


Abstract
The emergence of drug-resistant Mycobacterium tuberculosis strains highlights the need to discover anti-tuberculosis drugs with novel mechanisms of action. Here we discovered a mycobactericidal strategy based on the prodrug activation of selected chemical derivatives classified as nitronaphthofurans (nNFs) mediated by the coordinated action of the sigH and mrx2 genes. The transcription factor SigH is a key regulator of an extensive transcriptional network that responds to oxidative, nitrosative, and heat stresses in M. tuberculosis. The nNF action induced the SigH stress response which in turn induced the mrx2 overexpression. The nitroreductase Mrx2 was found to activate nNF prodrugs, killing replicating, non-replicating and intracellular forms of M. tuberculosis. Analysis of SigH DNA sequences obtained from spontaneous nNF-resistant M. tuberculosis mutants suggests disruption of SigH binding to the mrx2 promoter site and/or RNA polymerase core, likely promoting the observed loss of transcriptional control over Mrx2. Mutations found in mrx2 lead to structural defects in the thioredoxin fold of the Mrx2 protein, significantly impairing the activity of the Mrx2 enzyme against nNFs. Altogether, our work brings out the SigH/Mrx2 stress response pathway as a promising target for future drug discovery programs.
INTRODUCTION
Despite the centuries that humans have been suffering from tuberculosis (TB), Mycobacterium tuberculosis remains nowadays one of the most infectious killers worldwide due to a single infectious agent. In 2019, an estimated 10 million people fell ill with TB and 1.5 million people died due to the disease (Global tuberculosis report 2020. World Health Organization. https://apps.who.int/iris/handle/10665/336069). Both high incidence and high mortality due to TB are disproportionately registered in low-income countries, where poverty, overcrowding, malnutrition and a high HIV incidence are the main causes of the fatality. In addition, the emergence and spread of multidrug-resistant (MDR) and extensively drug-resistant (XDR) M. tuberculosis strains is hampering TB control worldwide and worsening the TB scenario for the foreseeable future. In fact, MDR and XDR forms of TB account for one-third of all worldwide deaths attributable to antimicrobial resistant pathogens (https://www.tballiance.org/why-new-tb-drugs/antimicrobial-resistance).
The TB global situation is exacerbated by the fact that an estimated two billion people may act as a persistent reservoir of TB (latent TB) (1), of which 10–15% are predicted to develop an active disease in their lifetime. Latent TB is generally assumed to be maintained by low metabolic, non-replicating or totally inactive M. tuberculosis forms, a phenotype that is considered to represent the refined and adaptive mechanism that M. tuberculosis developed to persist in the host (2–4). However, the current definition of latent TB covers a range of situations, from infections that have been totally cleared to others in which actively replicating bacteria are emerging in the absence of clinical symptoms (5,6). Similarly, active TB may be better understood as a heterogeneous and complex infection in which different M. tuberculosis phenotypes are represented, including non-replicating bacteria (7).
It is evident that new antibiotics to treat TB are urgently needed, and that they should preferably target new molecular pathways in order to avoid cross-resistance with conventional TB drugs. The majority of current anti-tuberculosis drugs act on molecular targets in actively replicating M. tuberculosis and are largely ineffective against non-replicating forms of the bacteria (3,8), which determine the need for extended multidrug treatment schedules in order to successfully clearance the infection—4 to 6 months in sensitive TB cases and up to 24 months in MDR/XDR TB cases (9,10). It is therefore of paramount importance to develop effective drugs against non-replicating M. tuberculosis. In this context, nitro heterocyclic compounds have demonstrated promising activity against both replicating and non-replicating M. tuberculosis in several studies (11–17), thus representing attractive compounds for the development of future anti-tuberculosis drugs. The nitroimidazoles OPC-67683 (Delamanid; (18)) and PA-824 (Pretomanid; (19)), both anti-TB drugs recently approved by FDA and EMA, were developed after extensive chemical structural optimization, providing acceptable administration safety along with high efficacy against TB. Both pretomanid and delamanid are pro-drugs that require bioactivation by the M. tuberculosis deazaflavin-dependent nitroreductase (Ddn; (20–23)), leading to the release of nitric oxide (NO) which is the main factor responsible for the killing of non-replicating M. tuberculosis bacteria by a respiratory poisoning mechanism. At least other four additional nitroreductases have been described in M. tuberculosis: Rv2032 (24,25), Rv3131 (25,26), Rv3127 (25) and Rv2466c (27–30). They are believed to be part of the anti-oxidative and anti-nitrosative arsenal that M. tuberculosis exhibits to survive the host killing mechanisms. Many such defence and repair mechanisms in M. tuberculosis are under a stringent gene expression control mediated by transcription factors, alternative sigma factors, and two component systems. These are in charge of translating the environmental stress signals into transcriptional changes that allow the pathogen to respond with necessary physiological adaptations (31–37).
In this study, we identify a series of nitronaphthofuran (nNF) derivatives that showed bactericidal activity against both actively replicating and non-replicating M. tuberculosis, and also a potent killing of the bacteria inside macrophages. The mechanism of action is dependent on the reduction of the nitroaromatic moiety. Spontaneous M. tuberculosis resistant mutants against nNFs were found to carry mutations in the rv2466c (mrx2) gene, which codifies for a recently annotated Mycoredoxin (Mrx2) described as a nitroreductase (27–30). Interestingly, spontaneous resistant mutants with an intact mrx2 gene, were found to carry mutations in the sigH gene. SigH is a sigma factor and a key regulator of the transcriptional response to oxidative, nitrosative, and heat stresses in M. tuberculosis. We show how SigH directly modulates the levels of Mrx2 expression and get induced by the nNF treatment. Our findings provide insights into the molecular mechanism underlying the anti-tuberculosis activity of nNFs and a promising basis for future drug development programs.
MATERIALS AND METHODS
Selection of nNF derivatives
A high-throughput whole-cell screening of a chemical library composed of 35 860 compounds was performed on a Freedom EVO platform (Tecan), using M. aurum ATCC23366 as a surrogate of M. tuberculosis (38). A nNF compound from the Institut Curie chemical library (part of the ‘Chimiothèque Nationale’ (39)) was identified as a potent anti-tuberculosis hit (nNF-C18; Figure 1 and Supplementary Table S1) and nineteen additional derivatives belonging to the same chemical library were selected (nNF-C1 to nNF-C20; Figure 1 and Supplementary Table S1). In addition, 13 desnitro analogues were also chosen from the same library (derivatives C1B to C13B; Supplementary Table S2). nNF-C1- nNF-C20 and C1B-C13B comprised the chemical set of compounds for analysis in our study. The chemical synthesis of all chemical derivatives was previously described (40–48). Compounds were dissolved in DMSO, protected from light, and stored at –20°C until use.
Figure 1.

Chemical structures of the nNF derivatives. The most active derivatives nNF-C2 and nNF-C4 displayed MIC90 values against actively-replicating (AR) M. tuberculosis of 0.062 μg/ml and 0.125 μg/ml respectively, and MBC90 values of 0.5 μg/ml against non-replicating (NR) M. tuberculosis H37Rv, respectively.
Nuclear magnetic resonance (NMR) determination and spectroscopic analysis
Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker Avance 300 (300 MHz for 1H; 75 MHz for 13C), using TMS as internal standard. Deuterated DMSO-d6 was purchased from SDS. Chemical shifts are given in parts per million (ppm) (δ relative to the residual solvent peak for 1H and 13C). The following abbreviations are used: singlet (s), doublet (d), triplet (t) and multiplet (m). The purity was determined by high performance liquid chromatography (HPLC) using an Alliance Waters system [Alliance Waters 2695 (pump) and Waters 2998 (Photodiode Array detector)] with a column Waters XBridge C-18, 3.5 μm particle size (3.0 mm × 100 mm). Low-resolution mass spectrometry (ESI-MS) was recorded on a micromass ZQ 2000 (Waters). High-resolution mass spectrometry (ESI-MS) was performed by the I.C.S.N. (Institut de Chimie des Substances Naturelles, Gif-sur-Yvette). The chemical formula and molecular weight of each derivative included in the study are depicted in Figure 1 and Supplementary Tables S1 and S2: 2,7-dinitronaphtho[1,2-b:6,5-b']difuran (nNF-C1)1H NMR (300 MHz, DMSO-d6) δ 9.00 (s, 1H), 8.57 (t, J = 9.7 Hz, 2H), 8.36 (s, 1H), 8.18 (dd, J = 18.4, 8.9 Hz, 2H); HPLC purity = 100%; LRMS (ESI-MS) m/z = 297.32 [M-H]−. 2,8-Dinitronaphtho[2,1-b:7,6-b']difuran (nNF-C2)1H NMR (300 MHz, CDCl3) δ 8.49 (s, 1H), 8.38 (s, 1H), 8.22 (s, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.87 (s, 1H), 7.76 (d, J = 9.3 Hz, 1H); HPLC purity = 100%; LRMS (ESI-MS) m/z = 297.22 [M-H]−. 2,7-Dinitronaphtho[2,1-b:6,5-b']difuran (nNF-C3)1H NMR (300 MHz, DMSO-d6) δ 9.04 (s, 2H), 8.83 (d, J = 9.1 Hz, 2H), 8.27 (d, J = 9.0 Hz, 2H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 299.21 [M+H]+. Methyl 2-nitronaphtho[2,1-b]furan-7-carboxylate (nNF-C4)1H NMR (300 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.85 (s, 1H), 8.65 (d, J = 8.4 Hz, 1H), 8.45 (d, J = 9.2 Hz, 1H), 8.23 (d, J = 8.6 Hz, 1H), 8.08 (d, J = 9.2 Hz, 1H), 3.95 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 241.07 [M-NO]+. 2-Nitronaphtho[2,1-b]furan-7-carbonitrile (nNF-C5)1H NMR (300 MHz, DMSO-d6) δ 9.00 (s, 1H), 8.79 (s, 1H), 8.71 (d, J = 8.5 Hz, 1H), 8.33 (d, J = 9.2 Hz, 1H), 8.19–8.05 (m, 2H); HPLC purity = 100%; LRMS (ESI-MS) m/z = 237.29 [M-H]−. 7-Bromo-2-nitronaphtho[2,1-b]furan (nNF-C6)1H NMR (300 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.47 (dd, J = 13.3, 5.3 Hz, 2H), 8.21 (d, J = 9.1 Hz, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.92 (dd, J = 8.7, 2.0 Hz, 1H); HPLC purity = 94%; LRMS (ESI-MS) m/z = 305.98 [M-H]−. 7-Methoxy-1-methyl-2-nitronaphtho[2,1-b]furan (nNF-C7)1H NMR (300 MHz, CDCl3) δ 8.35 (d, J = 8.8 Hz, 1H), 7.91 (d, J = 9.1 Hz, 1H), 7.63 (d, J = 9.1 Hz, 1H), 7.36 (d, J = 8.8 Hz, 2H), 3.97 (s, 3H), 3.12 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 258.00 [M+H]+. 7,8-Dimethoxy-2-nitronaphtho[2,1-b]furan (nNF-C8)1H NMR (300 MHz, DMSO-d6) δ 8.88 (s, 1H), 8.11–7.94 (m, 2H), 7.72 (d, J = 9.4 Hz, 1H), 7.56 (s, 1H), 3.98 (s, 3H), 3.91 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 273.03 [M+H]+. 2-Nitro-1-undecylnaphtho[2,1-b]furan (nNF-C9)1H NMR (300 MHz, CDCl3) δ 8.35 (d, J = 7.2 Hz, 1H), 8.08–7.55 (m, 5H), 3.64–3.48 (m, 2H), 1.91–1.83 (m, 2H), 1.67–1.56 (m, 2H), 1.41–1.26 (m, 14H), 0.88 (t, J = 6.6 Hz, 3H); HPLC purity = 78%. 1,7-Dimethoxy-2-nitronaphtho[2,1-b]furan (nNF-C10)1H NMR (300 MHz, CDCl3) δ 8.52 (d, J = 9.0 Hz, 1H), 7.92 (d, J = 9.2 Hz, 1H), 7.56 (d, J = 9.1 Hz, 1H), 7.34 (dd, J = 16.8, 5.7 Hz, 2H), 4.38 (s, 3H), 3.96 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 274.03 [M + H]+. 1-Butyl-2-nitronaphtho[2,1-b]furan (nNF-C11)1H NMR (300 MHz, CDCl3) δ 8.52 (d, J = 9 Hz, 1H), 8.02–7.99 (m, 2H), 7.73–7.61 (m, 3H), 4.50 (q, J = 6 Hz, 1H), 1.66 (s, 3H), 1.64 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 256.01 [M+H]+. 1-Butyl-2-nitronaphtho[2,1-b]furan (nNF-C12)1H NMR (300 MHz, CDCl3) δ 8.35 (d, J = 8.3 Hz, 1H), 8.17–7.83 (m, 2H), 7.81–7.47 (m, 3H), 3.76–3.27 (m, 2H), 2.01–1.75 (m, 2H), 1.62 (dd, J = 14.9, 7.4 Hz, 2H), 1.04 (t, J = 7.3 Hz, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 270.14 [M+H]+. 1-Methoxy-2-nitronaphtho[2,1-b]furan (nNF-C13)1H NMR (300 MHz, CDCl3) δ 8.63 (d, 1H), 8.03–7.97 (m, 2H), 7.75–7.70 (m, 1H), 7.65–7.57 (m, 2H), 4.40 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 244.06 [M+H]+. 1-Methyl-2-nitronaphtho[2,1-b]furan (nNF-C14)1H NMR (300 MHz, CDCl3) δ 8.46 (d, J = 8.3 Hz, 1H), 8.01 (d, J = 9.3 Hz, 2H), 7.77–7.40 (m, 3H), 3.16 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 228.13 [M+H]+. 2-Nitronaphtho[2,1-b]furan-1-amine (nNF-C15)1H NMR (300 MHz, DMSO-d6) δ 8.74 (d, J = 8.2 Hz, 13C H), 8.27 (d, J = 9.1 Hz, 1H), 8.15 (d, J = 8.1 Hz, 1H), 7.78 (dd, J = 8.0, 4.3 Hz, 2H), 7.66 (t, J = 7.4 Hz, 1H) 13C NMR (75, MHz, DMSO-d6) δ 152.42, 139.72, 135.49, 130.43, 130.26, 129.19, 128.24, 126.24, 123.63, 113.26; HPLC purity = 100%; LRMS (ESI-MS) m/z = 229.12 [M+H]+. HRMS : m/z calculated for C12 H8 N2 O3 + H+ [M+H]+: 229.0608. Found: 229.0253 and m/z calculated for C12 H7 N2 O3 [M-H]+: 227.0457. Found: 227.0453. 7-Methoxy-2-nitronaphtho[2,1-b]furan (nNF-C16)1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 10.2 Hz, 2H), 7.92 (d, J = 9.1 Hz, 1H), 7.66 (d, J = 9.1 Hz, 1H), 7.37 (dd, J = 8.8, 2.5 Hz, 1H), 7.32 (d, J = 2.3 Hz, 1H), 3.97 (s, 3H); HPLC purity = 100%. 1-Ethyl-2-nitronaphtho[2,1-b]furan (nNF-C17)1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 8.3 Hz, 1H), 8.11–7.93 (m, 2H), 7.82–7.49 (m, 3H), 3.60 (q, J = 7.5 Hz, 2H), 1.52 (t, J = 7.5 Hz, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 242.17 [M+H]+. 9-Bromo-8-methoxy-2-nitronaphtho[2,1-b]furan (nNF-C18)1H NMR (300 MHz, DMSO-d6) δ 9.07 (d, J = 0.7 Hz, 1H), 8.25 (dd, J = 9.1, 3.2 Hz, 2H), 7.88 (d, J = 9.1 Hz, 1H), 7.63 (d, J = 9.0 Hz, 1H), 4.05 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 325.20 [M+H]+. 5-Methoxy-2-nitronaphtho[2,1-b]furan (nNF-C19)1H NMR (300 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.45 (d, J = 7.9 Hz, 1H), 8.28 (d, J = 8.2 Hz, 1H), 7.78 (t, J = 7.0 Hz, 1H), 7.66 (dd, J = 11.3, 4.1 Hz, 1H), 7.48 (s, 1H), 4.10 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 244.06 [M+H]+. Ethyl 2-({2-nitronaphtho[2,1-b]furan-8-yl}oxy)acetate (nNF-C20)1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 8.11 (dd, J = 24.3, 9.1 Hz, 3H), 7.77 (d, J = 9.1 Hz, 1H), 7.33 (dd, J = 8.9, 2.5 Hz, 1H), 5.00 (s, 2H), 4.21 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 315.04 [M+H]+. Diethyl naphtho[2,1-b:7,6-b']difuran-2,8-dicarboxylate (C1B)1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.58 (d, J = 3.1 Hz, 2H), 8.21 (d, J = 9.3 Hz, 1H), 7.98 (s, 1H), 7.87 (d, J = 9.5 Hz, 1H), 4.41 (q, J = 7.1 Hz, 4H), 1.38 (t, J = 6.9 Hz, 6H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 353.07 [M + H]+. 2-(Ethoxycarbonyl)naphtho[1,2-b:6,5-b']difuran-7-carboxylic acid (C2B)1H NMR (300 MHz, DMSO-d6) δ 8.41–8.34 (m, 2H), 8.06–7.98 (m, 2H), 7.93 (s, 1H), 7.82 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.0 Hz, 3H); HPLC purity = 56%, LRMS (ESI-MS) m/z = 323.11 [M-H]−. Ethyl naphtho[1,2-b:6,5-b']difuran-2-carboxylate (C3B)1H NMR (300 MHz, CDCl3) δ 8.40 (d, J = 9.2 Hz, 1H), 8.16–7.90 (m, 2H), 7.88–7.71 (m, 3H), 6.96 (d, J = 2.1 Hz, 1H), 4.50 (q, J = 7.1 Hz, 2H), 1.47 (t, J = 7.1 Hz, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 280.99 [M+H]+. Naphtho[2,1-b:7,6-b']difuran-2,8-dicarboxylic acid (C4B)1H NMR (300 MHz, DMSO-d6) δ 8.76 (s, 1H), 8.52 (d, J = 14.7 Hz, 2H), 8.17 (d, J = 9.3 Hz, 1H), 7.96–7.62 (m, 2H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 295.13 [M-H]−. 1-{Naphtho[2,1-b]furan-2-yl}ethan-1-one (C5B)1H NMR (300 MHz, CDCl3) δ 8.18 (d, J = 8.1 Hz, 1H), 8.03–7.85 (m, 3H), 7.75–7.61 (m, 2H), 7.57 (d, J = 8.0 Hz, 1H), 2.68 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 211.20 [M + H]+. Naphtho[2,1-b]furan-8-yl acetate (C6B)1H NMR (300 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 1H), 7.84 (d, J = 2.2 Hz, 1H), 7.79–7.60 (m, 3H), 7.26–7.15 (m, 2H), 2.39 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 227.13 [M+H]+. 7-Methoxynaphtho[2,1-b]furan-2-carbonitrile (C7B)1H NMR (300 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.28 (d, J = 8.9 Hz, 1H), 8.03 (d, J = 9.1 Hz, 1H), 7.85 (d, J = 9.1 Hz, 1H), 7.58 (d, J = 2.4 Hz, 1H), 7.39 (dd, J = 8.9, 2.6 Hz, 1H), 3.91 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 222.25 [M-H]−. Naphtho[2,1-b]furan (C8B)1H NMR (300 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.78 (d, J = 2.0 Hz, 1H), 7.71 (dd, J = 19.0, 9.0 Hz, 2H), 7.64–7.55 (m, 1H), 7.54–7.43 (m, 1H), 7.28 (d, J = 1.3 Hz, 1H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 169.28 [M + H]+. 1-{7-Methoxy-1-methylnaphtho[2,1-b]furan-2-yl}ethan-1-one (C9B)1H NMR (300 MHz, DMSO-d6) δ 8.38 (d, J = 9.1 Hz, 1H), 7.99 (d, J = 9.1 Hz, 1H), 7.80 (d, J = 9.1 Hz, 1H), 7.59 (d, J = 2.6 Hz, 1H), 7.36 (dd, J = 9.1, 2.6 Hz, 1H), 3.90 (s, 3H), 2.95 (s, 3H), 2.60 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 255.11 [M+H]+. Methyl 8-methoxynaphtho[2,1-b]furan-2-carboxylate (C10B)1H NMR (300 MHz, CDCl3) δ 8.00 (d, J = 0.7 Hz, 1H), 7.83 (dd, J = 16.5, 9.0 Hz, 2H), 7.55 (d, J = 9.0 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 7.18 (dd, J = 8.9, 2.5 Hz, 1H), 4.00 (d, J = 6.1 Hz, 6H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 257.10 [M + H]+. 7-Methoxy-1-methylnaphtho[2,1-b]furan-2-carboxylic acid (C11B)1H NMR (300 MHz, DMSO-d6) δ 8.37 (d, J = 9.1 Hz, 1H), 7.94 (d, J = 9.1 Hz, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.57 (d, J = 2.6 Hz, 1H), 7.35 (dd, J = 9.0, 2.7 Hz, 1H), 3.90 (s, 3H), 2.94 (s, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 257.10 [M+H]+. 6-Bromo-7-methoxynaphtho[2,1-b]furan-2-carboxylic acid (C12B)1H NMR (300 MHz, DMSO-d6) δ 8.54 (t, J = 8.5 Hz, 1H), 8.38 (s, 1H), 8.20 (d, J = 9.7 Hz, 1H), 7.98 (t, J = 8.2 Hz, 1H), 7.67 (dd, J = 8.9, 5.4 Hz, 1H), 4.02 (s, 3H), 3.93 (s, 1H); HPLC purity = 54%, LRMS (ESI-MS) m/z = 318.92 [M-H]−. Ethyl 6-bromo-7-methoxynaphtho[2,1-b]furan-2-carboxylate (C13B)1H NMR (300 MHz, DMSO-d6) δ 8.63–8.44 (m, 2H), 8.23 (d, J = 9.4 Hz, 1H), 8.00 (d, J = 10.0 Hz, 1H), 7.68 (d, J = 9.0 Hz, 1H), 4.40 (d, J = 7.1 Hz, 2H), 4.02 (s, 2H), 1.37 (t, J = 7.1 Hz, 3H); HPLC purity = 100%, LRMS (ESI-MS) m/z = 348.99 + 350.98 [M + H]+.
Bacterial strains and growth conditions
Mycobacteria strains used in this study included: M. tuberculosis H37Rv (ATCC 27294),M. tuberculosis H37Rv sigH null mutant (ST49; (49)), M. tuberculosis H37Rv mrx2 null mutant (B461c; (30)) a clinical Beijing M. tuberculosis strain (GC1237) which was isolated from a patient who was infected in the reported TB outbreak in Gran Canaria Islands (Spain) (50), two different multidrug resistant M. tuberculosis clinical strains isolated from TB patients at the Padova University Hospital (Padova, Italy), Mycobacterium abscessus ATCC19977, Mycobacterium aurum ATCC23366,Mycobacterium avium subsp. hominissuis ATCC 25291, Mycobacterium bovis BCG 1173 P2, Mycobacterium marinum ATCC BAA-535 and Mycobacteriumsmegmatis mc2155. All mycobacterial strains were grown in Middlebrook 7H9 broth (Difco) supplemented with 10% albumin-dextrose-catalase (ADC; Difco), 0.2% glycerol and 0.05% Tyloxapol (Sigma-Aldrich). When necessary, Middlebrook 7H10 agar (Difco) plates supplemented with 10% oleic-albumin-dextrose-catalase (OADC; Difco) were used. Supplemented Middlebrook media is referred to as complete Middlebrook 7H9 or 7H10 media in the text. All mycobacteria strains were grown at 37°C, with the exception of M. marinum which was incubated at 32°C. Other bacterial clinical species as Escherichia coli, Enterobacter aerogenes, and Enterobacter cloacae, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus and Salmonella enterica were obtained from the biobank at the Bichat-Claude Bernard Hospital and growth at 37°C in Lysogeny broth (LB; Gibco) and/or Mueller Hinton broth (Sigma-Aldrich).
DH5-alpha electrocompetent E. coli cells were used for cloning procedures and grown in LB broth and/or LB agar (Gibco). They were prepared according to the protocol at the New England Biolabs website. When required, the antibiotics kanamycin or hygromycin B at concentrations of 25 and 100 μg/ml, respectively, when working with mycobacteria, and 50 and 200 μg/ml, respectively, when working with E. coli, were added to the growth medium.
Minimal inhibitory concentration (MIC) determination
The Resazurin Microtiter Assay (REMA) protocol used for Drug Susceptibility Testing (DST) was carried out as previously described (51) with some modifications. The microdilution test was performed in 96-well round-bottomed microtiter plates (Nunc, USA) using (i) Middlebrook 7H9 broth supplemented with 10% OADC and 0.1% Bacto Casitone (Difco) for all mycobacteria strains, and (ii) Muller Hinton and/or Lysogeny broth for the rest of the non-mycobacteria species. Two-fold dilutions were assayed starting from a final drug concentration of 32 μg/ml in a final volume of 200 μl of media. The final concentration of DMSO was kept below 1%. The bacterial inoculum was adjusted to 5 × 104 bacteria in the case of M. tuberculosis H37Rv ATCC 27294, M. tuberculosis Beijing GC1237 and M. bovis BCG 1173 P2, and 1–5 × 103 bacteria for the other bacterial species assayed. Inoculated plates were incubated in sealed plastic bags at 37°C, except plates containing M. marinum ATCC BAA-535 that required an incubation temperature of 32°C. Positive and negative growth controls were included in every assay. Rifampicin (RIF) was included as a reference antibiotic. Resazurin (Sigma-Aldrich) was added (i) at day 7 after bacterial inoculation in the case of M. tuberculosis H37Rv ATCC 27294, M. tuberculosis Beijing GC1237 and M. bovis BCG 1173 P2, (ii) at day 3 for M. marinum ATCC BAA-535 and M. avium subsp. hominissuis ATCC 25291, (iii) after 16 hs for M. abscessus ATCC19977 and M. aurum ATCC23366, and (iv) after 4 hs for M. smegmatis mc2155 and the rest of non-mycobacteria species. The final concentration of resazurin in the plates was adjusted to 0.003%. Readouts were taken 24 h post-resazurin. Fluorescence was measured at an excitation wavelength of 535 nm and emission wavelength of 590 nm using a microplate reader (Synergy H1, BioTek). The MIC90 was defined as the lowest concentration at which the fluorescence value was ≤10% of the value for the untreated control.
Assessment of nNFs bactericidal activity in non-replicating M. tuberculosis H37Rv
Derivatives that showed bactericidal activity against actively replicating M. tuberculosis were also tested against non-replicating M. tuberculosis. Non-replicating M. tuberculosis H37Rv were obtained by nutrient starvation (52) and low oxygen concentration conditions. Briefly, cultures were grown in complete Middlebrook 7H9 broth to an OD600 = 0.8 and then centrifuged using 50 ml falcon tubes. The pellets were washed twice with 30 ml of phosphate buffer saline (PBS) and resuspended in 45 ml of the same buffer. The falcon tubes were sealed with parafilm and let starve in standing conditions at 37°C for 6 weeks. After the starvation period, a microdilution test was performed in 96 well-plates using PBS. Two-fold dilutions of selected nNFs derivatives were assayed starting from a final drug concentration of 32 μg/ml. Ten microliters from the starved non-replicating M. tuberculosis H37Rv culture at OD600 = 0.2, which corresponds to ca. 105 bacterial cells, was used as inoculum for the microdilution plate test. The microtiter plates were sealed with parafilm and incubated at 37°C for 12 days inside sealed plastic bags. Then, serial dilutions were plated onto 7H10 agar plates supplemented with OADC to enumerate viable bacteria. The initial inoculum was also submitted to serial dilutions and plated in order to enumerate initial viable bacteria. The concentrations at which the tested compounds killed 90% of the non-replicating M. tuberculosis H37Rv at the initial inocula were expressed as the average of the results obtained in two independent experiments. Rifampicin (RIF), Isoniazid (INH) and Pretomanid (PA-824) were included in the assay.
To discard the possibility of a carry-over effect in our assay, we performed the same experiment on the selected derivatives nNF-C2 and nNF-C4, using the Charcoal Agar Resazurin Assay (CARA) as previously described (53). PA-824, a known anti-tuberculosis compound with activity against non-replicating M. tuberculosis, and INH, an antibiotic with demonstrated lack of activity against non-replicating M. tuberculosis bacteria, were included as controls in the assay. Briefly, at day 12 of incubation of non-replicating M. tuberculosis H37Rv bacteria in the microdilution plate (as described above), 10 μl of a homogenized suspension of each well were spotted on the surface of 7H11-OADC-charcoal agar microplates. Plates were incubated at 37°C and 5% CO2 for 7 days inside of zip plastic bags. On day 7, 40 μl of PBS first and then 50 μl of Alamarblue reagent (Invitrogen) prepared at a final concentration of 5% Tween 80 were added to the wells and fluorescence readouts were obtained after 24 h. As performed for the MIC determination in the REMA assay, fluorescence was measured at an excitation wavelength of 535 nm and emission wavelength of 590 nm and the MBC90 for non-replicating M. tuberculosis was defined as the lowest concentration at which the fluorescence value was ≤ 10% of the value for the untreated control. Additionally, independent charcoal containing agar plates in which bacterial cultures were spotted after treatment, were incubated for 2–3 weeks and visual inspection of mycobacterial growth was carried out.
Minimal bactericidal concentration (MBC) determination
MBC was defined as the lowest concentration of a compound at which 99% of the bacteria in the starting inoculum was killed. Determinations were done in duplicate from the REMA plates used to determine the MIC for M. tuberculosis H37Rv. The bacterial content of three independent wells at the beginning of the assay (day 1) was plated onto complete Middlebrook 7H10 agar plates to enumerate the viable bacteria (colony forming units; CFU) at the starting point of the experiment. The corresponding 10-fold serial dilutions were performed in order to count 30–300 CFU per plate. After reading the REMA plates, the four consecutive growth-negative wells (blue wells) after the MIC were diluted using 7H9 broth and plated onto complete Middlebrook 7H10 agar plates, in order to enumerate CFU. 7H10 agar plates were incubated at 37°C, up to 5 weeks. In order to rule out the possibility of a carry-over effect, we performed the same experiment using 0.4% activated charcoal agar containing plates as regrowth agar substrate (53) as described for non-replicating M.tuberculosis. Two selected derivatives, nNF-C2 and nNF-C4, were chosen to be tested by the charcoal agar experimental approach.
Assessment of nNFs cytotoxicity
The cellular viability was evaluated using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega), which determines the number of viable cells based on the quantification of molecules of ATP as an indicator of metabolically active cells. The assay generates a ‘glow-type’ luminescent signal, produced by the luciferase reaction. Cytotoxicity was assessed in Vero (Kidney African green monkey; ATCC CCL-81) and HepG2 (Human liver; ATCC HB-8065) cell lines. Both cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2mM glutamine and penicillin-streptomycin 100 U/ml (Invitrogen). Cells were dislodged, centrifuged, resuspended in fresh medium and seeded in 96 well-plates at densities of 25 000 Vero cells, and 40 000 HepG2 cells per well. After 24 h of incubation, independent 96 well-plates were prepared to contain two-fold serial dilutions of the selected nNFs derivatives using 100 μg/ml as the starting maximum concentration. The medium of the sub-confluent monolayer of cells that were grown overnight was removed and 100 μl of fresh media containing the different concentrations of test compounds was added to the plates. Plates were incubated at 37°C in a humidified atmosphere containing 5% CO2 for 72 h. To carry on the cellular viability assay, 50 μl of the CellTiter-Glo reagent was added to each well in the plate resulting in cell lysis and generation of a luminescent signal proportional to the amount of ATP present that was recorded using a luminometer (Victor3 plate reader, PerkinElmer). Cells were also visually examined daily under a phase-contrast microscope to determine the minimum concentration of compound that induced alterations in the cell morphology. The toxicity was referred to as the 50% cytotoxic concentration (CC50), defined as the compound's concentration required for the reduction of cell viability by 50%. The Selectivity Index (SI) for each compound was calculated by dividing the CC50 value obtained in the cytotoxicity assay using Vero cells by the MIC value obtained in the REMA using replicating M. tuberculosis H37Rv.
Intracellular activity of nNFs in M. tuberculosis infected bone marrow-derived macrophages
Wild-type C57BL/6JRj (WT) mice were purchased from Janvier Labs. Bone marrow-derived macrophages (BMDM) were prepared as described previously (54). Briefly, bone marrow was flushed from the tibias and femurs of 11-week-old mice and red blood cells were removed by incubation in the RBC lysis buffer. Bone marrow cells were then incubated in RPMI-medium supplemented with 10% FCS, 30% L929 conditioned medium and 50 μM β-mercaptoethanol in non-tissue culture treated dishes at 37°C for 7 days with medium changes after 3 and 6 days. Differentiated BMDM were then detached by incubating the cell monolayer in Ca+/Mg2+-free PBS at 4°C and harvested by centrifugation. Cells were then maintained in RPMI medium supplemented with 10% FCS, 10% L929-conditioned medium and 50 μM β-mercaptoethanol. For the infection assay, BMDM were seeded in 48-well plates at a density of 2 × 105 cells/well and overnight incubated in a humidified atmosphere of 5% CO2 at 37°C. The infection inocula was prepared as follow: 10 ml of an exponentially grown M. tuberculosis H37Rv culture was centrifuged and washed twice with PBS. The pellet was resuspended in 4 ml of PBS and passed through a 25-gauge syringe 15 times and then OD600 was adjusted to 0.8 using PBS, which corresponds to 1.2 × 108 bacteria/ml in our experimental settings. BMDM were infected with the M. tuberculosis H37Rv infection inocula at a MOI of 1 for 3 h in order to allow bacterial uptake. Right after the infection, cells were treated with 100 μg/ml of Amikacin for 1 h to kill all the extracellular mycobacteria. Then, cells were washed three times with PBS and 100 μl of fresh RPMI media supplemented with 10% FBS was replaced. Infected macrophages were incubated for 24 h before the treatment started. Test compounds were assayed at 2, 4, 8, 16 and 32 times of the in vitro MIC for M. tuberculosis H37Rv. To evaluate the treatment efficacy, cells were lysed with Triton 0.1% at day 1 before the treatment, and at day 7 after the treatment. CFU were enumerated by plating serial dilutions of the lysates on Middlebrook 7H10 agar plates supplemented with 10% OADC. Non-infected and no treated cells, infected but not treated cells and infected and rifampicin-treated cells were included as controls in the experiment.
Isolation of spontaneous nNF resistant mutants and identification of mutations by whole genome sequencing analysis
Three independent M. tuberculosis H37Rv cultures and two Beijing M. tuberculosis GC1237 cultures were grown in complete Middlebrook 7H9 broth to an OD600 ≅ 1.5–2 and then plated on independent Middlebrook 7H10 agar plates containing 4, 10 and 20 times the MIC of derivatives nNF-C1, nNF-C2, nNF-C3, nNF-C4, nNF-C6, nNF-C8, nNF-C18 and nNF-C20. Plates were incubated at 37°C for up to 5 weeks. When possible, two emergent resistant M. tuberculosis colonies from each plate were subcultured independently in complete Middlebrook 7H9 broth in presence of half of the compound concentration at which they were isolated in the agar plates. Cultures were grown to an OD600 of 0.8 and then processed for genomic DNA extraction (55). DNA from at least two resistant M. tuberculosis clones per assayed derivative were sequenced using the Illumina MiSeq pair-ended (2 × 300 bp) sequencing system (Illumina, San Diego, CA, USA). Raw sequencing reads produced for this study have been deposited in the European Nucleotide Archive (ENA) under accession number ENA: PRJEB48019. The analysis of Single Nucleotide Polymorphisms (SNPs) and Insertions/Deletions (INDELs) was performed using the genome of the M. tuberculosis H37Rv (NC000962.3) as a reference. Paired-end (PE) reads in fastq format were pre-processed by trimming Illumina adaptors and excluding low-quality reads using fastp (56). Thereafter, we used bwa-mem to align PE filtered reads against the reference strain. Duplicated reads were identified and removed using picard (http://broadinstitute.github.io/picard/). Local alignment of reads was performed around gapped regions before SNP calling using the GATK IndelRealigner module (https://software.broadinstitute.org/gatk/). Alignment files in BAM format were used as inputs for variants detection. SNP calling was performed using samtools and bcftools (https://samtools.github.io/bcftools/). SNPs located within a frame of 20 bp from indels were excluded with samtools. For each sample, all identified SNPs and INDELs were filtered and annotated using snpToolkit (https://github.com/Amine-Namouchi/snpToolkit). snpToolkit was used to filter and annotate SNPs from vcf files according to three criteria: quality score (≥30), depth of coverage (≥20) and allele frequency (≥90%).
nNF resistance frequency in M. tuberculosis
Three independent M. tuberculosis H37Rv cultures were grown in complete Middlebrook 7H9 broth to an OD600 ≅ 1.5 and then 50–500 μl of 10 times concentrated bacterial cultures were plated on independent complete Middlebrook 7H10 agar plates containing 5, 10 and 20 times of the MIC of derivatives nNF-C1, nNF-C2, nNF-C3, nNF-C4, nNF-C6, nNF-C8, nNF-C18 and nNF-C20. Plates were incubated at 37°C for up to 5 weeks and resistant emergent colonies were enumerated. The number of viable M. tuberculosis bacteria was obtained from plating serial dilutions of the concentrated cultures in Middelbrook 7H10 (nNF-free) agar plates. The frequency of mutation per each compound and each condition was calculated by dividing the number of resistant mutants and the number of viable bacteria that were initially plated.
Complementation strategies for sigH and mrx2 genes in the M. tuberculosis knockout mutants
The M. tuberculosis H37Rv mrx2 null mutant (Δmrx2; (27,30)) was complemented with a copy of either mrx2WT or the corresponding mrx2 mutant cloned into pFRA220, a derivative of the suicide plasmid pMV360, that carries a cassette of resistance to kanamycin. To this end, the mrx2 gene was amplified including a 603 bp upstream region using the oligonucleotides 5′-GATATCCGCCGTTGGAGATCACCTTC-3′ and 5′-GATATCCCGACATTGTGCCCGACA-3′, as forward and reverse primers. The mrx2 mutants mrx2S46R, mrx2D16G, mrx2H140D, mrx2G95S, mrx2R198G, mrx2L114P and mrx2T25R, plus three additional substitutions that were predicted to have an impact on the activity of the enzyme, mrx2W23R, mrx2L90P and mrx2Y91S, were cloned into pFRA220, using pFRA220-mrx2WT as template (GenScript, NJ, USA).
To complement the ΔsigH mutant strain, a 2022 bp PCR fragment containing a copy of the sigHWT gene and its anti-sigma factor rshA, was cloned into pMV360H (pMV360H-sigHWT), a suicide plasmid that contains a cassette conferring hygromycin resistance. sigH amplification was carried out using the oligonucleotides 5′-ATCGGCGGGGACAGCGTCAG-3′ and 5′-TCACTTCCGCGCAACCCATG-3′, as forward and reverse primers. The sigHV156D and sigHL58P mutants were synthesized by GenScript using the pMV360H-sigHWT construct as template (GenScript, NJ, USA).
The sigHWT and sigHV156D and sigHL58P mutants constructs were introduced in a pristinamycin inducible expression system (57). First the different sigH genes were amplified from the relative wt and mutant strains, and then cloned into pCR-Blunt II-TOPO (Invitrogen). The constructs were subsequently excised from the plasmid using EcoRI and subcloned into the plasmid pFRA171, downstream of a pristinamycin inducible promoter (58). A BglII cassette conferring resistance to Hygromicin was then inserted in the resulting plasmids.
Phenotypic analysis of sigH and mrx2 mutant variants in the ΔsigH and Δmrx2 mutant backgrounds
Independent plasmid constructions containing either a copy of the mrx2WT gene or a mrx2 mutant variant were transferred by electroporation to Δmrx2 M. tuberculosis H37Rv, followed by the selection of transformants in complete 7H10 agar plates containing 25 μg/ml kanamycin. Similarly, independent plasmid constructions containing either a copy of the sigHWT gene or the different mutant variants were transformed into ΔsigH M. tuberculosis H37Rv, followed by the selection of transformant colonies in complete 7H10 agar plates containing 50μg/ml of hygromycin. Three independent colonies were picked up for each plasmid construction transformation and then checked by PCR in order to confirm the plasmid insertion. Positive colonies were grown to an OD600 = 0.6 and used to determine the MIC90 for derivatives nNF-C2 and nNF-C4 using the REMA protocol previously described. An Infinite 200Pro microplate reader (Tecan Group) with excitation and emission wavelengths of 535 and 590 nm respectively was used to determine the MIC90 values.
Determination of the sigH and mrx2 expression levels
M. tuberculosis H37Rv, ΔsigH M. tuberculosis H37Rv, and ΔsigH M. tuberculosis H37Rv transformed with pFRA71-sigHWT, pFRA71-sigHV156D, pFRA71-sigHY157D, and pFRA71-sigHL58P plasmids were used to evaluate the expression levels of mrx2 when sigH was overexpressed under the pip-ON system. All strains were grown in 10 ml of complete Middlebrook 7H9 broth in standing at 37 °C to an OD600 = 0.4. Then cultures were divided into two experimental samples of 4 ml each, one received a 2 μg/ml of pristinamycin treatment in order to induce the sigH expression, and the other was kept as a negative control. After 24 h, 2 ml of the experimental (pristinamycin) and control M. tuberculosis samples were collected for RNA extraction (59).
Quantitative reverse transcription real-time PCR was performed using Sybr Green Master Mix (Applied Biosystems) as described (59). Results were normalized to the amount of sigA mRNA as internal housekeeping control (60). Fold change values of sigH and mrx2 were expressed as the ratio between the transcriptional level of the gene of interest in presence of 2 μg/ml pristinamycin and in its absence and were normalized to the values obtained with the reference M. tuberculosis H37Rv strain. The primers used for quantitative real-time PCR were (i) 5′-CCATCCCGAAAAGGAAGACC-3′ and 5′- AGGTCTGGTTCAGCGTCGAG-3′, forward and reverse, respectively (sigA fragment of 209 bp); (ii) 5′-CCGGATACTGACCAACACCT-3′ and 5′-CGCTTCTAACGCTTCGACTT-3′, forward and reverse, respectively (sigH fragment of 157 bp); and 5′-CTGGTTCGATCCGCTGTGCC-3′ and 5′-CATGCCCTCGCCATGCCTTC-3′, forward and reverse, respectively (mrx2 fragment of 168 bp). RNA samples that had not been reverse transcribed were included in all experiments to exclude significant DNA contamination. For each sample, melting curves were used to confirm the purity of the amplification products. Experiments were performed at least twice, starting from independent biological samples.
Transcriptional levels of sigH and mrx2 were also determined in presence of the nNF-C2. Briefly, M. tuberculosis H37Rv was grown in 40 ml of complete Middlebrook 7H9 broth in roller bottles at 37°C to an OD600 = 0.4. Then, culture was split in aliquots of 8 ml which were treated respectively with the corresponding MIC (0.0625 μg/ml), 2× MIC (0.125 μg/ml), 4× MIC (0.25 μg/ml), and 8× MIC (0.5 μg/ml) or kept untreated. Cultures were left in standing and samples were collected at 2, 6, 24 h after the addition of compound and total RNA was extracted (59). A positive control for the sigH expression induction was included in the experiment using 5, 10 and 20 μM of diamide and collecting RNA after 30 min. Q-PCR was performed as previously described and the transcriptional levels of sigH and mrx2 for all the treated conditions were calculated by normalizing them to the expression levels of those genes in non-treated M. tuberculosis H37Rv at the same time points. RNA samples that had not been reverse transcribed were included in all experiments to exclude significant DNA contamination. For each sample, melting curves were used to confirm the purity of the amplification products. Experiments were performed at least twice, starting from independent biological samples.
Cloning, expression and purification of Mrx2 mutants
pET29a-mrx2 vector, previously reported (27), was used as template to generate the following mutants: Mrx2D16G, Mrx2W23R, Mrx2T25R, Mrx2S46R, Mrx2L90P, Mrx2Y91S, Mrx2G95S, Mrx2L114P, Mrx2H140D and Mrx2R198G (GenScript, NJ, USA). The expression and purification of the wild type and mutant proteins were followed as previously described (27,28). Specifically, E. coli BL21(DE3) pLysS cells transformed with the corresponding mutant were grown in 500 ml of 2× YT medium supplemented with 25 μg/ml kanamycin at 37°C. When the culture reached an OD600 = 0.8, Mrx2 expression was induced by adding 1 mM isopropy-β-d-thiogalactopyranoside (IPTG, MIP). After ∼16 h at 18°C, cells were harvested and resuspended in 40 ml of 50 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, 10% glycerol at pH 7.5, (solution A), containing protease inhibitors (Thermo Scientific™, A32955) and 2.5 μl of Benzonase (Merck, 71205). Cells were disrupted by sonication (9 cycles of 10 s pulses with 59 s cooling intervals between the pulses, and 60% of amplitude) and the suspension was centrifuged at 59 000 × g for 30 min. The supernatant was applied to a HisTrap Chelating column (5 ml, GE HealthCare) equilibrated with solution A. The column was washed with solution A until no absorbance at 280 nm was detected. Elution was performed with a linear gradient of 10–500 mM imidazole in 50 ml of solution A at 1 ml/min, and fractions containing Mrx2WT and mutants. Fractions were pooled and loaded into a Superdex 75 26/60 column (120 ml; GE Healthcare), equilibrated in 50 mM Tris–HCl, 150 mM NaCl, 10% glycerol at pH 7.5 (solution B) and assessed for purity by SDS-PAGE. The purified Mrx2WT and mutants were concentrated to 1 mg/ml, flash-frozen, and stored at −80°C until ready for use.
Mrx2WT and Mrx2 mutants enzyme kinetics
Activity measurements were performed as previously described (27). nNF-C2 reduction was followed by change in absorbance of the compound in solution, in the presence of Mrx2WT and mutants. Absorption spectra were recorded in 100 mM NaH2PO4—NaOH pH 7.0, 1 mM DTT, 10% DMSO, 4% M. smegmatis methanol extract, 10 μM compound and 30 μM Mrx2WT or mutants. The concentration used for Mrx2Y91S was 6 μM due to the low expression levels of this mutant. Spectra was recorded at 412 nm for compound nNF-C2 (molar extinction coefficient 23 664 M/cm at 412 nm in DMSO) on a UV-Vis NanoDrop™ One/OneC at 25°C. The presence of the His-tag in the N-terminus did not affect the activity of Mrx2WT. All enzymatic activity measurements were determined in triplicates.
Molecular graphics and structural analysis
Molecular graphics and structural analysis were performed with the UCSF Chimera package (61).
Nitric oxide release assay
The determination of NO release was performed using both the 2,3-diaminonaphthalene (DAN) assay and the diaminofluorescein-FM diacetate (DAF-FM) assay in nNF-treated M. bovis BCG cultures. The last method was applied in combination with carboxy-PTIO potassium salt (C-PTIO; Sigma Aldrich) to study the NO scavenging dynamics in cultures exposed to nNF-C2 and nNF-C4.
For the DAN assay, the OD600 of an exponentially grown M. bovis BCG culture was adjusted to 0.8 and 250 μl were transferred into a 48 well-plate. Different concentrations of the nNF-C2 and nNF-C4 derivatives were added to each well resulting in a final DMSO concentration of 1%. A negative control containing just DMSO was included. Plates were incubated at 37°C for 24 h, centrifuged at 3500 for 10 min to pellet down the bacteria and then 100 μl were transferred into a black opaque 96-well plate. A serial dilution of NaNO2 in 7H9 broth supplemented with 10% ADC and 0.05% Tyloxapol was used to calculate a standard curve. The DAN assay was adapted from reports previously described (62,63). Briefly, 20 μl of 0.05 mg/ml DAN (Alfar Aesar) in 0.62 M HCl was added per well and incubated at RT for 10 min in the dark. Then, 100 μl of 0.28 M NaOH was added and fluorescence was measured at an excitation wavelength of 355 nm and an emission wavelength of 460 nm in a Wallac Victor 2 microplate reader (Perkin Elmer).
For the DAF-FM method, M. bovis BCG cultures (50ml) were grown in complete Middlebrook 7H9 broth up to OD600 of 1. Bacterial cultures were spin down and washed 3 times using PBS containing 0.05% tyloxapol, and then resuspended in 10ml of PBS. 5 μM of DAF-FM diacetate (Thermo Fisher Scientific) were added to the bacterial cultures and incubated overnight at 37°C in the dark. The next day, cultures were washed 2 times with PBS, resuspended in a final volume of 20 ml and then split onto 4 × 5 ml cultures. NO scavenger Carboxy-PTIO potassium salt (CPTIO; Sigma Aldrich) was tested at the following concentrations: No CPTIO, 75 μM, 150 μM and 300 μM, and incubated for 3 h at 37°C. Approx. 107 M.bovis BCG bacteria were dispensed in opaque 96-well plates containing serial dilutions of nNF-C2 and nNF-C4 in PBS. Plates were incubated overnight and the following day recorded in a microplate reader (Synergy H1, BioTek; Excitation:495nm; Emission: 515nm).
Separately, same M.bovis BCG cultures were used to inoculate ∼106 M.bovis BCG bacteria in 96-well plates containing serial dilutions of nNF-C2 and nNF-C4 in PBS. Plates were incubated for additional 7 days at 37°C inside of zip bags to avoid evaporation. 1:100 dilutions of each of the conditions assayed in the 96 well plate was done using fresh Middlebrook 7H9 broth containing 10% OADC in a new 96-well plate. Plates were incubated for 2 days and then resazurin was added at a final concentration of 0,003%. Readouts were taken at 24 and 48 h. In parallel, CFUs were enumerated by plating onto Middlebrook 7H10 agar plates.
RESULTS
Selected nNFs efficiently kill actively replicating and non-replicating M. tuberculosis
A high-throughput whole-cell screening of a chemical library composed of 35 860 compounds was performed using M. aurum ATCC23366 as a surrogate for M. tuberculosis (38). A nNF compound, further validated on both M. aurum ATCC23366 and M. tuberculosis H37Rv ATCC 27294, was identified as anti-tuberculosis lead (nNF-C18; Figure 1 and Supplementary Table S1) and nineteen additional chemical derivatives were chosen from the same chemical library (nNF-C1 to nNF-C20; Figure 1 and Supplementary Table S1). Among the twenty nNFs derivatives tested, ten compounds, nNF-C1, nNF-C2, nNF-C3, nNF-C4, nNF-C5, nNF-C6, nNF-C8, nNF-C16, nNF-C18 and nNF-C20, were highly active against replicating M. tuberculosis H37Rv and both sensitive and MDR clinical strains, showing MIC90 values ranging from 0.031 to 0.5 μg/ml (Table 1; Figure 1; Supplementary Tables S1 and S3). Five nNF derivatives, nNF-C7, nNF-C11, nNF-C12, nNF-C14 and nNF-C17, showed a moderate activity with MIC90 values of 2, 4 and 8 μg/ml (Table 1; Figure 1; Supplementary Tables S1 and S3). The five remaining compounds in the chemical set analysed were poorly active or inactive against M. tuberculosis H37Rv showing MIC90 values higher than 16 μg/ml. The ten nNFs that were classified as highly active in inhibiting the M. tuberculosis growth were found to be bactericidal at 1–4-fold of their corresponding MIC90 values (Supplementary Table S6). Strikingly, the nNF derivatives that were found to be highly effective against actively replicating M. tuberculosis, also efficiently killed non-replicating M. tuberculosis (Supplementary Figure S1). A carry over effect was ruled out for nNF-C4 since similar results were obtained when testing the MBC90 in non-replicating M. tuberculosis using the Charcoal Agar Resazurin Assay (CARA) (Supplementary Figure S1). This is an in vitro method that includes activated charcoal in the agar medium to mitigate the impact of compound carry-over. However, some discrepancies were found when testing the nNF-C2 using CARA compared to the CFU enumeration in complete Middlebrook 7H10 agar plates. While nNF-C2 showed a MBC90 of 0.5 μg/ml in the CAR assay, the CFU enumeration method in Middlebrook agar plates resulted in a MBC90 for non-replicating M. tuberculosis of 0.062 μg/ml. Despite these differences, the antimycobacterial activity of both nNF-C2 and nNF-C4 against non-replicating M. tuberculosis in the charcoal assay was in a range of 4–8 times with respect to the MIC90 against replicating bacteria, supporting that nNFs are powerful compounds against non-replicating forms of M. tuberculosis.
Table 1.
In vitro anti-tuberculosis activity of nNF derivatives (nNF-C1–nNF-C20) against actively replicating M. tuberculosis H37Rv
| MIC90 REPLICATING M. tuberculosis | ||||
|---|---|---|---|---|
| Compound | μg/ml (range) | μg/ml (median) | μM (median) | |
| High activity | nNF-C1 | 0.062–0.25 | 0.125 | 0.42 |
| nNF-C2 | 0.062–0.125 | 0.062 | 0.21 | |
| nNF-C3 | 0.25–0.5 | 0.5 | 1.68 | |
| nNF-C4 | 0.03–0.125 | 0.125 | 0.46 | |
| nNF-C5 | 0.25–0.5 | 0.25 | 1.05 | |
| nNF-C6 | 0.5 | 0.5 | 1.63 | |
| nNF-C8 | 0.062–0.25 | 0.125 | 0.46 | |
| nNF-C16 | 0.03–0.062 | 0.062 | 0.25 | |
| nNF-C18 | 0.125–0.25 | 0.25 | 0.78 | |
| nNF-C20 | 0.125 | 0.125 | 0.4 | |
| Moderate activity | nNF-C7 | 4–8 | 4 | 15.55 |
| nNF-C11 | 8–16 | 8 | 32.89 | |
| nNF-C12 | 2–8 | 4 | 14.85 | |
| nNF-C14 | 0.5–4 | 2 | 8.8 | |
| nNF-C17 | 2–4 | 2 | 8.29 | |
| Low/No activity | nNF-C9 | >32 | >50 | |
| nNF-C10 | >32 | >100 | ||
| nNF-C13 | >32 | >100 | ||
| nNF-C15 | 32 | >100 | ||
| nNF-C19 | 16 | >50 | ||
| TB antibiotics | INH | 0.01–0.04 | 0.03 | 0.218 |
| RIF | 0.062–0.125 | 0.125 | 0.35 | |
| PA-284 | 0.25–0.5 | 0.25 | 0.31 | |
MIC90 determination was carried out by REMA. According to their activity against replicating bacteria, nNF compounds were divided in three different groups: High activity (MIC ≤ 0.5 μg/ml), Moderate Activity (MIC ≤ 8 μg/ml) and No Activity (MIC ≥ 16 μg/ml). MIC90 values are expressed in μg/ml (range and median) and μM (median).
The family of active compounds here identified is based on condensed heteropolycyclic compounds consisting of a common scaffold 2-nitronaphtho[2,1-b]furan. The compounds nNF-C1, nNF-C2 and nNF-C3 have a second 2-nitrofuran ring. The compound nNF-C2 differs from nNF-C1 and nNF-C3 by the substitution position of the second 2-nitrofuran ring on the naphthalene core, respectively on the C6-C7 bond for nNF-C2 and on the C5-C6 bond of naphthalene, which likely explains its better inhibitory activity compared to nNF-C1 and nNF-C3. Compounds nNF-C4 to nNF-C20 have only the common scaffold 2-nitronaphtho[2,1-b]furan and differ from each other by different substituents grafted either on the furan or on the naphthalene ring. It can be noted that the presence of a substituent group in position 1 on the furan ring, in particular for compounds nNF-C7, nNF-C9, nNF-C10, nNF-C11, nNF-C12, nNF-C13, nNF-C14, nNF-C15 and nNF-C17, seems to cause a loss of activity against M. tuberculosis (Table 1; Figure 1; Supplementary Table S1). On the other hand, we studied the influence of the substituent group (ether, ester, nitrile, halogen) and its position on the naphthalene core (carbon 5, 7, 8 and 9). The methoxy group on carbon 5 position in the nNF-C19 compound caused a total loss of activity compared to the nNF-C16 compound, in which the methoxy group is carried by carbon 7. The other positions on the naphthalene core had no impact on the activity of the nNF derivatives. A bromine in position 7 or position 8 (nNF-C6 and nNF-C18 respectively) causes a decrease in the anti-tuberculosis activity. The nNF-C2 and nNF-C4 were the most active compounds, displaying growth inhibitory and bactericidal concentration values in the range of 0.0625–0.5 μg/ml against replicating and non-replicating M. tuberculosis, and were therefore selected for further studies (Table 1; Figure 1; Supplementary Table S1; Figure S1).
Antimicrobial spectrum
We further determined the ability of nNFs to inhibit the growth of other mycobacteria and non-mycobacteria species (Supplementary Tables S4 and S5). nNFs compounds, including nNF-C2, were found equally active/inactive in M. bovis BCG with respect to M. tuberculosis. M. aurum and M. avium were sensitive to the same set of compounds active against M. tuberculosis, but showing higher MIC90 values and resistance to compounds nNF-C5, nNF-C6 and nNF-C7. M. marinum was as sensitive as M. tuberculosis or even more for some compounds, including derivatives nNF-C13, nNF-C15 and nNF-C19, for which M. tuberculosis was found to be resistant. In contrast, M. abscessus and M. smegmatis were the most resistant bacteria to the action of nNFs. Among other bacterial species, only K. pneumoniae and S. enterica showed sensitive profiles similar to the ones found in some other mycobacteria species (Supplementary Table S5). The remaining bacteria tested, including E. aerogenes, E. cloacae, E. coli, P. aeruginosa and S. aureus, were resistant to almost all the compounds assayed.
The lead derivative nNF-C2 displays a high selectivity index
To determine the cytotoxicity selectivity properties of the nNF compounds, we determined their 50% cytotoxic concentration (CC50) in VERO and HepG2 cell lines and calculated their respective selectivity index (SI) values. A very sensitive assay based on the quantification of ATP was performed to indirectly determine the cellular viability in the presence of the nNF compounds. We established a very strict cut-off in the selection of ‘non-toxic’ in vitro compounds referred as those compounds whose SI was equal to or higher than 100. Based on this criteria, compounds nNF-C1, nNF-C2, nNF-C4 and nNF-C6 were the safest in our study showing CC50 values ≥ 64 μg/ml, and SI values ranging from 182.6 to 1 211.5 (Table 2). Despite showing a SI < 100, nNF-C11 and nNF-C14 were also included in the ‘safety set’ since no toxicity was observed in mammalian cells at the maximum concentration assayed (CC50 values ≥ 100 μg/ml). In contrast, compounds nNF-C3, nNF-C5, nNF-C20, nNF-C18, nNF-C8 and nNF-C16 were considered–in increasing order - the most toxic compounds, showing CC50 values ranging from 0.3 to 21.5 μg/ml and SI ≤ 21.5. The remaining derivatives, nNF-C7, nNF-C12 and nNF-C17, showed low cytotoxicity in mammalian cells displaying CC50 values ranging between 55.1 and 73.5 μg/ml, but with SI values lower than 100.
Table 2.
Cytotoxicity assessment of nNFs
| M. tuberculosis | VERO cells | |||
|---|---|---|---|---|
| Compound | MIC90 (μg/ml) | CC50 (μg/ml) | SI | |
| High activity | nNF-C1 | 0.125 | 69.2 | 553.6 |
| nNF-C2 | 0.0625 | 75.7 | 1211.5 | |
| nNF-C3 | 1 | 21.5 | 21.5 | |
| nNF-C4 | 0.125 | 64.9 | 519.2 | |
| nNF-C5 | 0.25 | 12.5 | 50.0 | |
| nNF-C6 | 0.5 | 91.3 | 182.6 | |
| nNF-C8 | 0.125 | 0.4 | 3.4 | |
| nNF-C16 | 0.0625 | 0.3 | 5.2 | |
| nNF-C18 | 0.25 | 2.2 | 8.8 | |
| nNF-C20 | 0.25 | 9.3 | 37.1 | |
| Moderate activity | nNF-C7 | 4 | 67.5 | 16.9 |
| nNF-C11 | 8 | >100 | >12,5 | |
| nNF-C12 | 4 | 73.5 | 18.4 | |
| nNF-C14 | 2 | >100 | >50 | |
| nNF-C17 | 2 | 55.1 | 27.5 | |
| RIF | 0.01 | 78.2 | 7824.1 |
Cytotoxicity of the nNF derivatives classified as highly or moderately active against M. tuberculosis was determined in Vero cells after 72 h of chemical exposition. Cellular viability was determined by indirectly measuring the amount of ATP. CC50 was calculated as the concentration at which 50% of the cells were viable compared to the untreated control. MIC90 and CC50 values are expressed in μg/ml. The selectivity index (SI) is expressed as CC50/MIC90.
nNF-C2 efficiently kills intracellular M. tuberculosis
Since M. tuberculosis is mainly an intracellular pathogen, anti-tuberculosis candidates must be able to penetrate and diffuse inside the cells, reaching the tuberculous bacilli in their active chemical structure, therefore maintaining their killing properties in the intracellular environment. Taking into account their in vitro killing activity and cytotoxicity selectivity properties, we tested the ability of compounds nNF-C2, nNF-C4, and nNF-C20 to kill M. tuberculosis H37Rv in infected mouse Bone Marrow-Derived Macrophages (BMDM). Specifically, concentrations ranging from 2 to 32 times of the corresponding in vitro MIC90 for each compound were assayed. As depicted in Figure 2, nNF-C2 and nNF-C4, reduced the number of intracellular M. tuberculosis bacteria by ca. 2 logs at 4 times of MIC90 (0.25 μg/ml) and 2 times of MIC90 (0.25 μg/ml), respectively. In contrast, the concentration that the first-line drug rifampicin needed to achieve a similar level of intracellular killing was at 32 times its MIC (0.32 μg/ml). Importantly, at that relative concentration (32 × MIC), compounds nNF-C2 and nNF-C4 were able to reduce the burden of intracellular bacteria by more than 4 logs. However, not all nNFs that displayed an in vitro potent killing activity were found to be as active in the intracellular assay. That was the case for compound nNF-C20, which needed 32 times of its MIC90 (4 μg/ml), to decrease by ≅1 log(31.8-fold times) the number of intracellular M. tuberculosis bacteria.
Figure 2.

Intramacrophage killing activity of selected nNF derivatives against M. tuberculosis H37Rv. BMDM were infected with M. tuberculosis H37Rv at a MOI of 1, and viable intracellular bacteria were enumerated by CFU after 6 days. The Y axis represents the total number of CFU calculated per well for the different experimental groups: untreated infected control (black), and infected and treated with nNF-C2 (blue), nNF-C4 (orange), nNF-C20 (yellow) and RIF (grey). Two to 32 times of the corresponding in vitro MIC90 obtained for each compound were the concentrations used for the intracellular bactericidal assessment. Exact assayed concentrations for each compound are displayed in μg/ml. CFU values are represented as the mean ± SD of 3 independent experiments.
sigH and mrx2 genes are involved in the anti-tuberculosis activity of nNF-C2
To elucidate the bactericidal mechanism of action of nNFs in M. tuberculosis, we isolated spontaneous resistant mutants using the M. tuberculosis laboratory strain H37Rv and a Beijing clinical strain (GC1237). A total of 28 nNF resistant M. tuberculosis mutants were isolated in the presence of different nNF-compounds at 5, 10 and/or 20 fold times of their in vitro MIC90. The frequency at which spontaneous resistant M. tuberculosis H37Rv mutants arose against the high active nNF derivatives varied from 1.4 × 10−6 to 4.5 × 10−8, depending on the compound and the concentration used for the mutants’ selection (Supplementary Table S7). No resistant mutants could be isolated for nNF-C5 and nNF-C16.
To identify the genetic mutations responsible for the nNF resistance in the corresponding M. tuberculosis resistant mutants, whole-genome sequencing comparative analysis was performed, revealing three and seven point mutations in the sigH (Rv3223c) and mrx2 (Rv2466c) genes, respectively (Table 3). In addition, four different insertions/deletions (INDELs) were detected in each of the genes (Table 3). Both sigH and mrx2 were reported to be non-essential genes by Himar1-based transposon mutagenesis in M. tuberculosis H37Rv strain (64). It is worth noting that simultaneous mutations in the sigH and mrx2 genes were not found, suggesting the importance of both genes in the mechanism of action of nNFs.
Table 3.
Mutations found in nNF spontaneous resistant M. tuberculosis mutants
| Gene | Mutation |
|---|---|
| rv2466c Mrx2 | DEL 1 bp at pos. 19 |
| Mycothiol-dependent cytoplasmic reductase | INS 1 bp at pos. 252 |
| DEL 2 bp at pos. 85 | |
| INS 1 bp at pos. 20 | |
| S 46 R | |
| D 16 G | |
| H 140 D | |
| G 95 S | |
| R 198 G | |
| L 114 P | |
| T 25 R | |
| rv3223c SigH | INS 1 bp at pos. 535 |
| Extracytoplasmic function (ECF) sigma | INS 1 bp at pos. 231 |
| factor | DEL 2 bp at pos. 577 |
| DEL 1 bp at pos. 495 | |
| V 156 D | |
| Y 157 D | |
| L 58 P |
Single nucleotide polymorphisms (SNPs) and Insertions/Deletions (INS/DELs) listed in the table, correspond to independent M. tuberculosis H37Rv and M. tuberculosis GC1237 Beijing resistant clones. Mutants were isolated using 4, 8 and/or 20 times the MIC90 of compounds nNF-C1, nNF-C2, nNF-C3, nNF-C4, nNF-C6, nNF-C8, nNF-C18 and nNF-C20.
To determine the role of sigH and mrx2 genes in the resistance mechanism to nNFs, the sigH and mrx2 M. tuberculosis H37Rv knockouts (ΔsigH and Δmrx2, respectively) were submitted to a sensitivity determination assay using two representative nNF compounds, nNF-C2 and nNF-C4. Specifically, Δmrx2 showed 8 and 16 times higher MIC values than M. tuberculosis H37Rv for nNF-C2 and nNF-C4 respectively, while ΔsigH showed at least 32 times higher resistance for nNF-C2 and 16 times in the case of nNF-C4 (Figure 3). In both cases, the sensitivity was recovered after ΔsigH and Δmrx2 were complemented with a full-length copy of the corresponding sigH and mrx2 wild type gene (Figure 3). Altogether, the experimental data support the notion that SigH and Mrx2 are involved in the mechanism of action of nNF-C2 and nNF-C4 against M. tuberculosis.
Figure 3.
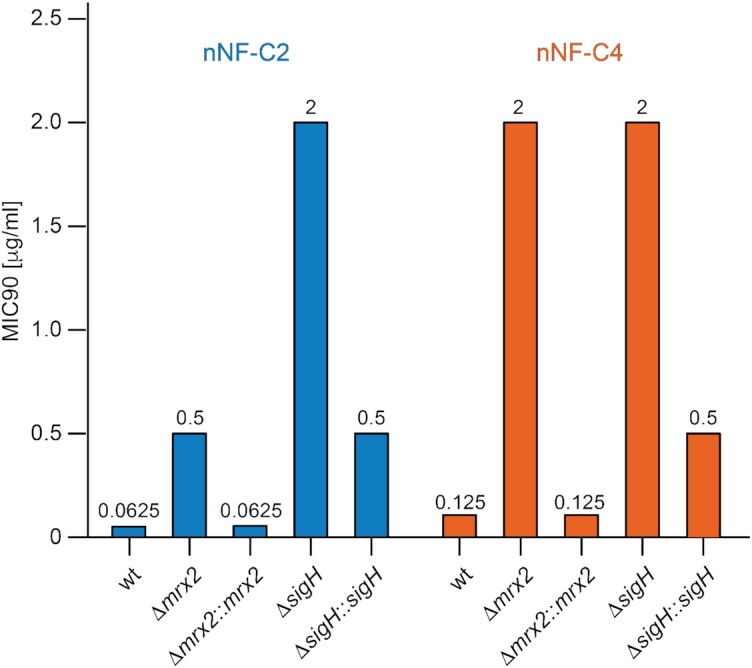
M. tuberculosis SigH and Mrx2 are involved in the prodrug activation of nNFs. MIC90 values (μg/ml) for the nNF derivatives nNF-C2 and nNF-C4 in the M. tuberculosis H37Rv-wild type strain (wt), M. tuberculosis mrx2 KO mutant (Δmrx2), complemented M. tuberculosis mrx2 KO (Δmrx2::mrx2), M. tuberculosis sigH KO (ΔsigH), and the complemented M. tuberculosis sigH KO (ΔsigH::sigH). mrx2 and sigH KO mutants showed higher resistance to both nNF-C2 and nNF-C4 than the H37RvWT, while their complementation with the corresponding wt version of the KO-gene, restored the sensitivity.
nNF-C2 prodrug is activated by Mrx2 conveying anti-tuberculosis activity
Mrx2 is a recently discovered mycothiol dependent reductase involved in the activation of the anti-tuberculosis prodrug TP053, leading to the release of nitric oxide (27,30,65). We therefore investigated whether Mrx2 is similarly involved in the activation of nNF compounds. For that aim, we expressed and purified the Mrx2 wild type (Mrx2WT) protein and studied its ability to metabolize the selected nNF-C2 compound using a previously reported enzyme kinetic assay in which the time-dependent decrease in nNF-C2 absorbance at 412 nm was monitored (27). As depicted in Figure 4A, nNF-C2 was completely metabolized when incubated with Mrx2WT.
Figure 4.

nNFs prodrugs are activated by Mrx2 conveying anti-tuberculosis activity. (A) In vitro enzymatic activity of Mrx2WT represented by the time-dependent decrease of nNF-C2 absorbance at 412 nm; (B) nitric oxide release in nNF treated M. bovis BCG cultures. NO release in BCG cultures exposed to different concentrations of nNF-C2, nNF-C4, PA-824, and RIF, was quantified in the culture supernatants using the DAN assay; (C, D) phenotypical characterization of mrx2 mutations cloned into pFRA220 and expressed in Δmrx2. MIC90 values (μg/ml) for the nNF derivatives nNF-C2 (C) and nNF-C4 (D) obtained in M. tuberculosis H37RvWT (wt), Δmrx2, and Δmrx2 transformants expressing the different mrx2 mutants (Δmrx2::mrx2MUT); (E) in vitro enzymatic activity of Mrx2 wild type and mutants against compound nNF-C2; (F) ribbon representation of the crystal structure of Mrx2 (PDB 4ZIL), showing the localization of the Mrx2 mutations.
To determine whether the mechanism of action of our set of nNFs was involving the NO production as suggested with other nitroaromatic compounds, we performed an experimental assay in which the generation of NO was measured in M. bovis BCG cultures exposed to different concentrations of nNF-C2 and nNF-C4. The nitroimizadol PA-824, which is known to release NO after its activation by the nitroreductase Ddn, and RIF whose mechanism of action is not related to NO production, were introduced in the assay as positive and negative controls, respectively. Both nNF-C2 and nNF-C4, as well as PA-824, showed significant NO release in a concentration dependent manner (Figure 4B). The levels of NO differed between compounds, with the maximum levels detected for PA-824, followed by nNF-C4, which generated almost 4 times higher nitrite levels than its analogue nNF-C2.
Additionally, we performed a complementary experiment in which the NO release in the presence of nNF-C2, nNF-C4, and PA-824, was measured intracellularly by using the fluorescent permeable cell probe DAF-FM together or not with the NO scavenger CPTIO (Supplementary Figure S2). The DAF-FM method was sensitive and allowed us to detect in a concentration dependent manner the NO production in the presence of compounds nNF-C2, nNF-C4 and PA-824. A prominent reduction in the NO detection was observed in M. bovis BCG cultures exposed to nNF-C4 and PA-824 after 48 h of treatment with the NO scavenger CPTIO. The reduction in the levels of NO was less significant in the case of nNF-C2 treatment (Supplementary Figure S2A). However, despite the reduction of intracellular NO levels in the presence of CPTIO, only discrete differences in the viability of the bacteria were observed (Supplementary Figure S2B and C).
Finally, we tested the in vitro activity of an array of 13 desnitro nNF analogues, carrying substituents other than a nitro group. None of those compounds showed any measurable antibacterial activity against M. tuberculosis in our assay (MIC90 > 32 μg/ml), pointing out the essentiality of the nitro moiety in the mechanism of action of the nNFs (Supplementary Table S2).
Mrx2 mutants show impaired enzyme activity leading to a nNF resistant M. tuberculosis phenotype
We further evaluated the effect on the nNF sensitivity of ten different mrx2 mutants (Methods section) that were expressed in a M. tuberculosis H37Rv mrx2-KO background. In the case a mutation was involved in the mechanism of nNF resistance, complementation of the KO strain would not be expected since a defective form of the gene would be introduced and, therefore, the strain would maintain its resistance to nNFs. In contrast, if the mutation is not involved in the resistance mechanism, the functionality of the protein would be restored and the KO strain would become sensitive to nNFs, as observed previously in the complementation assay using a wt copy of mrx2. Mrx2 mutations H140D, W23R, R198G, L90P, T25R, S46R Y91S and D16G, did not restore sensitivity to nNF-C2 and nNF-C4 in the Δmrx2 strain, showing MIC values from 4 to 16 times higher compared to the wt-complemented strain (Figure 4C, D). In contrast, mutations L114P and G95S reversed the resistant phenotype of Δmrx2. L114P was identified in a spontaneous resistant M. tuberculosis mutant that also carried the confirmed nNF resistant mutation R198G, thus L114P was not necessarily related to the loss of enzymatic function and consequent nNF resistance acquisition. The mutation G95S was identified in a Beijing spontaneous resistant clone isolated using the nNF-C6 derivative, and its relevance in the mechanism of resistance might be only associated to this specific derivative (e.g. interaction of Mrx2 with nNF-C6) and not to nNF-C2 or nNF-C4 that were used to perform the MIC determination and the enzyme kinetic assay.
In agreement with the phenotypic observations, the purified mutant proteins Mrx2T25R, Mrx2S46R, Mrx2Y91S, Mrx2H140D and Mrx2R198G displayed only residual activity against nNF-C2 in the enzyme kinetic assay previously described for the Mrx2WT (Figure 4E), while the Mrx2G95S mutant maintained its ability to metabolize nNF-C2. A careful inspection of the crystal structure of the reduced form of Mrx2 revealed that the mutants likely destabilize (i) the catalytic motif (Y91), (ii) the thioredoxin fold (T25, S46, H140) and (iii) the dimer architecture (R198) (Figure 4F; (27,28)). The Mrx2G95S mutant maintained the capacity to process nNF-C2 most likely due to the substitution of G95 by serine, a small and polar residue that can be accommodated in the α4 of the four α-helical bundle without destabilization of the enzyme. It is worth mentioning that Mrx2D16G, Mrx2W23G, Mrx2L90P and Mrx2L114P resulted in insoluble proteins, and we could not assess their activity. Specifically, residues D16, W23, L90 and L114 are located in four α-helical bundle and their replacement can certainly lead to structural defects.
SigH mediates nNF anti-tuberculosis activity via inducing the expression of Mrx2
We first analysed the phenotype of the SigH mutants that were found among the nNF spontaneous resistant M. tuberculosis mutants. We determined the MIC90 against nNF-C2 and nNF-C4 of ΔsigH H37Rv clones expressing the SigH mutants, SigHV156D and SigHL58P. Both mutants did not complement the resistant phenotype of the ΔsigH H37Rv, showing MIC90 values 16 and 32 times higher than H37RvWT against nNF-C2 and nNF-C4 (Figure 5A, B).
Figure 5.

SigH mediates anti-tuberculosis activity of nNFs via inducing the expression of mrx2, which is impaired in the sigH mutants. (A, B) Phenotypical characterization of the sigH mutations cloned into pMV306H and expressed in ΔsigH. MIC90 values (μg/ml) for the nNF compound nNF-C2 (A) and nNF-C4 (B) obtained in H37RvWT (wt), ΔsigH, and ΔsigH transformants expressing different sigH mutants; (C, D) sigH (C) and mrx2 (D) expression levels before and after pristinamycin sigH-induction. Fold change values are represented in the Y axis as the mean ± SD of the ratio between the transcriptional level of the gene of interest in the strain grown in presence of 2 μg/ml pristinamycin and the transcriptional level of that gene in the same strain grown in absence of pristinamycin, and normalized with the values obtained in M. tuberculosis H37Rv; (E) surface representation of the crystal structure of the transcription initiation complex of M. tuberculosis σH-RNAP (σHRPo) comprising promoter DNA and an RNA primer. Close up of the residue L58 on the σH2 domain (top) and the residues V156 and Y157D on the σH4 domain (bottom).
Taking into consideration that the alternative sigma factor sigH regulates the gene expression of major components involved in the oxidative, nitrosative and heat stress responses in M. tuberculosis, including mrx2 (49,66), we hypothesized that the sigH mutations we found in the spontaneous resistant M. tuberculosis clones could be affecting the SigH binding to the promoter region of mrx2, or to the RNA polymerase core binding, in both cases impairing the regulatory control on the expression of mrx2. As a consequence, low levels of mrx2 transcription would be translated in lower nNF prodrug-activation and therefore into the acquisition of a resistant phenotype. To test this hypothesis, we cloned the sigHWT gene and the two sigH mutant variants, sigHV156D and sigHL58P, into an integrative plasmid in which the expression of the sigH gene was under the control of a pristinamycin (Pip-ON) inducible promoter (57,67). The resultant plasmids were transformed into the ΔsigH M. tuberculosis H37Rv strain. After pristinamycin treatment, the expression levels of sigH and mrx2 in the ΔsigH clone expressing sigHWT under the inducible promoter (ΔsigH::Pptr sigHwt) were 9.8 and 43.7 fold times higher than the levels without pristinamycin, respectively (Figure 5C, D), confirming that the induction of sigH expression leads to a high expression of the mrx2 gene. Importantly, no mrx2 overexpression was detected after induction of the sigH mutant constructs. ΔsigH::Pptr sigHL58P and ΔsigH::Pptr sigHV156D increased the expression of sigH by 6.1- and 7.8-fold after the pristinamycin treatment, respectively, but showed basal levels of mrx2 expression (1.6 and 0.7 fold, respectively).
SigH is composed of a σH2 domain (residues 22–99), σH3.2 domain (residues 100–139), and σH4 domain (residues 140–195). Both σH2 and σH4 fold into independent helical domains (68). In a recent study, Li et al. obtained the crystal structure of a transcription initiation complex of M. tuberculosis σH-RNAP (σHRPo) comprising promoter DNA and an RNA primer. The authors showed that σH2 and σH4 domains stay on the surface of RNAP, and σH3.2 inserts into the active centre. The σH2 domain interact unwinds promoter DNA at the −11/−10 and interacts with the RNAP whereas σH4 binds to the major groove of dsDNA of the −35 element and makes base-specific polar interactions with nucleotides (68). The mutation L58P in SigH could disrupt the regularity of the α2 of the σH2 domain because proline cannot donate a hydrogen bond to stabilize the helix (Figure 5E). This could destabilize the σH2 domain composed of four helices that interact simultaneously with promoter DNA and the RNA polymerase. The structure weighted sequence alignment of ECF σ factors and primary σA factors (Supplementary Figure S4) revealed that the α helix containing L58 is the most conserved in the σH2 domain—a leucine or a small hydrophobic residue in other sigma factors. The mutation of this residue by proline can certainly induce a kink (69) in the helical axis leading to conformational changes of neighbouring residues that are also conserved and important for the interaction with the DNA and RNAP (68). Similarly, our structure weighted sequence alignment (Supplementary Figure S4) also shows that V156, located at σH4 domain, is highly conserved among the compared sigma factors, being a hydrophobic residue in other sigma factors. Thus, mutations of residues by aspartic acid (V156D and Y157D) could produce a similar destabilization effect on the α8 and the adjacent helices of this domain that interact with the dsDNA and RNAP (Figure 5E).
nNF-C2 induces Mrx2 overexpression via triggering SigH expression
Since the activation of nNF-C2 was linked to the NO release, and this has been shown to upregulate sigH expression (70), we aimed to know whether that reaction might be leading to the stimulation of the SigH response in our system, and in consequence the induction of Mrx2 (as it was observed for the oxidazing agent diamide, Supplementary Figure S3). To test this hypothesis, we studied the sigH and mrx2 expression levels at different time points in M. tuberculosis H37Rv cultures that were exposed to nNF-C2 at one time, two times, four times and eight times of the in vitro MIC90. Indeed, we could observe a time and concentration-dependant induction of both genes, even if the correlation between the mRNA increase of the two genes was not always linear, suggesting the possible existence of additional layers of regulation (Figure 6).
Figure 6.

sigH and mrx2 induction upon exposure to nNF-C2. Independent M. tuberculosis H37Rv cultures were treated with 1×, 2×, 4× or 8× MIC concentrations of nNF-C2. A non-treated control group was grown in parallel. Bacterial samples were taken after 6 or 24 h post treatment initiation and submitted to RNA extraction. Transcriptional levels of sigH and mrx2 were determined by qRT-PCR using sigA as internal invariant control. Fold change values are represented in the Y axis as the mean ± SD of the ratio between the transcriptional level of the gene of interest in the treated group and the transcriptional level of the same gene in the control untreated group at the same time point in 2 independent experiments.
DISCUSSION
The discovery of new anti-tuberculosis molecules has become an international health emergency, but also the identification of novel mechanisms of action and new cellular pathways that can be efficiently targeted to enable the killing of M. tuberculosis. In this study, we identified 14 nNF derivatives with bactericidal activity against actively replicating and non-replicating M. tuberculosis, being nNF-C2 the most active compound with highest Selective Index, followed by nNF-C4. The nNF compounds were also active against some other mycobacteria species such as M. bovis BCG and M. marinum, while were less active against others such as M. aurum, M. avium, M. abscessus and M. smegmatis. Among non-mycobacteria species, our selected nNF derivatives were especially active against S. enterica, an intracellular pathogen like M. tuberculosis (71,72), and K. pneumoniae, which has been shown to survive intracellularly in epithelial cells and macrophages (73,74). This may indicate a certain specificity of nNFs activity against intracellular bacterial pathogens. However, molecular basis supporting differences in nNF sensitivity profiles among bacterial species are unknown.
Like other nitroaromatic compounds, nNF derivatives represent a ‘red alert’ in medicinal chemistry due to the potential toxic and genotoxic properties mainly associated with the hydroxylamino metabolites that can result from the reduction of the NO2 groups (75). However, the recent approval of delamanid and pretomanid, both anti-tuberculosis nitroimidazole drugs that were developed from a refined and systematic optimization of a genotoxic lead with emphasis on reducing its genotoxicity, is encouraging (19,76,77). We found a broad range of toxicity levels in mammalian cells among the derivatives with a few compounds being highly toxic, while others were found to be less toxic or not toxic at the maximum concentration assayed (100 μg/ml). In a recent study, we evaluated the toxicity of a subset of these nNF derivatives in an in vivo zebrafish embryo model, a very sensitive animal model to study chemical toxicity (78), and similarly, we observed differential patterns of toxicity between derivatives, indicating that the chemical periphery of the furan ring could play an important role in the safety of these drugs.
The anti-tuberculosis nNF derivatives here discovered are prodrugs that need to undergo bioactivation inside the tuberculosis bacilli, as reported for other nitroaromatic compounds such as nitroimidazoles, nitrofurans and quinoxaline-di-N-oxides (22,23,79–81). It is worth noting that 13 desnitro naphthofuran analogues were found to be inactive against M. tuberculosis, pointing to the essentiality of the NO2 moiety for the activity of these compounds. The common antibacterial mechanism of nitro-drugs is believed to be the release of NO and intermediary metabolites upon their reduction by specific bacterial nitroreductases. These metabolites are known to be highly reactive and have multiple targets, including DNA and several enzymes, such cytochromes and cytochrome oxidase and thus, interfering with the electron flow and ATP homeostasis (22,82–85). Indeed, in our experimental setting, significant levels of NO were released from the nNF-C2 and nNF-C4 derivatives in a concentration and time-dependent manner when incubated with M. bovis BCG cultures. However, and despite observing a reduction in the levels of intracellular NO when introducing CPTIO as NO scavenger, bactericidal concentration values for nNF-C2 and nNF-C4 didn’t change and only a discrete increase of bacterial viability (from 5–12% compared to controls without CPTIO) was detected in the presence of the scavenger. Similar reductions in mycobacterial viability in the presence of CPTIO have been reported associated to antibiotics with a ‘recognised’ NO release based mechanism of action against non-replicating bacteria as it is PA-824 (20). In our opinion, NO is a highly reactive radical that could interact with bacterial structures with higher affinity that it does with the scavenger, and therefore, the evaluation of the protective role of CPTIO might be underestimated. In addition, other authors have claimed that the results when using CPTIO as scavenger should be taken cautiously due to many factors that directly affect the stichometry of the scavenging reaction (86). On the other hand, the role of secondary metabolites, not yet characterized, cannot be ignored as an important piece in the mechanism of action of nNFs.
We found that the bioactivation of our set of nNFs is mediated by Mrx2 (Rv2466c), a novel nitroreductase class that also plays a critical role in the activation mechanism of thienopyrimidine (27,30) and nitrofuranylcalanolides (29) to kill M. tuberculosis. By the isolation and characterization of independent spontaneous resistant mutants against different nNF derivatives in both M. tuberculosis H37Rv and Beijing-GC1237 strains, we could obtain a large and refined list of SNPs and INDELs targeting the mrx2 gene. Among the ten mrx2 mutations studied, eight (H140D, R198G, T25R, S46R, D16G, W23R, L90P and Y91S) were found to be directly involved in the mechanism of resistance to nNF-C2 and nNF-C4. These mutations clearly predict structural defects in the thioredoxin fold, including the destabilization of the dimerization core and the CPWC motif, thus impairing the activity of Mrx2. Interestingly, some specific mrx2 mutations (D16G) and codon positions (Y91 and R198) were also reported by Negri and colleagues in spontaneous resistant M. tuberculosis mutants against nitrofuranylcalanolides (29), highlighting the importance of those mutations in the structure of the enzyme for its activity over different chemical structures.
In addition to the role of the nitroreductase Mrx2 in the bioactivation of the nNF-prodrugs, we identified a novel alternative mechanism of resistance to our nNF compounds based on mutations and INDELs found in the sigH gene. Experimentally, both the inactivation of sigH (ΔsigH) and the expression of the different sigH mutants in M. tuberculosis were found to confer resistance to the nNF-C2 and nNF-C4. Strikingly, the mutations found in sigH and mrx2 were exclusive, meaning that both genes might be involved in the same resistance mechanism pathway. This observation was supported by previous works (66,87) and by our own experimental data showing direct transcriptional regulation of mrx2 by sigH.
Sigma factors are interchangeable RNA polymerase components able to drive the enzyme on specific promoters, thus promoting the expression of specific sets of genes (88). The sigma factor SigH is a key regulator of an extensive transcriptional network that responds to oxidative, nitrosative, and heat stresses in M. tuberculosis (49,66,89). It orchestrates the transcription of 31 genes (90), and indirectly may control about one-fifth of the entire M. tuberculosis transcriptome (49). The SigH regulon might play an important role in the intracellular survival of M. tuberculosis since its expression is induced inside macrophages after infection (91). Furthermore, SigH expression is induced under the ‘enduring hypoxia response’ (92), and nutrient starvation stress (93), highlighting the importance of the SigH regulon in the transition of an actively replicating to a non-replicating state and reactivation of latent infection (93,94). Similarly, Mrx2 was postulated to play an important role in the oxidative and nitrosative stress response of M. tuberculosis (49,66). It has been shown to be essential for the survival of M. tuberculosis under H2O2 stress (30), and to confer partial protection to redox stress induced by menadione (29). In addition, mrx2 overexpression is induced in non-replicating M. tuberculosis (92,95), therefore, likely playing a role in the M. tuberculosis pathogenesis. Thus, it might be that it is the full SigH-Mrx2 stress response which should be considered as a team player in facing the hostile environment in the host by increasing the pathogens antioxidant capabilities. In that sense, the reported overexpression of sigH and mrx2 in non-replicating (92,93,95) and intracellular M. tuberculosis (91), would likely explain the high bactericidal activity observed for the nNF in this bacterial phenotypes. The extraordinary killing activity observed in the macrophage infection assay for most of the tested nNF derivatives was only comparable to a few of the currently used anti-tuberculosis drugs, such as isoniazid (96,97) or bedaquiline (98,99).
We identified Mrx2 as a key player in the bioactivation of nNFs into anti-tuberculosis compounds, but also demonstrated that the alternative sigma factor SigH plays a major role in order nNFs to achieve bactericidal potency. Using a Pip-ON inducible promoter system (57), we showed that Mrx2 expression is directly induced by SigH. The failure of SigHL58P and SigHV156D resistant mutants in inducing the mrx2 expression in this system, despite unaffected SigH expression levels, strengthens the notion that the SigH-induced Mrx2 expression is necessary for bioactivation of nNF compounds. The deficient induction of Mrx2 expression can likely be explained by impaired binding of SigH to the promoter region of mrx2 (87). Based on the crystal structure of SigH, the mutation L58P may potentially disrupt the stability of the α2 helix in the σH2 domain due to the inability of the proline to form a hydrogen bond which contributes to the stability of the helix structure in the wt protein. This in turn can impair the simultaneous interaction of the σH2 domain with promoter DNA and the RNA polymerase. Similarly, the mutation V156D, located in the σH4 domain, may cause the destabilization of the α8 helix and adjacent helices and thereby disrupting the interaction of the σH4 domain with the dsDNA and RNAP.
We believe that this mechanism of action might be shared by bacteria in which nNFs were found to be active, representing an universal bacterial killing mechanism. In this sense, nNFs spontaneous resistant M. marinum mutants were found to carry mutations in MMAR_0119, an hypothetical protein predicted to be a nitroreductase, and that according the study of Abshire and colleagues (100), is overexpressed together with sigH in an experimental set up aimed at mimicking a nutrient-deprived environment similar to that expected within macrophage during infection.
The generation of intracellular oxidative stress has been proposed as a common mechanism of action of several bactericidal antibiotics which target cell wall, translational process or DNA replication (101,102). Little is known about the role of sigma factors in the response of M. tuberculosis to antibiotics, but Yoo and colleagues described the induction of sigR in Streptomyces, an homologue of sigH in M. tuberculosis, by translation-inhibiting antibiotics (103). As the expression of SigH had been reported to be induced in response to nitric oxide (70), we investigated if the axis SigH-Mrx2 could get induced in the presence of nNFs. Indeed, we found the expression of both SigH and Mrx2 significantly induced in M. tuberculosis when treated with nNF-C2. It is therefore conceivable that the initial NO release from the nNF activation by pre-existent Mrx2 molecules will in turn promote a pleiotropic reaction, including the oxidative stress response mediated - in part - by the induction of sigH expression (104) and, consequently, the overexpression of mrx2. Therefore, the reduction of nNF-C2 is enhanced by elevated levels of intracellular mycothiol, SigH and Mrx2 under oxidizing conditions. We propose a positive feedback loop (Figure 7) in which the nNFs get activated more efficiently over time, due to the presence of more Mrx2 molecules that lead to the release of NO and other not yet identified secondary metabolites that eventually kill the bacteria. This mode of action could be also considered as a model of activation for thienopyrimidine and nitrofuranyl calanolide compounds. We hypothesize that during the bioactivation process of the nNFs, secondary metabolites are released and display a complementary mechanism of killing, probably by targeting an essential (65) or environment conditioned pathway (32,105) in the mycobacteria. Likewise, we have not yet addressed whether other genes in the sigH regulon could also have a role in the mechanism of action of these compounds, but future transcriptomic studies in response to these drugs, may unravel other mechanisms and/or genes involved.
Figure 7.

Proposed mechanism of action for the nitronaphthofuran nNF-C2. The nNF-C2 is reduced by Mrx2 which uses mycothiol (MSH) as cofactor. The reduction of the nNF-C2 nitro group leads to the release of nitric oxide (NO) and yet unknown secondary metabolites, which target the viability of M. tuberculosis and also promote the activation of a stress response in M. tuberculosis. In this programmed stress response, the expression of the alternative sigma factor SigH and its antisigma factor RshA - which is co-transcribed with SigH - are induced. Oxidative stress causes the release of the antisigma factor RshA from SigH, thus enabling SigH to exert its action as a transcriptional factor inducing the sigH regulon, in which mrx2 is included. Higher expression levels of mrx2 increase the ratio of nNF-C2 activation, that in turn enhances the production of NO and reactive metabolites, which keep inducing the sigH expression. We propose that the reduction of nNF-C2 is enhanced by elevated levels of intracellular mycothiol, SigH, and Mrx2 under oxidizing conditions (highlighted in blue). This positive feedback loop would explain the killing activity of nNF-C2 on replicating, non-replicating, and intracellular M. tuberculosis.
Taken altogether, our study shows that the transcriptomic mediated stress response, specifically the node sigH-mrx2 can be exploited by nNF derivatives to exert bactericidal activity against both actively replicating and non-replicating M. tuberculosis. A better understanding of the M. tuberculosis transcriptional factors and their regulons, which are part of the refined adaptation of the pathogen to its host, will definitely represent a new way to explore in the drug discovery process in the future. In that sense, targeting the SigH stress response may represent a novel and effective anti-tuberculosis strategy, especially against persistent M. tuberculosis, and thus enabling the aimed goal of shortening TB treatment.
DATA AVAILABILITY
Raw sequencing reads have been deposited in the European Nucleotide Archive (ENA) under accession number ENA: PRJEB48019.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: N.A.R., R.M., B.G. and M.E.G. conceived the project. N.A.R., L.C.M., F.B., A.M., M.S., M.C., G.S., S.B. and B.T. performed the experiments. N.A.R., L.C.M., F.B., C.B., M.S., A.N, A.M., G.S., H.M.L., B.T. and M.E.G. analyzed the results. N.A.R. and M.E.G. wrote the original draft. N.A.R., R.M., M.S., G.G., C.B. and M.E.G. reviewed and edited the final version of the manuscript.
Contributor Information
Laura Cioetto-Mazzabò, Department of Molecular Medicine, University of Padova, Padova 35122, Italy.
Francesca Boldrin, Department of Molecular Medicine, University of Padova, Padova 35122, Italy.
Claire Beauvineau, Chemical Library Institut Curie/CNRS, CNRS UMR9187, INSERM U1196 and CNRS UMR3666, INSERM U1193, Université Paris-Saclay, Orsay 91405, France.
Martin Speth, Department Biosciences, Faculty of Mathematics and Natural Sciences, University of Oslo, Oslo 0371, Norway.
Alberto Marina, Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), Bizkaia Technology Park, Derio 48160 Spain.
Amine Namouchi, Génétique Mycobactérienne, Institute Pasteur, Paris 75015, France; Centre for Ecological and Evolutionary Synthesis (CEES), Department of Biosciences, University of Oslo, Oslo 0371, Norway.
Greta Segafreddo, Department of Molecular Medicine, University of Padova, Padova 35122, Italy.
Mena Cimino, Génétique Mycobactérienne, Institute Pasteur, Paris 75015, France.
Sandrine Favre-Rochex, Génétique Mycobactérienne, Institute Pasteur, Paris 75015, France.
Seetha Balasingham, Department of Microbiology. Oslo University Hospital, Oslo 0450, Norway.
Beatriz Trastoy, Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), Bizkaia Technology Park, Derio 48160 Spain; Structural Glycobiology Laboratory, Biocruces Bizkaia Health Research Institute, Cruces University Hospital, Bizkaia 48903, Spain.
Hélène Munier-Lehmann, Département de Biologie Structurale et Chimie, Institut Pasteur, CNRS UMR3523, Université de Paris, Paris 75015, France.
Gareth Griffiths, Department Biosciences, Faculty of Mathematics and Natural Sciences, University of Oslo, Oslo 0371, Norway.
Brigitte Gicquel, Génétique Mycobactérienne, Institute Pasteur, Paris 75015, France; Department of Tuberculosis Control and Prevention, Shenzhen Nanshan Centre for Chronic Disease Control, Shenzhen 518054, China.
Marcelo E Guerin, Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), Bizkaia Technology Park, Derio 48160 Spain; Structural Glycobiology Laboratory, Biocruces Bizkaia Health Research Institute, Cruces University Hospital, Bizkaia 48903, Spain; IKERBASQUE, Basque Foundation for Science, Bilbao 48009, Spain.
Riccardo Manganelli, Department of Molecular Medicine, University of Padova, Padova 35122, Italy.
Noelia Alonso-Rodríguez, Department Biosciences, Faculty of Mathematics and Natural Sciences, University of Oslo, Oslo 0371, Norway; Génétique Mycobactérienne, Institute Pasteur, Paris 75015, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Seventh Framework Program Nanotherapeutics against Resistant Emerging Bacterial Pathogens [NAREB Project 604237]; European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie [609020 to N.A.R., 844905 to B.T.]; Norwegian Research Council [275873, 273319]; MINECO/FEDER EU contracts [PID2019-105649RB-I00]; Severo Ochoa Excellence Accreditation [SEV-2016-0644]; Basque Government [KK-2019/00076]; NIH [R01AI149297 to M.E.G.]; Innovative Medicines Initiative 2 Joint Undertaking (JU) [853989 to R.M.]. Funding for open access charge: University of Oslo.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cohen A., Mathiasen V.D., Schön T., Wejse C.. The global prevalence of latent tuberculosis: a systematic review and meta-analysis. Eur. Respir. J. 2019; 54:1900655. [DOI] [PubMed] [Google Scholar]
- 2. Connolly L.E., Edelstein P.H., Ramakrishnan L.. Why is long-term therapy required to cure tuberculosis?. PLoS Med. 2007; 4:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y., Yew W.W., Barer M.R.. Targeting persisters for tuberculosis control. Antimicrob. Agents Chemother. 2012; 56:2223–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Evangelopoulos D., da Fonseca J.D., Waddell S.J.. Understanding anti-tuberculosis drug efficacy: rethinking bacterial populations and how we model them. Int. J. Infect. Dis. 2015; 32:76–80. [DOI] [PubMed] [Google Scholar]
- 5. Young D.B., Gideon H.P., Wilkinson R.J.. Eliminating latent tuberculosis. Trends Microbiol. 2009; 17:183–188. [DOI] [PubMed] [Google Scholar]
- 6. Salgame P., Geadas C., Collins L., Jones-López E., Ellner J.J.. Latent tuberculosis infection–revisiting and revising concepts. Tuberculosis. 2015; 95:373–384. [DOI] [PubMed] [Google Scholar]
- 7. Behr M.A., Edelstein P.H., Ramakrishnan L.. Revisiting the timetable of tuberculosis. BMJ. 2018; 362:k2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sarathy J.P., Via L.E., Weiner D., Blanc L., Boshoff H., Eugenin E.A., Barry C.E., Dartois V.A.. Extreme drug tolerance of mycobacterium tuberculosis in Caseum. Antimicrob. Agents Chemother. 2018; 62:e02266-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. WHO consolidated guidelines on tuberculosis Module 4: Treatment - Drug-resistant tuberculosis treatment Geneva: World Health Organization; 2020. ISBN-13: 978-92-4-000704-8ISBN-13: 978-92-4-000705-5. [PubMed] [Google Scholar]
- 10. Treatment of drug-susceptible tuberculosis: rapid communication. Global Tuberculosis Programme. ISBN: 9789240028678.
- 11. Murugasu-Oei B., Dick T.. Bactericidal activity of nitrofurans against growing and dormant mycobacterium bovis BCG. J. Antimicrob. Chemother. 2000; 46:917–919. [DOI] [PubMed] [Google Scholar]
- 12. Rakesh Bruhn, D.F., Scherman M.S., Woolhiser L.K., Madhura D.B., Maddox M.M., Singh A.P., Lee R.B., Hurdle J.G., McNeil M.R.et al.. Pentacyclic nitrofurans with in vivo efficacy and activity against nonreplicating Mycobacterium tuberculosis. PLoS One. 2014; 9:e87909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Carvalho L.P.S., Lin G., Jiang X., Nathan C.. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J. Med. Chem. 2009; 52:5789–5792. [DOI] [PubMed] [Google Scholar]
- 14. Darby C.M., Nathan C.F.. Killing of non-replicating Mycobacterium tuberculosis by 8-hydroxyquinoline. J. Antimicrob. Chemother. 2010; 65:1424–1427. [DOI] [PubMed] [Google Scholar]
- 15. Grant S.S., Kawate T., Nag P.P., Silvis M.R., Gordon K., Stanley S.A., Kazyanskaya E., Nietupski R., Golas A., Fitzgerald M.et al.. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem. Biol. 2013; 8:2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salina E., Ryabova O., Kaprelyants A.. New 2-thiopyridines as potential candidates for killing both actively growing and dormant Mycobacterium tuberculosis cells. Antimicrob. Agents Chemother. 2014; 58:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hurdle J.G., Lee R.B., Budha N.R., Carson E.I., Qi J., Scherman M.S., Cho S.H., McNeil M.R., Lenaerts A.J., Franzblau S.G.et al.. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J. Antimicrob. Chemother. 2008; 62:1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsumoto M., Hashizume H., Tomishige T., Kawasaki M., Tsubouchi H., Sasaki H., Shimokawa Y., Komatsu M.. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006; 3:e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stover C.K., Warrener P., VanDevanter D.R., Sherman D.R., Arain T.M., Langhorne M.H., Anderson S.W., Towell J.A., Yuan Y., McMurray D.N.et al.. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000; 405:962–966. [DOI] [PubMed] [Google Scholar]
- 20. Singh R., Manjunatha U., Boshoff H.I.M., Ha Y.H., Niyomrattanakit P., Ledwidge R., Dowd C.S., Lee I.Y., Kim P., Zhang L.et al.. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008; 322:1392–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cellitti S.E., Shaffer J., Jones D.H., Mukherjee T., Gurumurthy M., Bursulaya B., Boshoff H.I., Choi I., Nayyar A., Lee Y.S.et al.. Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure. 2012; 20:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Manjunatha U., Boshoff H.I., Barry C.E.. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun. Integr. Biol. 2009; 2:215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manjunatha U.H., Boshoff H., Dowd C.S., Zhang L., Albert T.J., Norton J.E., Daniels L., Dick T., Pang S.S., Barry C.E.. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu Y., Coates A.R.M.. Mycobacterium tuberculosis ACG gene is required for growth and virulence in vivo. PLoS One. 2011; 6:e20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Purkayastha A., McCue Lee A., McDonough Kathleen A.. Identification of a Mycobacterium tuberculosis putative classical nitroreductase gene whose expression is coregulated with that of the acr gene within macrophages, in standing versus shaking cultures, and under low oxygen conditions. Infect. Immun. 2002; 70:1518–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peddireddy V., Doddam S.N., Qureshi I.A., Yerra P., Ahmed N.. A putative nitroreductase from the DosR regulon of Mycobacterium tuberculosis induces pro-inflammatory cytokine expression via TLR2 signaling pathway. Sci. Rep. 2016; 6:24535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Albesa-Jové D., Chiarelli L.R., Makarov V., Pasca M.R., Urresti S., Mori G., Salina E., Vocat A., Comino N., Mohorko E.et al.. Rv2466c mediates the activation of TP053 to kill replicating and non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2014; 9:1567–1575. [DOI] [PubMed] [Google Scholar]
- 28. Albesa-Jové D., Comino N., Tersa M., Mohorko E., Urresti S., Dainese E., Chiarelli L.R., Pasca M.R., Manganelli R., Makarov V.et al.. The redox state regulates the conformation of Rv2466c to activate the antitubercular prodrug TP053. J. Biol. Chem. 2015; 290:31077–31089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Negri A., Javidnia P., Mu R., Zhang X., Vendome J., Gold B., Roberts J., Barman D., Ioerger T., Sacchettini J.C.et al.. Identification of a mycothiol-Dependent nitroreductase from Mycobacterium tuberculosis. ACS Infect Dis. 2018; 4:771–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosado L.A., Wahni K., Degiacomi G., Pedre B., Young D., de la Rubia A.G., Boldrin F., Martens E., Marcos-Pascual L., Sancho-Vaello E.et al.. The antibacterial prodrug activator Rv2466c is a mycothiol-dependent reductase in the oxidative stress response of Mycobacterium tuberculosis. J. Biol. Chem. 2017; 292:13097–13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zahrt T.C., Deretic V.. Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:12706–12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bretl D.J., Demetriadou C., Zahrt T.C.. Adaptation to environmental stimuli within the host: two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol. Mol. Biol. Rev. 2011; 75:566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warner D.F., Mizrahi V.. The survival kit of Mycobacterium tuberculosis. Nat. Med. 2007; 13:282–284. [DOI] [PubMed] [Google Scholar]
- 34. Manganelli R., Provvedi R., Rodrigue S., Beaucher J., Gaudreau L., Smith I.. Sigma factors and global gene regulation in Mycobacterium tuberculosis. J. Bacteriol. 2004; 186:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He H., Hovey R., Kane J., Singh V., Zahrt T.C.. MprAB is a stress-responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J. Bacteriol. 2006; 188:2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Flentie K., Garner A.L., Stallings C.L.. Mycobacterium tuberculosis transcription machinery: ready to respond to host attacks. J. Bacteriol. 2016; 198:1360–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leistikow R.L., Morton R.A., Bartek I.L., Frimpong I., Wagner K., Voskuil M.I.. The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J. Bacteriol. 2010; 192:1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buchieri M.V., Cimino M., Rebollo-Ramirez S., Beauvineau C., Cascioferro A., Favre-Rochex S., Helynck O., Naud-Martin D., Larrouy-Maumus G., Munier-Lehmann H.et al.. Nitazoxanide analogs require nitroreduction for antimicrobial activity in mycobacterium smegmatis. J. Med. Chem. 2017; 60:7425–7433. [DOI] [PubMed] [Google Scholar]
- 39. Hibert M.F. French/European academic compound library initiative. Drug Discov. Today. 2009; 14:723–725. [DOI] [PubMed] [Google Scholar]
- 40. Einhorn J., Demerseman P., Royer R., Cavier R., Gayral P.. Effect of a substituent in the β position of the heterocycle on the antibacterial and parasiticidal properties of 2-nitronaphtho[2.1-b]furans. Eur. J. Med. Chem. 1984; 19:405–410. [Google Scholar]
- 41. Arrault A., Touzeau F., Guillaumet G., Mérour J.-Y.. A straightforward synthesis of 1,2-Dihydronaphtho[2,1-b]furans from 2-Naphthols. Synthesis. 1999; 1999:1241–1245. [Google Scholar]
- 42. Einhorn J., Demerseman P., Royer R., Cavier R.. Research on nitro-derivatives of biological interest. XXX. Methylated 2-nitronaphthofurans, synthesis and activity against microorganisms. Eur. J. Med. Chem. 1983; 18:175–180. [Google Scholar]
- 43. Buisson J.P., Royer R., Gayral P.. Study of nitrated derivatives of biological interest. XXXV. Synthesis and preliminary tests against bacteria and protozoa of polymethoxy 2-nitronaphtho [2,1-b]furan derivatives. Eur. J. Med. Chem. 1984; 19:249–253. [Google Scholar]
- 44. Bilger C., Demerseman P., Buisson JP., Royer R., Gayral P., Fourniat J.. Modifications of biological activity following the addition of a second nitrofuran ring to nitroarenofuran homocycles. Eur. J. Med. Chem. 1987; 22:213–219. [Google Scholar]
- 45. Einhorn J., Demerseman P., Royer R.. Research on benzofurans. LXX. Synthesis of 2-nitro-3-methoxybenzofurans and the corresponding naphthofurans. J. Heterocycl. Chem. 1985; 22:1243–1247. [Google Scholar]
- 46. López-Ramos M., Prudent R., Moucadel V., Sautel C.F., Barette C., Lafanechère L., Mouawad L., Grierson D., Schmidt F., Florent J.-C.et al.. New potent dual inhibitors of CK2 and Pim kinases: discovery and structural insights. FASEB J. 2010; 24:3171–3185. [DOI] [PubMed] [Google Scholar]
- 47. Pierre R.R.A.B. Studies on nitro derivatives of biological interest. Synthesis of methoxy- or halo-2-nitronaphthofurans. Eur. J. Med. Chem. 1980; 15:275–278. [Google Scholar]
- 48. Einhorn J., Lamotte G., Buisson J. Studies on nitrated derivatives of biological interest. XXXIV. Synthesis and microorganism effect of 2-nitronaphtho[2,1-b]furancarboxylic acid derivatives. Eur. J. Med. Chem. 1984; 19:143–147. [Google Scholar]
- 49. Manganelli R., Voskuil M.I., Schoolnik G.K., Dubnau E., Gomez M., Smith I.. Role of the extracytoplasmic-function sigma factor sigma(H) in Mycobacterium tuberculosis global gene expression. Mol. Microbiol. 2002; 45:365–374. [DOI] [PubMed] [Google Scholar]
- 50. Caminero J.A., Pena M.J., Campos-Herrero M.I., Rodríguez J.C., García I., Cabrera P., Lafoz C., Samper S., Takiff H., Afonso O.et al.. Epidemiological evidence of the spread of a Mycobacterium tuberculosis strain of the Beijing genotype on Gran Canaria Island. Am. J. Respir. Crit. Care Med. 2001; 164:1165–1170. [DOI] [PubMed] [Google Scholar]
- 51. Palomino J.-C., Martin A., Camacho M., Guerra H., Swings J., Portaels F.. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002; 46:2720–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Betts J.C., Lukey P.T., Robb L.C., McAdam R.A., Duncan K.. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002; 43:717–731. [DOI] [PubMed] [Google Scholar]
- 53. Gold B., Roberts J., Ling Y., Quezada L.L., Glasheen J., Ballinger E., Somersan-Karakaya S., Warrier T., Warren J.D., Nathan C.. Rapid, semiquantitative assay to discriminate among compounds with activity against replicating or nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015; 59:6521–6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Speth M.T., Repnik U., Müller E., Spanier J., Kalinke U., Corthay A., Griffiths G.. Poly(I:c)-Encapsulating nanoparticles enhance innate immune responses to the tuberculosis vaccine Bacille Calmette-Guérin (BCG) via synergistic activation of Innate immune receptors. Mol. Pharm. 2017; 14:4098–4112. [DOI] [PubMed] [Google Scholar]
- 55. Larsen M.H., Biermann K., Tandberg S., Hsu T., Jacobs W.R. Jr. Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 2007; Chapter 10:Unit 10A.2. [DOI] [PubMed] [Google Scholar]
- 56. Chen S., Zhou Y., Chen Y., Gu J.. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boldrin F., Degiacomi G., Serafini A., Kolly G.S., Ventura M., Sala C., Provvedi R., Palù G., Cole S.T., Manganelli R.. Promoter mutagenesis for fine-tuning expression of essential genes in Mycobacterium tuberculosis. Microb. Biotechnol. 2018; 11:238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boldrin F., Casonato S., Dainese E., Sala C., Dhar N., Palù G., Riccardi G., Cole S.T., Manganelli R.. Development of a repressible mycobacterial promoter system based on two transcriptional repressors. Nucleic Acids Res. 2010; 38:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maciag A., Dainese E., Rodriguez G.M., Milano A., Provvedi R., Pasca M.R., Smith I., Palù G., Riccardi G., Manganelli R.. Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J. Bacteriol. 2007; 189:730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Manganelli R., Dubnau E., Tyagi S., Kramer F.R., Smith I.. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol. Microbiol. 1999; 31:715–724. [DOI] [PubMed] [Google Scholar]
- 61. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 62. Misko T.P., Schilling R.J., Salvemini D., Moore W.M., Currie M.G.. A fluorometric assay for the measurement of nitrite in biological samples. Anal. Biochem. 1993; 214:11–16. [DOI] [PubMed] [Google Scholar]
- 63. Neniskyte U., Brown G.C.. Analysis of microglial production of reactive oxygen and nitrogen species. Methods Mol. Biol. 2013; 1041:103–111. [DOI] [PubMed] [Google Scholar]
- 64. Sassetti C.M., Boyd D.H., Rubin E.J.. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003; 48:77–84. [DOI] [PubMed] [Google Scholar]
- 65. Chiarelli L.R., Salina E.G., Mori G., Azhikina T., Riabova O., Lepioshkin A., Grigorov A., Forbak M., Madacki J., Orena B.S.et al.. New insights into the mechanism of action of the thienopyrimidine antitubercular prodrug TP053. ACS Infect. Dis. 2020; 6:313–323. [DOI] [PubMed] [Google Scholar]
- 66. Raman S., Song T., Puyang X., Bardarov S., Jacobs W.R. Jr, Husson R.N. The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J. Bacteriol. 2001; 183:6119–6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Forti F., Crosta A., Ghisotti D.. Pristinamycin-inducible gene regulation in mycobacteria. J. Biotechnol. 2009; 140:270–277. [DOI] [PubMed] [Google Scholar]
- 68. Li L., Fang C., Zhuang N., Wang T., Zhang Y.. Structural basis for transcription initiation by bacterial ECF σ factors. Nat. Commun. 2019; 10:1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. von Heijne G. Proline kinks in transmembrane alpha-helices. J. Mol. Biol. 1991; 218:499–503. [DOI] [PubMed] [Google Scholar]
- 70. Voskuil M.I., Bartek I.L., Visconti K., Schoolnik G.K.. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2011; 2:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Malik-Kale P., Winfree S., Steele-Mortimer O.. The bimodal lifestyle of intracellular Salmonella in epithelial cells: replication in the cytosol obscures defects in vacuolar replication. PLoS One. 2012; 7:e38732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dandekar T., Astrid F., Jasmin P., Hensel M.. Salmonella enterica: a surprisingly well-adapted intracellular lifestyle. Front. Microbiol. 2012; 3:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cano V., March C., Insua J.L., Aguiló N., Llobet E., Moranta D., Regueiro V., Brennan G.P., Millán-Lou M.I., Martín C.et al.. Klebsiella pneumoniae survives within macrophages by avoiding delivery to lysosomes. Cell. Microbiol. 2015; 17:1537–1560. [DOI] [PubMed] [Google Scholar]
- 74. Bengoechea J.A., Sa Pessoa J.. Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol. Rev. 2019; 43:123–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Corbett M.D., Wei C., Corbett B.R.. Nitroreductase-dependent mutagenicity of p-nitrophenylhydroxylamine and its N-acetyl and N-formyl hydroxamic acids. Carcinogenesis. 1985; 6:727–732. [DOI] [PubMed] [Google Scholar]
- 76. Ashtekar D.R., Costa-Perira R., Nagrajan K., Vishvanathan N., Bhatt A.D., Rittel W.. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1993; 37:183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Machado D., Girardini M., Viveiros M., Pieroni M.. Challenging the drug-Likeness dogma for new drug discovery in tuberculosis. Front. Microbiol. 2018; 9:1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cassar S., Adatto I., Freeman J.L., Gamse J.T., Iturria I., Lawrence C., Muriana A., Peterson R.T., Van Cruchten S., Zon L.I.. Use of zebrafish in drug discovery toxicology. Chem. Res. Toxicol. 2020; 33:95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Grigor’ev N.B., Chechekin G.V., Arzamaztsev A.P., Levina V.I., Granik V.G.. Nitrogen oxide generation during reduction of nitrofuran antibacterial drugs. Chem. Heterocycl. Compd. 1999; 35:788–791. [Google Scholar]
- 80. Tangallapally R.P., Yendapally R., Daniels A.J., Lee R.E.B., Lee R.E.. Nitrofurans as novel anti-tuberculosis agents: identification, development and evaluation. Curr. Top. Med. Chem. 2007; 7:509–526. [DOI] [PubMed] [Google Scholar]
- 81. Vicente E., Villar R., Burguete A., Solano B., Pérez-Silanes S., Aldana I., Maddry J.A., Lenaerts A.J., Franzblau S.G., Cho S.-H.et al.. Efficacy of quinoxaline-2-carboxylate 1,4-di-N-oxide derivatives in experimental tuberculosis. Antimicrob. Agents Chemother. 2008; 52:3321–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brown G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta. 2001; 1504:46–57. [DOI] [PubMed] [Google Scholar]
- 83. Rhee K.Y., Erdjument-Bromage H., Tempst P., Nathan C.F.. S-nitroso proteome of Mycobacterium tuberculosis: enzymes of intermediary metabolism and antioxidant defense. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jamaati H., Mortaz E., Pajouhi Z., Folkerts G., Movassaghi M., Moloudizargari M., Adcock I.M., Garssen J.. Nitric oxide in the pathogenesis and treatment of tuberculosis. Front. Microbiol. 2017; 8:2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jones-Carson J., Yahashiri A., Kim J.-S., Liu L., Fitzsimmons L.F., Weiss D.S., Vázquez-Torres A.. Nitric oxide disrupts bacterial cytokinesis by poisoning purine metabolism. Sci Adv. 2020; 6:eaaz0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Goldstein S., Russo A., Samuni A.. Reactions of PTIO and carboxy-PTIO with *NO, *NO2, and O2-*. J. Biol. Chem. 2003; 278:50949–50955. [DOI] [PubMed] [Google Scholar]
- 87. Sharp J.D., Singh A.K., Park S.T., Lyubetskaya A., Peterson M.W., Gomes A.L.C., Potluri L.-P., Raman S., Galagan J.E., Husson R.N.. Comprehensive definition of the SigH regulon of Mycobacterium tuberculosis reveals transcriptional control of diverse stress responses. PLoS One. 2016; 11:e0152145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ishihama A. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol. Rev. 2010; 34:628–645. [DOI] [PubMed] [Google Scholar]
- 89. Coulson G.B., Johnson B.K., Zheng H., Colvin C.J., Fillinger R.J., Haiderer E.R., Hammer N.D., Abramovitch R.B.. Targeting Mycobacterium tuberculosis sensitivity to thiol stress at acidic pH kills the bacterium and potentiates antibiotics. Cell Chem Biol. 2017; 24:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kaushal D., Schroeder B.G., Tyagi S., Yoshimatsu T., Scott C., Ko C., Carpenter L., Mehrotra J., Manabe Y.C., Fleischmann R.D.et al.. Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative sigma factor, SigH. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:8330–8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Graham J.E., Clark-Curtiss J.E.. Identification of Mycobacterium tuberculosis rnas synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. U.S.A. 1999; 96:11554–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rustad T.R., Harrell M.I., Liao R., Sherman D.R.. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS One. 2008; 3:e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Matern W.M., Rifat D., Bader J.S., Karakousis P.C.. Gene enrichment analysis reveals major regulators of Mycobacterium tuberculosis gene expression in two models of antibiotic tolerance. Front. Microbiol. 2018; 9:610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Du P., Sohaskey C.D., Shi L.. Transcriptional and physiological changes during Mycobacterium tuberculosis reactivation from non-replicating persistence. Front. Microbiol. 2016; 7:1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rao S.P.S., Camacho L., Huat Tan B., Boon C., Russel D.G., Dick T., Pethe K.. Recombinase-based reporter system and antisense technology to study gene expression and essentiality in hypoxic nonreplicating mycobacteria. FEMS Microbiol. Lett. 2008; 284:68–75. [DOI] [PubMed] [Google Scholar]
- 96. Hartkoorn R.C., Chandler B., Owen A., Ward S.A., Bertel Squire S., Back D.J., Khoo S.H.. Differential drug susceptibility of intracellular and extracellular tuberculosis, and the impact of P-glycoprotein. Tuberculosis. 2007; 87:248–255. [DOI] [PubMed] [Google Scholar]
- 97. Christophe T., Jackson M., Jeon H.K., Fenistein D., Contreras-Dominguez M., Kim J., Genovesio A., Carralot J.-P., Ewann F., Kim E.H.et al.. High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog. 2009; 5:e1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dhillon J., Andries K., Phillips P.P.J., Mitchison D.A.. Bactericidal activity of the diarylquinoline TMC207 against Mycobacterium tuberculosis outside and within cells. Tuberculosis. 2010; 90:301–305. [DOI] [PubMed] [Google Scholar]
- 99. Giraud-Gatineau A., Coya J.M., Maure A., Biton A., Thomson M., Bernard E.M., Marrec J., Gutierrez M.G., Larrouy-Maumus G., Brosch R.et al.. The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. Elife. 2020; 9:e55692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Abshire C.F., Prasai K., Soto I., Shi R., Concha M., Baddoo M., Flemington E.K., Ennis D.G., Scott R.S., Harrison L.. Exposure of mycobacterium marinum to low-shear modeled microgravity: effect on growth, the transcriptome and survival under stress. NPJ Microgravity. 2016; 2:16038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dwyer D.J., Belenky P.A., Yang J.H., MacDonald I.C., Martell J.D., Takahashi N., Chan C.T.Y., Lobritz M.A., Braff D., Schwarz E.G.et al.. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2100–E2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kohanski M.A., Dwyer D.J., Hayete B., Lawrence C.A., Collins J.J.. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007; 130:797–810. [DOI] [PubMed] [Google Scholar]
- 103. Yoo J.-S., Oh G.-S., Ryoo S., Roe J.-H.. Induction of a stable sigma factor SigR by translation-inhibiting antibiotics confers resistance to antibiotics. Sci. Rep. 2016; 6:28628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Voskuil M.I., Schnappinger D., Visconti K.C., Harrell M.I., Dolganov G.M., Sherman D.R., Schoolnik G.K.. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 2003; 198:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schnappinger D., Ehrt S., Voskuil M.I., Liu Y., Mangan J.A., Monahan I.M., Dolganov G., Efron B., Butcher P.D., Nathan C.et al.. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 2003; 198:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing reads have been deposited in the European Nucleotide Archive (ENA) under accession number ENA: PRJEB48019.