

Abstract
Pyroptosis is a mechanism of programmed, necrotic cell death mediated by gasdermins, a family of pore-forming proteins. Caspase-1 activates gasdermin D (GSDMD) under inflammatory conditions, whereas caspase-3 activates GSDME under apoptotic conditions, such as those induced by chemotherapy. These pathways are thought to be separate. However, we found that they are part of an integrated network of gatekeepers that enables pyroptotic cell death. We observed that GSDMD was the primary pyroptotic mediator in cultured blood cells in response to doxorubicin and etoposide, two common chemotherapies for hematopoietic malignancies. Upon treatment, the channel protein pannexin-1 (PANX1), which is stimulated by the initiation of apoptosis, increased membrane permeability to induce K+ efflux-driven activation of the NLRP3 inflammasome and GSDMD. However, either PANX1 or GSDME could also be the primary mediator of chemotherapy-induced pyroptosis when present at higher amounts. The most abundant pore-forming protein in acute myeloid leukemias from patients predicted the cell death pathway in response to chemotherapy. This interconnected network is thus a multi-step switch that converts apoptosis to pyroptosis, which may be clinically desirable to stimulate antitumor immunity in patients but undesirable in those at risk of developing cytokine release syndrome.
INTRODUCTION
Pyroptosis is historically defined as caspase-1 (CASP1)-dependent, pro-inflammatory programmed cell death in response to Salmonella typhimurium, ischemia, or ATP depletion (1). The CASP1 substrate gasdermin D (GSDMD) was recently found to be a common downstream pyroptotic effector that causes plasma membrane rupture through pore formation (2–6). Upstream triggers of GSDMD pyroptosis converge at the inflammasome complex (7), a macromolecular signaling and effector platform that converts recognition of ligands or cellular dyshomeostasis into an activated inflammatory cascade initiated by transient CASP1 autoproteolytic activation (8). CASP1 cleavage of GSDMD releases an autoinhibited state to initiate GSDMD N-terminus pore formation at membranes. Hereupon, pyroptosis can result but is not guaranteed: hyperactivated, non-lytic GSDMD activity can instead act as a conduit for interleukin 1-beta (IL-1β) and normally membrane-impermeant dyes in a sublytic state (9–11).
Other gasdermins can also produce lytic cell death; in fact, pyroptosis is currently defined as any gasdermin-driven necrotic cell death (12). For example, the related family member GSDME was shown to be activated under normally apoptotic circumstances, such as upon chemotherapy treatment or immunogenic cell death (13–15), to similarly bring about pyroptosis after activating cleavage by the apoptotic executioner caspase CASP3. Whether GSDME cleavage always results in lysis is a matter of debate; multiple studies have found that GSDME cleavage did not associate with necrotic morphology or release of intracellular contents (16, 17) or that it was dispensable for such (18–20).
Many studies of gasdermin function are usually constrained by the assumption that each is the final effector of a cell death decision: GSDMD for inflammasome activation, GSDME for apoptotic cascade activation. Several studies have explored secondary CASP8-initiated GSDME cleavage at unproductive inflammasomes, for example (21–23), but GSDMD activation during apoptosis has been less well investigated for two-fold reasons: GSDME is usually assumed to be the driver of lytic death in such circumstances, and CASP3 performs inhibitory cleavage of GSDMD (24)
In this study, we found that in addition to executing distinct lytic pathways, pore-forming proteins and channels are woven in an interconnected lattice of sequential activation such that apoptotic cell death can be subverted to pyroptosis at multiple points. Whereas at high concentrations, GSDME exerted primacy in apoptosis-to-pyroptosis conversion, at medium to low concentrations it was neither necessary nor sufficient for pyroptosis, supporting the concept of a minimal pore concentration for pyroptosis. Further, we found that CASP1-cleaved GSDMD can substitute for lytic GSDME through a non-canonical inflammasome pathway mediated by induction of the channel pannexin-1 (PANX1). We additionally identified a distinct PANX1-dependent pathway to pyroptosis that did not require GSDMD. Finally, we classified patient samples of acute myeloid leukemia (AML) into GSDMD-, GSDME-, and PANX1-dominant lysis categories by respective protein abundance and demonstrated expected sensitivities to cell death-blocking agents, suggesting that this framework might be leveraged for personalized medicine. Our work thus establishes a model whereby any of these three proteins can mediate pyroptosis upon activation of general intrinsic apoptosis and therefore challenges the assumptions that GSDME is the sole executioner of chemotherapy-induced pyroptosis.
RESULTS
GSDME is sufficient but dispensable for chemotherapy-induced lysis
To address the role of GSDME in chemotherapy-induced pyroptosis, we deleted it with Cas9-mediated gene editing in megakaryoblastic MEG-01 cells and erythroblastic K-562 cells (both of which harbor the BCR-ABL1 oncogene) as well as in THP-1 monocytic leukemia cells. Extracellular accumulation of lactate dehydrogenase activity, an approximate measure of cell lysis, was ameliorated by GSDME ablation in MEG-01 cells (which express a relatively high amount of GSDME) in response to dasatinib, a tyrosine kinase inhibitor used in the treatment of BCR-ABL1-positive leukemias (Fig. 1, A and B). In contrast, K-562 cells (which express a relatively low amount of GSDME) did not exhibit GSDME-dependent lysis (Fig. 1A, B). Unexpectedly, GSDME deletion in THP-1 cells did not affect cell lysis (Fig. 1C) in response to doxorubicin or etoposide, two clinically used topoisomerase-targeting chemotherapy drugs that cause both DNA and chromatin damage leading to initiation of intrinsic apoptosis through the CASP9 apoptosome (25, 26). Despite this, the CASP9-CASP3 apoptotic cascade was active in both control THP-1 and GSDME−/− cells, and GSDME was cleaved to its active N-terminal p30 fragment in wild-type cells (Fig. 1D). Slight activation of CASP8 was also detected (Fig. 1D).
Figure 1. GSDME is sufficient but dispensable for chemotherapy-induced lysis.
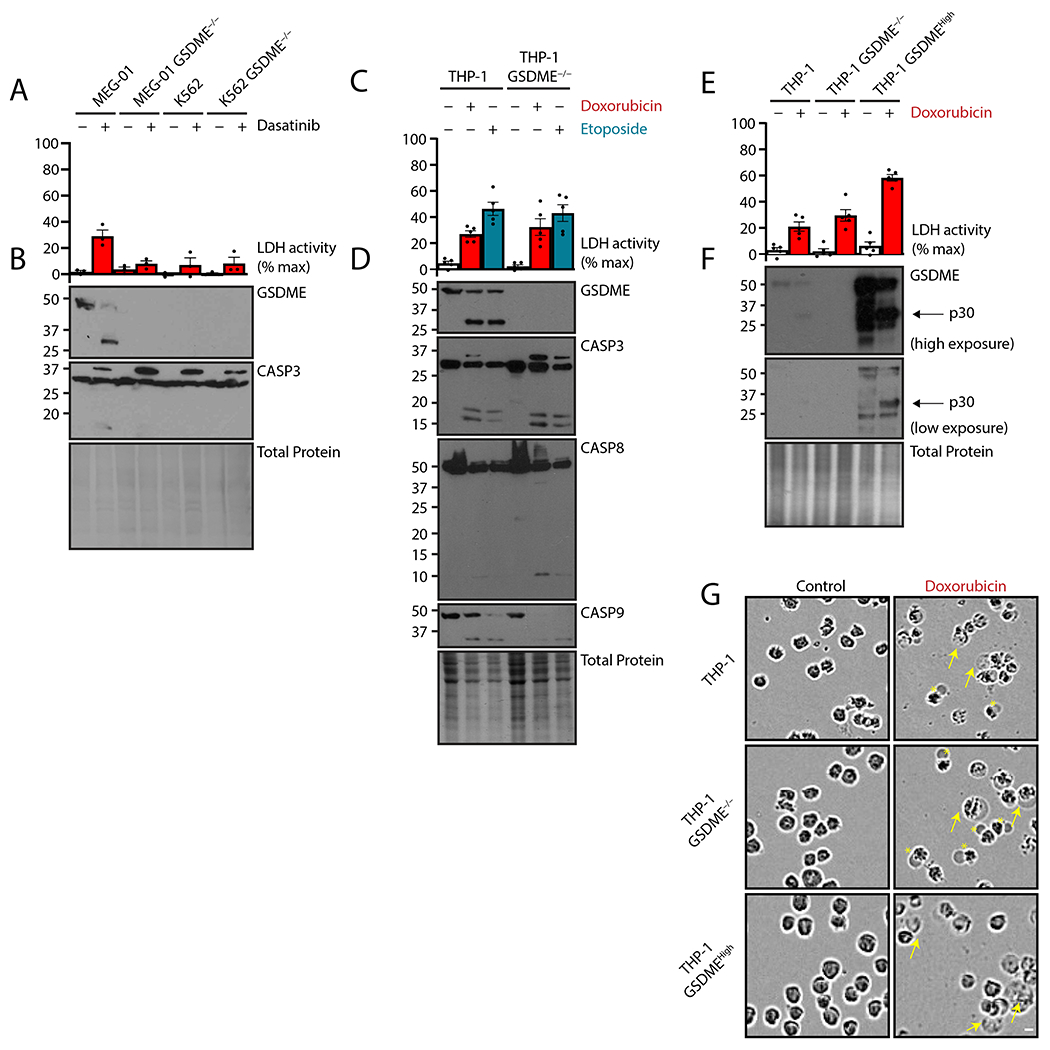
(A to G) MEG-01 megakaryoblastic cells, K-562 erythroblastic cells, or respective GSDME−/− cells were treated with tyrosine kinase inhibitor dasatinib (50 nM) for 24 hours (A and B), and naive human THP-1 monocytic cells, GSDME−/− cells, or GSDME−/− cells overexpressing GSDME (GSDMEHigh) were treated with topoisomerase poisons doxorubicin (2 μM) or etoposide (50 μM) for 24 hours (C to G). Plasma membrane rupture was quantified by lactate dehydrogenase assay of cell-free supernatant (A, C, and E), and lysates were assessed by immunoblot (B, D, and F). Micrographs of THP-1 cells described in (C to F) are representative of ≥ 3 biological replicates (G). Yellow arrows indicate traditionally pyroptotic cells. Asterisks indicate atypical cells with large single membrane blebs. Scale bar represents 10 μm.Graph dots represent mean of ≥ 2 technical replicates per biological replicate; graph bars represent mean ± standard error of ≥ 3 biological replicates per experiment; and immunoblots are representative of at least 3 biological replicates.
To confirm that GSDME is capable of mediating pyroptotic conversion at higher protein concentrations, we reintroduced GSDME in null THP-1 cells with lentivirus (23). Reconstitution of GSDME expression to supra-endogenous levels doubled the extent of cell lysis triggered by the chemotherapy drugs (Fig. 1E, F). Imaging of treated cells showed typical pyroptotic morphology in these GSDMEHigh cells, as expected (Fig. 1G). In both wild-type cells and GSDME−/− cells, however, we observed fewer of these pyroptotic structures and the presence of single cell-sized balloon morphology. These data suggested that although GSDME was able to induce pyroptosis, another source of lytic cell death was engaged by these cells in response to chemotherapeutic drug challenge.
CASP1-NLRP3-GSDMD activation mediates chemotherapy-induced pyroptosis
The study of inflammatory, lytic cell death centers on GSDMD, which is typically activated by CASP1 after stimulation of an inflammasome or by CASP4 after intracellular LPS detection (2–4). Based on the cell morphology above (Fig. 1G), we tested whether GSDMD was activated after chemotherapy treatment in THP-1 cells (Fig. 2, A and B). Immunoblotting revealed a majority presence of GSDMD p43 (Asp87-His484), a deactivated fragment generated by CASP3-mediated cleavage (24). However, we also observed a p30 fragment, albeit at lower stoichiometric ratios that would be expected from direct inflammasome-induced pyroptosis. Notably, we observed a p20 fragment with a polyclonal antibody to an N-terminal epitope, indicating that this was likely the Asp87-Asp275 fragment resulting from double CASP3 and CASP1 cleavage. Cell death in response to both topoisomerase inhibitors in GSDMD−/− cells was substantially lower than that in wild-type cells (Fig. 2A), whereas the CASP3-GSDME axis was unaffected (Fig. 2B). Further, doxorubicin-treated cells lacking GSDMD did not display pyroptotic morphology like wild-type cells (Fig. 2C), although they did manifest the small bubble morphology seen in a subset of wild-type and GSDME−/− cells (Fig. 1G). These data suggested that GSDMD, rather than GSDME, was the ultimate effector of pyroptosis in THP-1 cells treated with chemotherapeutic agents.
Figure 2. CASP1-NLRP3-GSDMD activation licenses chemotherapy pyroptosis.
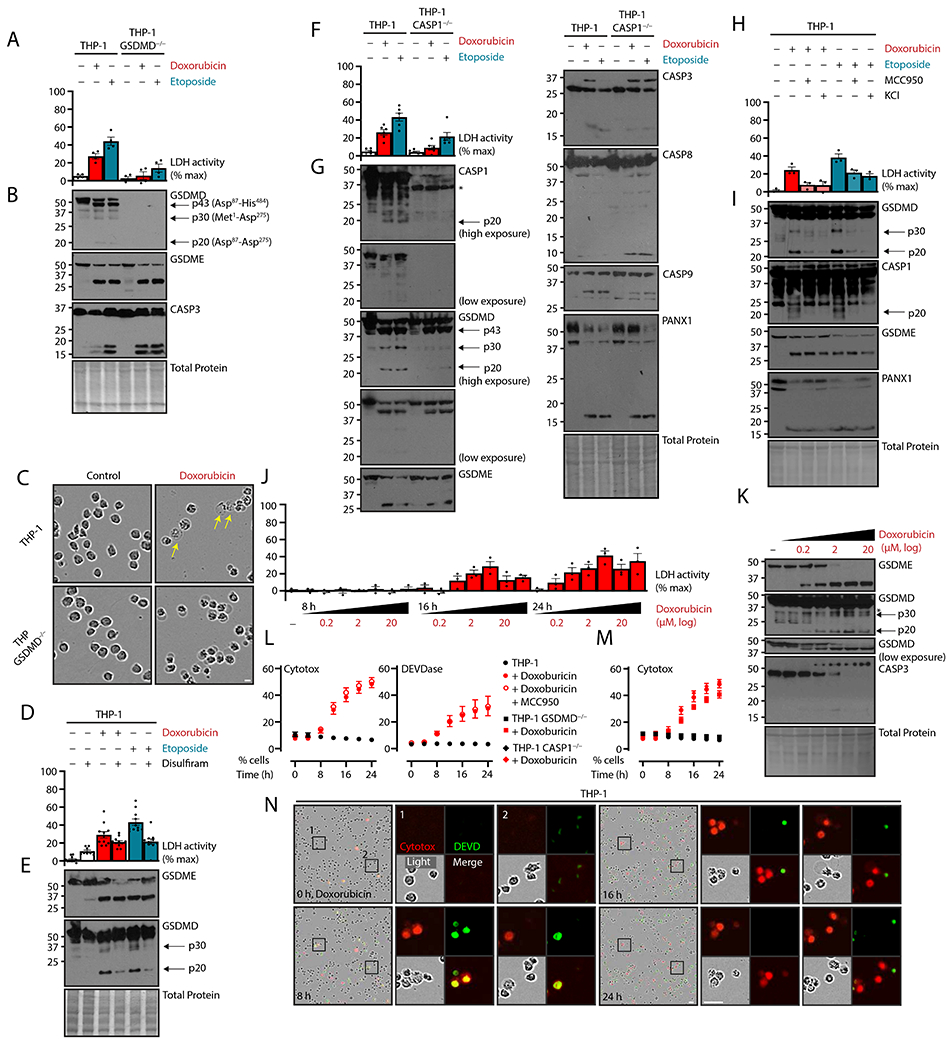
(A to I) Naive human THP-1 monocytic cells or indicated deletion mutants were treated with topoisomerase poisons doxorubicin (2 μM) or etoposide (50 μM) for 24 hours. Concurrent GSDMD inhibitor disulfiram (30 μM), NLRP3 inflammasome inhibitor MCC950 (10 μM), NLRP3 inflammasome antagonism by extracellular potassium chloride (50 mM) were added as indicated. Plasma membrane rupture was quantified by lactate dehydrogenase assay of cell-free supernatant (A, D, F, H), and lysates were assessed by immunoblot (B, E, G, I). Micrographs of THP-1 cells described in (A, B) are representative of ≥ 3 biological replicates (C). Yellow arrows indicate traditionally pyroptotic cells. Scale bar, 10 μm. (J and K) THP-1 cells treated at half-logarithm titrations of doxorubicin at 8, 16, and 24 h were assessed for plasma membrane rupture by lactate dehydrogenation assay and assessed by immunoblot. (L to N) THP-1 cells and indicated knockouts were treated with doxorubicin and inhibitors as in (A to I) and visualized with live cell imaging. Plasma membrane permeability was quantified by cell-impermeable Cytotox dye uptake, and apoptotic caspase activity was assayed by a dye with caspase-cleavable DEVD motif (L, M). Control and doxorubicin-treated THP-1 data are repeated in these panels. True-color micrographs of treated cells are shown (N) and representative of ≥ 2 biological replicates. Scale bar, 10 μm. Scatter plot points represent mean ± standard error of ≥ 3 technical replicates across ≥ 2 biological replicates per experiment; graph dots represent mean of ≥ 2 technical replicates per biological replicate; graph bars represent mean ± standard error of ≥ 3 biological replicates per experiment; and immunoblots are representative of ≥ 3 biological replicates.
To further test the primacy of GSDMD as an effector of pyroptosis in these conditions, we co-treated with the cysteine-reactive drug disulfiram, which was shown to inhibit GSDMD pore formation, sometimes with concurrent caspase inhibition like other cysteine-reactive molecules (27, 28). Disulfiram decreased cell lysis associated with etoposide more than with doxorubicin (Fig. 2D), with some likely intrinsic toxicity of cysteine-reactive drugs, as shown by disulfiram treatment alone. In our system, disulfiram co-treatment also decreased production of GSDMD p30 and p20 (Fig. 2E), while apoptotic cleavage of GSDME was unaffected. Together, these data further support the role of GSDMD as the actual driver of the milder GSDME-independent cell death associated with topoisomerase inhibitor drugs. Additionally, this mild phenotype was similar to the low GSDMD cleavage and lysis previously reported in cells treated with Val-boroPro (also known as talabostat), a small-molecule activator of the CARD8 or NLRP1 inflammasomes (23, 29).
To determine the source of GSDMD activation, we individually deleted known caspase activators of GSDMD. First, we ablated CASP8, because we observed its activation by chemotherapeutics, and others have shown it to mediate pyroptotic GSDMD cleavage upon Yersinia pestis infection (18, 30). In the context of topoisomerase inhibition, however, CASP8 deletion changed neither cell lysis (fig. S1A) nor GSDMD p30 production (fig. S1B). Previous studies implicated the ripoptosome complex, comprising receptor-interacting serine/threonine-protein kinase 1 (RIPK1), CASP8, and similar proteins, in etoposide-mediated apoptosis (31). Alternatively, deficiency in CASP8 processing of RIPK1 shunts the process to cell lysis by another pore-forming protein, mixed lineage kinase domain-like protein (MLKL). Here, we found that the RIPK1 inhibitor necrostatin-1s had no effect on the lytic death caused by doxorubicin (fig. S1C). As a control for necrostatin-1s activity, we confirmed that the compound was able to inhibit necroptosis induced pharmacologically by the combination of tumor necrosis factor alpha (TNF-α), second mitochondria-derived activator of caspase- (SMAC-)mimetic GDC-1052, and caspase inhibitor z-VAD-fmk (fig. S1D).
Having excluded apoptotic CASP8 as the GSDMD-activating protease, we next tested deletion of CASP1, the inflammasome-based activator of GSDMD. This, however, recapitulated the lysis reduction seen in GSDMD−/− THP-1 cells (Fig. 2F). In this case, apoptotic caspase activation as well as GSDME cleavage was not affected, but cleavage of GSDMD into p30 and p20 (Asp87-Asp275) fragments was greatly diminished (Fig. 2G). These findings suggested the possibility of a signal transduction cascade resulting in secondary inflammasome activation during chemotherapy-induced apoptosis.
Multiple inflammasome detectors can initiate pyroptosis. Because our system involved no pathogens, we hypothesized that NLRP3, a sensor of general cellular dyshomeostasis and ion flux (32), was the responsible inflammasome sensor downstream apoptosis caspase activation. Involvement of an inflammasome was further supported by the higher activation of CASP8 p10 in CASP1−/− cells (Fig. 2G), as CASP8 is known to aggregate at the inflammasome after unproductive or absent pyroptosis (17, 21, 33). To test the involvement of NLRP3, we used both the specific molecular inhibitor MCC950 (34) and extracellular potassium, a retardant of the potassium efflux driven-pathway for NLRP3 activation (35). Both types of inhibition reduced cell lysis upon chemotherapy treatment (Fig. 2, H and I) and were able to reduce GSDMD p30 production and p20 production (Fig. 2I), a finding consistent with NLRP3-driven CASP1 activation.
Because the presence of p43, p30, and p20 GSDMD fragments suggested enzymatic competition between CASP1 activation and CASP3 deactivation, we assessed the kinetics of cell lysis and GSDMD cleavage versus both time and chemotherapeutic dose (Fig. 2, J and K). Appreciable lysis was evident by between 16 and 24 hours of doxorubicin treatment, but a drop-off between 6.7 μM and 20 μM of doxorubicin was apparent (Fig. 2J). Cleavage analysis indicated that, in correlation with reduced cell lysis, less p30 GSDMD was present at higher concentrations of doxorubicin, whereas GSDME cleavage was only increased (Fig. 2K). These data suggested the existence of an optimal dosage for cell lysis depending on the relative activity of apoptotic and pyroptotic cascades.
To explore the relationship between these pathways, we live-imaged doxorubicin-treated cells. Apoptotic caspase activity was monitored by cell-permeable green fluorescent DNA-binding gated by a DEVD caspase cleavage motif, and pore formation was measured by the cell-impermeable DNA-binding dye Cytotox Red (Fig. 2, L to N). Binding of nucleic acids coupled to DEVDase activity almost always preceded Cytotox intercalation (Fig. 2, L and M). Double-positive cells were only transient, suggesting displacement or quenching of DEVD dye by Cytotox Red.
We expected that inhibition of the inflammasome would reduce cell-impermeable dye uptake concurrently with a decrease in cell lysis, but instead we observed that MCC950 inhibition of NLRP3 inflammasome did not decrease membrane permeability to Cytotox dye (Fig. 2M). This finding indicated that enhanced plasma membrane permeability occurred upstream of NLRP3 and GSDMD activation. Consistent with this idea was the limited effect of either CASP1 or GSDMD deletion on membrane permeability (Fig. 2N), although both had decreased cell lysis (Fig. 2, A and F).
Chemotherapy-activated pannexin-1 channels induce NLRP3-driven GSDMD cleavage
The inability of GSDMD or CASP1 deletion to attenuate plasma membrane permeability to Cytotox dye indicated that another mediator of leakage upstream of NLRP3 activation. Several studies have shown that CASP3-activated PANX1 hexameric channels are able to facilitate NLRP3 activation (20, 34, 35) and, here, we similarly observed PANX1 cleavage in response to both doxorubicin and etoposide but independent of NLRP3 inflammasome inhibitors (above; Fig. 2, G and I). Therefore, we hypothesized that PANX1 channel activity preceded inflammasome activation of GSDMD in our system.
Analysis of our knockout lines indicated that chemotherapy-induced PANX1 cleavage was upstream or independent of both gasdermins, CASP1, and CASP8 (Fig. 3A). To test whether PANX1 activity was responsible for downstream inflammasome activation, we co-treated with three therapeutics with known PANX1 inhibitory activity: the antibiotic trovafloxacin (36), the aldosterone antagonist spironolactone (37), and the anion transporter inhibitor probenecid (38). All three drugs were able to lower cell lysis (Fig. 3B) and decreased the pyroptotic processing of GSDMD but did not affect PANX1 or GSDME cleavage (Fig. 3C).
Figure 3.
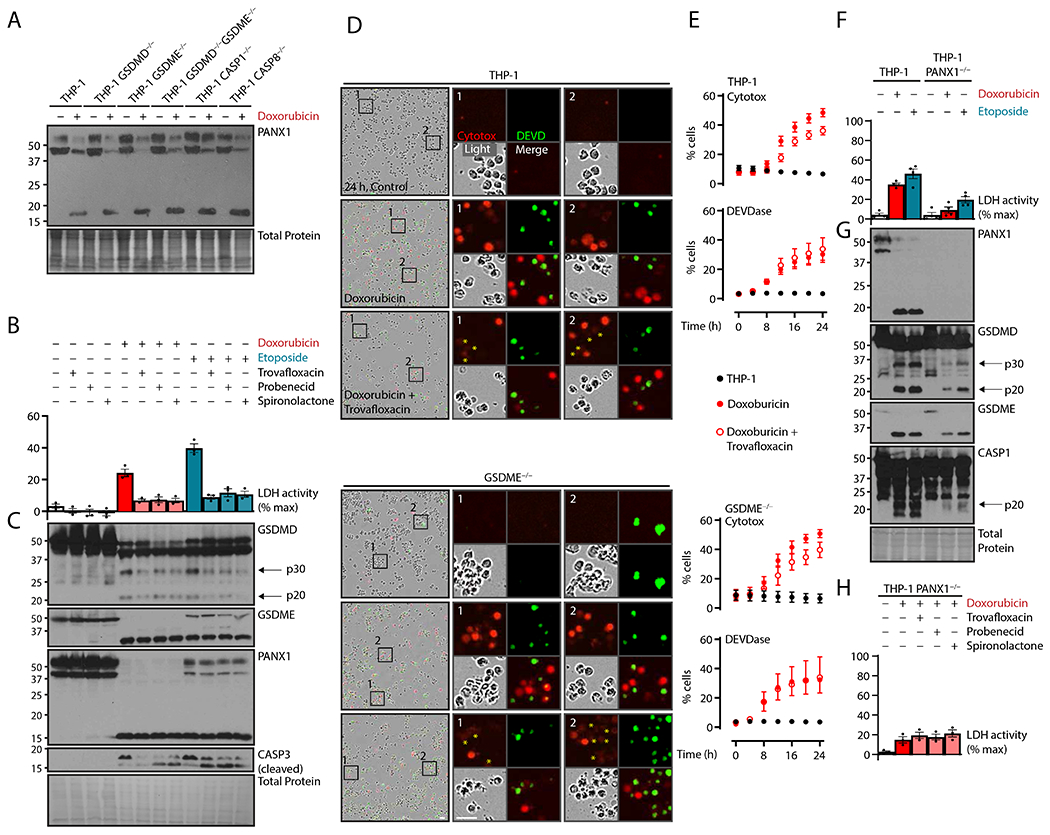
Chemotherapy-activated pannexin-1 channels induce NLRP3-driven GSDMD cleavage. (A) Naive human THP-1 monocytic cells or indicated deletion mutants were treated with topoisomerase poisons doxorubicin (2 μM) or etoposide (50 μM) for 24 hours and assayed by immunoblot. (B and C) THP-1 cells were treated with topoisomerase poisons as in (A), and concurrent PANX1 inhibitors trovafloxacin (20 μM), probenecid (1 mM), or spironolactone (20 μM) were added as indicated. Plasma membrane rupture was quantified by lactate dehydrogenase assay of cell-free supernatant (B), and lysates were assessed by immunoblot (C). (D and E) THP-1 cells and GSDME−/− cells were treated with doxorubicin plus trovafloxacin as in (B) and visualized with live cell imaging. True-color micrographs are shown in (D) and are representative of ≥ 2 biological replicates. Asterisks indicate moderately Cytotox-positive cells. Scale bar, 10 μm. Plasma membrane permeability was quantified by cell-impermeable Cytotox dye uptake, and apoptotic caspase activity was assayed by a dye with caspase-cleavable DEVD motif (E). (F and G) THP-1 cells or PANX1−/− cells were treated as in (A), and plasma membrane rupture and immunoblots were performed as in (B, C). (H) THP-1 PANX1−/− cells were treated with PANX1 inhibitors as in (B, C) and assayed with plasma membrane rupture by lactate dehydrogenase assay of cell-free supernatant. Scatter plot points represent mean ± standard error of ≥ 3 technical replicates across ≥ 2 biological replicates per experiment; graph dots represent mean of ≥ 2 technical replicates per biological replicate; graph bars represent mean ± standard error of ≥ 3 biological replicates per experiment; and immunoblots are representative of ≥ 3 biological replicates.
Further, whereas CASP3 activity was not affected by PANX1 inhibitors, trovafloxacin reduced Cytotox dye uptake in wild-type cells (Fig. 3, D and E). Although we expected incomplete inhibition (38), this mild effect was unexpected, and we observed a moderate proportion of low-staining cells which could explain these results (Fig. 3D). Because GSDME is also a pore-forming protein that is activated concurrently with PANX1 by CASP3, we hypothesized that both proteins would allow for the potassium efflux to trigger NLRP3-mediated GSDMD release. However, PANX1 inhibition in GSDME−/− cells did not further reduce membrane permeability, indicating that GSDME is dispensable for membrane permeability when PANX1 is active (Fig. 3E).
To further test whether PANX1 was indeed an upstream driver for lytic GSDMD cell death, we generated PANX1 knockouts, which showed decreased cell lysis, GSDMD cleavage, and CASP1-mediated p20 formation in response to both chemotherapy drugs (Fig. 3, F and G). GSDME cleavage remained and thus could be responsible for the remnant cell lysis. This remaining lysis in PANX1−/− THP-1 cells was unresponsive to PANX1 inhibitor treatment (Fig. 3H), further supporting both the specificity of these inhibitors as well as the primary role for PANX1 here.
PANX1 and GSDME-dominant lysis are divergent, redundant nodes for lytic death
The above data have demonstrated a linear pathway from CASP9-CASP3-PANX1 pathway to potassium efflux that triggers canonical NLRP3 inflammasome activation and subsequent pyroptosis from GSDMD in THP-1 cells in response to chemotherapy. However, aggregate scientific literature had not specifically observed this phenotype, and some of our earlier experiments, for example, with dasatinib in other myeloid leukemia lines (Fig. 1, A and B), did not directly support these findings as universal. Our data (Fig. 1, E and F) and others in the literature have also imputed GSDME as the major apoptosis-to-pyroptosis converter (14, 39).
To address this discrepancy, we compared pore-forming protein expression in THP-1 cells with acute monocytic leukemia MOLM-14, B-cell leukemia BLaER1, megakaryoblastic MEG-01, and erythroleukemia K-562 cell lines (40). We initially hypothesized that differences in the relative contributions of each terminal lysis effector – GSDMD for THP-1 cells and GSDME for MEG-01 cells – depended solely on protein expression (Fig. 4A). Unexpectedly, MOLM-14 cells, which were similar to THP-1 cells except for the lack of GSDME, seemed to rely on PANX1 but not GSDMD for lytic cell death (Fig. 4, B to D). Membrane rupture in MOLM-14 cells treated with doxorubicin was MCC950-insensitive, and GSDMD deletion did not alter lysis (Fig. 4, B and C); lysis was ameliorated, however, by the PANX1 inhibitors (Fig. 4D), and PANX1 cleavage was present in both wild-type and GSDMD−/− MOLM-14 lines (Fig. 4B). These data positioned PANX1 both as the gatekeeper of GSDMD-dependent lysis for THP-1 cells and as the intermediary or executory of lysis in MOLM-14 cells. The PANX1 high-expressing BLaER1 cells revealed a similar inhibition profile to MOLM-14 cells (fig. S2, A to C).
Figure 4. PANX1 and GSDME-dominant lysis are divergent, redundant nodes for lytic death.
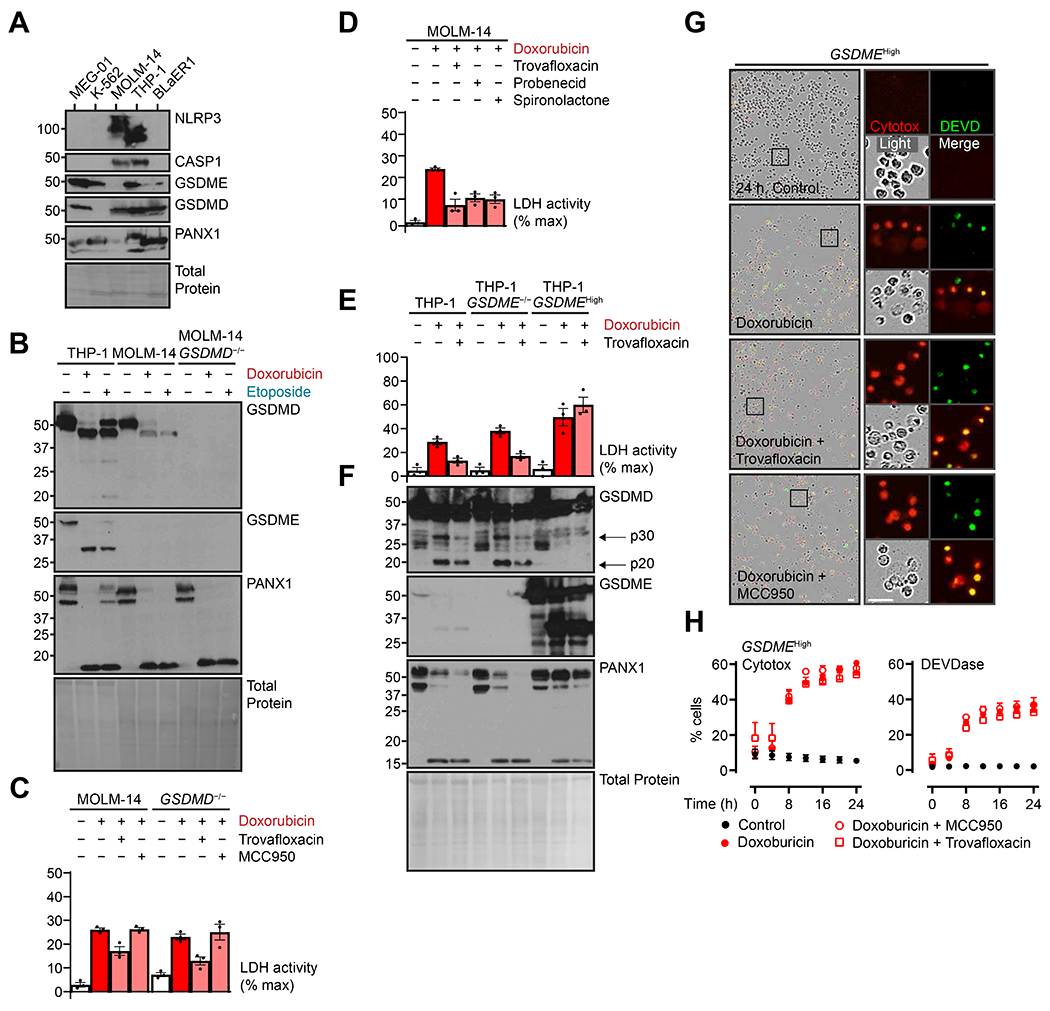
(A) MEG-01, K-562, MOLM-14, THP-1, and BLaER1 cells representative of multiple immune lineages were assessed by immunoblot for baseline protein expression of pore-forming proteins. (B and C) Indicated cells were treated with topoisomerase poisons doxorubicin (2 μM) or etoposide (50 μM) for 24 hours with concurrent PANX1 inhibitor trovafloxacin (20 μM) or NLRP3 inflammasome inhibitor MCC950 (10 μM). Lysates were assayed by immunoblot (B), and plasma membrane rupture was quantified by lactate dehydrogenase assay of cell-free supernatant (C). (D) MOLM-14 cells were treated as above and with additional PANX1 inhibitors probenecid (1 mM) or spironolactone (20 μM) then assessed for plasma membrane rupture as in (C). (E and F) THP-1 cells, GSDME−/− cells, and deletion cells reconstituted with high constitutive expression (GSDMEHigh) were treated with doxorubicin (2 μM) with or without PANX1 inhibitor trovafloxacin (20 μM) for 24 hours then assessed as in (B, C). (G and H) GSDMEHigh THP-1 cells were treated as in (C) and visualized with live-cell imaging. True-color micrographs are shown in (G) and are representative of ≥ 2 biological replicates. Scale bar, 10 μm. Plasma membrane permeability was quantified by cell-impermeable Cytotox dye uptake, and apoptotic caspase activity was assayed by a dye with caspase-cleavable DEVD motif (H). Scatter plot points represent mean ± standard error of ≥ 3 technical replicates across ≥ 2 biological replicates per experiment; graph dots represent mean of ≥ 2 technical replicates per biological replicate; graph bars represent mean ± standard error of ≥ 3 biological replicates per experiment; and immunoblots are representative of ≥ 3 biological replicates.
Finally, we examined the contribution of the PANX1-CASP1-GSDMD pathway to chemotherapy pyroptosis in the context of high GSDME expression. The GSDMEHigh cells described earlier (Fig. 1E) treated with doxorubicin were not sensitive to PANX1 inhibition by trovafloxacin (Fig. 4, D and E). Furthermore, protein analysis of these cells showed no presence of GSDMD p30. Live-cell imaging revealed similarly that neither membrane permeability nor CASP3 activation was affected by either trovafloxacin or MCC950 (Fig. 4, F and G). Compared to wild-type cells, GSDMEHigh THP-1 cells also showed more rapid kinetics of cell lysis (Fig. 4H), consistent with the more direct CASP3-GSDME pathway relative to secondary inflammasome-mediated pyroptosis (compare to Fig. 2L). These experiments indicated that overwhelming GSDME expression superseded both the PANX1 and GSDMD lysis pathways, and in conjunction with MOLM-14 and BLaER1 cell data showed that either GSDME or PANX1 could trigger cell lysis upstream and/or independent from the inflammasome-mediated GSDMD pathway in various cell types.
Pore-forming proteins sensitize AML to chemotherapeutic lysis
The above experiments delineated three modes of lytic cell death in response to chemotherapy in leukemia cells: the previously described GSDME-mediated lysis, a lysis mechanism dependent on PANX1 independent of gasdermin, and a pathway connecting PANX1-mediated potassium efflux to—unexpectedly—the well-established pyroptosis effector GSDMD through the NLRP3 inflammasome. Our data support a model wherein the relative abundance of pore-forming proteins determines the effector of apoptosis-triggered cell lysis (Fig. 5A), which can be envisioned as a basic electrical circuit diagram (Fig. 5B, discussed below).
Figure 5. Pore-forming proteins sensitize acute myeloid leukemias to chemotherapy lysis.
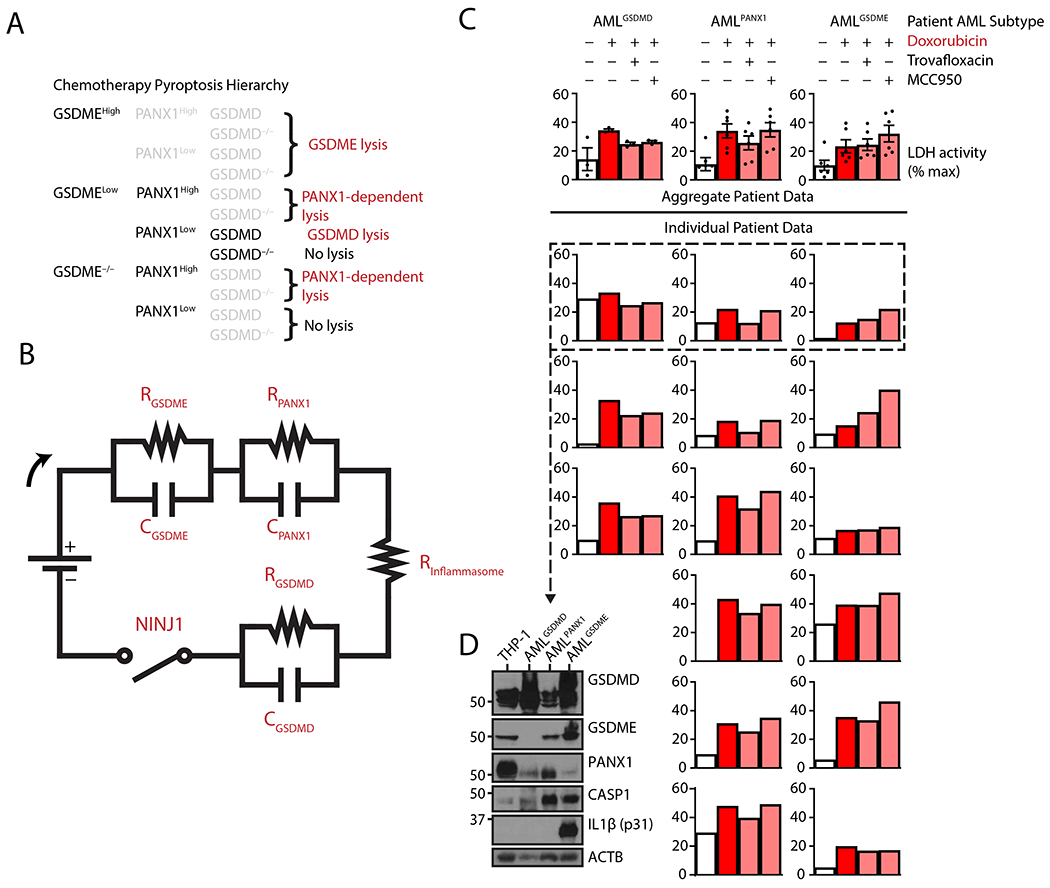
(A) Hierarchical representation of plasma membrane rupture following exposure to chemotherapy drugs. Greyed out decision points indicate irrelevance of specific pathways once a higher priority pathway determines cell fate. (B) Electrical circuit approximation of gasdermin and PANX1-mediated pyroptosis. Apoptosis activation induces a current that is sensed by resistors correlating to the concentration of the pore-forming protein. A higher resistance (higher pore-forming protein concentration) induces an equal voltage drop across parallel capacitors, which represent pore-forming and lytic ability of their cognate gasdermin. An inflammasome-dependent resistor represents the delay to inflammasome activation before GSDMD is cleaved by CASP1, and the transmembrane protein NINJ1 may act as a final common pathway switch to realize full cell lysis. (C and D) Quantification of plasma membrane rupture by lactate dehydrogenase assay of cell-free supernatant from mononuclear cells acquired from diagnostic peripheral blood and bone marrow aspirates from fifteen patients with acute myeloid leukemia (AML) – classified as GSDMD-high, PANX1-high, or GSDME-high [shown at baseline from a representative 3 samples (D)] – treated with topoisomerase poison doxorubicin (2 μM) and concurrent PANX1 inhibitor trovafloxacin (20 μM) or NLRP3 inflammasome inhibitor MCC950 (10 μM) as indicated (C). Due to limited sample availability, individual data bars represent means of ≥ 2 technical replicates per single experiment per patient, and the immunoblot is a single experiment showing full-length protein. ACTB: β-actin loading control.
To confirm the existence of these distinct pathways in primary patient leukemic blasts, we purified mononuclear cells from diagnostic peripheral blood and bone marrow aspirate specimens from fifteen AML patients (median age 66.7 years old, range 21-88 years old, 12 of 15 male). We then analyzed cell lysis in response to doxorubicin with or without inhibition of NLRP3 and PANX1 activity. Of the primary AML lines, 20% exhibited dual sensitivity to PANX1 and NLRP3 inhibition, suggestive of the PANX1-GSDMD pathway, 40% exhibited only PANX1 inhibitor sensitivity, and 40% exhibited no sensitivity to either inhibitor, implying GSDME-dependent lysis (Fig. 5, C and D). Protein analysis of a subset of these malignancies showed correlations with, respectively, high GSDMD expression with no GSDME and low PANX1 expression (AMLGSDMD), high PANX1 protein concentration with lower GSDMD and GSDME expression (AMLPANX1), and highest GSDME expression (AMLGSDME). These data provide primary cell evidence of that this trichotomy of lysis routes may be representative of multiple subsets of clinical disease and challenge the well supported previous notion that GSDME holds primacy in chemotherapy-induced lytic cell death.
DISCUSSION
In this work, we used genetic and pharmacologic methods to dissect the interrelations among pore-forming proteins with respect to regulated death of human leukemia cells treated with the topoisomerase poisons doxorubicin and etoposide. We identified GSDMD as the primary pyroptosis effector in the monocytic THP-1 cell line, contrary to previous studies in this field that have inferred GSDME-mediated pore formation with pyroptosis.
GSDME was initially characterized as an apoptosis-to-pyroptosis converter in the context of diverse chemotherapeutics (14), whereby intrinsic apoptosis activation of CASP3 led to cleavage and activation of GSDME, resulting in assumed pore activity akin to GSDMD. This initial study had surveyed the NCI-60 cancer cell line collection and reported GSDME-related pyroptosis selectively in high-expressing cell lines and subpopulations. Although a few studies have found cases in which GSDME cleavage did not correlate with cell lysis (16, 17), most have assumed that GSDME cleavage concurrent with cell lysis (usually approximated by extracellular supernatant lactate dehydrogenase activity measurement) had a direct causal link. However, to our knowledge, only two studies have attempted to analyze whether GSDME self-association or pore formation occurs post-activation (23, 41). Thus, many studies of GSDME and pyroptosis do not explicitly demonstrate that GSDME is the de facto pore-forming protein to terminally drive membrane rupture.
The idea that GSDME cleavage is associated with obligatory cell lysis following apoptotic activation has been challenged by other studies. Similar to our findings, the Broz group showed that murine Panx1 deletion was able to partially decrease GSDMD cleavage and cell lysis in extrinsic apoptosis (20). They also found that Panx1 deletion in the context of primary mitochondrial intrinsic apoptosis by BH3-mimetic ABT-737 and MCL-1 inhibitor S63845 was able to decrease IL-1β release, and a follow-up study indicated that PANX1 deficiency did not decrease cell lysis (42), suggesting another activation method for the NLRP3–GSDMD axis, namely through GSDME-mediated potassium efflux. Similarly, Vince et al. found that ABT-737/S63845-induced intrinsic apoptosis did not rely on either gasdermin but was able to drive NLRP3 activation and both CASP1- and CASP8-dependent IL-1β activation (19). Unexplained in that study, however, was why GSDME deficiency lessened IL-1β secretion but not cleavage. We hypothesize that PANX1 is the missing link, as its allowance of potassium efflux would permit NLRP3 activation and IL-1β cleavage and release through GSDME (23). Notably, most of these studies were undertaken in murine cells, and our study may highlight an alternative preference in human cells, which have many differences, such as a discovered necessity of NLRP11 for NLRP3 inflammasome activation (43).
In our model, however, we determined instead that GSDMD activation could be downstream of NLRP3 activation following PANX1 cleavage. Thus, studies placing GSDME as the sole pyroptotic effector, even with knockout validation, should be scrutinized closely, particularly if the cell type in question also expresses GSDMD. Most studies focused on GSDME do not assess (or have not reported) GSDMD cleavage. For example, the initial study with chemotherapy drugs found that GSDME deletion ameliorated cell death in SH-SY5Y cells (14), but a later study showed that GSDMD was concurrently cleaved upon similar stimulation (44) in SH-SY5Y cells, which express the full NLRP3 inflammasome pathway (45). In that system, then, one cannot rule out that is merely a potassium conduit for GSDMD-mediated secondary pyroptosis.
One additional aspect of these cell death pathways that is seldom monitored but critical for drawing generalizable conclusions is timing of cell death and cellular events, even more so when apoptosis- and inflammasome-driven pyroptosis are competing in a first-past-the-post system. GSDMD was shown to be deactivated by CASP3 by Asp87 cleavage when apoptosis is active (24), and our current study mirrored their incidental finding that there was a quantitatively low but, as we demonstrated, important activation of GSDMD cleavage by CASP1. However, the current and above-mentioned studies described an inactive p20 Asp87-Asp275 fragment generated by dual cleavage by CASP1 and CASP3 (19, 20). We found that the benchmark for productive GSDMD p30 pyroptosis in these systems was bimodally proportional to strength of apoptosis activation, as monitored by disappearance of full-length gasdermins. In other words, as apoptotic strength increased from doxorubicin or etoposide (present study), to TNF-α and TAK1 inhibitor (20), to ABT-737/S63845 treatment (19), less full-length GSDMD was available to effect cell death from CASP1 or CASP8 cleavage.
In conjunction with prior works, our findings suggested a model that explains the contribution of three pore-forming proteins to pyroptosis after apoptosis activation. CASP3-mediated cleavage of PANX1 and perhaps GSDME in low amounts was enough to actuate NLRP3 inflammasome activation through potassium efflux. However, if either apoptotic channel/pore was present in sufficient quantities, it alone was sufficient for pyroptosis. This model could be approximated with a basic electrical circuit diagram (Fig. 5B), in which “resistance” of each pore-forming protein is correlated with protein expression: the more protein, the more voltage drop across a resistor-capacitor pair, and the more “charge” the capacitor for each protein builds up, representing stress on plasma membrane-dependent ion homeostatic pathways. At a threshold of charge accumulation, that protein lyses the cell. If resistance (concentration) of PANX1 or GSDME is not sufficient to drive lysis themselves, all of the “current” in the system can dissipate GSDMD lysis. Finally, the transmembrane protein NINJ1 seems to act as the final common regulator of lytic cell death (46) and may act as a de facto on-off switch for any and all lytic pathways. Our model captures the order of pore-forming protein activity downstream of apoptosis and reasonably approximates what happens with cell-specific expression of these three proteins in different relative concentrations.
Future apoptosis-to-pyroptosis studies should, at a minimum, monitor cleavage of GSDMD, GSDME, and PANX1 and use inhibitors at different steps of the suspected pathway to pinpoint the actual effector of pyroptosis. A hierarchy for intrinsic apoptosis-induced cell lysis shows which mode of lysis may occur and how it can be blocked (Fig. 5A). For example, we found that pyroptosis in BLaER1 and MOLM-14 cells is inhibitable at the PANX1 but not GSDMD stage, suggesting PANX1 as the actual trigger for plasma membrane rupture despite the presence of GSDMD (Fig. 4C and fig. S2). Notably, how cell lysis occurs in a sole PANX1 system is undetermined, although it can be hypothesized either to trigger another gasdermin or pore-forming protein or to induce a common upstream cellular milieu for activation of the NINJ1 pathway for plasma membrane rupture (46).
Our AML patient cohort provided reassuring evidence that this hierarchy established by studies from immortalized human cell lines could be extrapolated to clinical disease, since individual samples recapitulated inhibitor sensitivity profiles representing each of the three pyroptosis effectors. These findings have immediate ramifications for translational decision-making: Each pore-forming protein represents an interventional node for triggering or preventing lytic cell death. The crux of this decision could lay in whether triggering lytic cell death and concurrent immunogenicity is favorable (for example, to stimulate anti-tumor immunity) or whether a non-lytic, silent cell death per the traditional apoptosis definition is desired (for example, to prevent cytokine storm or tumor lysis syndrome). As treatment for hematopoietic malignancies are further refined as for GSDMD inhibition (27, 28), clinicians will have more tools to inhibit and, hopefully, induce certain pathways. Such a personalized medicine approach will need to encompass many variables to make the optimal clinical decision. As an example, our AMLGSDME cells exhibited baseline IL-1β as well as, unexpectedly, susceptibility to MCC950 as a lysis instigator: early intervention with dual chemotherapy/MCC950, in this case, could generate a highly immunogenic cancer with a pro-inflammatory milieu. Separately, recent data show that high expression of CASP1 or NLRP3 in AML correlate with inferior survival (47, 48). Although expression does not always recapitulate pathway activity, one could hypothesize that the NLRP3-CASP1-GSDMD pathway may promote AML relapse and/or therapeutic resistance. Thus, specific GSDMD or NLRP3 inhibitors might improve patient outcomes by preventing release of leukemia-promoting factors (49). Moving forward, it will be critical to carefully dissect the role of pore-forming proteins, including those not yet implicated, in hematologic and perhaps other malignancies to tailor an appropriate therapeutic response.
MATERIALS AND METHODS
Cell Lines
Human THP-1 (TIB-202; American Type Culture Collection), K-562 (CCL-243; American Type Culture Collection), and BLaER1 (SCC165; Millipore Sigma) cells were cultured in RPMI with 10% fetal bovine serum (Gemini Bio) without antibiotics. THP-1 and K-562 cells were passaged with fresh media every three to four days and kept under 0.8 × 106 cells mL−1 and BLaER1 cells under 2 × 106 cells mL−1 every three days.
Knockouts were generated with nucleoporation of Cas9 protein (Integrated DNA Technologies) coupled with Alt-R tracrRNA and Alt-R crRNA (Integrated DNA Technologies) according to manufacturer protocols. The 4D-Nucleofector, SG cell line kit, and default THP-1 program (Lonza) as well as the Neon electroporation system with program #24 (Fisher ThermoScientific) were used for transient Cas9 transfection and gene editing. Single cell clones per line were obtained by limiting dilution and verified with immunoblot. At least three clones per cell line were verified and pooled for experiments.
For knockouts, targeted protospacer adjacency motifs (NGG nucleotides after the 20 cognate crRNA base pairs) were based on manufacturer-suggested sites located at 653CCT(−) for GSDME, 154CCC(−) for GSDMD, 731TGG(+) for CASP1, 274AGG(+) for CASP8, and 20TGG(−) and 517TGG(−) for PANX1.
GSDME-overexpressing THP-1, denoted GSDMEHigh, were generated previously (23): we subcloned the open reading frame of the more common 142Pro human GSDME variant (NCBI reference sequence NM_004403.2) into a lentiviral expression plasmid with constitutive expression driven by the mammalian elongation factor-1α short promoter. Transduced cells were selected with G418/geneticin (InvivoGen) at 0.5 μg mL−1 for three weeks, and total populations were used for experiments.
Experimental conditions
For experiments, cells were centrifuged at 200 × g for 4 min and resuspended in fresh media. THP-1 and K-562 experiments were carried out at 0.5 × 106 mL−1 in 5 mL in flat-bottom six-well plates or 1 mL in 24-well plates. BLaER1 cells were carried out at 1-2 × 106 mL−1 similarly.
Live-cell imaging
Flat-bottom 96-well plates were coated with 50 μL of 0.01% poly-L-ornithine solution. After aspiration and drying, 2.5 × 104 cells in 100 μL of media with Incucyte Cytotox red (250 nM) and Caspase-3/7 Green (5 μM) dyes (Essen BioScience) were added to coated plates and cells allowed to adhere for 30-45 min. Then, an equal volume of the dye-containing media with treatment or DMSO vehicle were added. After 1 hour of equilibration in an Incucyte Zoom S3 (Essen BioScience), phase, red, and green channel images were captured every four hours over a day using a 10× objective lens.
Image quantification
Four 2.15 mm2 areas were monitored per each of three wells per condition per biological replicate. Cells were counted using AI learning with ImageJ plugin Trainable Weka Segmentation (50). Cell percentages were generated using numbers of Cytotox-positive and caspase activity-positive cells as quantified with Incucyte Zoom software. Green channel images were all modified by setting a brightness/contrast minimum value of 125 in ImageJ to remove background seen in almost all untreated cells, and automatic quantification settings in Incucyte Zoom were modified to match these outputs. Values from at least eight technical replicates per used per biological replicate.
Drug treatment
Doxorubicin (2 μM) or etoposide (50 μM) were used to activate intrinsic apoptosis. These dosages achieved 80-100% maximal response for THP-1 cells over one day at used cell/media conditions (data not shown). Concurrently given inhibitors included NLRP3 inhibitor MCC950 (10 μM; Invivogen); GSDMD inhibitor disulfiram (30 μM); PANX1 inhibitors trovafloxacin (20 μM), probenecid (1 mM), and spironolactone (20 μM); RIPK1 inhibitor necrostatin-1s (50 μM); TNF-α (10 ng mL−1); pan-caspase and cathepsin inhibitor z-VAD-fmk (20 μM); and SMAC-mimetic GDC-0152 (400 nM). DMSO was used for 1000× stock solutions. Drugs were purchased from Millipore Sigma unless otherwise noted.
Plasma membrane rupture quantification
Cell lysis was measured using the CyQUANT LDH Cytotoxicity Assay kit per instructions (ThermoFisher Scientific). Cellular supernatant was cleared at 21,000 × g for 1 min and used undiluted, and maximum signal was obtained by lysing cells for 30 min at 37 °C in kit lysis buffer. Individual values were calculated as slope of absorbance change in linear ranges. Warmed media was used as baseline and subtracted from all other samples. Final percentages were calculated by dividing by the maximum signal for each condition to account for differences in cell number from drugs.
Immunoblots
Cells were centrifuged at 21,000 × g for 1 min, and then pellets were washed with PBS, re-centrifuged, and disrupted with lysis/sample buffer (2% SDS, 1 mM EDTA, 0.06 M Tris, 10% glycerol, 150 mM NaCl, and 1% 2-mercaptoethanol) at 25 °C, and then held for 5-10 minutes at 98 °C. Lysates of equal starting cell numbers were separated using SDS-PAGE, and proteins were wet-transferred onto 0.22 μm nitrocellulose membranes. Total protein staining was performed with InstantBlue Coomassie (Abcam). Membranes were washed with Tris-buffered saline with 0.01% Tween-20 (TBST) and incubated with primary antibody diluted in 5% bovine serum albumin in TBST for 16-20 hours at 4 °C. Membranes were washed thrice in TBST for 5 min and then incubated with HRP-conjugated secondary antibody diluted in 5% milk in TBST for 1-2 hours at 25 °C. Membranes were again washed then developed with enhanced chemiluminescence.
Primary antibodies were used at 1:1000-1:500 dilution: GSDMD (HPA044487, Atlas Antibodies, RRID:AB_2678957), GSDME (ab215191, Abcam; RRID:AB_2737000), PANX1 (91137, Cell Signaling Technology [CST]; RRID:AB_2800167) CASP1 p20 (AG-20B-0048-C100, Adipogen; RRID:AB_2490257), CASP3 (9662, CST; RRID:AB_331439), CASP8 (4790, CST; RRID:AB_10545768), CASP9 (9508, CST; RRID:AB_2068620). Secondary antibodies were used at 1:5000-1:2000 dilution: anti-mouse (7076, CST; RRID:AB_330924) and anti-rabbit (7074, CST; RRID:AB_2099233).
Data plotting and analysis
Experiments were performed with at least three independent biological replicates unless otherwise noted. Micrographs were representative of at least three independent biological replicates. Quantitative experiments were performed with at least two technical replicates per biological replicate. Data were compiled in Microsoft Excel and plotted using GraphPad Prism 9 as mean ± standard error of the mean. Statistical analysis was omitted due to sample size vs. number of conditions.
Primary AML patient samples
Patient specimens were collected as part of routine clinical care at University Hospitals Cleveland Medical Center and used as discarded tissue per institutional review board-approved protocol. Patient diagnosis was determined by a hematopathologist during routine clinical practice according to World Health Organization 2017 classification criteria. After completion of all clinical studies, and no longer than 72 hours after specimen collection, mononuclear cells were isolated by density-gradient centrifugation and allowed to recover in RPMI with 10% FBS overnight or were immediately used for experimentation.
Supplementary Material
Acknowledgements:
We are grateful to James Ahad (Department of Biomedical Engineering, Case Western Reserve University, for discussions on electrical circuits.
Funding:
This work was supported by grants from the National Institutes of Health (T32 GM007250 and T32 AI089474 for BZ; P01 AI141350 for DWA and GRD; and R35 GM141603 to DWA).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. All cell lines generated are available upon reasonable request.
References and Notes
- 1.Cookson BT, Brennan MA, Pro-inflammatory programmed cell death [2], Trends Microbiol. 9, 113–114 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death, Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling, Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 4.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J, Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion, Cell Res. 25, 1285–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, Hiller S, GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death , EMBO J. 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J, Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores, Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinon F, Burns K, Tschopp J, The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta., Mol. Cell 10, 417–26 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, Yap AS, Bezbradica JS, Schroder K, Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity, J. Exp. Med , jem.20172222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC, The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages, Immunity 48, 35–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P, The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice, Eur. J. Immunol 48, 584–592 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC, An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells, Science (80-. ). 352, 1232–1236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broz P, Pelegrín P, Shao F, The gasdermins, a protein family executing cell death and inflammation., Nat. Rev. Immunol (2019), doi: 10.1038/s41577-019-0228-2. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, Sengupta S, Yao Y, Wu H, Lieberman J, Gasdermin E suppresses tumour growth by activating anti-tumour immunity, Nature 579, 415–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F, Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin, Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, Liu M, Zhou N, Lv J, Tang K, Xie J, Gao Y, Cheng F, Zhou Y, Zhang Z, Hu Y, Zhang X, Gao Q, Zhang Y, Huang B, Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome, Sci. Immunol 5, eaax7969 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Tixeira R, Shi B, Parkes MAF, Hodge AL, Caruso S, Hulett MD, Baxter AA, Phan TK, Poon IKH, Gasdermin E Does Not Limit Apoptotic Cell Disassembly by Promoting Early Onset of Secondary Necrosis in Jurkat T Cells and THP-1 Monocytes, Front. Immunol 9, 2842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, Haley B, Cuellar TL, Reichelt M, Kayagaki N, ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages., Sci. Rep 8, 3788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A, Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection, Proc. Natl. Acad. Sci. U. S. A 115, E10888–E10897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, Rizzacasa MA, Man SM, Silke J, Masters SL, Lessene G, Huang DCS, Gray DHD, Kile BT, Shao F, Lawlor KE, The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and −7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation, Cell Rep. 25, 2339–2353.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P, Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP 3 inflammasome assembly, EMBO J. 38, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, Kinoshita T, Kushiyama H, Sakurai M, Shiroishi T, Suda T, Caspase-1 initiates apoptosis in the absence of gasdermin D, Nat. Commun 10, 2091 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aizawa E, Karasawa T, Watanabe S, Komada T, Kimura H, Kamata R, Ito H, Hishida E, Yamada N, Kasahara T, Mori Y, Takahashi M, GSDME-dependent incomplete pyroptosis permits selective IL-1α release under caspase-1 inhibition, iScience 23, 101070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou B, Abbott DW, Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases., Cell Rep. 35, 108998 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taabazuing CY, Okondo MC, Bachovchin DA, Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages, Cell Chem. Biol 24, 507–514.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao X, Van Der Zanden SY, Wander DPA, Borràs DM, Song JY, Li X, Van Duikeren S, Van Gils N, Rutten A, Van Herwaarden T, Van Tellingen O, Giacomelli E, Bellin M, Orlova V, Tertoolen LGJ, Gerhardt S, Akkermans JJ, Bakker JM, Zuur CL, Pang B, Smits AM, Mummery CL, Smit L, Arens R, Li J, Overkleeft HS, Neefj J, Uncoupling DNA damage from chromatin damage to detoxify doxorubicin, Proc. Natl. Acad. Sci. U. S. A 117, 15182–15192 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montecucco A, Zanetta F, Biamonti G, Molecular mechanisms of etoposide, EXCLI J. 14, 95–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, Xiao TS, Li X, Abbott DW, Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis., Sci. Immunol 3 (2018), doi: 10.1126/sciimmunol.aat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, Wang J, Hollingsworth LR, Magupalli VG, Zhao L, Luo HR, Kim J, Lieberman J, Wu H, FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation, Nat. Immunol (2020), doi: 10.1038/s41590-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okondo MC, Johnson DC, Sridharan R, Bin Go E, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH, Sanford DG, Arciprete MO, Golub TR, Bachovchin WW, Bachovchin DA, DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis, Nat. Chem. Biol 13, 46–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E, Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death, Science (80-. ). 362, 1064–1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P, The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs, Mol. Cell 43, 432–448 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Gong T, Yang Y, Jin T, Jiang W, Zhou R, Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes., Trends Immunol. 14, 1590–604 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, Mocarski ES, Dubyak GR, Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling., J. Biol. Chem 290, 20167–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, Arostegui JI, Pelegrin P, MCC950 closes the active conformation of NLRP3 to an inactive state, Nat. Chem. Biol 15, 560–564 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J, Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration, Cell Death Differ. 14, 1583–1589 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Poon IKH, Chiu YH, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS, Unexpected link between an antibiotic, pannexin channels and apoptosis, Nature 507, 329–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Good ME, Chiu YH, Poon IKH, Medina CB, Butcher JT, Mendu SK, DeLalio LJ, Lohman AW, Leitinger N, Barrett E, Lorenz UM, Desai BN, Jaffe IZ, Bayliss DA, Isakson BE, Ravichandran KS, Pannexin 1 channels as an unexpected new target of the anti-hypertensive drug spironolactone, Circ. Res 122, 606–615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silverman W, Locovei S, Dahl G, Probenecid, a gout remedy, inhibits pannexin 1 channels, Am. J. Physiol. - Cell Physiol 295, 761–767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES, Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death, Nat. Commun 8, 14128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AAB, Cooper MA, Graf T, Hornung V, Human Monocytes Engage an Alternative Inflammasome Pathway, Immunity 44, 833–846 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES, Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation, Nat. Commun 10, 1689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen KW, Demarco B, Broz P, Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation, Eur. J. Immunol 50, 170–177 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Gangopadhyay A, Devi S, Tenguria S, Carriere J, Nguyen H, Jäger E, Khatri H, Chu LH, Ratsimandresy RA, Dorfleutner A, Stehlik C, NLRP3 licenses NLRP11 for inflammasome activation in human macrophages, Nat. Immunol 23, 892–903 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Chen Y, He Q, Distinct characteristics of dasatinib-induced pyroptosis in gasdermin E-expressing human lung cancer A549 cells and neuroblastoma SH-SY5Y cells, Oncol. Lett 20, 145–154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, Son J, Yu JW, MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration, Cell Death Differ. 26, 213–228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, Sandoval W, Yan D, Kang J, Xu M, Zhang J, Lee WP, McKenzie BS, Ulas G, Payandeh J, Roose-Girma M, Modrusan Z, Reja R, Sagolla M, Webster JD, Cho V, Andrews TD, Morris LX, Miosge LA, Goodnow CC, Bertram EM, Dixit VM, NINJ1 mediates plasma membrane rupture during lytic cell death, Nature 591, 131–136 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A, Bachovchin DA, DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia, Nat. Med 24, 1151–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luciano M, Blöchl C, Vetter J, Urwanisch L, Neuper T, Elmer DP, Bauer R, Dang H-H, Strandt H, Neureiter D, Krenn P, Tesanovic S, Rieser S, Bergsleitner O, Zell L, Binder S, Schaller S, Strunk D, Pleyer L, Greil R, Winkler S, Hartmann TN, Huber CG, Aberger F, Horejs-Hoeck J, The NLRP3/eIF2 axis drives cell cycle progression in acute myeloid leukemia, bioRxiv , 2021.06.25.449862 (2021). [Google Scholar]
- 49.Carey A, Edwards DK, Eide CA, Newell L, Traer E, Medeiros BC, Pollyea DA, Deininger MW, Collins RH, Tyner JW, Druker BJ, Bagby GC, McWeeney SK, Agarwal A, Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia., Cell Rep. 18, 3204–3218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arganda-Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, Sebastian Seung H, Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification., Bioinformatics 33, 2424–2426 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. All cell lines generated are available upon reasonable request.