαS by ExoN. Data were fit to a single exponential to determine the observed rate of cleavage and half-life (t1/2)
| NMP | Rate (min−1) | t 1/2 (min) |
|---|---|---|
| CMP | 0.22 ± 0.010 | 3 |
| CMPαS (Rp,Sp) | 0.0016 ± 0.0003 | 350 |
| AMP | 0.13 ± 0.02 | 5 |
| AMPαS (Rp) | 0.0015 ± 0.0003 | 460 |
Another interesting possibility for use of the P-S substitution is the creation of non-hydrolyzable antiviral ribonucleotide analogs. Most clinically used monophosphorylated ribonucleotide analogs are readily delivered to cells as prodrugs that require intracellular activation by histidine triad nucleotide-binding protein 1 (HINT-1) (78,79), the so-called ProTide strategy (7,80–82). Similar strategies can be used for P-S-substituted ribonucleoside monophosphates. It is clear that HINT-1 can activate such compounds in cells, although the kinetics of activation are slower than conventional ProTides (78). HINT-1 is known to convert P-S-substituted NMPs to the natural NMP (78,79). The efficiency of this side reaction is likely tunable as the nature of the base and sugar contribute to the rate of sulfur removal (78). Side reactions such as these could also be alleviated by using P-S-substituted di- or triphosphorylated prodrugs (83). The pursuit of non-hydrolyzable ribonucleotide analogs is a guaranteed solution to the problem of excision of antiviral ribonucleotides by ExoN.
The majority of our studies focused on excision independent of RNA synthesis. An early report on ExoN from SARS-CoV suggested that co-transcriptional repair could occur in vitro (46,48). These studies used primed-templates of insufficient length to form a stable elongation complex (46,48). So, just as we were able to see ‘co-transcriptional’ repair using POLRMT (e.g. Figure 9), this repair required dissociation of POLRMT to permit ExoN access to the 3′-end. Formation of a stable elongation complex using the SARS CoV-2 RTC requires a duplex of 50-bp or greater (30,57,58). The ability to split the primer into 40-nt and 10-nt fragments permitted us to monitor extension by the RTC and excision by ExoN at single-nucleotide resolution in the same reaction (Figure 10). Interestingly, assembling the SARS CoV-2 RTC on these primed templates and forcing the complex to make errors was insufficient to get the complex to dissociate and expose the 3′-end to ExoN (Figure 10). ExoN removed these same termini in the presence of POLRMT (Figure 9).
We conclude that the SARS-CoV-2 RTC alone does not sense the presence of a mispair, a non-natural nucleotide, or the pause caused by a chain terminator (panel 1 in Figure 11B). A more active mechanism must exist to remove the RTC from the 3′-end, perhaps another viral factor pushes the RTC out of the way (panel 2 in Figure 11B). The nsp13-encoded 5′-3′ RNA helicase may function in this capacity. nsp13 associates with the RTC (57,58). While incorporating nucleotides, the RTC may overcome helicase action. When stalled, however, the helicase is situated to push the RTC backwards (57,58). We suggest two potential scenarios: one where the replicase backtracks and one where the replicase is displaced backward. In the former, the product strand exits through the NTP channel, revealing the product strand 3′-end for mismatch correction by ExoN (57,58,84). In the latter, the displacement of the replicase leaves the duplex strand annealed and accessible to ExoN for mismatch correction (panel 3 in Figure 11B). In both cases, establishing a tug of war between the polymerase and helicase in the RTC would favor exposure of any 3′-end that impedes forward motion of the polymerase. In such a scenario, access would regulate processing by ExoN and may explain the lack of a strict specificity for a mispaired end. So, what would happen in the presence of a catalytically inactive ExoN? It is likely that the complex would associate with the 3′-end, as illustrated in panel 3 of Figure 11B, and create a physical block to synthesis by the RTC. It would be interesting to determine the extent to which substitutions that impair association of ExoN with the RNA duplex phenocopy those that preclude catalysis.
Remdesivir is one nucleotide analog that shows efficacy against SARS-CoV-2 in humans (1). The mechanism of action clearly relates to perturbed dynamics of the RTC (8,31,33,85). Whether or not some perturbations can occur immediately or only when present at the n + 4 position has not been completely resolved. If remdesivir-terminated RNA were ever present, then our results would suggest that attempts at excision by ExoN would not be as facile as other nucleotide pairs (Figure 7). This particular observation highlights the utility of monitoring hydrolysis by ExoN with single-nucleotide resolution.
The structure of ExoN reveals a complex of nsp10 and nsp14 at a 1:1 stoichiometry (19,22,29,46,47,49). However, such an observation does not infer picomolar affinity. We make this statement because the vast majority of the published studies on SARS-CoV-2 ExoN emphasize the stoichiometry of nsp10:nsp14 used without any consideration of the concentrations of each protein present. Our studies suggest that the dissociation constant for the nsp10–nsp14 complex is in the micromolar range (Figure 4). Because our major conclusions derive from studies of the steady state at saturating concentrations of RNA, knowledge of the precise concentration of ExoN is not essential. However, conclusions from single-turnover experiments would be compromised. Direct studies of the nsp10–nsp14 binding equilibrium (Step 1 in Figure 11A) will be essential to elucidation of the kinetic and chemical mechanisms.
Together, we describe a robust system for evaluating co-transcriptional proofreading of the SARS-CoV-2 polymerase and associated factor by the exoribonuclease. The framework established here will hopefully help the field at large by highlighting some of the key gaps in our understanding of the mechanism of ExoN-catalyzed nucleotide excision. Addressing these gaps will yield a clear picture of the structure-dynamics-function relationship of ExoN that can be used to guide design of ExoN-resistant nucleotide analogs.
DATA AVAILABILITY
All data are incorporated into the article and its online supplementary material. Constructs and data sets presented in this study are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
Our thanks to Roel Fleuren and Efra Rivera-Serrano for their contributions to the production of figures and models. We thank Stewart Shuman for providing the expression plasmid for RtcA.
Contributor Information
Rukesh Chinthapatla, Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599, USA.
Mohamad Sotoudegan, Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599, USA.
Pankaj Srivastava, Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599, USA.
Thomas K Anderson, Department of Biochemistry and Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA.
Ibrahim M Moustafa, Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA.
Kellan T Passow, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Samantha A Kennelly, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Ramkumar Moorthy, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
David Dulin, Department of Physics and Astronomy, and LaserLaB Amsterdam, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands; Junior Research Group 2, Interdisciplinary Center for Clinical Research, Friedrich-Alexander-University Erlangen-Nürnberg (FAU), Cauerstr. 3, 91058 Erlangen, Germany.
Joy Y Feng, Gilead Sciences, Inc, Foster City, CA 94404, USA.
Daniel A Harki, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Robert N Kirchdoerfer, Department of Biochemistry and Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA.
Craig E Cameron, Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599, USA.
Jamie J Arnold, Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01AI161841 to C.E.C., J.J.A., D.D.; P01CA234228, R01GM110129 to D.A.H., R01AI158463 to R.K.]; University of Minnesota, Office of the Vice President for Research, Grant-in-Aid (to D.A.H.); Interdisciplinary Center for Clinical Research (IZKF) at the University Hospital of the University of Erlangen-Nuremberg, the German Research Foundation [DFG-DU-1872/4-1 to D.D.]; Netherlands Ministry of Education, Culture and Science (OCW); Netherlands Organization for Scientific Research (NWO) [024.003.019 to D.D.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S.et al.. Remdesivir for the treatment of Covid-19 - Final report. N. Engl. J. Med. 2020; 383:1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jayk Bernal A., Gomes da Silva M.M., Musungaie D.B., Kovalchuk E., Gonzalez A., Delos Reyes V., Martin-Quiros A., Caraco Y., Williams-Diaz A., Brown M.L.et al.. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022; 386:509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lamb Y.N. Nirmatrelvir plus Ritonavir: first approval. Drugs. 2022; 82:585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seley-Radtke K.L., Yates M.K.. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antiviral Res. 2018; 154:66–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson K.A., Dangerfield T.. Mechanisms of inhibition of viral RNA replication by nucleotide analogs. Enzymes. 2021; 49:39–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maheden K., Todd B., Gordon C.J., Tchesnokov E.P., Gotte M.. Inhibition of viral RNA-dependent RNA polymerases with clinically relevant nucleotide analogs. Enzymes. 2021; 49:315–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slusarczyk M., Serpi M., Pertusati F.. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother. 2018; 26:2040206618775243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seifert M., Bera S.C., van Nies P., Kirchdoerfer R.N., Shannon A., Le T.T., Meng X., Xia H., Wood J.M., Harris L.D.et al.. Inhibition of SARS-CoV-2 polymerase by nucleotide analogs from a single-molecule perspective. Elife. 2021; 10:e70968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swanstrom R., Schinazi R.F.. Lethal mutagenesis as an antiviral strategy. Science. 2022; 375:497–498. [DOI] [PubMed] [Google Scholar]
- 10. Dulin D., Arnold J.J., van Laar T., Oh H.S., Lee C., Perkins A.L., Harki D.A., Depken M., Cameron C.E., Dekker N.H.. Signatures of nucleotide analog incorporation by an RNA-Dependent RNA polymerase revealed using high-Throughput magnetic tweezers. Cell Rep. 2017; 21:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malone B., Urakova N., Snijder E.J., Campbell E.A.. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022; 23:21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S.. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011; 8:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robson F., Khan K.S., Le T.K., Paris C., Demirbag S., Barfuss P., Rocchi P., Ng W.L.. Coronavirus RNA proofreading: molecular basis and therapeutic targeting. Mol. Cell. 2020; 79:710–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogando N.S., Ferron F., Decroly E., Canard B., Posthuma C.C., Snijder E.J.. The Curious case of the nidovirus exoribonuclease: its role in RNA synthesis and replication fidelity. Front. Microbiol. 2019; 10:1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Minskaia E., Hertzig T., Gorbalenya A.E., Campanacci V., Cambillau C., Canard B., Ziebuhr J.. Discovery of an RNA virus 3'→5' exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Nat. Acad. Sci. U.S.A. 2006; 103:5108–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E.. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003; 331:991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beese L.S., Steitz T.A.. Structural basis for the 3'-5' exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991; 10:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen P., Jiang M., Hu T., Liu Q., Chen X.S., Guo D.. Biochemical characterization of exoribonuclease encoded by SARS coronavirus. J. Biochem. Mol. Biol. 2007; 40:649–655. [DOI] [PubMed] [Google Scholar]
- 19. Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y., Lou Z., Yan L., Zhang R., Rao Z.. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Nat. Acad. Sci. U.S.A. 2015; 112:9436–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E.. RNA 3'-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Nat. Acad. Sci. U.S.A. 2012; 109:9372–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bouvet M., Lugari A., Posthuma C.C., Zevenhoven J.C., Bernard S., Betzi S., Imbert I., Canard B., Guillemot J.C., Lecine P.et al.. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 2014; 289:25783–25796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Czarna A., Plewka J., Kresik L., Matsuda A., Karim A., Robinson C., O’Byrne S., Cunningham F., Georgiou I., Wilk P.et al.. Refolding of lid subdomain of SARS-CoV-2 nsp14 upon nsp10 interaction releases exonuclease activity. Structure. 2022; 30:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ogando N.S., Zevenhoven-Dobbe J.C., van der Meer Y., Bredenbeek P.J., Posthuma C.C., Snijder E.J.. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J. Virol. 2020; 94:e01246-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becares M., Pascual-Iglesias A., Nogales A., Sola I., Enjuanes L., Zuniga S.. Mutagenesis of coronavirus nsp14 reveals its potential role in modulation of the Innate immune response. J. Virol. 2016; 90:5399–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith E.C., Blanc H., Surdel M.C., Vignuzzi M., Denison M.R.. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 2013; 9:e1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deval J., Gurard-Levin Z.A.. Opportunities and challenges in targeting the proofreading activity of SARS-CoV-2 polymerase complex. Molecules. 2022; 27:2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reha-Krantz L.J. DNA polymerase proofreading: multiple roles maintain genome stability. Biochim. Biophys. Acta. 2010; 1804:1049–1063. [DOI] [PubMed] [Google Scholar]
- 28. Carvajal-Maldonado D., Drogalis Beckham L., Wood R.D., Doublié S.. When DNA polymerases multitask: functions beyond nucleotidyl transfer. Front. Mol. Biosci. 2021; 8:815845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu C., Shi W., Becker S.T., Schatz D.G., Liu B., Yang Y.. Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science. 2021; 373:1142–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P.. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020; 584:154–156. [DOI] [PubMed] [Google Scholar]
- 31. Gordon C.J., Lee H.W., Tchesnokov E.P., Perry J.K., Feng J.Y., Bilello J.P., Porter D.P., Gotte M.. Efficient incorporation and template-dependent polymerase inhibition are major determinants for the broad-spectrum antiviral activity of remdesivir. J. Biol. Chem. 2022; 298:101529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kabinger F., Stiller C., Schmitzova J., Dienemann C., Kokic G., Hillen H.S., Hobartner C., Cramer P.. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021; 28:740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tchesnokov E.P., Gordon C.J., Woolner E., Kocinkova D., Perry J.K., Feng J.Y., Porter D.P., Gotte M.. Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J. Biol. Chem. 2020; 295:16156–16165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das U., Shuman S.. 2'-Phosphate cyclase activity of RtcA: a potential rationale for the operon organization of RtcA with an RNA repair ligase RtcB in Escherichia coli and other bacterial taxa. RNA. 2013; 19:1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moorthy R., Kennelly S.A., Rodriguez D.J., Harki D.A.. An efficient synthesis of RNA containing GS-441524: the nucleoside precursor of remdesivir. RSC Adv. 2021; 11:31373–31376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Passow K.T., Caldwell H.S., Ngo K.A., Arnold J.J., Antczak N.M., Narayanan A., Jose J., Sturla S.J., Cameron C.E., Ciota A.T.et al.. A chemical strategy for intracellular arming of an endogenous broad-Spectrum antiviral nucleotide. J. Med. Chem. 2021; 64:15429–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnold J.J., Bernal A., Uche U., Sterner D.E., Butt T.R., Cameron C.E., Mattern M.R.. Small ubiquitin-like modifying protein isopeptidase assay based on poliovirus RNA polymerase activity. Anal. Biochem. 2006; 350:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fairhead M., Howarth M.. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 2015; 1266:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raducanu V.S., Tehseen M., Shirbini A., Raducanu D.V., Hamdan S.M.. Two chromatographic schemes for protein purification involving the biotin/avidin interaction under native conditions. J. Chromatogr. A. 2020; 1621:461051. [DOI] [PubMed] [Google Scholar]
- 40. Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005; 41:207–234. [DOI] [PubMed] [Google Scholar]
- 41. Studier F.W. Stable expression clones and auto-induction for protein production in E. coli. Methods Mol. Biol. 2014; 1091:17–32. [DOI] [PubMed] [Google Scholar]
- 42. Smidansky E.D., Arnold J.J., Reynolds S.L., Cameron C.E.. Human mitochondrial RNA polymerase: evaluation of the single-nucleotide-addition cycle on synthetic RNA/DNA scaffolds. Biochemistry. 2011; 50:5016–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arnold J.J., Ghosh S.K., Bevilacqua P.C., Cameron C.E.. Single-nucleotide resolution of RNA strands in the presence of their RNA complements. BioTechniques. 1999; 27:450–452. [DOI] [PubMed] [Google Scholar]
- 44. Maio N., Lafont B.A.P., Sil D., Li Y., Bollinger J.M. Jr, Krebs C., Pierson T.C., Linehan W.M., Rouault T.A.. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science. 2021; 373:236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baddock H.T., Brolih S., Yosaatmadja Y., Ratnaweera M., Bielinski M., Swift L.P., Cruz-Migoni A., Fan H., Keown J.R., Walker A.P.et al.. Characterization of the SARS-CoV-2 ExoN (nsp14ExoN-nsp10) complex: implications for its role in viral genome stability and inhibitor identification. Nucleic Acids Res. 2022; 50:1484–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ferron F., Subissi L., Silveira De Morais A.T., Le N.T.T., Sevajol M., Gluais L., Decroly E., Vonrhein C., Bricogne G., Canard B.et al.. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Nat. Acad. Sci. U.S.A. 2018; 115:E162–E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin S., Chen H., Chen Z., Yang F., Ye F., Zheng Y., Yang J., Lin X., Sun H., Wang L.et al.. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 2021; 49:5382–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma Z., Pourfarjam Y., Kim I.K.. Reconstitution and functional characterization of SARS-CoV-2 proofreading complex. Protein Expression Purif. 2021; 185:105894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moeller N.H., Shi K., Demir O., Belica C., Banerjee S., Yin L., Durfee C., Amaro R.E., Aihara H.. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc. Nat. Acad. Sci. U.S.A. 2022; 119:e2106379119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Riccio A.A., Sullivan E.D., Copeland W.C.. Activation of the SARS-CoV-2 NSP14 3'-5' exoribonuclease by NSP10 and response to antiviral inhibitors. J. Biol. Chem. 2022; 298:101518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saramago M., Barria C., Costa V.G., Souza C.S., Viegas S.C., Domingues S., Lousa D., Soares C.M., Arraiano C.M., Matos R.G.. New targets for drug design: importance of nsp14/nsp10 complex formation for the 3'-5' exoribonucleolytic activity on SARS-CoV-2. FEBS J. 2021; 288:5130–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Subissi L., Posthuma C.C., Collet A., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Decroly E., Snijder E.J., Canard B., Imbert I.. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Nat. Acad. Sci. U.S.A. 2014; 111:E3900–E3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deng X., Baker S.C.. An “old” protein with a new story: coronavirus endoribonuclease is important for evading host antiviral defenses. Virology. 2018; 517:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J.. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Nat. Acad. Sci. USA. 2004; 101:12694–12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hackbart M., Deng X., Baker S.C.. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Nat. Acad. Sci. U.S.A. 2020; 117:8094–8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kindler E., Gil-Cruz C., Spanier J., Li Y., Wilhelm J., Rabouw H.H., Zust R., Hwang M., V’Kovski P., Stalder H.et al.. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog. 2017; 13:e1006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen J., Malone B., Llewellyn E., Grasso M., Shelton P.M.M., Olinares P.D.B., Maruthi K., Eng E.T., Vatandaslar H., Chait B.T.et al.. Structural basis for helicase-Polymerase coupling in the SARS-CoV-2 replication-Transcription complex. Cell. 2020; 182:1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Malone B., Chen J., Wang Q., Llewellyn E., Choi Y.J., Olinares P.D.B., Cao X., Hernandez C., Eng E.T., Chait B.T.et al.. Structural basis for backtracking by the SARS-CoV-2 replication-transcription complex. Proc. Nat. Acad. Sci. U.S.A. 2021; 118:e2106379119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Canal B., McClure A.W., Curran J.F., Wu M., Ulferts R., Weissmann F., Zeng J., Bertolin A.P., Milligan J.C., Basu S.et al.. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp14/nsp10 exoribonuclease. Biochem. J. 2021; 478:2445–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arnold J.J., Cameron C.E.. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+. Biochemistry. 2004; 43:5126–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnson K.A. The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta. 2010; 1804:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Griffiths A.D., Potter B.V., Eperon I.C.. Stereospecificity of nucleases towards phosphorothioate-substituted RNA: stereochemistry of transcription by T7 RNA polymerase. Nucleic Acids Res. 1987; 15:4145–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu J., Tsai M.D.. DNA polymerase beta: pre-steady-state kinetic analyses of dATP alpha S stereoselectivity and alteration of the stereoselectivity by various metal ions and by site-directed mutagenesis. Biochemistry. 2001; 40:9014–9022. [DOI] [PubMed] [Google Scholar]
- 64. Romaniuk P.J., Eckstein F.. A study of the mechanism of T4 DNA polymerase with diastereomeric phosphorothioate analogues of deoxyadenosine triphosphate. J. Biol. Chem. 1982; 257:7684–7688. [PubMed] [Google Scholar]
- 65. Brautigam C.A., Steitz T.A.. Structural principles for the inhibition of the 3'-5' exonuclease activity of Escherichia coli DNA polymerase I by phosphorothioates. J. Mol. Biol. 1998; 277:363–377. [DOI] [PubMed] [Google Scholar]
- 66. Eckstein F. Nucleoside phosphorothioates. Annu. Rev. Biochem. 1985; 54:367–402. [DOI] [PubMed] [Google Scholar]
- 67. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014; 24:374–387. [DOI] [PubMed] [Google Scholar]
- 68. Putney S.D., Benkovic S.J., Schimmel P.R.. A DNA fragment with an alpha-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc. Nat. Acad. Sci. U.S.A. 1981; 78:7350–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang Z., Sismour A.M., Benner S.A.. Nucleoside alpha-thiotriphosphates, polymerases and the exonuclease III analysis of oligonucleotides containing phosphorothioate linkages. Nucleic Acids Res. 2007; 35:3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Elfiky A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 2020; 253:117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lim Y.S., Nguyen L.P., Lee G.H., Lee S.G., Lyoo K.S., Kim B., Hwang S.B.. Asunaprevir, a potent Hepatitis C virus protease inhibitor, blocks SARS-CoV-2 propagation. Mol. Cells. 2021; 44:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lo H.S., Hui K.P.Y., Lai H.M., He X., Khan K.S., Kaur S., Huang J., Li Z., Chan A.K.N., Cheung H.H.et al.. Simeprevir potently suppresses SARS-CoV-2 replication and synergizes with Remdesivir. ACS Cent Sci. 2021; 7:792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gizzi A.S., Grove T.L., Arnold J.J., Jose J., Jangra R.K., Garforth S.J., Du Q., Cahill S.M., Dulyaninova N.G., Love J.D.et al.. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature. 2018; 558:610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jones A.N., Mourao A., Czarna A., Matsuda A., Fino R., Pyrc K., Sattler M., Popowicz G.M.. Characterization of SARS-CoV-2 replication complex elongation and proofreading activity. Sci. Rep. 2022; 12:9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bera S.C., Seifert M., Kirchdoerfer R.N., van Nies P., Wubulikasimu Y., Quack S., Papini F.S., Arnold J.J., Canard B., Cameron C.E.et al.. The nucleotide addition cycle of the SARS-CoV-2 polymerase. Cell Rep. 2021; 36:109650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rivera-Serrano E.E., Gizzi A.S., Arnold J.J., Grove T.L., Almo S.C., Cameron C.E.. Viperin reveals its true function. Annu Rev Virol. 2020; 7:421–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jahns H., Taneja N., Willoughby J.L.S., Akabane-Nakata M., Brown C.R., Nguyen T., Bisbe A., Matsuda S., Hettinger M., Manoharan R.M.et al.. Chirality matters: stereo-defined phosphorothioate linkages at the termini of small interfering rnas improve pharmacology in vivo. Nucleic Acids Res. 2022; 50:1221–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chou T.F., Baraniak J., Kaczmarek R., Zhou X., Cheng J., Ghosh B., Wagner C.R.. Phosphoramidate pronucleotides: a comparison of the phosphoramidase substrate specificity of human and Escherichia coli histidine triad nucleotide binding proteins. Mol Pharm. 2007; 4:208–217. [DOI] [PubMed] [Google Scholar]
- 79. Ozga M., Dolot R., Janicka M., Kaczmarek R., Krakowiak A.. Histidine triad nucleotide-binding protein 1 (HINT-1) phosphoramidase transforms nucleoside 5'-O-phosphorothioates to nucleoside 5'-O-phosphates. J. Biol. Chem. 2010; 285:40809–40818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li Y., Yang B., Quan Y., Li Z.. Advancement of prodrug approaches for nucleotide antiviral agents. Curr. Top. Med. Chem. 2021; 21:2909–2927. [DOI] [PubMed] [Google Scholar]
- 81. Serpi M., Madela K., Pertusati F., Slusarczyk M.. Synthesis of phosphoramidate prodrugs: proTide approach. Curr. Protoc. Nucleic Acid Chem. 2013; Chapter 15:Unit15 15. [DOI] [PubMed] [Google Scholar]
- 82. Serpi M., Pertusati F.. An overview of ProTide technology and its implications to drug discovery. Expert Opin. Drug Discov. 2021; 16:1149–1161. [DOI] [PubMed] [Google Scholar]
- 83. Meier C. Nucleoside diphosphate and triphosphate prodrugs - An unsolvable task. Antivir. Chem. Chemother. 2017; 25:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yan L., Ge J., Zheng L., Zhang Y., Gao Y., Wang T., Huang Y., Yang Y., Gao S., Li M.et al.. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. 2021; 184:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kokic G., Hillen H.S., Tegunov D., Dienemann C., Seitz F., Schmitzova J., Farnung L., Siewert A., Hobartner C., Cramer P.. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021; 12:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are incorporated into the article and its online supplementary material. Constructs and data sets presented in this study are available upon request.