


Abstract
Analytical tools for gene expression profiling of individual cells are critical for studying complex biological systems. However, the techniques enabling rapid measurements of gene expression on thousands of single-cells are lacking. Here, we report a high-throughput RNA cytometry for digital profiling of single-cells isolated in liquid droplets enveloped by a thin semi-permeable membrane (microcapsules). Due to the selective permeability of the membrane, the desirable enzymes and reagents can be loaded, or replaced, in the microcapsule at any given step by simply changing the reaction buffer in which the microcapsules are dispersed. Therefore, complex molecular biology workflows can be readily adapted to conduct nucleic acid analysis on encapsulated mammalian cells, or other biological species. The microcapsules support sequential multi-step enzymatic reactions and remain intact under different biochemical conditions, freezing, thawing, and thermocycling. Combining microcapsules with conventional FACS provides a high-throughput approach for conducting RNA cytometry of individual cells based on their digital gene expression signature.
INTRODUCTION
The reverse-transcription polymerase chain reaction (RT-PCR) remains one of the most commonly used molecular biology techniques for a quick evaluation of gene expression levels in cells and tissues (1,2). The extraordinary sensitivity down to one nucleic acid molecule per 1 μl reaction volume (3), combined with high specificity, accuracy, and fast readout offered by RT-PCR assays, have propelled a broad range of biological and diagnostic applications (2,4–6). However, a comprehensive characterization of heterogeneous biological samples (7–9), such as tumors requires the isolation of each individual member in a population so as to study it separately. As a result, numerous efforts have focused on developing single-cell RT-PCR (scRT-PCR) approaches using microtiter plates (10–12), microfluidics (13–25), and other reaction formats (26–28). For example, conventional fluorescence-activated cell sorting (FACS) into microtiter plates followed by RT-PCR (29–33), enables the analysis of gene expression of sorted cells relative to their surface markers. While clearly useful for some biological and clinical applications (32,33), unfortunately, such an approach has not found broader use due to the limited number of single-cells that can be simultaneously processed in 96-well plates and the relatively high costs associated with large reaction volumes (34). Moreover, the unbiased characterization of complex populations may require the profiling of thousands of cells individually at a scale that is beyond the practical scope of plate-based assays (35–37). FISH-flow enables high-throughput single-cell profiling, yet the sample preparation process is time-consuming (>30 h) and is prone to cell loss and RNA degradation (38). Alternative techniques based on microwells and valve-operated microfluidics (39,40) offer the throughput of scRT-PCR assays of up to 1000 reactions per run (41), but such assay formats often rely on sophisticated microfluidic operations (18) that are difficult to implement, prone to errors and expensive (42,43).
Droplet microfluidics technology provides an alternative for performing ultra-high-throughput assays at a scale of 106 reactions per run (39,44). Individual cells can be isolated in pico- to nano-liter volume range droplets, lysed, and their genetic makeup can be assessed using various molecular biology techniques (21,22,35). Therefore, combining the high sensitivity of the RT-PCR technique with the scalability of droplet microfluidics technology opens the possibility of developing digital assays for high-throughput quantification and analysis of the transcriptional profile of single-cells at a molecular level. However, a digital scRT-PCR assay that is broadly applicable in biological and biomedical research must fulfill several criteria. First, it should provide an efficient approach to isolate and retain encapsulated cells and nucleic acids throughout analytical procedures. Second, the method should enable multi-step biochemical reactions on thousands of single-cells simultaneously, including efficient cell lysis, nucleic acid amplification, and analysis, in a straightforward manner, such as using a regular laboratory pipette and tubes. Third, the digital scRT-PCR signal should efficiently differentiate true positive events (cells), from false positive events (e.g. cell-free nucleic acids), and other confounding outcomes. Finally, although not critical, the compartments should provide an option to release the amplified genetic material for further analysis, without damaging it. To the best of our knowledge, no high-throughput, single-cell RT-PCR technique reported to date fulfills the above requirements, although several noticeable examples that rely on sophisticated microfluidic operations have been reported (15,17,22).
Here, we introduce a novel nucleic acid analysis approach for the digital profiling of single-cells isolated in liquid droplets with a semi-permeable membrane (microcapsules). The concept is easy to appreciate: the cells are isolated in water-in-oil droplets, comprising dextran and chemically-modified gelatin, whereby each droplet, on average, contains no more than one cell. Upon liquid-liquid phase separation, the dextran forms a liquid core, while the polypeptide forms a shell, which is cross-linked into a thin membrane by a brief photo-polymerization. Once the microcapsules having a concentric core/shell structure are forrmed, the subsequent analytical procedures are carried out using a regular pipette and tubes (i.e. no need for expensive equipment). We showed that using microcapsules as reaction vessels enables efficient isolation and retention of mammalian cells. The microcapsules sustain multi-step analytical procedures required for the extraction, purification, amplification, and digital analysis of nucleic acids. In addition, the microcapsules readily withstand various chemical environments (e.g. solvents), freezing, and thermocycling, and are compatible with conventional FACS used to conduct high-throughput, single-cell RNA cytometry, and to accurately quantify the gene expression of thousands of individual mammalian cells.
MATERIALS AND METHODS
Fabrication and use of microfluidic devices
The polydimethylsiloxane (PDMS) microfluidic devices having microchannels 30 and 40 μm heights were obtained from Droplet Genomics.
Liquid polymer solution preparation
The stock of shell-forming solution comprising 10% (w/w) gelatin methacrylate, GMA (Sigma-Aldrich) dissolved in 1× DPBS (Gibco) was stored at 4°C in 200 μl aliquots for up to 6 months. Before the experiment, the GMA was melted at 40°C for 30 min, diluted down to 3% (w/v) in 1× DPBS buffer, and centrifuged at 10 000 × g for 10–15 min (at 37°C). The warm GMA solution was loaded onto a microfluidic device using a 1 ml syringe connected via 0.56 mm inner diameter PTFE tubing (Droplet Genomics). The stock of core-forming solution comprising 30% (w/w) dextran, MW ∼500k (Sigma-Aldrich) dissolved in 1× DPBS was stored at 4°C in 1 ml aliquots for up to 6 months. Before the experiment, the dextran solution was equilibrated at room temperature and diluted with cell suspension down to 15% (w/v). The dextran solution was loaded onto a microfluidic device using a 1 ml syringe connected via 0.56 mm inner diameter PTFE tubing.
Microcapsule generation
Following cell encapsulation, the resulting aqueous two-phase system (ATPS) droplets quickly formed a core-shell structure comprising a liquid core enriched in dextran, and a liquid shell enriched in GMA. The ATPS droplets were subjected to a two-step polymerization procedure. At first, the water-in-oil droplets were incubated at 4°C for 15–30 min to solidify the GMA phase. The resulting intermediate-microcapsules, having a liquid core and physically cross-linked GMA shell, were recovered from the oil phase by applying an emulsion breaker (Droplet Genomics), and released into Cell Washing Buffer (1× DPBS and 0.1% (w/v) Pluronic F-68). The suspension of intermediate-microcapsules was equilibrated at room temperature for 5 min, mixed with a photo-initiator (0.1% (w/v) lithium phenyl-2,4,6-trimethylbenzoylphosphinate (Sigma-Aldrich) and exposed to low-energy 405 nm light emitting diode (LED) device (Droplet Genomics) for 20 seconds. The resulting microcapsules contained a liquid core enriched in dextran and a thin (3.0 ± 0.2 μm) membrane comprising chemically cross-linked polypeptides. Continuing procedures on ice, photopolymerized microcapsules were rinsed twice in Cell Washing Buffer and subjected to cell lysis or cell fixation in ethanol (see below).
Preparation of cells
K-562 (ATCC), HEK293 (ATCC), and NB-4 (kind gift by Dr V.V. Borutinskaite) cells were cultured in Iscove's modified Dulbecco's medium (Gibco), Dulbecco's modified Eagle's medium (Gibco), and Roswell Park Memorial Institute (RPMI) 1640 Medium (Gibco), respectively, supplemented with 10% fetal bovine serum (Gibco) and 1× penicillin-streptomycin (Gibco) at 37°C in the presence of 5% CO2. Cells were collected from a culture dish, washed once in an ice-cold Cell Washing Buffer (1× DPBS and 0.1% w/v Pluronic F-68), and then in 1× DPBS. Before the encapsulation, cells were re-suspended in 15% (w/v) dextran solution at a concentration of 0.1–0.2M cells/100 μl when a 40 μm height microfluidic device was used. Using a 30 μm height microfluidic device, the final cell concentration in 15% dextran solution was 0.6M cells/100 μl. All centrifugation steps were performed at 300 × g for 5 min at 4°C.
Preparation of PBMCs
PBMCs (ATCC, PCS-800–011) were thawed from liquid nitrogen using RPMI 1640 Medium supplemented with 10% fetal bovine serum. Thawed cells were washed two times in an ice-cold Cell Washing Buffer and resuspended in 1× DPBS at a concentration of 12M cells/ml. All centrifugation steps were performed at 300 × g for 7–10 min at 4°C.
Cell encapsulation
Cell isolation in microcapsules was performed on a microfluidics platform Onyx (Droplet Genomics) using a microfluidic device having a nozzle 40 μm deep and 40 μm wide (Supplementary Figure S1). Alternatively, cell encapsulation in microcapsules can be performed on a custom-built microfluidics platform such as reported previously (45). Typical flow rates used were 250 μl/h for GMA solution, 100 μl/h for dextran solution with cells, and 700 μl/h for droplet stabilization oil (Droplet Genomics). NB-4 and PBMC isolation in microcapsules was performed using a microfluidic device having a nozzle 30 μm deep and 20 μm wide. Typical flow rates used were 125 μl/h for GMA solution, 50 μl/h for dextran solution with cells, and 700–800 μl/h for droplet stabilization oil. The dilution of cells was chosen such that majority of microcapsules would contain either 0 or 1 cell (occupancy ∼ 0.1). The encapsulations were performed at room temperature for up to 30 min. The encapsulated cells were collected in a 1.5 ml tube prefilled with 200 μl of light mineral oil (Sigma-Aldrich).
Cell fixation in ethanol
To fix the encapsulated cells, the microcapsules were suspended in 70% (v/v) ice-cold ethanol and stored at –20°C until further analysis. To rehydrate the fixed cells, the tube with microcapsules was equilibrated on ice for 5 min, centrifuged at 2000 × g for 2 min at 4°C, and then washed once in ice-cold Rehydration Buffer (3× SSC buffer supplemented with 0.04% BSA, 1 mM DTT and 0.2 U/μl RiboLock RNase Inhibitor (Thermo Fisher Scientific, TFS). Cells were permeabilized in a mild-lysis buffer as described below.
Cell lysis
The harsh-lysis of encapsulated cells was performed by suspending microcapsules in 1 ml GeneJET RNA Purification Kit Lysis Buffer (TFS) supplemented with 40 mM DTT. Microcapsules were washed in GeneJET Lysis Buffer 3-times, with 1 to 5 min of incubation between the washes. After lysis, the microcapsules were rinsed 5-times in a Washing Buffer (10 mM Tris–HCl [pH 7.5] with 0.1% (v/v) Triton X-100). During washes, the centrifugation steps were performed at 2000 × g for 2 min at 4°C.
The mild-lysis of encapsulated cells was performed by suspending microcapsules in 1 ml buffer comprising 10 mM Tris–HCl [pH 7.5], 0.6% (v/v) IGEPAL CA-630, 40 mM DTT and 10 mM EDTA. The microcapsules were incubated at room temperature for 15 min, rinsed 3-times in a Washing Buffer, and then added to the RT reaction mix. During washes, the centrifugation steps were performed at 2000 × g for 2 min at 4°C.
Genomic DNA depletion
Genomic DNA depletion was performed in a 200 μl DNase I reaction mix containing 100 μl close-packed microcapsule suspension, 10 U DNase I (TFS), 40 U RiboLock RNase Inhibitor, and 1× DNase I Buffer with MgCl2, at 37°C for 20 min. Then, additional 5 U of DNase I enzyme were added and incubated for 10 min at 37°C. The microcapsules were rinsed 3–5 times in Washing Buffer and then subjected to a reverse transcription reaction.
Reverse transcription
cDNA synthesis was performed in 200 μl Maxima H Minus RT reaction mix comprising 100 μl close-packed microcapsule suspension, 5 μM Oligo(dT)21 primer (IDT), 0.5 mM dNTP Mix, 1000 U Maxima H Minus Reverse Transcriptase, 40 U RiboLock RNase Inhibitor, and 1× RT Buffer, at 50°C for 60 min. Every 20 min microcapsules were briefly dispersed. After cDNA synthesis, the RT enzyme was heat inactivated at 85°C for 5 min. Then, microcapsules were rinsed three times in Washing Buffer and subjected to multiplex PCR. For capturing low abundance transcripts (e.g. PML-RARα), 2.5 μM Oligo(dT)21 primer was combined with 2.5 μM random hexamer primer (TFS). The cDNA reaction was initiated by pre-incubating the reaction mixture at room temperature for 10 min followed by 50°C for 60 min.
Multiplex PCR
PCR was performed in a 100 μl reaction mix comprising ∼50 μl close-packed microcapsule suspension, 0.5 μM of each PCR primer (see primer sequences in Supplementary Material), and 1× Phire Tissue Direct PCR Master Mix (TFS). Samples were thermally cycled through the following program: 98°C (5 min), 98°C (5 s)/64°C (5 s)/72°C (20 s) for 30 cycles, 72°C (1 min). Following thermal cycling, microcapsules were treated with 100 U Exonuclease I (NEB) for 15 min at 37°C, rinsed three times in Washing Buffer, and used for the subsequent microscopy and FACS analysis. For capturing low abundance transcripts, at first, the pre-amplification was performed by 10 cycles of PCR, then microcapsules were washed twice in Washing Buffer and subjected to 30 cycles of PCR using identical conditions as indicated above. The post-PCR microcapsules were treated with 100 U Exonuclease I (NEB) for 15 min at 37°C, rinsed three times in Washing Buffer, and used for the subsequent microscopy and FACS analysis.
Post-PCR microcapsule staining with DAPI
To identify microcapsules with isolated cells, post-RT-PCR microcapsules were immersed in a Washing Buffer containing 300 nM of 4',6-diamidino-2-phenylindole, dihydrochloride-DAPI (Invitrogen) and incubated in the dark on ice for 10 min. Then microcapsules were washed three times in Washing Buffer and used for the subsequent epifluorescence microscopy and FACS analysis.
Fluorescence microscopy analysis
The fluorescence intensity of microcapsules was recorded by layering the microcapsules on a standard hemocytometer (Sigma-Aldrich) and imaging under Nikon Eclipse Ti-E microscope with DAPI, GFP, RFP, and Cy5 fluorescence filter sets. Imaging settings were kept the same for each experiment with an exposure time of 400 ms, and the gain value set at 1.0. The microscope objective used for imaging was CFI Plan Fluor 10X (N.A. 0.30, W.D. 16.0 mm). For each analysis, at least ten brightfield and fluorescence images (∼200 microcapsules per image), were recorded with Nikon DS-Qi2 digital camera.
Flow cytometry
The microcapsules were washed twice in Washing Buffer, filtered through a 100 μm size cell strainer (Corning), and loaded onto the Partec CyFlow Space (Figure 4) and BD FACSAria III (Figure 6) instruments. The microcapsules were detected using forward scatter, side scatter, and fluorescence channels. Isolated cells in microcapsules were detected by measuring signal height in the DAPI channel on the BD FACSAria III instrument. Note, that due to the spillover of Alexa Fluor 488 to the Alexa Fluor 555 channel and the imperfect compensation process, the PTPRC-positive population showed increased intensity in Alexa Fluor 555 channel when measurements were performed using Partec CyFlow Space (Figure 4).
Figure 4.

Flow cytometry of individual cells based on intrinsic cell-specific marker expression. The FACS plots show three microcapsule samples containing a mixture of K-562 and HEK293 cells (A), K-562 cells (B) or HEK293 cells (C). The microcapsules were first gated based on forward vs. side scatter signal (Microcapsule gate). The resulting sub-population was then gated on ACTB marker expression (Actin gate) and finally, the expression of PTPRC and YAP markers was evaluated on the fluorescence scatter plot (Cell marker gene). The percentage indicates the total events.
Figure 6.

RNA cytometry of acute promyelocytic leukemia cells and human peripheral blood mononuclear cells. Upper and bottom rows exhibit image-based and FACS-based RNA cytometry of human PBMCs spiked with different dilutions of NB-4 cells, respectively. The multiplex scRT-PCR targeted PTPRC (CD45) and PML-RARα transcripts. The PBMC cells were detected by expression of marker gene PTPRC, the NB-4 cells were quantified by recording the co-expression of PTPRC and PML-RARα. The percentages depict the fraction of total counts. Refer to accompanying Supplementary Figure S13 for the gating strategy.
RT-qPCR
To evaluate marker expression in cell lines bulk RT-qPCR was conducted using QuantStudio-1 real-time PCR system (TFS). The total RNA from K-562, HEK293, NB-4, and PBMC were extracted using the GeneJET RNA Purification kit (TFS). The gDNA traces were depleted by RapidOut DNA Removal Kit (TFS). cDNA synthesis was performed in 50 μl Maxima H Minus RT reaction mix comprising 2 μg total RNA, 5 μM Oligo(dT)21 primer (IDT), 0.5 mM dNTP Mix (TFS), 250 U Maxima H Minus Reverse Transcriptase (TFS), 100 U RiboLock RNase Inhibitor and 1× RT Buffer (TFS), at 50°C for 30 min. After cDNA synthesis, the RT enzyme was heat inactivated at 85°C for 5 min. Then, cDNA material was diluted 10-fold in nuclease-free water and used directly for qPCR. qPCR was performed in 10 μl reaction volume comprising 2 μl cDNA, 5 μl 2× Maxima SYBR Green/ROX qPCR Master Mix (TFS), and 3 μl of 1 μM forward/reverse primer mix (Supplementary Table S1). Samples were thermally cycled through the following program: 95°C (10 min), 95°C (15 s)/60°C (30 s)/72°C (60 s) for 40 cycles, and the number of threshold cycles (Ct) for each marker gene was recorded (Supplementary Table S2) using software provided with the QuantStudio-1 instrument.
Double-stranded DNA retention in microcapsules
GeneRuler Low Range DNA Ladder (TFS) was mixed with 30% (w/w) dextran at a ratio 1:1 and encapsulated using the standard procedure described above. Following DNA encapsulation, the emulsion was transferred to 4°C for 60 min. A 30 μl aliquot of the emulsion was taken from the tube, broken and treated with 0.5 μl of 20 mg/ml proteinase K (TFS) for 10 min at 37°C and then 10 μl was combined with 2 μl of Gel Loading Dye, Purple (6x) (NEB) and analyzed on 3% agarose gel in 1× TAE buffer (Supplementary Figure S3). This sample was considered as a control since no DNA loss was expected (Well #1). The remaining (150 μl) of the emulsion was converted to microcapsules as follows: the physically cross-linked (intermediate) microcapsules (Well #2) were released from the water-in-oil emulsion by adding 50 μl of emulsion breaker, washed once in 1 ml ice-cold Cell Washing Buffer and pelleted at 500 × g for 2 min at 4°C. A 30 μl aliquot of physically cross-linked microcapsules was combined with 1 ml of ice-cold Cell Washing Buffer and incubated on ice for 30 min. Then, the microcapsules were rinsed twice in ice-cold Cell Washing Buffer, pelleted by centrifugation, and treated with 0.5 μl of 20 mg/ml proteinase K at 37°C for 10 min. Next, 10 μl of the treated sample was combined with 2 μl of Gel Loading Dye, Purple (6x) and analyzed on 3% agarose gel in 1× TAE buffer (Well#2). The remaining (physically cross-linked) microcapsules were dispersed in 1 ml of ice-cold Cell Washing Buffer supplemented with the photo-initiator (0.1% (w/v) LAP). The intermediate microcapsules were cross-linked by a 20 s exposure to a 405 nm LED device (Droplet Genomics) to obtain microcapsules having a covalently cross-linked membrane. The microcapsules were rinsed once in ice-cold Cell Washing Buffer and then divided into 4 tubes at equal 30 μl portions. 1 ml of Cell Washing Buffer was added to each tube, and microcapsule suspensions were incubated for 30 min at different temperatures: 4°C (Well #3), 22°C (Well #4), 50°C (Well #5), and 70°C (Well #6). Then, the microcapsules that were incubated on ice (4°C) were rinsed twice in ice-cold Cell Washing Buffer and pelleted by centrifugation. The microcapsules that were incubated at 22–70°C were rinsed twice in room temperature Cell Washing Buffer and pelleted by centrifugation. The 30 μl of microcapsule suspension of each tube was treated with 0.5 μl of 20 mg/ml proteinase K and incubated at 37°C for 10 min. Finally, 10 μl of each treated sample was combined with 2 μl of Gel Loading Dye, Purple (6×) and analyzed on 3% agarose gel in 1× TAE buffer.
RNA leakage among microcapsules
To investigate RNA leakage, blank microcapsules (without cells) were immersed in 200 μl Maxima H Minus RT reaction mixture comprising 5 μM oligo(dT)21 primer (IDT), 0.5 mM dNTP Mix, 1000U Maxima H Minus Reverse Transcriptase, 40 U RiboLock RNase Inhibitor, 1× RT Buffer, 1 μg total RNA purified from K-562 cells and 1 μg total RNA purified from HEK293 cells. Microcapsules occupied half of the final reaction volume (100 μl). RT step was conducted at 50°C for 60 min. Every 20 min, microcapsules were briefly dispersed. After cDNA synthesis, the RT enzyme was heat inactivated at 85°C for 5 min. Then, microcapsules were rinsed three times in Washing Buffer and subjected to multiplex PCR targeting ACTB, PTPRC and YAP. The post-RT-PCR microcapsules (n ∼ 4000) were analyzed under the epifluorescence microscope.
Data analysis
Cell retention in microcapsules
To evaluate K-562 cell encapsulation and retention, 36 digital images were recorded: 18 images for water-in-oil droplets and 18 images for microcapsules. Each image contained at least 100 compartments. For each image, the occupancy (lambda value, λ) of cells was estimated. Then, distribution normality was verified by applying Lilliefors (Kolmogorov–Smirnov) normality test. The calculated P-value was 0.6481, which confirmed a normal data distribution. Then F-test for homogeneity of variance was applied. The calculated P-value was 0.372. Assuming equal variance between cell occupancy in droplets and microcapsules, an independent sample t-test was performed (under a two-sided alternative hypothesis). Occupancy measurements were visualized using Python 3.7.6, Pandas framework and Seaborn library.
DNA retention in microcapsules
DNA fragment retention was quantified by measuring the DNA band intensity on an agarose gel. Each DNA fragment retention value was averaged from three independent measurements and analyzed using a Fiji software package. The measurements were visualized using Python 3.7.6, Pandas framework and Seaborn library.
Microscopy analysis of post-RT-PCR microcapsules
Microcapsules from bright-field images were analyzed using a Python script (provided in the GitHub repository). Microcapsules were outlined using Hough circle transform and the masks were used to crop and measure mean and max fluorescence from corresponding images taken using DAPI, GFP, RFP and Cy5 filters. Measurements were processed and visualized using Python 3.7.6, Pandas framework and Seaborn library.
Flow cytometry data analysis
Flow cytometry data were analyzed and visualized using FCS Express 7 software (version 7.12.0005). The post-RT-PCR microcapsules after harsh lysis were analyzed using the Partec CyFlow Space FACS instruments. The gating process was performed in the following manner: (i) gating the microcapsules based on forward versus side scatter signal, (ii) gating ACTB positive events based on Alexa Fluor 647 versus side scatter signal, (iii) analyzing PTPRC and YAP marker abundance based on Alexa Fluor 488 versus Alexa Fluor 555 signal. All measurements were performed by analyzing the signal area.
The post-RT-PCR microcapsules after mild lysis on NB-4 and PBMCs were analyzed using the BD FACSAria III FACS instrument. The gating process was performed in the following manner: (i) gating the microcapsules with isolated cells based on Alexa Fluor 488 signal area vs. DAPI-stained nuclei fluorescence signal height, (ii) gating PML-RARα positive events based on Alexa Fluor 488 signal area vs. Alexa Fluor 555 signal area.
Sensitivity, specificity and positive/negative predictive values
Sensitivity was defined as 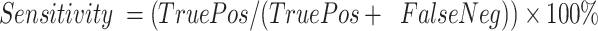 . Specificity was defined as
. Specificity was defined as 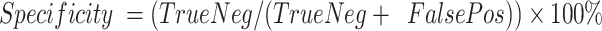 . Positive predictive value was defined as
. Positive predictive value was defined as 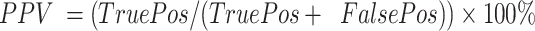 . Negative predictive value was defined as
. Negative predictive value was defined as 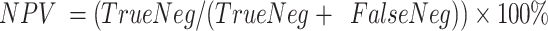 . Microcapsules carrying a cell and being positive for one of the marker genes (ACTB, B2M, TBP, PTPRC, YAP) were counted as true positives. Microcapsules carrying no cells and showing no fluorescence were counted as true negatives. The false positives were microcapsules lacking cells but being fluorescent for one of the marker genes. The false negatives were microcapsules with cells displaying no signal for any marker gene.
. Microcapsules carrying a cell and being positive for one of the marker genes (ACTB, B2M, TBP, PTPRC, YAP) were counted as true positives. Microcapsules carrying no cells and showing no fluorescence were counted as true negatives. The false positives were microcapsules lacking cells but being fluorescent for one of the marker genes. The false negatives were microcapsules with cells displaying no signal for any marker gene.
RESULTS
Microcapsules for high-throughput nucleic acid analysis of individual cells
The overall concept of the microcapsule-based approach for conducting single-cell RNA cytometry is summarized in Figure 1. At first, the mixture of cells is isolated in uniform and concentric microcapsules having a liquid core surrounded by a thin semi-permeable membrane. The encapsulated cells are then lysed by dispersing the microcapsules in a lysis mix and processed through a series of washing steps to purify the cells’ genetic material. The size-selective permeability of a membrane prevents cellular nucleic acids from escaping the compartments, while simultaneously enabling the intracellular proteins (e.g. RNases) and other low-molecular-weight compounds to leave the microcapsule. Therefore, RNA and DNA molecules encoded by individual cells can be efficiently purified and retained within the compartments. Once the nucleic acids are purified, the microcapsules are transferred to the RT reaction mix to initiate cDNA synthesis, followed by multiplex PCR. During PCR, the fluorescently labelled primers in the reaction mix cross the membrane by diffusion and are incorporated into the PCR amplicons, rendering the compartments with a cell fluorescent. Thus, using a multiplex panel of fluorescently labelled PCR primers, the expression of the selected genes of interest—in hundreds to tens of thousands of individual cells—can be digitally profiled and quantified using conventional flow cytometry, or epifluorescence microscope.
Figure 1.

The concept of performing single-cell multiplex RT-PCR using microcapsules. The individual cells are isolated (compartmentalized) in semi-permeable microcapsules and are lysed by dispersing microcapsules in an appropriate lysis buffer. Upon lysis, the nucleic acid molecules longer than 300 bp. are retained within the microcapsules, while low molecular weight biomolecules are removed by dialysis. Following cell lysis step, the microcapsules carrying purified nucleic acids are dispersed in RT-PCR reaction mix containing fluorescently labelled oligonucleotides. Once dispersed in a reaction mix, the reaction components quickly diffuse to the microcapsule enabling multiplex RT-PCR reaction on encapsulated nucleic acids. After RT-PCR the microcapsules carrying a particular cell type, and the transcript(s) of interest, become fluorescent thus enabling differentiation of encapsulated cells based on their digital transcriptional profile.
Generation of uniform microcapsules for efficient mammalian cell isolation and retention
To realize the experimental strategy presented in Figure 1, we created microcapsules with a semi-permeable membrane composed of a gelatin derivative, a thermo-responsive polypeptide that solidifies at lower temperatures and can be crosslinked into a porous gel. To produce uniform microcapsules, we first generated aqueous two-phase system (ATPS) droplets in the carrier oil on a 40-μm deep co-flow microfluidics device at a throughput of 540 droplets per second, by infusing gelatin methacrylate (GMA), and dextran solutions, with the cells suspended in the latter at a desirable density (Figure 2A and Supplementary Figure S1). Following droplet generation at room temperature, the GMA and dextran phases quickly phase-separated (within minutes), forming a shell enriched in GMA and a liquid core enriched in dextran (Figure 2B). The shell surrounding the liquid core was converted to a semi-permeable membrane in a two-step procedure. First, the shell was solidified by cooling the water-in-oil droplets to 4°C and then covalently cross-linked under a brief exposure to a light-activated photo-initiator (see Materials and Methods). This two-step procedure, where physical gelation is followed by a covalent cross-linking, ensured a highly reproducible generation of microcapsules comprising a well-centered liquid core surrounded by a thin layer of chemically cross-linked polypeptide. We confirmed that microcapsules remain intact under a variety of experimental conditions, such as freezing, thawing and centrifugation at high speeds (e.g. 20 000 × g), and in the presence of different salts and solvents (Supplementary Figure S2).
Figure 2.

Microcapsule generation, single-cell encapsulation and DNA retention. (A) Cell encapsulation and collection off-chip. A mixture of cells is encapsulated in aqueous two-phase system (ATPS) droplets comprising a liquid core and a liquid shell and collected off-chip in the tube. (B) The liquid shell of collected droplets is converted to a semi-permeable membrane in a two-step process: at first, the liquid shell is gelled by cooling, and then covalently cross-linked by photo-polymerization. (C) The boxplots represent mammalian cell retention in droplets and microcapsules. (D) DNA retention in microcapsules at different temperatures after 30 min incubation. The DNA fragments 100 bp. and smaller freely transverse the microcapsule's membrane, while DNA fragments 300 bp. and longer are fully retained. Solid lines serve only for visual guidance. Scale bars, 100 μm.
To evaluate the mammalian cell encapsulation and retention efficiency, we generated 75.0 ± 1.5 μm size microcapsules with a 3.0 ± 0.2 μm thick membrane. We loaded human cells (K-562) and quantified cell retention microscopically immediately after encapsulation, and after forming the microcapsules. No significant difference was detected (two-tailed t-test (n = 18) t = 1.22, P = 0.2314), confirming that compartmentalized cells are efficiently retained during the microcapsule generation process (Figure 2C). These results sharply contrast with hydrogel bead-based assays, which experience uncontrolled cell loss due to the cell's tendency to adhere at the water/oil interface (46–48). Hence, the use of microcapsules, having a liquid core and a thin polyampholyte membrane, ensures minimal or no cell loss.
Nucleic acid retention and permeability of microcapsules
While efficient cell isolation and retention are necessary for conducting single-cell assays, to be broadly applicable in RT-PCR the microcapsules must also ensure efficient retention of nucleic acid molecules during the analytical procedures. To experimentally verify the permeability and retention of the nucleic acid molecules within microcapsules, we encapsulated dsDNA fragments ranging from 25 to 700 bp. and incubated the microcapsules at temperatures ranging from 4 to 70ºC for 30 min. We then washed the microcapsules in a neutral pH buffer to remove all unretained DNA molecules and extracted the retained DNA within the microcapsules by dissolving the membrane (see Materials and Methods). The results, shown in Figure 2D and Supplementary Figure S3, clearly indicate that DNA fragments ≥300 bp. were efficiently retained inside the microcapsules, the molecular weight of which approximately corresponds to ∼180 kDa. Considering the typical oligonucleotide length of 20–50 nt. (12–30 kDa), and the molecular weight of M-MLV reverse transcriptase (71 kDa) and Taq DNA polymerase (94 kDa), we postulated that microcapsules with such permeability are well-suited for conducting massively parallel biochemical reactions on thousands of single cells by simply dispersing them in a suitable reaction mixture.
Digital multiplex RT-PCR enables accurate cell type identification
To illustrate the digital profiling of individual human cells based on their gene expression, we mixed human leukemia cell line (K-562) and human embryonic kidney cells (HEK293) at a 1:1 ratio, and encapsulated the mixture at a dilution level such that each microcapsule, on average, would contain no more than one cell. As a reference, we also separately encapsulated either K-562 or HEK293 cells. We first lysed the encapsulated cells by dispersing the microcapsules in a chaotropic lysis mix followed by extensive washes and genomic DNA (gDNA) digestion with DNase I to obtain the microcapsules with purified total RNA derived from single cells. The microcapsules were then dispersed in the RT reaction mix to convert the cellular mRNA to copy DNA (cDNA) using poly(dT) primers (see Materials and Methods for further details). To identify individual cells based on their digital expression signature, the post-RT microcapsules were transferred into a PCR reaction mix supplemented with fluorescently labelled primers targeting cell-specific markers (Supplementary Table S1). The cDNA of transcripts that encode the protein tyrosine phosphatase receptor type C (PTPRC) and Yes-associated protein 1 (YAP) were chosen as targets of interest for the K-562 and HEK293 cells, respectively, whereas the cDNA of β-actin (ACTB) served as a reference (Supplementary Table S2). The target-specific PCR primers were fluorescently labelled at 5’-end with fluorophores emitting light at different wavelengths, to enable the identification of amplified nucleic acids based solely on the fluorescence signal. During the PCR, the fluorescently labeled oligonucleotides diffuse from the bulk solution into the interior of the microcapsules and get incorporated into the PCR amplicon upon binding to the target DNA template, thereby turning the amplified DNA into a fluorescent product (Figure 3 and Supplementary Figure S4). Only the microcapsules carrying a target template (e.g. PTPRC, YAP or ACTB) could incorporate the fluorescent probes into newly synthesized amplicons rendering them fluorescent (Figure 3B). The microcapsules lacking nucleic acid template remained blank. As a result, the microcapsules containing a cell of interest could be distinguished by a cell-type-specific fluorescence signal. For example, given the differential expression of PTPCR and YAP genes (49) and the ubiquitous expression of ACTB, the post-RT-PCR microcapsules harboring K-562 and HEK293 cells turned positive in two channels, PTPCR-ACTB and YAP-ACTB, respectively (Figure 3C, cyan and magenta). Altogether these results demonstrate that microcapsule-based scRT-PCR assay enables accurate cell-type identification based on their digital gene expression signature.
Figure 3.

Digital gene expression profiling using microcapsule-based RT-PCR. (A) Schematic of the experimental design. The mixture of K-562 and HEK293 cells is loaded in microcapsules and subjected to uniplex RT-PCR or multiplex RT-PCR assay using fluorescently labelled primers targeting the transcripts of interest (PTPRC, YAP, ACTB). The digital gene expression profile of individual cells is then analyzed microscopically by layering the microcapsules on a hemocytometer and recording their fluorescence. (B) Epifluorescence images of microcapsules after RT-PCR reaction targeting one transcript of interest: PTPRC (green), YAP (red), or ACTB (blue). (C) Epifluorescence images of microcapsules after multiplex RT-PCR reaction simultaneously targeting two or three transcripts of interest. The microcapsules carrying both PTPRC and ACTB amplicons appear as cyan partitions; microcapsules carrying both YAP and ACTB amplicons appear as magenta partitions; microcapsules with ACTB amplicons appear as blue partitions. Scale bars, 100 μm.
Flow cytometry for high-throughput analysis of intrinsic cell markers
To validate the microscopy results presented in Figure 3 and eliminate potential under-sampling artifacts, we explored the high-throughput capabilities offered by flow cytometry instruments. We loaded the post-RT-PCR microcapsules carrying K-562 cells, HEK293 cells, or a mixture of both, onto the FACS instrument and performed RNA cytometry (Figure 4 and Supplementary Figure S5). As a demonstration, up to 30 000 microcapsules were analyzed per experiment, although the total microcapsule count was not limited and could be easily scaled up. The distribution of flow cytometry events in the forward vs. side scatter plot (Figure 4, Capsule gate), the fluorescence of Alexa Fluor 647 vs. side scatter plot (Figure 4, Actin gate) and the Alexa Fluor 488 vs. Alexa Fluor 555 intensity plot (Figure 4, Cell marker gate) enabled precise quantification of cell-type-specific marker expression and identification of different cell types in the population. The results, summarized in Supplementary Table S3, show a very close agreement between the flow cytometry and epifluorescence microscopy measurements. The microcapsules carrying K-562 cells were PTPRC positive (3.19% and 3.63% events), whereas the microcapsules carrying HEK293 cells were YAP positive (4.84% and 5.46% events), approaching the theoretical frequency expected from a Poisson distribution (∼5–6%). The microcapsules prepared with a mixture of HEK293 and K-562 cells showed a positive signal either in the PTPRC or YAP channel, while K-562 and HEK293 co-encapsulation events were rare (0.02–0.15%), and followed the Poisson distribution. Measuring the co-expression of one marker gene and one ubiquitously expressed gene, facilitated the digital profiling of single cells. For instance, 99.07% of PTPRC-positive events were also positive for the ACTB signal in the sample containing K-562 cells, and 94.57% of YAP-positive events were also positive for the ACTB signal in the sample containing HEK293 cells. Hence, the microcapsules are fully compatible with FACS instruments and enable a high-throughput cytometry of cells based on their gene activity rather than surface proteins based on antibody staining.
Unexpectedly, the comparative flow cytometry and microscopy analysis revealed the presence of a third subpopulation with a fluorescent signal corresponding to the ACTB target alone and lacking PTPCR and YAP expression. A fraction of ACTB-positive events (Supplementary Table S3, third column) was approximately 3-times higher than the number of target-specific YAP (fourth column) or PTPRC (sixth column) events. Considering the large excess of ACTB-positive events as compared to the number of loaded cells, we postulated that these events may represent ambient RNA molecules that were present in the initial cell suspension due to premature cell lysis. This notion is supported by the previous reports showing that scRT-PCR in water-in-oil droplets leads to an increased fraction of false positives than the actual number of cells (15,22), and that the ambient RNA released during cell preparation is the most likely source of these events (24). To rule out the possibility that ACTB-positive events are a by-product of nucleic acid molecule exchange during RT-PCR (e.g. RNA or cDNA molecules escape a microcapsule containing a cell and then enter a neighboring empty microcapsule where it gets amplified), we performed a control experiment in which 2 μg of purified total RNA from K-562:HEK293 cell mixture was added to the RT-PCR mixture supplemented with ∼200 000 blank (empty) microcapsules. In case there is nucleic acid leakage, the blank microcapsules should turn fluorescent upon completion of the RT-PCR. We analyzed ∼4000 randomly picked microcapsules and found no fluorescent events (Supplementary Figure S6), thus indicating no migration of the target mRNA or cDNA into the microcapsules occurred during the RT-PCR.
Accurate differentiation of true positive and false positive events
Accurate differentiation of the fluorescent microcapsules that carry a cell (true positive events) from the fluorescent microcapsules that lack a cell (false positive events) might be challenging since the end-point fluorescent readout can be indistinguishable between these events. We reasoned that using mild-lysis conditions the cell nucleus will retain its compact structure and, as a result, could be used as a reference to correctly identify the microcapsules containing cells irrespectively of their transcriptional activity. To test this approach, we loaded the mixture of K-562 and HEK293 cells in microcapsules, fixed them in ice-cold ethanol, permeabilized cells in mild-lysis conditions and performed multiplex RT-PCR (see Materials and Methods). We confirmed that our approach for freezing and preservation of ethanol-fixed cells had no significant impact on the RT-PCR accuracy (Supplementary Figure S7), albeit the mean fluorescence signal intensity was somewhat lower for preserved samples (Supplementary Figure S8). In addition, we compared the RT-PCR output of ACTB, PTPRC and YAP markers when encapsulated cells were disrupted using mild- and harsh-lysis conditions, and found no significant difference between the two cell-lysis protocols (Supplementary Figure S9).
Digital analysis of post-RT-PCR samples revealed that using mild lysis conditions, the cell nuclei not only remained compact but also emitted a characteristic, spatially confined fluorescence in all channels (ACTB, YAP, PTPRC), presumably due to non-specific incorporation of fluorescently labelled multiplex PCR probes (Figure 5B and Supplementary Figure S10). The fluorescence emitting from the nuclei could be enhanced by the addition of DAPI dye. In contrast, the fluorescence of the PCR amplicons was uniformly distributed across the entire volume of the microcapsule. Therefore, measuring the fluorescence profile of the entire microcapsule provided a straightforward approach to correctly identify the partitions that carry cells and separate them from the partitions with cell-free nucleic acid molecules (Figure 5C). Taking advantage of this feature, we evaluated the cell-free RNA levels of a freshly prepared mixture of cells targeting different housekeeping genes. We anticipated that highly-abundant transcripts such as ACTB would result in increased contamination levels as more RNA molecules will be released prematurely by the compromised cells. In agreement with this notion, we found that the number of cell-free microcapsules displaying a fluorescent signal constituted 16% of positive counts, but dropped down to 0.8% when targeting the TBP gene, which is being expressed at ∼10 copies per cell (25) (Supplementary Figure S11). Furthermore, being able to correctly identify and quantify the true/false positives and true/false negatives, we found the microcapsule-based multiplex scRT-PCR assay specificity to be very high, in the range of 97.71–99.93%. The sensitivity was estimated to be 98.65% for ACTB and 92.57% for YAP on HEK293 cells, and 98.37% for ACTB and 71.74% for PTPRC on K-562 cells. The cell-type specific markers, YAP and PTPRC, displayed excellent positive predictive values (PPVs), 98.56 and 99.25%, respectively. Likewise, negative predictive values (NPVs) were also very high, 99.60% and 98.23%, respectively.
Figure 5.

Detection of true positive and false positive events using microcapsule-based multiplex RT-PCR assay. (A) Depiction of an experimental scheme where a typical biological analyte comprising cells and ambient RNA is partitioned in microcapsules, subjected to multiplex RT-PCR and evaluated by fluorescence imaging. (B) Representative microscopy images of the microcapsules after multiplex scRT-PCR targeting ACTB, PTPRC and YAP markers. Note, the cell nucleus exhibits spatially localized fluorescence in all channels, whereas the PCR amplicons show diffusive fluorescence signal occupying the entire core of a microcapsule. (C) Image-based characterization of a biological sample comprising a mixture of K-562 and HEK293 cells. Grey color depicts microcapsules that exhibit a fluorescent signal; orange – microcapsules that contain a cell; blue – microcapsules that exhibit RT-PCR signal lacking a cell; purple, green, and red colors depict microcapsules positive for ACTB, PTPRC and YAP signal, respectively. Each plot is represented by >300 counts. Local minimum was used to set the thresholds separating negative and positive events.
Interestingly, digital profiling of cell mixture uncovered a subpopulation of cells that lacked detectable levels of PTPRC or YAP marker, yet expressed high levels of a housekeeping gene. To better understand the origin of this unusual gene expression signature, we performed a separate set of experiments, where multiplex scRT-PCR was performed on each cell type independently. In concordance with FACS (50), these experiments revealed a bimodal gene expression distribution in K-562 cells (Supplementary Figure S12), which are known to differentiate into the erythroid lineage that lacks PTPRC (51). However, bimodality may also arise due to transcriptional bursts (52), cell cycle dependence (53), and stochastic effects (54), amongst others, all of which may influence the appearance of cells that lack a transcript of interest at a given moment in time. Therefore, the microcapsule-based scRT-PCR output is highly sensitive to the transcriptional state of a cell and captures the individual gene activity at a molecular level.
Identification of leukemia cells in the mixture of peripheral blood mononuclear cells
To further demonstrate the potential use of the developed microcapsule technology tailored for biomedical applications, we sought to digitally profile primary human cells. To this end we encapsulated previously-frozen peripheral blood mononuclear cells (PBMCs) and performed scRT-PCR targeting commonly used marker PTPRC (CD45). While we initially expected to achieve >95% detection rate, the image-based, as well as the FACS-based analysis of the post-RT-PCR microcapsules, revealed 75.4–78.6% of the encapsulated cells being positive for PTPRC, respectively. Profiling PBMCs for another ubiquitous marker (B2M) showed a similar detection rate. These numbers, however, matched very closely the fraction of viable cells (∼77%), as determined by trypan blue staining, pointing out that cell detection rate by scRT-PCR correlates with cell viability. We then spiked PMBCs with acute promyelocytic leukemia cells (NB-4) and attempted to identify the leukemic cells based on the expression of the fused gene PML-RARα. As it was shown previously, weak PML-RARα expression in the NB-4 cells is within the range detected in primary leukemia samples (55). We applied single-cell multiplex RT-PCR as an analogy to the standard clinical practice where instead of individual cells, the total RNA is extracted from the blood and subjected to cancer diagnostics by bulk RT-PCR (55,56). The results presented in Figure 6 indicate that individual leukemic cells were successfully identified in the mixture of PBMCs, albeit the fraction of detected NB-4 cells appears ∼2-fold lower than the theoretical projections. To better understand this deviation, we quantified the expression levels of PML-RARα transcript in NB-4 cells using qPCR and found it to be ∼200-fold lower than for PTPRC or TBP (Supplementary Table S2), which roughly translates to less than 1 copy of PML-RARα transcript per cell, on average. Therefore, it is reasonable to assume that not all NB-4 cells are expressing PML-RARα fusion at a given moment in time, thus explaining the difference in the observed counts. In this context, it is worth noting that digital profiling of another fusion transcript (BCR-ABL) in K-562, which is expressed at approximately 40 copies per cell on average (57), resulted in a 98.6% detection rate, and targeting transcripts expressed at ∼10 copies per cell (e.g. TBP (25)) resulted in a 97.54% detection rate. Altogether, the RNA cytometry concept based on the semi-permeable microcapsules presented in this work offers a simple, easily customizable approach for rapid and highly sensitive digital profiling of thousands of individual cells at ultra-high-throughput rates, and is advantageous over existing microfluidic and plate-based platforms.
DISCUSSION
Over the past few years, single-cell gene expression analysis has become one of the essential tools for studying complex biological systems. Single-cell RNA-Seq technology has been particularly instrumental in delineating cellular functions and the discovery of novel biomarkers (35,58–60). However, the scRNA-Seq approach still remains a relatively slow and expensive process, and runs counter to the situations built on a fast and accurate assessment of well-characterized genetic markers. For example, assessing the expression of clinically relevant biomarkers using flow cytometry and RT-PCR remains a standard procedure for blood cancers (55,56,61,62) and can be completed within a day. While flow cytometry can efficiently discriminate individual cells based on their surface markers, the analysis of intracellular markers often relies on bulk RT-PCR assays, which blur the genotypic differences between the cells, and mask clinically relevant cell populations. Therefore, a fast and accurate digital assay for profiling single cells based on their gene activity provides a valuable and effective option for characterizing heterogeneous biological samples in biomedical research.
In this work, we describe, for the first time, a high-throughput RNA cytometry in semi-permeable microcapsules (shells) that enables digital gene expression profiling of tens of thousands of single cells in a matter of 4–6 h. The biocompatible microcapsules, comprised of a liquid core with polysaccharide and a thin membrane made of covalently cross-linked polypeptide, efficiently retain the encapsulated cells and nucleic acids during multi-step procedures. The microcapsules remain intact under different handling conditions, when frozen, thawed or thermocycled, and in the presence of various salts and solvents (e.g. 4M guanidinium isothiocyanate, 90% methanol, 90% acetone). The semi-permeable membrane supports the passive exchange of assay reagents such as enzymes and oligonucleotides, yet simultaneously prevents larger biomolecules such as mRNA, cDNA or gDNA molecules from escaping the microcapsules. We showed that the microcapsules efficiently retain dsDNA molecules longer than 300 bp in a broad range of different temperatures, yet remain permeable to shorter DNA fragments (Figure 2). Strikingly, the post-RT-PCR microcapsules stored at 4°C for 10 months retained PCR amplicons, as confirmed by epifluorescence microscopy (Supplementary Figure S14). While the current permeability cut-off is well suited for conducting a large variety of molecular biology assays, the permeability of the microcapsule's membrane may be further tuned by altering the cross-linking conditions, adjusting the concentration and modification degree of polypeptides, thereby providing flexibility for creating microcapsules tailored for specific biological and biomedical applications. This selective permeability simplifies the implementation and use of sequential biochemical reactions on encapsulated species (e.g. cells, microorganisms, viruses, nucleic acids) since the desirable enzymes and reagents can be loaded, or replaced, in the microcapsule by simply changing the reaction buffer that holds the microcapsules. Finally, the complete release of encapsulated nucleic acids is achieved by simply exposing the microcapsules to protease enzyme, which hydrolyzes the peptide bonds.
We demonstrate the utility of the microcapsule-based RNA cytometry approach via digital gene expression profiling of human leukemia cells, embryonic kidney cells and primary PBMC using multiplex scRT-PCR assay. At first, we applied harsh lysis conditions to obtain the microcapsules with a purified total RNA derived from single cells. Subjecting such microcapsules to multiplex RT-PCR generated bright fluorescent partitions specific to a given cell type. However, since microcapsules with cells and microcapsules with ambient RNA exhibited nearly the same fluorescence intensity (Figure 3B), obtained results also highlighted the danger of quantifying the cells based on the expression of a single gene marker alone. This limitation could be overcome by targeting two genes simultaneously (e.g. one ubiquitously and one differentially expressed gene) to correctly identify the partitions that carry cells (Figure 3C), yet at the cost of increased assay multiplexing. We then showed that scRT-PCR assay using mild-lysis conditions provides a convenient approach for quantifying the cells based on a characteristic, spatially confined fluorescence emanating from a cell nucleus (Figure 5). As a result, the microcapsules with cells and microcapsules with ambient RNA could be clearly differentiated irrespectively of the cell's transcriptional state even when the RT-PCR assay is designed to target a single marker gene.
While the microcapsule-based scRT-PCR assay specificity was extremely high (99.12 ± 0.89%) with all target probes used in this work, the sensitivity varied from 71 to 98% depending on the gene marker, cell type and primers used. Targeting transcripts expressed at low to medium levels (10–40 copies per cell) showed reduced cell-free RNA levels (<1%) while retaining high assay sensitivity (98.09 ± 0.77%). We also successfully profiled leukemia-associated marker PML-RARα expressed at extremely low levels (i.e. <1 copy per cell on average), although further optimizations of reaction conditions and the use of targeted RT primers (63) might improve the detection rate of low abundance transcripts. Nonetheless, the presented results show that multiplex scRT-PCR based on semi-permeable microcapsules provides a reliable approach to profile single cells rapidly, with high specificity and sensitivity.
The entire process, from the initial cell suspension to digital quantification, takes a few hours to complete, making the microcapsule-based RNA cytometry particularly attractive for rapid testing and diagnostics that require fast turnaround and are based on genotypic/transcriptional signatures of individual cells rather than their surface markers. Potential biomedical applications of the developed technique may include circulating tumor cells (CTCs), that often lack proper surface markers, yet their gene expression signatures have been shown to adequately predict the therapy response in multiple cancers (64–66). This also applies to biomarkers for which commercial antibodies are limited (e.g. G protein-coupled receptors), or targets for which antibodies cannot be made (e.g. viral transcripts, lncRNA). Another advantage of the microcapsule-based technique reported here is that nucleic acids encoded by single cells can be efficiently purified under a variety of lysis conditions leading to improved enzymatic reactions. The ability to entirely replace the reagents and easily alter the content of the microcapsules at any given step in a protocol should make microcapsules broadly applicable to different formats of multiplexing (67), including in situ hybridization and imaging (68).
In this work, the post-RT-PCR microcapsules were analyzed using flow cytometry and epifluorescence microscopy. Both approaches generated similar results (Supplementary Table S3 and Figure 6). In practice, using a multi-color FACS provides the most appealing option for high-throughput profiling of the individual cells, yet for samples comprising fewer cells (n < 10 000), the image-based analysis may represent a faster and simpler option. By conducting RNA cytometry of individual cells, we accurately identified different cell types and quantified their distribution in the heterogeneous cell population. When using established state-of-art droplet microfluidics platforms, the so-called ‘signal rain’ artifacts (i.e. when positive events display a trail of low and intermediate fluorescence values overlapping with a signal of negative and false positive events), create a significant challenge for correctly setting the detection thresholds for separating true positive from false positive events (67,69). Our flow cytometry results, corroborated by microscopy analyses, show that this problem can be overcome by using a microcapsule-based multiplex fluorescence readout. A multiplex scRT-PCR assay based on three simultaneous readouts, i) expression of a ubiquitous marker, ii) expression of a cell-type specific marker(s), and iii) a cell-based marker that does not rely on RNA (e.g. cell nucleus), not only enables accurate identification of distinct cell types present in a heterogeneous mix, but also provides an absolute cell count estimate irrespectively of the transcriptional state of a cell, and the ability to detect ambient nucleic acid molecules (false positives) present in the analyte.
We believe that the semi-permeable microcapsule concept we developed holds great potential for a broad range of clinical and biological applications. The multiplex scRT-PCR assay can be designed to target gene isoforms (70), differentially expressed markers of clinical relevance (71), or genetic aberrations (55,56,72,73). It can quantitively assess the distribution and frequency of somatic mutations in a panel of cancer-driving genes and benefit cancer diagnostics (74–76). Microcapsule-based nested-PCR could be applied for high-throughput screening of B- and T-cells, and for the next-gen sequencing of the immunoglobulin-encoding genes (16,21,23,77,78). Indeed, the applicability of microcapsules can be extended beyond the RT-PCR measurements: the gDNA or cDNA of single cells in microcapsules could be barcoded and sequenced to obtain the genome-encoded or transcriptome-encoded information. This could contribute greatly to complex disease characterization where genetic aberrations manifest on both DNA and RNA levels. Moreover, the microcapsules presented in this work may provide a very appealing option for single-cell combinatorial indexing using a split-pool synthesis (79,80). Due to a selective permeability, the delivery and removal of short barcoding-DNA oligonucleotides during the sequential nucleic acid barcoding reactions becomes straightforward, while simultaneously ensuring efficient retention of longer (barcoded) nucleic acids inside the microcapsules. In addition, because in microcapsules the genetic material released from the lysed cells can be purified and retained through multiple rounds of treatment and washing, it should provide an advantage over droplet-based and plate-based scRNA-Seq approaches, where cell lysis and mRNA barcoding are typically performed in the same reaction mix under suboptimal conditions, and in the presence of intracellular inhibitors (e.g. RNases). Finally, the possibility to completely replace reaction buffers and exchange reagents at any step in the workflow should provide further flexibility for performing multi-step molecular barcoding reactions. In conclusion, the semi-permeable microcapsule concept that we developed and report here provides a foundation for a variety of single-cell assays that could be built and innovated upon for various goals. We anticipate that this approach will eventually benefit diagnostics, cell biology and biomedicine, where the rapid turnaround is of foremost importance.
DATA AVAILABILITY
Raw data and analysis scripts have been deposited on the Github repository: https://github.com/L-Greta/RNA_Cytometry_Data.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are especially grateful to Dr Karolis Leonavičius (Droplet Genomics, Lithuania) for image analysis consulting and assistance, Indrė Dalgėdienė (Vilnius University, Lithuania) for assistance with Partec CyFlow Space FACS instrument and Dr Vytautas Kašėta (CIM, Lithuania) for assistance with BD FACSAria III instrument. Authors are grateful to Jonas Gasparavičius (Droplet Genomics, Lithuania) for supplying microfluidic devices used in this work, Suji Kim (Vilnius University, Lithuania) for assistance with cell preparation and Dr Veronika Viktorija Borutinskaitė (Vilnius University, Lithuania) for kindly providing NB-4 cell line. Authors express their gratitude to Arūnas Stirkė (FTMC, Lithuania) for kindly providing an access to epifluorescence microscope.
Contributor Information
Greta Leonaviciene, Institute of Biotechnology, Life Sciences Centre, Vilnius University, 7 Sauletekio av., Vilnius, LT-10257, Lithuania.
Linas Mazutis, Institute of Biotechnology, Life Sciences Centre, Vilnius University, 7 Sauletekio av., Vilnius, LT-10257, Lithuania.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work received funding from European Regional Development Fund [01.2.2-LMT-K-718-04-0002] under grant agreement with the Research Council of Lithuania. Funding for open access charge: Vilnius University.
Conflict of interest statement. G.L. and L.M. are listed as co-inventors on a US provisional patent application that includes the results of this work. L.M. is a shareholder of Droplet Genomics.
REFERENCES
- 1. Valasek M.A., Repa J.J.. The power of real-time PCR. Adv. Physiol. Educ. 2005; 29:151–159. [DOI] [PubMed] [Google Scholar]
- 2. Murphy J., Bustin S.A.. Reliability of real-time reverse-transcription PCR in clinical diagnostics: gold standard or substandard. Expert Rev. Mol. Diagn. 2009; 9:187–197. [DOI] [PubMed] [Google Scholar]
- 3. Wang X., Yao H., Xu X., Zhang P., Zhang M., Shao J., Xiao Y., Wang H.. Limits of detection of 6 approved RT-PCR kits for the novel SARS-Coronavirus-2 (SARS-CoV-2). Clin. Chem. 2020; 66:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manzotti M., Dell’Orto P., Maisonneuve P., Zurrida S., Mazzarol G., Viale G.. Reverse transcription-polymerase chain reaction assay for multiple mRNA markers in the detection of breast cancer metastases in sentinel lymph nodes. Int. J. Cancer. 2001; 95:307–312. [DOI] [PubMed] [Google Scholar]
- 5. Andergassen U., Kolbl A.C., Mahner S., Jeschke U.. Real-time RT-PCR systems for CTC detection from blood samples of breast cancer and gynaecological tumour patients (Review). Oncol. Rep. 2016; 35:1905–1915. [DOI] [PubMed] [Google Scholar]
- 6. Craney A.R., Velu P.D., Satlin M.J., Fauntleroy K.A., Callan K., Robertson A., La Spina M., Lei B., Chen A., Alston T.et al.. Comparison of two high-throughput reverse Transcription-PCR systems for the detection of severe acute respiratory syndrome coronavirus 2. J. Clin. Microbiol. 2020; 58:e00890-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kulkarni A., Anderson A.G., Merullo D.P., Konopka G.. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019; 58:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu B., Xu P., Ma L., Chen D., Wang J., Dai X., Huang L., Du W.. One cell at a time: droplet-based microbial cultivation, screening and sequencing. Mar. Life Sci. Technol. 2021; 3:169–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y., Jin R., Shen B., Li N., Zhou H., Wang W., Zhao Y., Huang M., Fang P., Wang S.et al.. High-throughput functional screening for next-generation cancer immunotherapy using droplet-based microfluidics. Sci. Adv. 2021; 7:eabe3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mar J.C., Rubio R., Quackenbush J.. Inferring steady state single-cell gene expression distributions from analysis of mesoscopic samples. Genome Biology. 2006; 7:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Phetsouphanh C., Xu Y., Amin J., Seddiki N., Procopio F., Sekaly R.P., Zaunders J.J., Kelleher A.D.. Characterization of transcription factor phenotypes within antigen-specific CD4+ t cells using qualitative multiplex single-cell RT-PCR. PLoS One. 2013; 8:e74946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Durruthy-Durruthy R., Gottlieb A., Hartman B.H., Waldhaus J., Laske R.D., Altman R., Heller S.. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell. 2014; 157:964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gong Y., Ogunniyi A.O., Love J.C.. Massively parallel detection of gene expression in single cells using subnanolitre wells. Lab Chip. 2010; 10:2334–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanchez-Freire V., Ebert A.D., Kalisky T., Quake S.R., Wu J.C.. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat. Protoc. 2012; 7:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eastburn D.J., Sciambi A., Abate A.R.. Ultrahigh-throughput mammalian single-cell reverse-transcriptase polymerase chain reaction in microfluidic drops. Anal. Chem. 2013; 85:8016–8021. [DOI] [PubMed] [Google Scholar]
- 16. DeKosky B.J., Ippolito G.C., Deschner R.P., Lavinder J.J., Wine Y., Rawlings B.M., Varadarajan N., Giesecke C., Dorner T., Andrews S.F.et al.. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 2013; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eastburn D.J., Sciambi A., Abate A.R.. Identification and genetic analysis of cancer cells with PCR-activated cell sorting. Nucleic Acids Res. 2014; 42:e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun H., Olsen T., Zhu J., Tao J., Ponnaiya B., Amundson S.A., Brenner D.J., Lin Q.. A bead-based microfluidic approach to integrated single-cell gene expression analysis by quantitative RT-PCR. RSC Adv. 2015; 5:4886–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu Y., Zhang Y.X., Liu W.W., Ma Y., Fang Q., Yao B.. Printing 2-dimentional droplet array for single-cell reverse transcription quantitative PCR assay with a microfluidic robot. Sci. Rep. 2015; 5:9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pellegrino M., Sciambi A., Yates J.L., Mast J.D., Silver C., Eastburn D.J.. RNA-Seq following PCR-based sorting reveals rare cell transcriptional signatures. BMC Genomics. 2016; 17:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDaniel J.R., DeKosky B.J., Tanno H., Ellington A.D., Georgiou G.. Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat. Protoc. 2016; 11:429–442. [DOI] [PubMed] [Google Scholar]
- 22. Kim S.C., Clark I.C., Shahi P., Abate A.R.. Single-Cell RT-PCR in microfluidic droplets with integrated chemical lysis. Anal. Chem. 2018; 90:1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanno H., McDaniel J.R., Stevens C.A., Voss W.N., Li J., Durrett R., Lee J., Gollihar J., Tanno Y., Delidakis G.et al.. A facile technology for the high-throughput sequencing of the paired VH:VL and TCRbeta:TCRalpha repertoires. Sci. Adv. 2020; 6:eaay9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma J., Tran G., Wan A.M.D., Young E.W.K., Kumacheva E., Iscove N.N., Zandstra P.W.. Microdroplet-based one-step RT-PCR for ultrahigh throughput single-cell multiplex gene expression analysis and rare cell detection. Sci. Rep. 2021; 11:6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eddington D.T., VanInsberghe M., Zahn H., White A.K., Petriv O.I., Hansen C.L.. Highly multiplexed single-cell quantitative PCR. PLoS One. 2018; 13:e0191601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esumi S., Kaneko R., Kawamura Y., Yagi T.. Split single-cell RT-PCR analysis of purkinje cells. Nat. Protoc. 2006; 1:2143–2151. [DOI] [PubMed] [Google Scholar]
- 27. Bengtsson M., Hemberg M., Rorsman P., Stahlberg A.. Quantification of mRNA in single cells and modelling of RT-qPCR induced noise. BMC Mol. Biol. 2008; 9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y., Thompson H., Hemphill C., Hong F., Forrester J., Johnson R.H., Zhang W., Meldrum D.R.. An improved one-tube RT-PCR protocol for analyzing single-cell gene expression in individual mammalian cells. Anal. Bioanal. Chem. 2010; 397:1853–1859. [DOI] [PubMed] [Google Scholar]
- 29. Gaynor E.M., Mirsky M.L., Lewin H.A.. Use of flow cytometry and RT-PCR for detecting gene expression by single cells. BioTechniques. 1996; 21:286–291. [DOI] [PubMed] [Google Scholar]
- 30. Tiller T., Busse C.E., Wardemann H.. Cloning and expression of murine ig genes from single b cells. J. Immunol. Methods. 2009; 350:183–193. [DOI] [PubMed] [Google Scholar]
- 31. Ho V., Yeo S.Y., Kunasegaran K., De Silva D., Tarulli G.A., Voorhoeve P.M., Pietersen A.M.. Expression analysis of rare cellular subsets: direct RT-PCR on limited cell numbers obtained by FACS or soft agar assays. BioTechniques. 2013; 54:208–212. [DOI] [PubMed] [Google Scholar]
- 32. Saadatpour A., Guo G., Orkin S.H., Yuan G.C.. Characterizing heterogeneity in leukemic cells using single-cell gene expression analysis. Genome Biol. 2014; 15:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ludwig J., Huber A.K., Bartsch I., Busse C.E., Wardemann H.. High-throughput single-cell sequencing of paired TCRα and TCRβ genes for the direct expression-cloning and functional analysis of murine T-cell receptors. Eur. J. Immunol. 2019; 49:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Auld D.S.P.D., Coassin P.B.S., Coussens N.P.P.D., Hensley P., Klumpp-Thomas C., Michael S., Sittampalam G.S.P.D., Trask O.B.S., Wagner B.K.P.D., Weidner J.R.P.D.et al.. Markossian S., Grossman A., Brimacombe K., Arkin M., Auld D., Austin C.P., Baell J., Chung T.D.Y., Coussens N.P., Dahlin J.L.et al.. Assay Guidance Manual. 2004; Bethesda (MD). [Google Scholar]
- 35. Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W.. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015; 161:1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J.et al.. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017; 8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matula K., Rivello F., Huck W.T.S.. Single-Cell analysis using droplet microfluidics. Adv Biosyst. 2020; 4:e1900188. [DOI] [PubMed] [Google Scholar]
- 38. Arrigucci R., Bushkin Y., Radford F., Lakehal K., Vir P., Pine R., Martin D., Sugarman J., Zhao Y.L., Yap G.S.et al.. FISH-Flow, a protocol for the concurrent detection of mRNA and protein in single cells using fluorescence in situ hybridization and flow cytometry. Nat. Protoc. 2017; 12:1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hummer D., Kurth F., Naredi-Rainer N., Dittrich P.S.. Single cells in confined volumes: microchambers and microdroplets. Lab Chip. 2016; 16:447–458. [DOI] [PubMed] [Google Scholar]
- 40. Caen O., Lu H., Nizard P., Taly V.. Microfluidics as a strategic player to decipher single-cell omics. Trends Biotechnol. 2017; 35:713–727. [DOI] [PubMed] [Google Scholar]
- 41. White A.K., VanInsberghe M., Petriv O.I., Hamidi M., Sikorski D., Marra M.A., Piret J., Aparicio S., Hansen C.L.. High-throughput microfluidic single-cell RT-qPCR. Proc. Natl. Acad. Sci. USA. 2011; 108:13999–14004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fung C.W., Chan S.N., Wu A.R.. Microfluidic single-cell analysis—toward integration and total on-chip analysis. Biomicrofluidics. 2020; 14:021502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qian J.Y., Hou C.W., Li X.J., Jin Z.J.. Actuation mechanism of microvalves: a review. Micromachines (Basel). 2020; 11:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo M.T., Rotem A., Heyman J.A., Weitz D.A.. Droplet microfluidics for high-throughput biological assays. Lab Chip. 2012; 12:2146–2155. [DOI] [PubMed] [Google Scholar]
- 45. Mazutis L., Gilbert J., Ung W.L., Weitz D.A., Griffiths A.D., Heyman J.A.. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013; 8:870–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lienemann P.S., Rossow T., Mao A.S., Vallmajo-Martin Q., Ehrbar M., Mooney D.J.. Single cell-laden protease-sensitive microniches for long-term culture in 3D. Lab Chip. 2017; 17:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fornell A., Pohlit H., Shi Q., Tenje M.. Acoustic focusing of beads and cells in hydrogel droplets. Sci. Rep. 2021; 11:7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leonaviciene G., Leonavicius K., Meskys R., Mazutis L.. Multi-step processing of single cells using semi-permeable capsules. Lab Chip. 2020; 20:4052–4062. [DOI] [PubMed] [Google Scholar]
- 49. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 50. Yao C.J., Works K., Romagnoli P.A., Austin G.E.. Effects of overexpression of HBP1 upon growth and differentiation of leukemic myeloid cells. Leukemia. 2005; 19:1958–1968. [DOI] [PubMed] [Google Scholar]
- 51. Hietakangas V., Poukkula M., Heiskanen K.M., Karvinen J.T., Sistonen L., Eriksson J.E.. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol. Cell. Biol. 2003; 23:1278–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Raj A., Peskin C.S., Tranchina D., Vargas D.Y., Tyagi S.. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006; 4:e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhong J.F., Chen Y., Marcus J.S., Scherer A., Quake S.R., Taylor C.R., Weiner L.P.. A microfluidic processor for gene expression profiling of single human embryonic stem cells. Lab Chip. 2008; 8:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwabe A., Rybakova K.N., Bruggeman F.J.. Transcription stochasticity of complex gene regulation models. Biophys. J. 2012; 103:1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gabert J., Beillard E., van der Velden V.H.J., Bi W., Grimwade D., Pallisgaard N., Barbany G., Cazzaniga G., Cayuela J.M., Cavé H.et al.. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – a europe against cancer program. Leukemia. 2003; 17:2318–2357. [DOI] [PubMed] [Google Scholar]
- 56. van Dongen J.J., Macintyre E.A., Gabert J.A., Delabesse E., Rossi V., Saglio G., Gottardi E., Rambaldi A., Dotti G., Griesinger F.et al.. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 concerted action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999; 13:1901–1928. [DOI] [PubMed] [Google Scholar]
- 57. Semrau S., Crosetto N., Bienko M., Boni M., Bernasconi P., Chiarle R., van Oudenaarden A.. FuseFISH: robust detection of transcribed gene fusions in single cells. Cell Rep. 2014; 6:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M.et al.. Highly parallel Genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015; 161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Galen P., Hovestadt V., Wadsworth Ii M.H., Hughes T.K., Griffin G.K., Battaglia S., Verga J.A., Stephansky J., Pastika T.J., Lombardi Story J.et al.. Single-Cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019; 176:1265–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gohil S.H., Iorgulescu J.B., Braun D.A., Keskin D.B., Livak K.J.. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021; 18:244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Freeman S.D., Virgo P., Couzens S., Grimwade D., Russell N., Hills R.K., Burnett A.K.. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J. Clin. Oncol. 2013; 31:4123–4131. [DOI] [PubMed] [Google Scholar]
- 62. Döhner H., Estey E., Grimwade D., Amadori S., Appelbaum F.R., Büchner T., Dombret H., Ebert B.L., Fenaux P., Larson R.A.et al.. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129:424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mercer T.R., Clark M.B., Crawford J., Brunck M.E., Gerhardt D.J., Taft R.J., Nielsen L.K., Dinger M.E., Mattick J.S.. Targeted sequencing for gene discovery and quantification using RNA captureseq. Nat. Protoc. 2014; 9:989–1009. [DOI] [PubMed] [Google Scholar]
- 64. Hong X., Sullivan R.J., Kalinich M., Kwan T.T., Giobbie-Hurder A., Pan S., LiCausi J.A., Milner J.D., Nieman L.T., Wittner B.S.et al.. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:2467–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kwan T.T., Bardia A., Spring L.M., Giobbie-Hurder A., Kalinich M., Dubash T., Sundaresan T., Hong X., LiCausi J.A., Ho U.et al.. A digital RNA signature of circulating tumor cells predicting early therapeutic response in localized and metastatic breast cancer. Cancer Discov. 2018; 8:1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Miyamoto D.T., Lee R.J., Kalinich M., LiCausi J.A., Zheng Y., Chen T., Milner J.D., Emmons E., Ho U., Broderick K.et al.. An RNA-Based digital circulating tumor cell signature is predictive of drug response and early dissemination in prostate cancer. Cancer Discov. 2018; 8:288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Whale A.S., Huggett J.F., Tzonev S.. Fundamentals of multiplexing with digital PCR. Biomol. Detect. Quantif. 2016; 10:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pichon X., Lagha M., Mueller F., Bertrand E.. A growing toolbox to image gene expression in single cells: sensitive approaches for demanding challenges. Mol. Cell. 2018; 71:468–480. [DOI] [PubMed] [Google Scholar]
- 69. Gegevicius E., Goda K., Mazutis L.. Ren C., Lee A.. Droplet gene analysis – digital PCR. Droplet Microfluidics. 2020; Royal Society of Chemistry; 89–121. [Google Scholar]
- 70. Vandenbroucke I.I., Vandesompele J., Paepe A.D., Messiaen L.. Quantification of splice variants using real-time PCR. Nucleic Acids Res. 2001; 29:E68–e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weisser M., Kern W., Rauhut S., Schoch C., Hiddemann W., Haferlach T., Schnittger S.. Prognostic impact of RT-PCR-based quantification of WT1 gene expression during MRD monitoring of acute myeloid leukemia. Leukemia. 2005; 19:1416–1423. [DOI] [PubMed] [Google Scholar]
- 72. Gorello P., Cazzaniga G., Alberti F., Dell’Oro M.G., Gottardi E., Specchia G., Roti G., Rosati R., Martelli M.F., Diverio D.et al.. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia. 2006; 20:1103–1108. [DOI] [PubMed] [Google Scholar]
- 73. Chen X., Wang F., Zhang Y., Ma X., Cao P., Yuan L., Wang L., Chen J., Zhou X., Wu Q.et al.. Fusion gene map of acute leukemia revealed by transcriptome sequencing of a consecutive cohort of 1000 cases in a single center. Blood Cancer J. 2021; 11:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cheng D.T., Mitchell T.N., Zehir A., Shah R.H., Benayed R., Syed A., Chandramohan R., Liu Z.Y., Won H.H., Scott S.N.et al.. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015; 17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhao Y., Murciano-Goroff Y.R., Xue J.Y., Ang A., Lucas J., Mai T.T., Da Cruz Paula A.F., Saiki A.Y., Mohn D., Achanta P.et al.. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021; 599:679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Martincorena I., Campbell P.J.. Somatic mutation in cancer and normal cells. Science. 2015; 349:1483–1489. [DOI] [PubMed] [Google Scholar]
- 77. Rajan S., Kierny M.R., Mercer A., Wu J., Tovchigrechko A., Wu H., Dall Acqua W.F., Xiao X., Chowdhury P.S.. Recombinant human b cell repertoires enable screening for rare, specific, and natively paired antibodies. Commun. Biol. 2018; 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Seah Y.F.S., Hu H., Merten C.A.. Microfluidic single-cell technology in immunology and antibody screening. Mol. Aspects Med. 2018; 59:47–61. [DOI] [PubMed] [Google Scholar]
- 79. Vitak S.A., Torkenczy K.A., Rosenkrantz J.L., Fields A.J., Christiansen L., Wong M.H., Carbone L., Steemers F.J., Adey A.. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods. 2017; 14:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rosenberg A.B., Roco C.M., Muscat R.A., Kuchina A., Sample P., Yao Z., Graybuck L.T., Peeler D.J., Mukherjee S., Chen W.et al.. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018; 360:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data and analysis scripts have been deposited on the Github repository: https://github.com/L-Greta/RNA_Cytometry_Data.