

Abstract
Antivirulence agents targeting endospore-transmitted Clostridioides difficile infections are urgently needed. C. difficile-specific DNA adenine methyltransferase (CamA) is required for efficient sporulation and affects persistence in the colon. The active site of CamA is conserved and closely resembles those of hundreds of related S-adenosyl-l-methionine (SAM)-dependent methyltransferases, which makes the design of selective inhibitors more challenging. We explored the solvent-exposed edge of the SAM adenosine moiety and systematically designed 42 analogs of adenosine carrying substituents at the C6-amino group (N6) of adenosine. We compare the inhibitory properties and binding affinity of these diverse compounds and present the crystal structures of CamA in complex with 14 of them in the presence of substrate DNA. The most potent of these inhibitors, compound 39 (IC50 ∼ 0.4 μM and KD ∼ 0.2 μM), is selective for CamA against closely related bacterial and mammalian DNA and RNA adenine methyltransferases, protein lysine and arginine methyltransferases, and human adenosine receptors.
Highlights
Synthesis of 23 analogs of the adenosine moiety of S-adenosyl-l-methionine (SAM) and their testing against CamA along with 19 already available analogs.
Determination of 14 structures of CamA-DNA-inhibitor complexes to resolutions between 2.19 and 2.81 Å.
Identification of compound 39 as a potent CamA inhibitor, with a half maximal inhibitory concentration (IC50) of ∼0.4 μM and a dissociation constant (KD) of ∼0.2 μM.
Determination that compound 39 is selective for CamA over closely related bacterial and mammalian DNA and RNA adenine methyltransferases, protein lysine and arginine methyltransferases, and human adenosine receptors.
1. Introduction
Clostridioides difficile infections (CDI) have been identified as an urgent threat as they are a leading cause of hospital-acquired diarrhea in older patients and community-associated infection in children (https://www.cdc.gov/drugresistance/biggest-threats.html).1,2 Older patients with dysbiosis from broad-spectrum antibiotics are most vulnerable, and those with cancer are also quite vulnerable to CDI.3 CDI pathogenesis involves epithelial damage by toxins while the production of drying- and heat-resistant endospores during therapy facilitates further transmission and recurrence.4 Furthermore, C. difficile forms biofilms in the infected colon, which serve as a reservoir for continued recrudescence of fulminant disease.5,6 In theory, drugs that reduce C. difficile carriage would be tremendously useful, whether or not they also inhibit growth of the bacteria.7,8 This critical medical need is unmet by currently available therapeutic strategies,9−12 and novel targeted therapeutics are urgently needed to combat CDI13−16 for two key reasons. First, there is increasing resistance of C. difficile to commonly used antibiotics,17 including the mainline CDI drugs vancomycin18 and metronidazole.19,20 Second, antibiotics vary in how rapidly, post treatment, they allow recovery of the normal, C. difficile-resistant microbiome.21
To address the unmet critical need for CDI therapeutics, several screens of small-molecule libraries, or screens for repurposing currently approved drugs, are being carried out by several groups.22,23 We report here our complementary approach to such studies: we aim to discover drug candidates for C. difficile that reduce sporulation and biofilm formation and minimize intestinal carriage by inhibiting a unique DNA adenine methyltransferase (MTase).
A potential avenue to epigenetically targeted treatment of C. difficile infections is provided by the recently discovered C. difficile adenine methyltransferase A (CamA), which in one study was present in all C. difficile genomes sequenced (>300 at that time), but was rarely found in other bacteria.24 This specificity still holds as of fall 2022, as the top BlastP match to CamA when C. difficile is excluded (not including “uncharacterized clostridia”) is the clostridial genus Romboutsia, with 67% amino acid identity; and when Clostridiales are excluded altogether, the best current hit is in the Bacteroidiales with just 41% identity to CamA (not shown).
The activity of this enzyme was first seen nearly two decades ago,25,26 but its significance was understood only 2 years ago.24 Most importantly, CamA-mediated DNA adenine methylation at CAAAAA (underlining indicates the target A) is required for normal sporulation and biofilm production by C. difficile, key steps in the transmission of this agent as well as (in an animal model) for gastrointestinal tract colonization.24 In a mouse model of C. difficile infection, the level of bacteria declined to near the limit of detection in the feces 6 days after inoculation for the CamA catalytic mutant, whereas the WT strain remained readily detectable at days 6 and 7. In a clindamycin-treated hamster model of infection, which includes vulnerability to the special effects of the C. difficile toxins, the strains of WT or CamA-mutant of C. difficile caused diarrhea symptoms and weight loss in hamsters, but no difference in the survival times of hamsters after inoculation. Together, these observations indicate that CamA controls the ability of C. difficile to persist within the host intestinal tract, but it does not directly influence toxin-mediated pathogenesis of C. difficile infection. Finding CamA inhibitors would thus specifically complement ongoing searches for other antivirulence and/or antimicrobial agents targeting C. difficile,27 could greatly reduce the spread of this pathogen in clinical settings, and could yield synergistic therapeutic approaches.
We recently characterized the catalytic mechanism of CamA and its structures bound with substrate DNA in the absence and presence of S-adenosyl-l-homocysteine (SAH).28 SAH is one of the reaction products, being the demethylated form of the co-substrate S-adenosyl-l-methionine (SAM). In addition, we found that a few SAM/SAH analogs, some of which are already in clinical trials as inhibitors of human epigenetic enzymes, can also inhibit CamA enzymatic activity in vitro at low micromolar concentrations.29 One particular observation, that a brominated adenosyl moiety enhanced inhibitory potency against CamA, led us to the systematic design of 42 analogs of the adenosine moiety of SAM/SAH. The most potent of these inhibitors, compound 39, exhibited a half-maximal inhibitory concentration (IC50) of ∼0.4 μM and a dissociation constant (KD) of ∼0.2 μM. Further, compound 39 is selective for CamA over closely related bacterial and mammalian DNA and RNA MTases, protein lysine and arginine MTases, and human adenosine receptors. We suggest that 39 represents the archetype of a class of small molecule inhibitors that may eventually be used to depress C. difficile carriage specifically by interfering with CamA activity.
2. Results
2.1. Chemistry
In this study, we characterized 42 compounds (Figure 1); among them, 19 compounds 1–9, 13, and 22–30 were commercially available. Compounds 10, 11,30,3112,3014,32,3315, 16, 17,3418,32,3319, 20,3521, 31 (patents EP0417999A1 and EP0423777A2), and 32–41 were prepared through an aromatic nucleophilic substitution reaction between the commercially available 6-chloropurin-9-riboside (43) and the appropriate primary or secondary amines, in the presence of triethylamine in dry ethanol under reflux conditions (Scheme 1). Compound 42 was obtained by removal of the tert-butoxycarbonyl protection from the piperidine moiety of compound 39 using a mixture of trifluoroacetic acid and water (Scheme 1). All amines used were commercially available, except for N1-(quinolin-4-yl)ethane-1,2-diamine, used for the preparation of compound 32, which was synthesized as previously reported.36 Chemical–physical data and elemental analyses for compounds 10–12, 14–21, and 31–42 are reported in Table 1 and Table S1 (Supporting Information), respectively.
Figure 1.
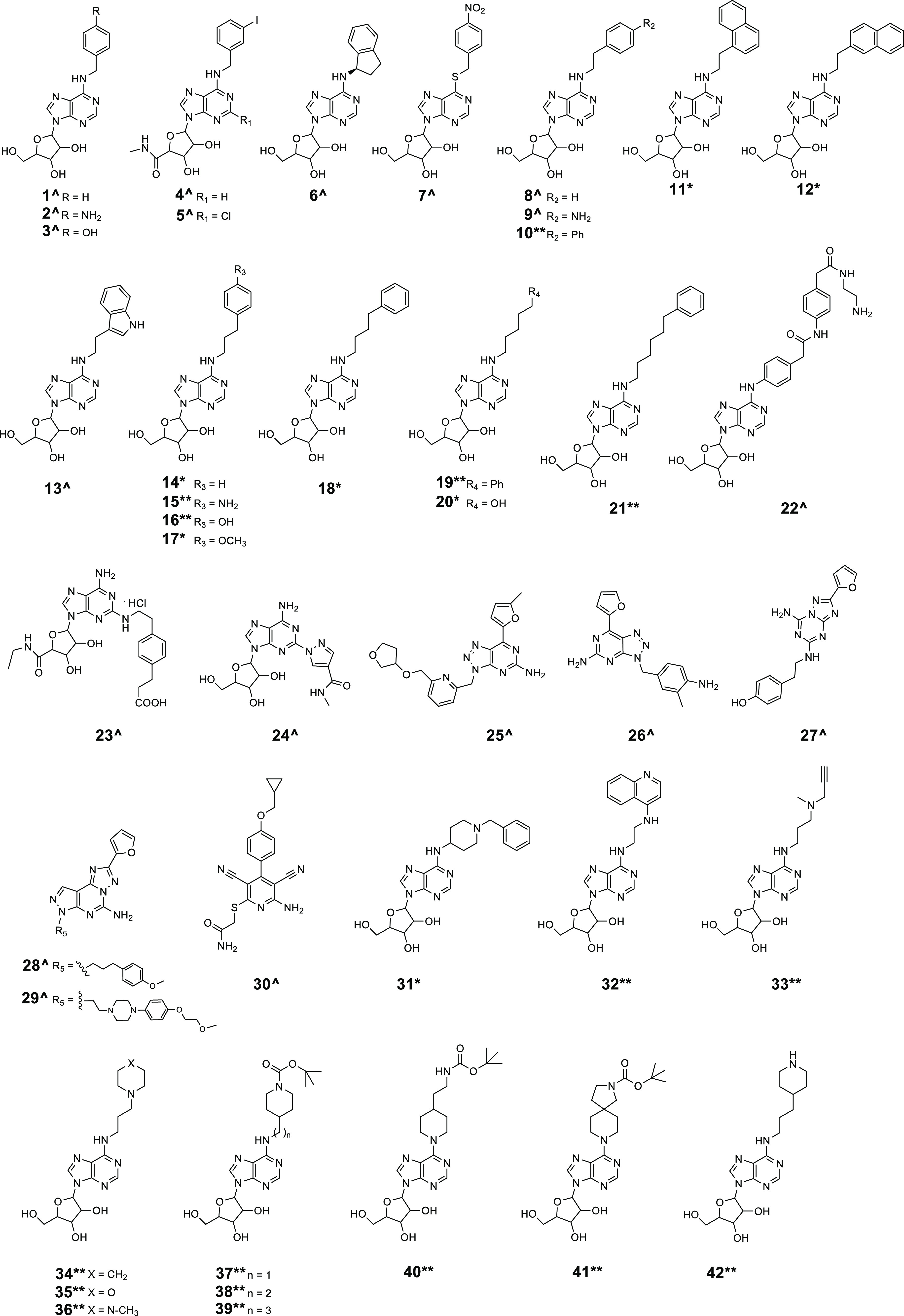
Compounds 1–42 (^commercially available, *previously reported and resynthesized, and **newly designed and synthesized) screened against CamA.
Scheme 1. Synthesis of Compounds 10–12, 14–21, and 31–42.
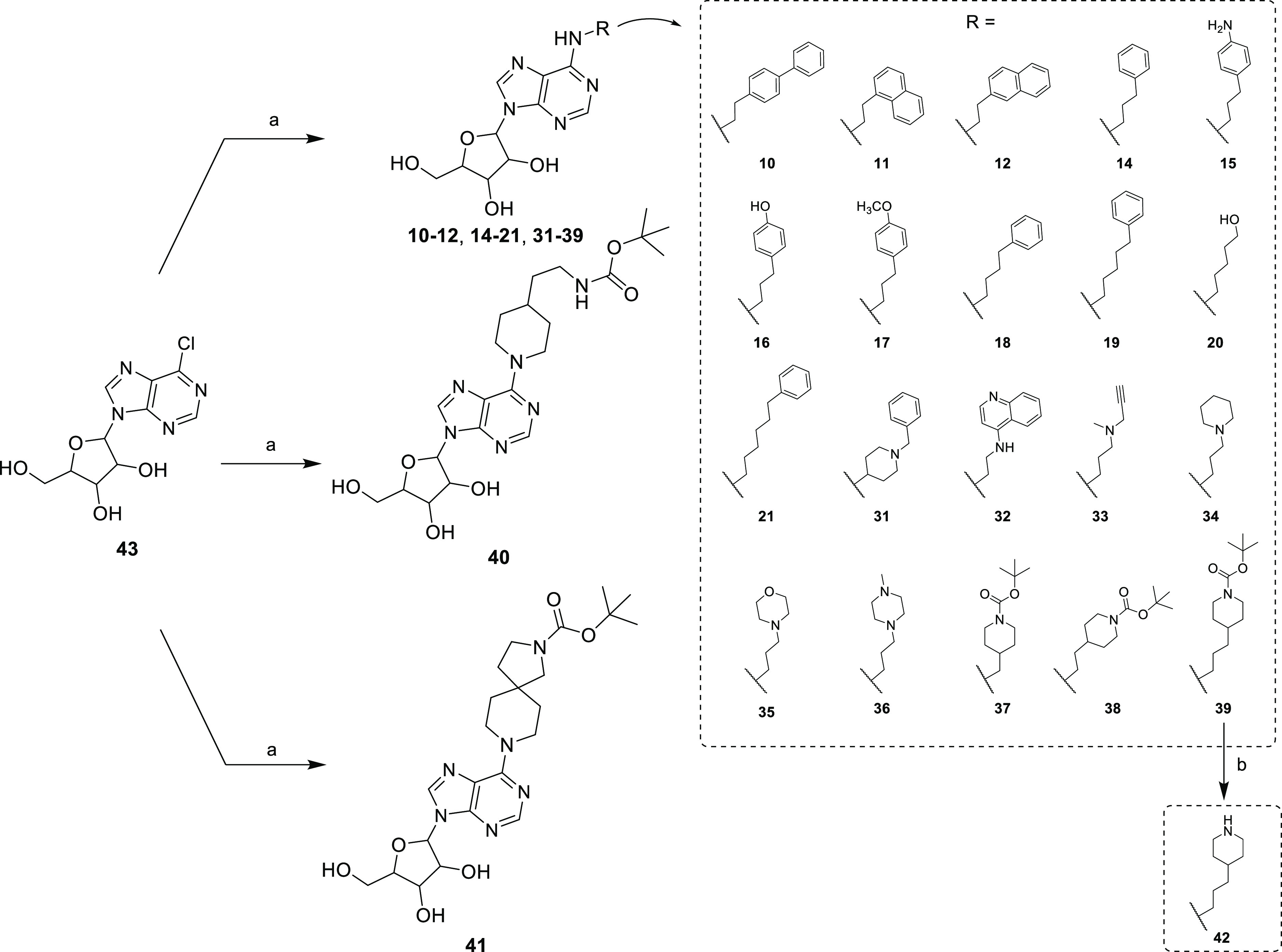
Reagents and conditions: (a) appropriate primary or secondary amine, triethylamine, dry ethanol, reflux. (b) Trifluoroacetic acid/water 9:1 (v/v), 0 °C to room temperature.
Table 1. Melting Point, Recrystallization System, and Yield for Compounds 10–12, 14–21, and 31–42.
| compound | Mp (°C) | recrystallization systema | yield (%) |
|---|---|---|---|
| 10 | 137–140 | A | 67 |
| 11 | 185–188 | B | 72 |
| 12 | 150–153 | B | 75 |
| 14 | 133–135 | C | 89 |
| 15 | 186–187 | B | 54 |
| 16 | 177–180 | B | 65 |
| 17 | 133–135 | C | 57 |
| 18 | 135–137 | C | 91 |
| 19 | 112–115 | C | 86 |
| 20 | 180–181 | B | 69 |
| 21 | 107–110 | C | 87 |
| 31 | 103–105 | D | 56 |
| 32 | 236–237 | B | 51 |
| 33 | 102–104 | D | 54 |
| 34 | 120–122 | C | 68 |
| 35 | 150–153 | A | 61 |
| 36 | 127–129 | C | 57 |
| 37 | 140–141 | C | 79 |
| 38 | 101–102 | D | 71 |
| 39 | 128–129 | C | 68 |
| 40 | 112–113 | D | 81 |
| 41 | 131–132 | D | 70 |
| 42 | 125–127 | C | 88 |
A: methanol; B: ethanol; C: acetonitrile; D: cyclohexane.
Most of these compounds (1–22, 31–39, 42) carried substituents at the C6-amino group (N6) of adenosine, including arylalkyl groups with a growing carbon-number alkyl chain (1–21), a N-(2-aminoethyl)-2-(4-(2-phenylacetamido)phenyl)acetamide structure (22), or variously substituted aminoalkyl groups (31–39, 42). Compounds 23 and 24 displayed substitution at the C2 position of the adenosine (or adenosine-like) scaffold, while 25–29 shared the 7-(furan-2-yl)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine or 2-(furan-2-yl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine core typical of some adenosine receptor ligands. Finally, 40 and 41 were two adenosine mimetics in which the adenosine C6 amino group is replaced by a substituted piperidine ring (Figure 1).
2.2. Identification of a Potential Site for Adenosine Modification in CamA Inhibitors
CamA inhibition by SGC0946, a brominated derivative of EPZ004777, exhibited stronger inhibition and improved binding (∼9× in IC50 and ∼3× in KD values) (Figure 2A),29 and this observation led us to examine some SAM-competitive inhibitors of the human histone H3 lysine 79 MTase DOT1L. These inhibitors included FED1, which has the same tert-butyl phenyl urea moiety37 as SGC0946 and EPZ004777 but features a nitrogen atom instead of carbon at the ring 7 position of the adenine ring (purine instead of 7-deazapurine as the central core; Figure 2A). The changes of nitrogen-to-carbon and brominated carbon increase the hydrophobicity of adenine moiety. However, FED1 is a poor inhibitor of CamA, which retains ∼75% activity at an inhibitor concentration of 50 μM, whereas SGC0946 inhibits CamA activity completely under the same conditions (Figure 2B). Unexpectedly, 5′-methylthioadenosine (MTA), 5′-dimethylthioadenosine (dMTA), and sinefungin, identified from a screen of SAM analogues (Figure S1), have better inhibition potency than that of FED1 (Figure 2B). This is surprising as sinefungin, MTA, and dMTA have the exact same adenosine moiety, suggesting that the tert-butyl phenyl urea of FED1 antagonizes CamA inhibition. We also compared the binding affinities of sinefungin, MTA, and adenosine (Figure 2C). MTA, which is naturally produced by SAM catabolism and is present in all mammalian tissues,38 showed ∼2× enhanced binding compared to sinefungin and >7× stronger binding than adenosine.
Figure 2.

Structure of CamA-DNA-MTA (5′-methylthioadenosine). (A) Chemical structures of SGC0946, a brominated analogue of EPZ004777, with a substitution of the purine nitrogen (N7) with carbon atom (indicated by a symbol *). Both modifications increase the hydrophobicity of the purine moiety. (B) Relative inhibition of CamA activity at a single inhibitor concentration of 50 μM, in the presence of the standard reaction condition of 40 μM SAM. Comparison of sinefungin, MTA, and adenosine (see Figure S1 for MTA related compounds). (C) KD measurement by isothermal titration calorimetry. (D) Structure comparison of the CamA-DNA complex in the absence of cofactor (left; PDB 7LNJ), bound with SAH (middle; PDB 7LT5), or MTA (right; PDB 8CXS). The MTA bound structure adopts the cofactor-free conformation along with disordered N-terminal residues prior to Gly28. (E) Omit electron density map (contoured at 5.0 σ above the mean) for the bound MTA (PDB 8CXS). An ethylene glycol (colored in yellow) binds to the edge of adenine moiety. (F) Superimposition of SAH (magenta) and MTA (gray).
We crystallized MTA in complex with CamA in the presence of a DNA substrate (Figure 2D, right panel). The N-terminal residues prior to Gly28 are disordered, as previously observed in the binary complex of CamA-DNA (PDB 7LNJ; left panel). However, these N-terminal residues do become ordered upon the binding of S-adenosyl-l-homocysteine (SAH) (PDB 7LT5; colored magenta in the middle panel) due to the interactions with the homocysteine moiety of SAH.28 Thus, the MTA-bound structure, which lacks a homocysteine moiety, adopts the cofactor-free conformation, and the common components (residue Gly28 and beyond) among the three structures are highly similar, with a root-mean-square deviation (RMSD) of <0.3 Å. MTA superimposed well onto the adenosine moiety of SAH, and the exposed edges of N6 and N7 interact with an ethylene glycol molecule (Figure 2E,F). We reasoned that the space occupied by the ethylene glycol could be explored for modifications of the adenosine, and below is a description of modifications we tested at the adenine N6 position and their effects on CamA inhibition. In particular, we focused on the interactions with the Asp149-Tyr178 pair (these two residues form an intramolecule hydrogen bond) and Glu175.
2.3. Design of Adenosine Analogs as CamA Inhibitors
We carried out a CamA inhibition screen, using 42 compounds originally designed as analogs of adenosine or adenosine receptors’ ligands (Figure 1). These compounds all contain a common adenosyl moiety. SGC0946 and sinefungin were utilized as positive controls. The screen started with compound 1 (N6-benzyladenosine), which showed similar inhibition strength to that of sinefungin, and reached the most potent inhibition with compound 39, which showed 12× higher potency than SGC0946 (Figure 3A). Of these 42 compounds, we chose 14 for a detailed inhibition study, quantifying the half-maximal inhibitory concentration (IC50) in the presence of 40 μM co-substrate SAM, and the dissociation constant (KD) as measured by isothermal titration calorimetry (Figure S2). The co-crystals of CamA with bound inhibitors were formed under the same conditions as for the MTA-bound complex, and their structures were determined at resolutions of 2.19–2.81 Å (Table S2). All structures were isomorphous, with RMSD of 0.1–0.3 Å for pairwise comparisons over 1505 pairs of Cα atoms (three complexes per crystallographic asymmetric unit, with the first ordered residue being Gly28 for each CamA protein).
Figure 3.

Structures of N6-benzyladenosine (1) and its related one-carbon analogs. (A) Relative inhibition of CamA activity at a single inhibitor concentration of 10 μM, in the presence of 40 μM co-substrate SAM. Compounds indicated by a red asterisk were chosen for further study. (B) Summary of inhibition (IC50 and Hill slope), dissociation constant (KD) measured by isothermal titration calorimetry, and X-ray information (PDB accession numbers and corresponding resolutions). (C) Additions at the N6 position of adenosine: benzyl (1), 4-aminobenzyl (2), 4-hydroxybenzyl (3), 3-iodobenzyl (4), and (1R)-indanyl ring (6). (D) N6-benzyl moiety (1) is surrounded by hydrophobic residues (PDB 8CXT). (E) Superimposition of benzyl-(PDB 8CXT), 4-aminobenzyl-(PDB 8CXU), 4-hydroxybenzyl-(PDB 8CXV), and 3-iodobenzyl-adenosines bound with CamA (PDB 8CXW). (F) Superimposition of benzyl- (PDB 8CXT) and indanyl-adenosines (PDB 8CXX). Tyr178 adopts two alternative conformations with the indane moiety.
We first compared five modifications with differently substituted benzyl groups at the C6-amino function of adenosine: benzyl (1), 4-aminobenzyl (2), 4-hydroxybenzyl (3), 3-iodobenzyl (4, in which also a N-methylcarboxamide replaced the 5-(hydroxymethyl) moiety at the tetrahydrofuran portion), and (1R)-indanyl ring (6) as a constrained form of the benzyl group (Figure 3C). We made the following observations: (a) In general, the IC50 and KD values for 1 and 2 are similar to those of MTA (IC50 values in the range 10–18 μM and KD values in the range 11–14 μM), while 3 and 4 displayed slightly higher inhibition potency (IC50 = 4.8 and 8 μM and KD = 5.3 and 3.9 μM, respectively) (Figure 3B). (b) Structurally, the benzyl ring is bordered by hydrophobic residues (Ile115, Pro167, Ile169, Leu174, and Leu196) (Figure 3D). The same hydrophobic environment applies to 4-aminobenzyl, 4-hydroxybenzyl, and 3-iodobenzyl rings, which form a face-to-face stacking interaction with the Tyr178-Asp149 pair (Figure 3E). (c) Interestingly, the 4-hydroxybenzyl ring (compound 3) demonstrated ∼2× improved IC50 or 2.6× tighter binding in the KD value compared to the 4-aminobenzyl counterpart (compound 2). The two compounds differ only at the tip of the moiety, having either a hydroxyl (OH) or an amino group (NH2). (d) The non-planar indan-1-yl ring (compound 6), which gave decreased inhibition, pushes toward Tyr178 and yields two alternative Tyr178 conformations, one of which has lost the H-bond with Asp149 (Figure 3F).
2.4. Linear Homology in the N6-Benzyl Portion: Effects of Varying Carbon Spacer between Adenine-N6 and the Phenyl Ring
Next, we tested the effect of the aliphatic carbon chain length connecting the adenosyl N6 position with the phenyl ring (1 in Figure 4A). It appears that elongating the N6-benzyl moiety to the −2-phenylethyl (8), −3-phenylpropyl (14), −4-phenylbutyl (18), and −5-phenylpentyl (19) portion led to improved CamA inhibitory potency of the compounds, while further elongation to a −6-phenylhexyl group (21) decreased the inhibition potency (see Figure 3A for its inhibition at 10 μM). Compound 14, with the three-carbon aliphatic chain, showed the highest potency as it has the lowest values of IC50 (0.7 μM) and KD (0.5 μM) values, while either decreasing or increasing the chain length resulted in lower inhibition (i.e., increased IC50 and KD values) (Figure 4B). Compared to compound 1 (1C-length), 14 with its 3C-length has markedly stronger inhibition (∼21× lower IC50 value) and improved binding (∼26× lower KD value) (Figure 4B). Structurally, superimposition of inhibitors with 1C-, 2C-, and 3C-length chains revealed that (i) the phenyl ring rotated from face-to-face (1C) to edge-to-face (2C and 3C) conformation with respect to Tyr178 (Figure 4C), and (ii) the 3C-length allowed the 3-phenylpropyl ring to reach Glu175, whereas 1C- or 2C-length chains are too short to make that contact (Figure 4D). Furthermore, the 4C-length chain positioned the phenyl ring pointing away from Tyr178 and Glu175 (Figure 4E), whereas the 5C-length chain put the phenyl ring too close to Glu175 and forced the side chain of Glu175 to rotate away from the position observed in which it interacts with the 3C-phenylpropyl ring (Figure 4F).
Figure 4.

Effects of varying the carbon spacer between adenine-N6 and the phenyl ring. (A) Chemical structures of five related compounds with varied carbon length from one to five carbons. (B) Summary of inhibition (IC50), dissociation constant (KD), and X-ray information (PDB). (C, D) Two views of superimposition of chains of 1C- (PDB 8CXT), 2C- (PDB 8CXY), and 3C-length with the 3C-phenylpropyl moiety reaching to Glu175 (PDB 8CXZ). (E) 4-Phenylbutyl moiety points away from Glu175 and Tyr178 (PDB 8CY0). (F) 5C-Phenyl ring pushes the side chain of Glu175 away (PDB 8CY1).
We observed the same trend of improved inhibition potency from 1C-to-3C-length using the 4-aminobenzyl moiety (compounds 2, 9, and 15 in Figure 5A,B). Compound 9 (also known as APNEA) was purchased and contains >5% impurity, which interferes with the MTase-Glo assay (Figure S3). Nevertheless, the 1C- and 2C-lengths between adenosine and the 4-aminophenyl ring (2 and 9) exhibited similar dissociation constants (KD = 14 or 18 μM), whereas 3C-lengthed 4-aminophenyl ring (15) has 9–12× enhanced binding (KD = 1.5 μM), according to the above observation. Indeed, the terminal amino group (NH2) of compound 15 makes a H-bond with one of the carboxylate oxygen atoms of Glu175 (Figure 5C). Interestingly, replacing the terminal amino group of 15 with a hydroxyl group (compound 16) resulted in stronger inhibition (∼2.7X lower IC50 value) and improved binding (∼6.5X lower KD value). This difference, between NH2 (compound 15) and OH groups (compound 16), is the same as we observed earlier between compounds 2 and 3 in Figure 3. The O–H···O H-bond observed with compound 16 and Glu175 might be analogous to that of the H-bond between Tyr178 and Asp149 (Figure 5D).
Figure 5.

Differences between 3C-length 4-aminobenzyl and 4-hydroxybenzyl moieties. (A) Chemical structures of three related compounds of amino benzyl with varied carbon length from 1–3-carbons. (B) Summary of inhibition (IC50), dissociation constant (KD), and X-ray information (PDB). (C) Superimposition of chains of 1C- (PDB 8CXU), 2C- (PDB 8CY2), and 3C-length with the 3C-aminobenzyl moiety forming a H-bond with the carboxylate group of Glu175 (PDB 8CY3). (D) Superimposition of 3C-aminobenzyl (PDB 8CY3) and 3C-hydroxybenzyl moiety (PDB 8CY4) with the terminal amino group or hydroxy oxygen atom forming a H-bond with Glu175.
2.5. Compound 39 Has the Most Potent Inhibition against CamA
As a further step, to increase the chemical flexibility of the 4-aminobenzyl moiety of 2 and its analogs, we transformed this portion into a 4-methylpiperidine 1-tert-butyl carbamate and related groups, obtaining the compounds 37–39 (Figure 6A). Again, we observed improved inhibition with chain length from 1C to 3C, with decreasing IC50 values from 2.0 μM (1C in compound 37), 0.71 μM (2C in 38), to 0.39 μM (3C in 39) (Figure 6B). The dissociation constant (KD = 0.14 μM) was not distinguishable between 2C and 3C compounds (38 and 39), but both were improved by ∼8× from that of 1C compound 37 (KD = 1.1 μM) (Figure 6B). In the structure of CamA bound with compound 39, the 4-n-propylpiperidine forms extensive van der Waals contacts with Asp149, Tyr178, Lys196, Ile169, and Leu174 (Figure 6C, D). The compound sits on a largely hydrophobic surface (colored green in Figure 6E). Instead of interacting with the side chain carboxylate group of Glu175, the terminal carbonyl oxygen atom of 39 forms a H-bond with the main chain amide nitrogen of Glu175, and two of the terminal methyl groups (tert-butyl substituent) form a C–H···O type of H-bond with the main chain carbonyl oxygen atom of Lys173 (Figure 6F). These interactions are unique to compound 39, which combines aromatic, van der Waals, and polar interactions. The gained polar interactions give rise to favorable binding of compound 39 as reflected by the change of the binding entropy factor (−TΔS in Figure 6G). Similar changes were observed for compounds 15, 16 (which interact with Glu175; Figure 5D), and 38.
Figure 6.

Compound 39 is the most potent inhibitor of CamA tested to date. (A) Chemical structures of compounds 37–39 with varied carbon length from 1- to 3-carbon atoms and of the related compounds 40–42. (B) Summary of inhibition (IC50), dissociation constant (KD), and X-ray information (PDB). (C, D) Two views of compound 39 showing the extensive hydrophobic contacts (PDB 8CY5). (E) Surface presentation (green for carbons, red for oxygens, and blue for nitrogen atoms) showing the binding site of 39, superimposed with the omit electron density map (contoured at 5.0 σ above the man). Note that the sulfur atom of Cys65 is visible (colored in yellow, towards the lower left). (F) Compound 39 forms H-bonds with the main-chain amide nitrogen atom of E175 and main-chain carbonyl oxygen atom of Lys173 (PDB 8CY5). (G) Thermodynamic parameters derived from ITC measurements (free energy ΔG, binding enthalpy ΔH, and entropy −TΔS). (H) CamA inhibition of five related compounds (37–41) at [I] = 1 μM, in the presence of 40 μM SAM co-substrate. (I) Compound 42 (without carbamate moiety) has reduced inhibition potency.
To gain insight into the structure–activity relationships, we prepared two further molecules, 40 and 41, the first of which (40) is an isomer of 38, bearing at the purine C6-position a (1-piperidin-4-yl)ethyl tert-butyl carbamate instead of the tert-butyl-4-(2-aminoethyl)piperidine-1-carboxylate while the other (41) is a constrained version of 40, with a tert-butyl-8-(2,8-diazaspiro[4.5]decane)-2-carboxylate at the purine C6-position (Figure 6A). Despite the similarity of 40 and 41 with 38 (38 and 40 have the exact same compositions of atoms, with the positions of piperidine ring and aliphatic carbon chains switched), compounds 40 and 41 resulted in loss of inhibition at the two concentrations tested (1 μM in Figure 6H or 10 μM Figure 3A). Finally, to assess the stability (Figure S4) and crucial role of the N-tert-butyl carbamate in 39, we prepared and tested 42, in which this moiety has been removed (Figure 6A). Compound 42 showed ∼10-fold lower potency than 39 in inhibiting CamA (IC50 = 3.8 μM vs 0.39 μM) (Figure 6I), confirming the importance of the carbamate moiety for CamA binding and inhibition.
2.6. Selectivity
Finally, we examined three of our most promising compounds, 16, 38, and 39, against a group of related SAM-dependent nucleic acid MTases, including two bacterial DNA adenine MTases (Caulobactor crescentus CcrM and Escherichia coli Dam; Figure 7A–C), two mammalian DNA cytosine MTases (human DNMT1 and mouse Dnmt3a-Dnmt3L; Figure 7D,E), and four human RNA (or dual RNA/DNA) adenine MTases (PCIF1, MettL5-Trm112, MettL16, and MettL3-MettL14; Figure 7F–I). We also tested the potency of these three compounds against four human protein (lysine or arginine) MTases (DOT1L, EZH2 complex, G9a, and PRMT1; Figure 7K–N). While we had to use a few different reaction conditions as appropriate for these diverse enzymes, none of them showed any significant inhibition by these three compounds at a concentration of 10 μM. In contrast, sinefungin, a pan inhibitor of SAM-dependent MTases, showed measurable inhibition against CcrM, Dam, Dnmt3a-3L, and PCIF1 as well as inhibiting CamA while SAH (the reaction product) exhibited measurable inhibition of DOT1L, G9a, and PRMT1. As a control, the RNA methylation activity of MettL3-MettL14 was inhibited by known selective MettL3 inhibitors (the two pure enantiomers R-UZH1 and S-UZH139 and STM245740), and these MettL3 inhibitors had no effect on CamA activity (Figure 7J).
Figure 7.

Selectivity of compounds 16, 38, and 39 against (A–C) three bacterial DNA adenine MTases (CamA, Caulobactor crescentus CcrM, and Escherichia coli Dam); (D, E) two mammalian DNA cytosine MTases (human DNMT1 and mouse Dnmt3a-3L); (F–I) four human RNA adenine MTases (PCIF1, MettL5-Trm112 and MettL16 and MettL3-MettL14); (J) MettL3-selective inhibitors [the two pure enantiomers R-UZH1 and S-UZH1 and STM2457] had no effect on CamA. All compounds were used at a concentration of [I] = 10 μM, in the presence of varied amounts of co-substrate SAM and individual buffer conditions, depending on the MTase (see Methods). The reaction turnover rates ([SAH]/[E]) for nucleic acid MTases were estimated to be 20 (EcoDam), 8 (CamA), 4 (CcrM), 3 (MettL5-Trm112), 2 (PCIF1, MettL16 and MettL3–14), and 1.5 (DNMT1 and Dnmt3a-3L). The DMSO control of Dnmt3a-3L (E) used the same reaction buffer which included 20% glycerol. (K–N) Four human protein lysine MTases (DOT1L, EZH2, and G9a) and one protein arginine MTase (PRMT1). (O,P) Two human adenosine receptors (A1 and A3).
We further evaluated the affinity of compounds 16, 38, and 39 for the human A1 and A3 adenosine receptors (A1AR and A3AR, respectively) via agonist radioligand binding assays (Figure 7O,P). In both assays, the three compounds displayed minimal inhibition of specific binding of the employed radioligand ([3H]2-chloro-N6-cyclopentyladenosine for A1AR and [125I]AB-MECA for A3AR). In contrast, the selective A1AR agonist N6-cyclopentyladenosine and the selective A3AR agonist piclidenoson (also known as IB-MECA, compound 4 in this study) completely abolished radioligand binding in their respective assays.
2.7. Discussion
We have sought here to identify potent and selective inhibitors targeting CamA, a Clostridioides difficile-specific DNA adenine MTase required for efficient sporulation and, more profoundly, persistence in the gastrointestinal tract.24 The basis for CamA’s role in CDI persistence is not yet known, but there is the potential for synergies by also inhibiting other pathways that reduce CDI carriage, such as cyclic-di-AMP metabolism.41
The inhibitors identified here are likely to have very similar effects on the CamA orthologs in all sequence-characterized C. difficile strains. We base this statement on the complete conservation of the 12 inhibitor-proximal residues (G28, Y30, D114, I115, D149, P167, Y168, I169, L174, Y178, L196, and F200; see Figure 3D), in the set of 58 nonidentical CamA sequences examined (see Figure S7 of ref (28)). Our efforts to depict the interactions of CamA with diverse adenosine analogs of inhibitors provide fundamental knowledge into structure–activity relationships of inhibitors against CamA.
Here, we present 39 [tert-butyl-4-(3-((9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)propyl)piperidine-1-carboxylate] as the first potent and specific inhibitor of CamA. In vitro, the compound binds in the adenosine binding site (like SAM/SAH) with the N6-addition pointing outward and sitting against a largely hydrophobic surface, forming a unique combination of aromatic, van der Waals, and polar interactions (Figure 6). We note that there is additional space that can be further explored to fully occupy the hydrophobic CamA surface (Figure 6E).
Next, we will turn our attention to the homocysteine moiety of SAH, which induced conformational change of N-terminal residues prior to Gly28 (Figure 3D). One interesting but puzzling observation is that the tert-butyl-phenyl urea of FED1 – which presumably occupies the site of homocysteine moiety – antagonized CamA inhibition (Figure 2B). The observation of weak inhibition by FED1 together with the cofactor-induced conformational change might provide avenues for designing 39-based inhibitors to engage in additional interactions with the N-terminal residues. The ability of future compounds to induce this movement may contribute substantially to optimizing compound potency and selectivity as CamA inhibitors. Interestingly, the movement of N-terminal residues exposes the buried Cys65 to solvent (Figure 6E). Cys65 is near the binding site of the homocysteine moiety and could be explored for interaction with the cysteine-reactive electrophile acrylamide as has been done for other enzymes including protein kinases,42−44 histone lysine methyltransferases,45,46 and a histone lysine demethylase.47
A lasting challenge is to improve our lead compound(s)’s ability to cross the cell wall of Gram-positive bacteria (the group to which C. difficile belongs) (Table S3). This cell wall is dominated by the peptidoglycan layer, presenting a landscape of large (up to 60 nm in diameter), deep (up to 23 nm) pores constituting a disordered gel.48 Furthermore, the peptidoglycan is covered by a proteinaceous S-layer49,50 and underlaid by a fairly typical lipid bilayer membrane.51 Finally, we note that a compound inhibiting CamA activity without killing the bacteria would represent an essential research tool for uncovering the possible mechanism(s) by which CamA epigenetically regulates gene expression for C. difficile sporulation and colonization.
3. Experimental Section
3.1. Chemistry
Melting points were determined on Buchi 530 melting point apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded at 400 and 100 MHz, respectively, with a Bruker AC400 spectrometer. Chemical shifts are reported in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). All compounds were analyzed by thin-layer chromatography (TLC) and 1H-NMR. TLC was performed on aluminum-backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254) with spots visualized by UV light. Yields of all reactions refer to the purified products. All chemicals were from Sigma-Aldrich srl, Milan (Italy), and were of the highest available purity. Low-resolution mass spectra were recorded with an API-TOF Mariner by a Perspective Biosystem (Stratford, TX); samples were injected by a Harvard pump using a flow rate of 5–10 μL/min with electrospray ionization (ESI). High-resolution mass spectrometry (HR-MS) was performed using an Exactive Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) at a mass range of 100–2000 m/z. The mass acquisition was set as follows: the resolution of 100.000 at m/z 200, positive ESI mode and mass spectra were acquired by recording 30 scans, sheath gas flow of 5 arbitrary units, spray voltage of 3.5 kV, capillary voltage 77.5 V, capillary temperature 250 °C, and tube lens voltage at 250 V. Software tools were from Thermo Fisher Scientific (Waltham, MA, USA). Specifically, mass spectra were processed using Xcalibur. Solutions for flow injection analysis were constituted by dissolving each compound in MeOH MS grade (c = 10–5 M). Elemental analysis was used to determine the purity of compounds; all analytical results were within ±0.40% of the theoretical values (Table S1).
The purity of compounds 1–42 was also analyzed by high-performance liquid chromatography (HPLC) (Supporting Information). The HPLC system consisted of a Dionex UltiMate 3000 UHPLC (Thermo Fisher) system equipped with an automatic injector, column heater and coupled with a Diode Array Detector DAD-3000 (Thermo Fisher). For compounds 1–24 and 31–42, the analytical controls were performed on an Adamas HILIC 3 μm (4.6 × 150 mm) column (SepaChrom) in gradient elution. Eluents: (A) CH3CN/H2O, 95/5 (v/v) + 5 mM ammonium acetate; (B) H2O/CH3CN, 95/5 (v/v) + 5 mM ammonium acetate. A 20 min linear gradient elution from 0% to 60% solvent B was followed by a 1 min ramp to 100% B and 4 min at 100% B. The flow rate was 1.0 mL/min, and the column was kept at a constant temperature of 30 °C. Samples were dissolved in solvent A at a concentration of 1 mg/mL, and the injection volume was 2 μL. For compounds 25–30, the analytical controls were performed on an XTERRA MS C18 3.5 μm (4.6 × 150 mm) column (Waters) in gradient elution. Eluents: (A) H2O/CH3CN, 95/5 (v/v) + 0.01% TFA; (B) CH3CN/H2O, 95/5 (v/v) + 0.01% TFA. A 20 min linear gradient elution from 0% to 60% solvent B was followed by a 1 min ramp to 100% B and 4 min at 100% B. The flow rate was 1.0 mL/min, and the column was kept at a constant temperature of 30 °C. Samples were dissolved in solvent A at a concentration of 1 mg/mL, and the injection volume was 2 μL. In some cases, it was necessary to add DMSO to increase compound solubility. Purity in all cases was >95% except for commercial compound 9 (APNEA), the purity of which was 92–94%.
All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulfate.
3.1.1. General Procedure for the Synthesis of Final Compounds 10, 11,(30,31)12,(30)14,(32,33)15, 16, 17,(34)18,(32,33)19, 20,(35)21, 31 (Patents EP0417999A1 and EP0423777A2), and 32–41
A mixture containing the properly substituted amine (3 equiv), 6-chloropurin-9-riboside (0.349 mmol, 100 mg, 1 equiv), and triethylamine (0.419 mmol, 42.36 mg, 0.058 mL, 1.2 equiv) in dry ethanol (3 mL) was stirred under reflux conditions for 1.5 h. The reaction mixture was then quenched with ethyl acetate (30 mL) and washed with water (3 mL) and brine (2 × 3 mL). The organic phase was then dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. Alternatively, the reaction mixture was diluted with 3 mL ethanol, cooled down to r.t., and stirred for a further 40 min allowing the formation of a precipitate, which was filtered and washed over the filter with ethanol, hexane, and diethyl ether. In both cases, the resulting crude product was purified by silica gel column chromatography eluting with the appropriate chloroform/methanol or chloroform/methanol/ammonia mixture to afford the desired final compounds 10–12, 14–21, and 31–41.
3.1.2. (2R,3R,4S,5R)-2-(6-((2-([1,1’-Biphenyl]-4-yl)ethyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol (10)
1H-NMR (400 MHz; DMSO-d6) δ 2.98 (t, 2H, NH-CH2-CH2-Ph-Ph), 3.53–3.59 (m, 1H, CH ribose), 3.66–3.71 (m, 1H, CH ribose), 3.76 (br s, 2H, NH-CH2-CH2-Ph-Ph), 3.97 (q, 1H, CH ribose), 4.16 (br s, 1H, CH ribose), 4.62 (br d, 1H, CH ribose), 5.17–5.24 (m, 1H, OH ribose), 5.37–5.44 (m, 1H, OH ribose), 5.47 (br s, 1H, OH ribose), 5.90 (d, 1H, CH ribose), 7.33–7.37 (m, 3H, benzene ring), 7.46 (t, 2H, benzene ring), 7.59–7.66 (m, 4H, benzene ring), 7.98 (br s, 1H, NH), 8.26 (s, 1H, C2-H adenine), 8.36 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 35.0, 41.7, 62.1, 71.1, 73.9, 86.4, 88.4, 120.3, 127.0 (2C), 127.1 (2C), 127.7, 129.4 (2C), 129.8 (2C), 138.5, 139.3, 140.4, 140.5, 148.8, 152.9, 155.0. MS (ESI), m/z: 448 [M + H]+. HR-MS (ESI) m/z: calculated for C24H26N5O4+ [M + H]+, 448.1979; found, 448.1971.
3.1.3. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((2-(naphthalen-1-yl)ethyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (11)30,31
1H-NMR (400 MHz; DMSO-d6) δ 3.39 (t, 2H, NH-CH2-CH2-naphthalene), 3.54–3.60 (m, 1H, CH ribose), 3.67–3.72 (m, 1H, CH ribose), 3.81 (br s, 2H, NH-CH2-CH2-naphthalene), 3.98 (q, 1H, CH ribose), 4.16 (d, 1H, CH ribose), 4.63 (d, 1H, CH ribose), 5.18–5.23 (m, 1H, OH ribose), 5.37–5.43 (m, 1H, OH ribose), 5.47 (d, 1H, OH ribose), 5.91 (d, 1H, CH ribose), 7.43–7.47 (m, 2H, CH naphthalene ring), 7.54 (t, 1H, CH naphthalene ring), 7.61 (t, 1H, CH naphthalene ring), 7.81 (d, 1H, CH naphthalene ring), 7.94 (d, 1H, CH naphthalene ring), 8.11 (br s, 1H, NH), 8.32 (s, 1H, C2-H adenine), 8.39–8.41 (m, 2H, C8-H adenine + CH naphthalene ring). 13C-NMR (100 MHz; DMSO-d6) δ 33.0, 41.4, 62.1, 71.1, 74.0, 86.4, 88.4, 120.4, 124.5, 126.1 (2C), 126.6, 127.1, 127.2, 129.0, 132.2, 133.9, 136.1, 140.3, 148.8, 153.0, 155.1. MS (ESI), m/z: 422 [M + H]+. HR-MS (ESI) m/z: calculated for C22H24N5O4+ [M + H]+, 422.1823; found, 422.1814.
3.1.4. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((2-(naphthalen-2-yl)ethyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (12)30
1H-NMR (400 MHz; DMSO-d6) δ 3.11 (t, 2H, NH-CH2-CH2-naphthalene), 3.53–3.59 (m, 1H, CH ribose), 3.66–3.71 (m, 1H, CH ribose), 3.83 (br s, 2H, NH-CH2-CH2-naphthalene), 3.97 (d, 1H, CH ribose), 4.15–4.17 (m, 1H, CH ribose), 4.63 (q, 1H, CH ribose), 5.16–5.22 (m, 1H, OH ribose), 5.37–5.43 (m, 1H, OH ribose), 5.45 (d, 1H, OH ribose), 5.89 (d, 1H, CH ribose), 7.44–7.50 (m, 3H, CH naphthalene ring), 7.77 (s, 1H, CH naphthalene ring), 7.85–7.88 (m, 3H, CH naphthalene ring), 8.00 (br s, 1H, NH), 8.27 (s, 1H, C2-H adenine), 8.35 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 35.2, 41.2, 61.8, 70.7, 73.6, 86.0, 88.0, 119.9, 125.4, 126.1, 126.8, 127.4, 127.6, 127.7, 127.8, 131.8, 133.2, 137.3, 139.9, 148.4, 152.5, 154.6. MS (ESI), m/z: 422 [M + H]+. HR-MS (ESI) m/z: calculated for C22H24N5O4+ [M + H]+, 422.1823; found, 422.1813.
3.1.5. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-phenylpropyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (14)32,33
1H-NMR (400 MHz; DMSO-d6) δ 1.91 (p, 2H, NH-CH2-CH2-CH2-Ph), 2.65 (t, 2H, NH-CH2-CH2-CH2-Ph), 3.52–3.59 (m, 3H, NH-CH2-CH2-CH2-Ph + CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15–4.16 (br m, 1H, CH ribose), 4.62 (q, 1H, CH ribose), 5.17–5.23 (m, 1H, OH ribose), 5.38–5.48 (m, 2H, OH ribose), 5.89 (d, 1H, CH ribose), 7.16–7.30 (m, 5H, benzene ring), 7.96 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.35 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) 30.8, 32.7, 40.7, 61.7, 70.7, 73.5, 85.9, 88.0, 125.7, 128.30 (2C), 128.32 (2C), 130.1, 139.7, 141.9, 148.9, 152.4, 154.7. MS (ESI), m/z: 386 [M + H]+. HR-MS (ESI) m/z: calculated for C19H24N5O4+ [M + H]+, 386.1823; found, 386.1817.
3.1.6. (2R,3R,4S,5R)-2-(6-((3-(4-Aminophenyl)propyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol (15)
1H-NMR (400 MHz; DMSO-d6) δ 1.79–1.91 (m, 2H, NH-CH2-CH2-CH2-Ph), 2.45–2.47 (m, 2H, NH-CH2-CH2-CH2-Ph), 3.48 (br s, 2H, NH-CH2-CH2-CH2-Ph), 3.53–3.59 (m, 1H, CH ribose), 3.66–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15 (d, 1H, CH ribose), 4.62 (q, 1H, CH ribose), 4.79 (br s, 2H, NH2), 5.13–5.19 (m, 1H, OH ribose), 5.38–5.45 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 6.48 (d, 2H, benzene ring), 6.86 (d, 2H, benzene ring), 7.89 (br s, 1H, NH), 8.20 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 31.7, 32.4, 40.5, 62.2, 71.2, 73.9, 86.4, 88.4, 114.5 (2C), 120.2, 129.1 (2C), 129.2, 140.1, 146.8, 148.7, 152.9, 155.1. MS (ESI), m/z: 401 [M + H]+. HR-MS (ESI) m/z: calculated for C19H25N6O4+ [M + H]+, 401.1932; found, 401.1923.
3.1.7. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-(4-hydroxyphenyl)propyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (16)
1H-NMR (400 MHz; DMSO-d6) δ 1.81–1.88 (m, 2H, NH-CH2-CH2-CH2-Ph), 2.51–2.55 (m, 2H, NH-CH2-CH2-CH2-Ph), 3.48 (br s, 2H, NH-CH2-CH2-CH2-Ph), 3.51–3.58 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (q, 1H, CH ribose), 4.15 (d, 1H, CH ribose), 4.62 (d, 1H, CH ribose), 5.16–5.22 (m, 1H, OH ribose), 5.39–5.47 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 6.66 (d, 2H, benzene ring), 7.00 (d, 2H, benzene ring), 7.93 (br s, 1H, NH), 8.20 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine), 9.11 (s, 1H, Ph-OH). 13C-NMR (100 MHz; DMSO-d6) δ 31.1, 31.8, 40.7, 61.7, 70.7, 73.5, 85.9, 88.0, 115.1 (2C), 129.0, 129.1 (2C), 131.8, 139.7, 148.2, 152.4, 154.7, 155.3. MS (ESI), m/z: 402 [M + H]+. HR-MS (ESI) m/z: calculated for C19H24N5O5+ [M + H]+, 402.1772; found, 402.1759.
3.1.8. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-(4-methoxyphenyl)propyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (17)34
1H-NMR (400 MHz; DMSO-d6) δ 1.83–1.91 (m, 2H, NH-CH2-CH2-CH2-Ph), 2.59 (t, 2H, NH-CH2-CH2-CH2-Ph), 3.49 (br s, 2H, NH-CH2-CH2-CH2-Ph), 3.53–3.58 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.72 (s, 3H, Ph-OCH3), 3.97 (q, 1H, CH ribose), 4.13–4.17 (br m, 1H, CH ribose), 4.59–4.64 (br m, 1H, CH ribose), 5.17–5.23 (m, 1H, OH ribose), 5.38–5.47 (m, 2H, OH ribose), 5.89 (d, 1H, CH ribose), 6.84 (d, 2H, benzene ring), 7.13 (d, 2H, benzene ring), 7.94 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.35 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 31.0, 31.7, 40.8, 54.9, 61.7, 70.7, 73.4, 85.9, 87.9, 113.7 (2C), 119.8, 129.2 (2C), 133.7, 139.7, 148.2, 152.4, 154.7, 157.4. MS (ESI), m/z: 416 [M + H]+. HR-MS (ESI) m/z: calculated for C20H26N5O5+ [M + H]+, 416.1928; found, 416.1918.
3.1.9. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((4-phenylbutyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (18)32,33
1H-NMR (400 MHz; DMSO-d6) δ 1.62 (s, 4H, NH-CH2-CH2-CH2-CH2-Ph), 2.59–2.63 (m, 2H, NH-CH2-CH2-CH2-CH2-Ph), 3.52–3.58 (m, 3H, NH-CH2-CH2-CH2-Ph + CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (q, 1H, CH ribose), 4.15 (br s, 1H, CH ribose), 4.61 (br s, 1H, CH ribose), 5.17–5.23 (m, 1H, OH ribose), 5.39–5.49 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.14–7.28 (m, 5H, benzene ring), 7.92 (br s, 1H, NH), 8.20 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 28.6, 28.8, 35.0, 40.0, 61.8, 70.8, 73.5, 86.0, 88.1, 119.8, 125.7, 128.3 (2C), 128.4 (2C), 139.7, 142.3, 148.3, 152.5, 154.7. MS (ESI), m/z: 400 [M + H]+. HR-MS (ESI) m/z: calculated for C20H26N5O4+ [M + H]+, 400.1979; found, 400.1971.
3.1.10. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((5-phenylpentyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (19)
1H-NMR (400 MHz; DMSO-d6) δ 1.30–1.38 (m, 2H, NH-CH2-CH2-CH2-CH2-CH2-Ph), 1.56–1.66 (m, 4H, NH-CH2-CH2-CH2-CH2-CH2-Ph), 2.57 (t, 2H, NH-CH2-CH2-CH2-CH2-CH2-Ph), 3.47 (br s, 2H, NH-CH2-CH2-CH2-CH2-CH2-Ph), 3.53–3.58 (m, 1H, CH ribose), 3.66–3.70 (m, 1H, CH ribose), 3.97 (q, 1H, CH ribose), 4.15 (br s, 1H, CH ribose), 4.62 (br s, 1H, CH ribose), 5.16–5.24 (m, 1H, OH ribose), 5.39–5.48 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.13–7.18 (m, 3H, benzene ring), 7.25 (t, 2H, benzene ring), 7.88 (br s, 1H, NH), 8.20 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 26.0, 28.9, 30.8, 35.1, 40.7, 61.7, 70.7, 73.5, 86.0, 88.0, 119.8, 125.6, 128.21 (2C), 128.28 (2C), 139.7, 142.3, 148.2, 152.4, 154.7. MS (ESI), m/z: 414 [M + H]+. HR-MS (ESI) m/z: calcd for C21H28N5O4+ [M + H]+, 414.2136; found, 414.2126.
3.1.11. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((5-hydroxypentyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (20)35
1H-NMR (400 MHz; DMSO-d6) δ 1.31–1.37 (m, 2H, NH-CH2-CH2-CH2-CH2-CH2-OH), 1.42–1.48 (m, 2H, NH-CH2-CH2-CH2-CH2-CH2-OH), 1.56–1.63 (m, 2H, NH-CH2-CH2-CH2-CH2-CH2-OH), 3.38 (q, 2H, NH-CH2-CH2-CH2-CH2-CH2-OH), 3.48 (br s, 2H, NH-CH2-CH2-CH2-CH2-CH2-OH), 3.53–3.58 (m, 1H, CH ribose), 3.66–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15 (br s, 1H, CH ribose), 4.32 (t, 1H, NH-CH2-CH2-CH2-CH2-CH2-OH), 4.61 (t, 1H, CH ribose), 5.15–5.24 (m, 1H, OH ribose), 5.37–5.49 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 6.84 (d, 2H, benzene ring), 7.86 (br s, 1H, NH), 8.20 (s, 1H, C2-H adenine), 8.33 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 23.0, 23.2, 29.0, 32.3, 41.0, 60.7, 61.7, 70.7, 73.5, 85.9, 88.0, 118.9, 139.6, 148.2, 152.4, 154.6. MS (ESI), m/z: 354 [M + H]+. HR-MS (ESI) m/z: calculated for C15H24N5O5+ [M + H]+, 354.1772; found, 354.1764.
3.1.12. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((6-phenylhexyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (21)
1H-NMR (400 MHz; DMSO-d6) δ 1.33–1.34 (m, 4H, NH-CH2-CH2-CH2-CH2-CH2-CH2-Ph), 1.53–1.60 (m, 4H, NH-CH2-CH2-CH2-CH2-CH2-CH2-Ph), 2.58 (t, 2H, NH-CH2-CH2-CH2-CH2-CH2-CH2-Ph), 3.47 (br s, 2H, NH-CH2-CH2-CH2-CH2-CH2-CH2-Ph), 3.52–3.58 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (q, 1H, CH ribose), 4.15 (br d, 1H, CH ribose), 4.61 (br d, 1H, CH ribose), 5.17–5.22 (m, 1H, OH ribose), 5.39–5.47 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.14–7.18 (m, 3H, benzene ring), 7.24–7.28 (t, 2H, benzene ring), 7.87 (br s, 1H, NH), 8.19 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 26.2, 28.5, 29.0, 31.0, 35.1, 40.5, 61.7, 70.7, 73.5, 85.9, 88.0, 119.8, 125.6, 128.22 (2C), 128.26 (2C), 139.7, 142.3, 148.2, 152.4, 154.7. MS (ESI), m/z: 428 [M + H]+. HR-MS (ESI) m/z: calculated for C22H30N5O4+ [M + H]+, 428.2292; found, 428.2284.
3.1.13. (2R,3R,4S,5R)-2-(6-((1-Benzylpiperidin-4-yl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol (31) (Patents EP0417999A1 and EP0423777A2)
1H-NMR (400 MHz; DMSO-d6) δ 1.65–1.68 (br m 2H, 2 × CH piperidine), 1.79–1.88 (br m 2H, 2 × CH piperidine), 2.04 (t, 2H, 2 × CH piperidine), 2.84 (t, 2H, 2 × CH piperidine), 3.47 (s, 2H, NH-CH2-Ph), 3.52–3.58 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (q, 1H, CH ribose), 4.11 (br s, 1H, NH-CH piperidine), 4.13–4.16 (m, 1H, CH ribose), 4.61 (q, 1H, CH ribose), 5.16–5.21 (m, 1H, OH ribose), 5.38–5.46 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.24–7.27 (m, 1H, CH benzene ring), 7.30–7.35 (m, 4H, CH benzene ring), 7.73 (br s, 1H, NH), 8.20 (s, 1H, C2-H adenine), 8.35 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 31.4 (2C), 47.3, 52.4 (2C), 61.7, 62.2, 70.7, 73.5, 85.9, 88.0, 119.9, 126.9, 128.2 (2C), 128.9 (2C), 139.1, 139.7, 150.6, 152.4, 154.0. MS (ESI), m/z: 441 [M + H]+. HR-MS (ESI) m/z: calculated for C22H29N6O4+ [M + H]+, 441.2245; found, 441.2236.
3.1.14. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((2-(quinolin-4-ylamino)ethyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (32)
1H-NMR (400 MHz; DMSO-d6) δ 3.41–3.48 (br m, 3H, CH ribose + NH-CH2-CH2-NH), 3.67–3.70 (br m, 1H, CH ribose), 3.81 (br s, 2H, NH-CH2-CH2-NH), 3.97 (d, 1H, CH ribose), 4.16 (d, 1H, CH ribose), 4.61 (q, 1H, CH ribose), 5.17–5.23 (m, 1H, OH ribose), 5.37–5.40 (m, 1H, OH ribose), 5.46 (d, 1H, OH ribose), 5.91 (d, 1H, CH ribose), 6.68 (d, 1H, C3-H quinoline ring), 7.33 (br s, 1H, NH-CH2-CH2-NH), 7.43 (t, 1H, C6-H quinoline ring), 7.61 (t, 1H, C7-H quinoline ring), 7.78 (d, 1H, C8-H quinoline ring), 8.12 (br s, 1H, NH-CH2-CH2-NH), 8.17 (d, 1H, C5-H quinoline ring), 8.33 (s, 1H, C2-H adenine), 8.39 (s, 1H, C8-H adenine), 8.42 (d, 1H, C2-H quinoline ring). 13C-NMR (100 MHz; DMSO-d6) δ 42.1, 42.2, 61.7, 70.6, 73.6, 85.9, 87.9, 98.4, 118.8, 121.5, 123.9, 128.8, 129.1, 139.1, 140.0, 148.3, 148.4, 149.9, 150.8, 152.4, 154.7. MS (ESI), m/z: 438 [M + H]+. HR-MS (ESI) m/z: calculated for C21H24N7O4+ [M + H]+, 438.1884; found, 438.1876.
3.1.15. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-(methyl(prop-2-yn-1-yl)amino)propyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (33)
1H-NMR (400 MHz; DMSO-d6) δ 1.68–1.75 (m, 2H, NH-CH2-CH2-CH2-N), 2.20 (s, 3H, NCH3), 2.41 (t, 2H, NH-CH2-CH2-CH2-N), 3.09 (br s, 1H, N-CH2-C≡CH), 3.32 (br s, 2H, N-CH2-C≡CH), 3.50–3.58 (m, 3H, CH ribose + NH-CH2-CH2-CH2-N), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15 (q, 1H, CH ribose), 4.61 (q, 1H, CH ribose), 5.16–5.20 (m, 1H, OH ribose), 5.38–5.46 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.89 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 26.9, 38.2, 41.2, 44.9, 52.7, 61.7, 70.7, 73.5, 75.9, 78.9, 85.9, 87.9, 119.8, 139.7, 148.2, 152.4, 154.7. MS (ESI), m/z: 377 [M + H]+. HR-MS (ESI) m/z: calculated for C17H25N6O4+ [M + H]+, 377.1932; found, 377.1922.
3.1.16. 2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-(piperidin-1-yl)propyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (34)
1H-NMR (400 MHz; DMSO-d6) δ 1.39 (br s, 2H, CH2 piperidine), 1.54 (br s, 4H, 2 × CH2 piperidine), 1.74–1.78 (m, 2H, NH-CH2-CH2-CH2-N), 2.36 (br s, 6H, 2 × CH2 piperidine + NH-CH2-CH2-CH2-N), 3.52–3.58 (m, 3H, CH ribose + NH-CH2-CH2-CH2-N), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15 (q, 1H, CH ribose), 4.61 (q, 1H, CH ribose), 5.16–5.20 (m, 1H, OH ribose), 5.38–5.44 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 8.05 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 24.0, 25.3 (2C), 25.7, 40.9, 54.0 (2C), 56.6, 61.7, 70.7, 73.5, 86.0, 88.0, 119.9, 139.8, 148.2, 152.4, 154.6. MS (ESI), m/z: 393 [M + H]+. HR-MS (ESI) m/z: calculated for C18H29N6O4+ [M + H]+, 393.2245; found, 393.2232.
3.1.17. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-morpholinopropyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (35)
1H-NMR (400 MHz; DMSO-d6) δ 1.73–1.79 (m, 2H, NH-CH2-CH2-CH2-N), 2.35–2.39 (m, 6H, 2 × CH2 morpholine + NH-CH2-CH2-CH2-N), 3.52–3.60 (m, 7H, 2 × CH2 morpholine + CH ribose + NH-CH2-CH2-CH2-N), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.14–4.15 (m, 1H, CH ribose), 4.61 (q, 1H, CH ribose), 5.17–5.21 (m, 1H, OH ribose), 5.38–5.47 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 8.02 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.35 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 25.5, 40.7, 53.4 (2C), 56.4, 61.7, 66.2 (2C), 70.7, 73.5, 85.9, 87.9, 119.8, 139.7, 148.2, 152.4, 154.6. MS (ESI), m/z: 395 [M + H]+. HR-MS (ESI) m/z: calculated for C17H27N6O5+ [M + H]+, 395.2037; found, 395.2038.
3.1.18. (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-(4-methylpiperazin-1-yl)propyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (36)
1H-NMR (400 MHz; DMSO-d6) δ 1.75–1.78 (m, 2H, NH-CH2-CH2-CH2-N), 2.25 (s, 3H, NCH3), 2.42 (br s, 10H, 4 × CH2 piperazine + NH-CH2-CH2-CH2-N), 3.53–3.58 (m, 3H, CH ribose + NH-CH2-CH2-CH2-N), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15 (q, 1H, CH ribose), 4.61 (q, 1H, CH ribose), 5.16–5.21 (m, 1H, OH ribose), 5.38–5.45 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.98 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.33 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 25.8, 38.5, 44.94 51.9 (2C), 54.0 (2C), 55.4, 61.6, 70.7, 73.5, 85.9, 87.9, 119.9, 139.7, 148.2, 152.4, 154.6. MS (ESI), m/z: 408 [M + H]+. HR-MS (ESI) m/z: calculated for C18H30N7O4+ [M + H]+, 408.2354; found, 408.2353.
3.1.19. tert-Butyl-4-(((9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)methyl)piperidine-1-carboxylate (37)
1H-NMR (400 MHz; DMSO-d6) δ 1.06–1.09 (m, 2H, 2 × CH piperidine) 1.39 (s, 9H, 3 × CH3), 1.66 (br d, 2H, 2 × CH piperidine), 1.85 (br s, 1H, CH piperidine), 2.68 (br s, 2H, 2 × CH piperidine), 3.38 (br s, 2H, NH-CH2-piperidine), 3.53–3.58 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.90 (br s, 1H, CH piperidine), 3.93 (br s, 1H, CH piperidine), 3.97 (q, 1H, CH ribose), 4.15–4.16 (br m, 1H, CH ribose), 4.61–4.62 (br m, 1H, CH ribose), 5.14–5.20 (m, 1H, OH ribose), 5.36–5.45 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.96 (br s, 1H, NH), 8.21 (s, 1H, C2-H adenine), 8.35 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 28.1 (3C), 31.0 (2C), 35.5, 43.1 (2C), 45.0, 61.7, 70.7, 73.5, 78.4, 85.9, 88.0, 119.9, 139.7, 148.3, 152.3, 153.9, 154.8. MS (ESI), m/z: 465 [M + H]+. HR-MS (ESI) m/z: calculated for C21H33N6O6+ [M + H]+, 465.2456; found, 465.2455.
3.1.20. tert-Butyl-4-(2-((9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)ethyl)piperidine-1-carboxylate (38)
1H-NMR (400 MHz; DMSO-d6) δ 0.98–1.03 (m, 2H, 2 × CH piperidine), 1.39 (s, 9H, 3 × CH3), 1.53–1.55 (m, 3H, NH-CH2-CH2-piperidine + CH piperidine), 1.70 (d, 2H, 2 × CH piperidine), 2.68 (br s, 2H, 2 × CH piperidine), 3.53–3.58 (m, 3H, NH-CH2-CH2-piperidine + CH ribose), 3.66–3.70 (m, 1H, CH ribose), 3.91 (d, 1H, 2 × CH piperidine), 3.97 (q, 1H, CH ribose), 4.13–4.17 (br m, 1H, CH ribose), 4.59–4.65 (br m, 1H, CH ribose), 5.18–5.23 (m, 1H, OH ribose), 5.38–5.46 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.88 (br s, 1 H, NH), 8.20 (s, 1H, C2-H adenine), 8.34 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 28.1 (3C), 31.7, 32.8 (2C), 35.6, 37.1, 42.5 (2C), 61.7, 70.7, 73.5, 78.4, 85.9, 87.9, 118.8, 139.7, 148.3, 152.4, 153.9, 154.7. MS (ESI), m/z: 479 [M + H]+. HR-MS (ESI) m/z: calculated for C22H35N6O6+ [M + H]+, 479.2613; found, 479.2611.
3.1.21. tert-Butyl-4-(3-((9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)propyl)piperidine-1-carboxylate (39)
1H-NMR (400 MHz; DMSO-d6) δ 0.92–0.98 (m, 2H, 2 × CH piperidine), 1.23–1.28 (m, 3H, NH-CH2-CH2-CH2-piperidine + CH piperidine), 1.39 (s, 9H, 3 × CH3), 1.57–1.64 (m, 4H, NH-CH2-CH2-CH2-piperidine +2 × CH piperidine), 2.61–2.70 (br m, 2H, 2 × CH piperidine), 3.42–3.48 (br m, 2H, NH-CH2-CH2-CH2-piperidine), 3.53–3.58 (m, 1H, CH ribose), 3.66–3.69 (m, 1H, CH ribose), 3.91 (d, 2H, 2 × CH piperidine), 3.97 (d, 1H, CH ribose), 4.15 (m, 1H, CH ribose), 4.62 (m, 1H, CH ribose), 5.15–5.20 (m, 1H, OH ribose), 5.37–5.45 (m, 2H, OH ribose), 5.88 (d, 1H, CH ribose), 7.87 (br s, 1 H, NH), 8.20 (s, 1H, C2-H adenine), 8.32 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 26.1, 28.1 (3C), 31.8 (2C), 33.2, 35.0, 43.2 (2C), 43.4, 61.7, 70.7, 73.5, 78.4, 85.9, 88.0, 120.0, 139.7, 148.2, 152.4, 153.9, 154.7. MS (ESI), m/z: 493 [M + H]+. HR-MS (ESI) m/z: calculated for C23H37N6O6+ [M + H]+, 493.2769; found, 493.2763.
3.1.22. tert-Butyl-(2-(1-(9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)piperidin-4-yl)ethyl)carbamate (40)
1H-NMR (400 MHz; DMSO-d6) δ 1.06–1.16 (m, 2H, 2 × CH piperidine), 1.31–1.35 (m, 2H, CH-CH2-CH2-NH), 1.38 (s, 9H, 3 × CH3), 1.65 (br s, 1H, CH-CH2-CH2-NH), 2.78 (d, 2H, 2 × CH piperidine), 2.95–3.05 (m, 4H, CH-CH2-CH2-NH + 2 × CH piperidine), 3.52–3.58 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15 (d, 1H, CH ribose), 4.58 (d, 1H, CH ribose), 5.15–5.20 (m, 1H, OH ribose), 5.28–5.49 (m, 4H, 2 × CH piperidine +2 × OH ribose), 5.91 (d, 1H, CH ribose), 6.78 (br s, 1H, NH-Boc), 8.21 (s, 1H, C2-H adenine), 8.38 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) δ 28.3 (3C), 31.9 (2C), 33.1, 36.1, 37.3, 45.0 (2C), 61.6, 70.5, 73.5, 77.4, 85.8, 87.8, 119.6, 138.6, 150.2, 151.8, 153.2, 155.6. MS (ESI), m/z: 479 [M + H]+. HR-MS (ESI) m/z: calculated for C22H35N6O6+ [M + H]+, 479.2613; found, 479.2604.
3.1.23. tert-Butyl-8-(9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)-2,8-diazaspiro[4.5]decane-2-carboxylate (41)
1H-NMR (400 MHz; DMSO-d6) δ 1.41 (s, 9H, 3 × CH3), 1.52–1.62 (br m, 4H, 4 × CH piperidine), 1.79 (t, 2H, 2 × CH pyrrolidine), 3.17 (s, 2H, 2 × CH pyrrolidine), 3.33 (br s, 2H, 2 × CH pyrrolidine), 3.53–3.59 (m, 1H, CH ribose), 3.65–3.70 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.15–4.44 (br m, 5H, 1 × CH ribose + 4 × CH piperidine), 4.58 (q, 1H, CH ribose), 5.15–5.20 (m, 1H, OH ribose), 5.29–5.36 (m, 1H, OH ribose), 5.44 (d, 1H, OH ribose), 5.92 (d, 1H, CH ribose), 8.23 (s, 1H, C2-H adenine), 8.39 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) 28.2 (3C), 34.1 (2C), 35.3, 40.8, 43.9, 44.1 (2C), 55.4, 61.5, 70.5, 73.5, 78.2, 85.8, 87.8, 119.6, 138.7, 150.2, 151.8, 153.1, 153.8. MS (ESI), m/z: 491 [M + H]+. HR-MS (ESI) m/z: calculated for C23H35N6O6+ [M + H]+, 491.2613; found, 491.2610.
3.1.24. Procedure for the Synthesis of (2R,3S,4R,5R)-2-(Hydroxymethyl)-5-(6-((3-(piperidin-4-yl)propyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diol (42)
Compound 39 (90 mg, 0.183 mmol) was dissolved at 0 °C in 3 mL of a mixture trifluoroacetic acid:water 9:1 (v/v), stirred at 0 °C for 30 min, and then at room temperature for 7 h until completion of the reaction. The mixture was then concentrated under vacuum and co-evaporated three times with methanol. The crude was then treated with sodium carbonate saturated solution (5 mL) at 0 °C and extracted with ethyl acetate (3 × 5 mL). After washing with brine (2 × 2 mL), the organic phase was evaporated, and the resulting crude was purified by column chromatography eluting with a mixture chloroform/methanol/ammonia (33%) 9:1:0.1 (v/v) to provide compound 42 as a white powder. 1H-NMR (400 MHz; DMSO-d6) δ 0.90–0.99 (m, 2H, 2 × CH piperidine), 1.23–1.28 (m, 3H, NH-CH2-CH2-CH2-piperidine + CH piperidine), 1.54–1.60 (m, 4H, NH-CH2-CH2-CH2-piperidine + 2 × CH piperidine), 2.87–2.96 (m, 4H, 4 × CH piperidine), 3.41–3.49 (br m, 2H, NH-CH2-CH2-CH2-piperidine), 3.52–3.58 (m, 1H, CH ribose), 3.66–3.69 (m, 1H, CH ribose), 3.97 (d, 1H, CH ribose), 4.13–4.16 (m, 1H, CH ribose), 4.34 (br s, 1H, NH piperidine), 4.59–4.63 (m, 1H, CH ribose), 5.14–5.23 (m, 1H, OH ribose), 5.39–5.49 (m, 2H, 2 × OH ribose), 5.88 (d, 1H, CH ribose), 7.87 (br s, 1 H, NH-CH2-CH2-CH2-piperidine), 8.20 (s, 1H, C2-H adenine), 8.33 (s, 1H, C8-H adenine). 13C-NMR (100 MHz; DMSO-d6) 24.0, 25.3 (2C), 27.7, 38.1, 46.7 (2C), 48.4, 62.2, 71.1, 73.5, 84.9, 88.0, 118.7, 138.6, 149.9, 152.4, 154.7. MS (ESI), m/z: 393 [M + H]+. HR-MS (ESI) m/z: calculated for C18H29N6O4+ [M + H]+, 393.2245; found, 393.2243.
3.1.25. Procedure for the Synthesis of N6-(S-(5’-Thioadenosyl)-propyl)-2’-deoxyadenosine (Compound 239 in Figure S1)
3′,5’-O-Diacetyl-O6-(triisopropylbenzenesulfonyl)-2′-deoxyinosine (62 mg, 103 μmol)52 and decarboxylated SAH 208 (70 mg, 206 μmol)53 were dissolved in dry DMF (4 mL). Triethylamine (72 μL, 0.51 mmol) was added dropwise, and the reaction mixture stirred at room temperature for 22 h. Solvent was removed under reduced pressure, and the crude product was suspended in an acetonitrile/water mixture (6 mL, 7:3, v/v). Sodium hydroxide (1 M, 4 mL) was added dropwise under ice cooling, and the reaction mixture was stirred at room temperature for 30 min. The reaction mixture was neutralized with acetic acid (1 M, 4 mL) and microfiltered. Purification of the product was performed by reverse-phase HPLC (SNC Prontosil C-18, 5 μm, 120 Å, 250 × 8 mm, Bischoff, Leonberg, Germany). Compounds were eluted with acetonitrile (10% for 10 min followed by linear gradients from 10% to 20% in 5 min, from 20% to 25% in 25 min, and from 25% to 100% in 5 min) in water and a flow of 3 mL/min. Compounds were detected at 260 and 280 nm, and the product 239 was eluted with a retention time of 29.5 min. Product containing fractions were collected, and the solvents were removed under reduced pressure to yield the product 239 (19.8 mg, 34.5 μmol, 34%) as a colorless solid after dying at high vacuum. 1H-NMR (400 MHz; DMSO-d6) δ 1.83 (pent, br, 3J = 7.2 Hz, 2H, linker-H), 2.22–2.31 (m, 1H, deoxyadenosyl 2’-Ha), 2.58 (t, br, 3J = 7.2 Hz, 2H, linker-H), 2.68–2.76 (m, 1H, deoxyadenosyl 2’-Hb), 2.83 and 2.93 (AB part of ABX system, 2J = 13.8 Hz, 3J = 6.4 Hz, 2H, adenosyl 5’-H), 3.45–3.55 (m, 3H, deoxyadenosyl 5’-Ha und linker-H), 3.55–3.65 (m, 1H, deoxyadenosyl 5’-Hb), 3.86–3.90 (m, 1H, deoxyadenosyl 4’-H), 4.02 (dt, br, 3J = 6.4 Hz, 3J = 3.8 Hz, 1H, adenosyl 4’-H), 4.15 (dd, br, 3J = 3.8 Hz, 3J = 4.9 Hz, 1H, adenosyl 3’-H), 4.38–4.44 (m, 1H, deoxyadenosyl 3’-H), 4.75 (dd, br, 3J = 4.9 Hz, 3J = 5.6 Hz, 1H, adenosyl 2’-H), 5.28 (s, br, 1H, deoxyadenosyl 5’-OH), 5.37 (s, br, 1H, deoxyadenosyl 3’-OH), 5.41 (s, br, 1H, adenosyl 3’-OH), 5.58 (s, br, 1H, adenosyl 2’-OH), 5.89 (d, 3J = 5.6 Hz, 1H, adenosyl 1’-H), 6.32–6.37 (m, 1H, deoxyadenosyl 1’-H), 7.29 (s, br, 2H, 6-NH2), 7.90 (s, br, 1H, NH), 8.15 (s, 1H, adenosyl 8-H), 8.20 (s, br, 1H, deoxyadenosyl 8-H), 8.33 (s, 1H, adenosyl 2-H), 8.36 (s, 1H, deoxyadenosyl 2-H). UV: λmax = 261.5 nm. ESI-MS m/z (%): 597.4 (100) [M + Na]+, 575.5 (16) [M + H]+.
3.2. Inhibition Assay of CamA-Mediated Methylation
The CamA enzyme (pXC2184) was prepared as in previous studies.28,29 The methylation inhibition assay of CamA was performed with a final volume of 20 μL in buffer 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 0.1 mg/mL BSA, and 0.25% DMSO. Typically, a 50 nM final concentration of CamA was preincubated with varied compound concentration at room temperature (∼22 °C) for 5 min followed by addition of 2.5 μM final concentration of double-stranded (ds) DNA (5′-CGA TTC AAA AAG TCC CAA G-3′ and 3′-GCT AAG TTT TTC AGG GTT C-5′ where the underlined A is the methylation target) and 40 μM final concentration of SAM. Reaction was incubated at room temperature (∼22 °C) for 3 min and quenched by adding TFA to 0.1%. 5 μL of the final reaction mixture was transferred to a low-volume 384-well plate, the enzyme activity was measured by a Promega luminescence assay (MTase-Glo), and the luminescence signal was detected using a Synergy 4 Multi-Mode Microplate Reader (BioTek). To correlate luminescence and SAH concentration, the standard SAH (staring from 8 μM) within the Promega assay kit was subjected to serial twofold dilution, and a linear regression of the SAH standard was plotted against luminescence (Figure S3B).
3.3. Selectivity Assays
The following enzymes used in the selectivity assays were biochemically characterized in our previous studies: Caulobacter crescentus CcrM (pXC2121),54Escherichia coli Dam (pXC1612),55,56 human DNMT1 (residues 351–1600; pXC915),57,58 mouse Dnmt3a2-Dnmt3L (pXC462 and pXC391),57,59 human PCIF1 (pXC2055),60,61 human MettL3-MettL14,62,63 human MettL5-Trm112 (pXC2062-pXC2076),60 and human MettL16 (pXC2210).60 Oligonucleotides were synthesized by Integrated DNA Technologies (IDT), and the mRNA cap analog was purchased from TriLink BioTechnologies (catalog number N-7113).
The inhibition reactions of CamA, CcrM, Dam, PCIF1, and MettL5-Trm112 were carried out under the same conditions at ∼22 °C for varying time under [E] = 0.1 μM, [S] = 5 μM, [SAM] = 30 μM, [I] = 10 μM at 50 mM TRIS–HCl pH 7.5, 100 mM NaCl, 1 mM DTT, and 0.25% DMSO (Figure 6A–C,F,G). The substrate concentrations of DNA (or RNA) and SAM were above the known KM values of these enzymes. For CcrM, the dsDNA substrate was (5′-CGA TTC AAA AAG TCC CAA G-3′ and 3′-GCT AAG TTT TTC AGG GTT C-5′) and assays were performed for 5 min. For Dam, the dsDNA substrate was (5′-CCG CGG ATC CTG CT-3′ and 3′-GGC GCC TAG GAC GA-5′) and assays were performed for 5 min. For PCIF1, the RNA substrate was capped RNA (m7GpppAmG) and the assays were performed for 15 min. For MettL5-Trm112, the ssRNA substrate was (5′-UCG UAA CAA GGU UU-3′) and the assays were performed for 1 h.
For MettL16, the ssRNA hairpin substrate was (5′-GGU UGG CGU AGG CUA CAG AGA AGC CAA CC-3′) and the assays were performed under two different conditions: for 30 min under the conditions of [E] = 0.4 μM, [S] = 5 μM, [SAM] = 160 μM, [I] = 10 μM at 20 mM Tris–HCl pH 8.0, 400 mM NaCl, 1 mM DTT, and 0.25% DMSO and assayed for 15 min under the CamA conditions ([SAM] = 30 μM). The MettL16 activity is affected by the SAM concentration used in the assay because of its high KM value for SAM.60
For MettL3-MettL14, the ssRNA substrate was (5′-AAC AGA AUG GGA CUG UUC-3′) and the assays were performed under two different conditions: for 10 min under the conditions of [E] = 0.1 μM, [S] = 10 μM, [SAM] = 20 μM, [I] = 10 μM at 50 mM HEPES pH 7.5, 5 mM NaCl, 1 mM DTT, and 0.25% DMSO and assayed for 15 min under the CamA conditions ([NaCl] = 100 mM). The activity of MettL3-MettL14 is sensitive to ionic strength.
For DNMT1, the dsDNA substrate was (5′-CCT GCG GAG GCT MGT CAT GA-3′ where M = 5-methylcytosine, and 3′-GGA CGC CTC CGA GCA GTA CT-5′) and assays were performed at 37 °C for 1 h under the conditions of [E] = 0.1 μM, [S] = 1 μM, [SAM] = 5.5 μM, [I] = 10 μM at 50 mM Tris pH 7.5, and 1 mM EDTA.
For Dnmt3a-Dnmt3L, the dsDNA substrate was (5′-G CAT GCG TTC TAA TTA GAA CGC ATG-3′ and 3′-GTA CGC AAG ATT AAT CTT GCG TAC G-5′) and the assays were performed at 37 °C for 1 h under the conditions of [E] = 0.5 μM, [S] = 5 μM, [SAM] = 25 μM, [I] = 10 μM at 20 mM Tris pH 7.5, 40 mM NaCl, 4 mM DTT, and 20% glycerol.59
For DOT1L, EZH2, G9a, and PRMT1, the enzymes were expressed and purified at Reaction Biology Corporation (USA) following previously reported procedures: DOT1L,64 EZH2,65 G9a,66 and PRMT1.67 The appropriate substrate [0.05 mg/mL oligonucleosomes for DOT1L, 0.05 mg/mL chicken core histone for EZH2 complex, 5 μM histone H3 (residues 1–21) peptide for G9a, and 5 μM histone H4 for PRMT1] was initially added to the freshly prepared reaction buffer [50 mM NaCl, 50 mM Tris–HCl (pH 8.5), 5 mM MgCl2, 0.01% Brij35, 1 mM DTT, 1% DMSO for DOT1L, G9a, and PRMT1; 50 mM Tris–HCl (pH 8.0), 0.01% Brij35, 1 mM EDTA, 1 mM DTT, 1% DMSO for EZH2]. Each MTase was then added to the substrate buffer, and the solution was mixed gently. Final enzyme concentrations: [DOT1L] = 1 nM, [EZH2] = 5 nM, [G9a] = 5 nM, [PRMT1] = 5 nM. The compounds were then added to the enzyme/substrate reaction mixture to a final concentration of 10 μM and incubated for 20 min at room temperature. Finally, 3H-SAM (1 μM) was added to the reaction mixture and this solution was incubated for 1 h at 30 °C. The reaction mixture was then delivered to filter paper for detection. The data were analyzed using Microsoft Excel and GraphPad Prism 8.0. The shown values are average of three replicates ± SD.
For human A1AR and A3AR, the affinity of the compounds was evaluated at Eurofins-Cerep SA (France) via agonist radioligand binding assays using membrane homogenates obtained from transfected CHO cells (A1AR)68 or HEK-293 cells (A3AR).69 For A1AR, cell membrane homogenates were incubated at 22 °C for 60 min incubation with [3H]CCPA (1 nM) in the absence or presence of each compound (10 μM) in a buffer consisting of 50 mM Tris–HCl (pH 7.4), 5 mM MgCl2, 1 mM EDTA, 2 UI/mL ADA, 1 μg/mL Leupeptin, 1 μM Pepstatin, and 10 μg/mL trypsin inhibitor. Nonspecific binding was determined in the presence of 10 μM CPA. For A3AR, cell membrane homogenates were incubated at 22 °C for 120 min with [125I]-AB-MECA (0.15 nM) in the absence or presence of each compound (10 μM) in a buffer consisting of 50 mM Tris–HCl (pH 7.4), 5 mM MgCl2, 1 mM EDTA, and 2 UI/mL ADA. Nonspecific binding was determined in the presence of 1 μM piclidenoson (compound 4 in this study). In both cases, following incubation, the reaction mixture was filtered under vacuum using glass fiber filters (GF/B, Packard) pre-soaked with 0.3% PEI and rinsed several times with ice-cold 50 mM Tris–HCl using a 96-sample cell harvester (Unifilter, Packard). The filters were dried, and radioactivity was detected in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). The results are expressed as a percent inhibition of the control radioligand specific binding of. The data was analyzed using Microsoft Excel and GraphPad Prism 8.0. The shown values are average of three replicates ± SD.
3.4. Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetry (ITC) experiments were performed with a MicroCal PEAQ-ITC automated system (Malvern). Experiments were conducted at 25 °C with a reference power of 8 μcal/s. Typically, 19 injections of 200–900 μM compound with an initial injection of 0.2 μL followed by 18 injections (each of 2 μL) were titrated into 20–60 μM CamA protein in buffer 325 mM NaCl, 20 mM Tris–HCl pH 7.5, 0.5 mM TCEP, and 10% DMSO. The reverse titrations were performed for compounds 6 and 19 to overcome the low solubility of compounds. ITC data were fitted as “one site” with the offset subtracted, and binding constants were calculated using the ITC analysis module supplied by the manufacturer.
3.5. X-ray Crystallography
For crystallization, CamA-DNA-inhibitor complexes were prepared as previously described.29 Briefly, all crystals grew under the similar conditions of 21–24% (w/v) polyethylene glycol 3350, 0.1 M Tris–HCl pH 7.0–7.5, and 0.28 M potassium citrate at room temperature (∼19 °C) after 3–4 days, via the sitting drop vapor diffusion method, using an Art Robbins Gryphon Crystallization Robot for a 0.4 μL drop setup. Crystals were flash frozen in liquid nitrogen after momentary soaking in reservoir solution supplemented with 20% (v/v) ethylene glycol.
Diffraction data were collected at the SER-CAT beamline 22ID of the Advanced Photon Source at Argonne National Laboratory with crystals in a 100 K cryostream and typically rotated 0.5° for each of 800 frames. Crystallographic datasets were processed with HKL2000.70 Structures of the CamA-DNA-inhibitor ternary complex were solved by the difference Fourier method using our previously determined binary CamA-DNA structure (PDB ID: 7LNJ).28 All crystals were essentially isomorphous, rigid body refinement was used for positioning the new structures in the unit cell in the first refinement cycle, and difference electron density maps (2Fo-Fc and Fo-Fc) were used for locating bound inhibitor molecules. A SMILES string for each inhibitor compound was submitted to the Grade web server (http://grade.globalphasing.org) to obtain geometrical restraints in the form of a CIF file that was applied in subsequent refinements and to supply its structure in PDB format. PHENIX REFINE71 was used for all refinements, which had 5% randomly chosen reflections for validation by the R-free value.72 Structure quality was analyzed during PHENIX refinements together with manual inspection using Coot.73 Final structure models were validated by the PDB validation server. Structure images were prepared by PyMol (Schrödinger, LLC).
Acknowledgments
We thank Ms. Yu Cao of MDACC and Mr. Sascha Peters of RWTH Aachen University for technical assistance. We thank the beamline scientists of Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source (APS), Argonne National Laboratory, USA. The work was supported by U.S. National Institutes of Health grant R35GM134744 (to X.C.), Cancer Prevention and Research Institute of Texas grant RR160029 (to X.C., who is a CPRIT Scholar in Cancer Research), FISR2019_00374 MeDyCa (to A.M.), Sapienza Ateneo Project 2021 RM12117A61C811CE (to D.R.). D.R. acknowledges Regione Lazio PROGETTI DI GRUPPI DI RICERCA 2020, project ID: A0375-2020-36597. The use of SER-CAT is supported by its member institutions and equipment grants (S10_RR25528, S10_RR028976, and S10_OD027000) from the US National Institutes of Health. Use of the APS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract W-31-109-Eng-38. Structural Genomics Consortium is a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant 875510), Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer, and Takeda.
Glossary
Abbreviations
- CamA
Clostridioides difficile-specific DNA adenine methyltransferase
- CDI
Clostridioides difficile infections
- IC50
half maximal inhibitory concentration
- KD
dissociation constant
- MTA
5′-methylthioadenosine
- MTase
methyltransferase
- SAM
S-adenosyl-l-methionine
- SAH
S-adenosyl-l-homocysteine
Data Availability Statement
The experimental data that support the findings of this study are contained within the article. The authors have deposited the X-ray structure (coordinates) and the source data (structure factor file) of CamA-DNA with bound inhibitors to the PDB and will be released upon article publication under accession numbers PDB 8CXS (MTA), PDB 8CXT (1), 8CXU (2), 8CXV (3), 8CXW (4), 8CXX (6), 8CXY (8), 8CXZ (14), 8CY0 (18), 8CY1 (19), 8CY2 (9), 8CY3 (15), 8CY4 (16) and 8CY5 (39).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01789.
Molecular formula strings (SMILES) and associated data (XSLX)
Elemental analyses for final compounds 10–12, 14–21, and 31–42; summary of X-ray data collection and refinement statistics; predicted cell permeability of compounds 16, 38, and 39; MTA-related compounds 201–241; Raw data of MTase-Glo inhibition assay and ITC measurements; compound 9 (APNEA) contains an impurity; chemical stability of compound 39; HR-MS spectra and HPLC traces of compounds 1–42; HPLC traces of compounds 201–241 (PDF)
Author Contributions
J.Z. performed protein purification, inhibition assays, ITC measurements, and crystallization. J.Z. and J.R.H. performed X-ray crystallography experiments. M.M, F.F., and D.R. performed compound synthesis. G.M. and A.C. performed HR-MS spectra registration and HPLC analytical controls. D.R. and A.M. designed the compounds and collaborated in the preparation of the manuscript. R.R. performed selectivity assays using mammalian DNA cytosine MTases (DNMTs). Y.D. purified human RNA adenine MTases (MettL5-Trm112 and MettL16). T.H. and M. V. provided purified MettL3-MettL14. E.W. provided compounds 201–241, and M.H. synthesized compound 239. R.M.B. participated in discussion and bioinformatic analyses and assisted in preparing the manuscript. X.Z. and X.C. organized and designed the scope of the study.
The authors declare no competing financial interest.
Supplementary Material
References
- Guh A. Y.; et al. Trends in U.S. Burden of Clostridioides difficile Infection and Outcomes. N. Engl. J. Med. 2020, 382, 1320–1330. 10.1056/NEJMoa1910215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams D. J.; Barone J. B.; Nylund C. M. Community-Associated Clostridioides difficile Infection in Children: A Review of Recent Literature. J. Pediatr. Infect. Dis. Soc. 2021, 10, S22–S26. 10.1093/jpids/piab064. [DOI] [PubMed] [Google Scholar]
- Yepez Guevara E. A.; et al. Clostridioides difficile infection in cancer and immunocompromised patients: Relevance of a two-step diagnostic algorithm and infecting ribotypes on clinical outcomes. Clin. Infect. Dis. 2021, 72, e460–e465. 10.1093/cid/ciaa1184. [DOI] [PubMed] [Google Scholar]
- Nibbering B.; Gerding D. N.; Kuijper E. J.; Zwittink R. D.; Smits W. K. Host Immune Responses to Clostridioides difficile: Toxins and Beyond. Front. Microbiol. 2021, 12, 804949 10.3389/fmicb.2021.804949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza-Torres J.; Auria E.; Dupuy B.; Tremblay Y. D. N. Wolf in Sheep’s Clothing: Clostridioides difficile Biofilm as a Reservoir for Recurrent Infections. Microorganisms 2021, 9, 1922. 10.3390/microorganisms9091922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay Y. D.; Dupuy B. The blueprint for building a biofilm the Clostridioides difficile way. Curr. Opin. Microbiol. 2022, 66, 39–45. 10.1016/j.mib.2021.12.002. [DOI] [PubMed] [Google Scholar]
- Stephenson B.; et al. Comparing intervention strategies for reducing Clostridioides difficile transmission in acute healthcare settings: an agent-based modeling study. BMC Infect. Dis. 2020, 20, 799. 10.1186/s12879-020-05501-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzas C.; Jara M.; Tucker R.; Curtis S.; A review of epidemiological models of Clostridioides difficile transmission and control (2009-2021). Anaerobe 2022, 74, 102541 10.1016/j.anaerobe.2022.102541. [DOI] [PubMed] [Google Scholar]
- Gil F.; Calderon I. L.; Fuentes J. A.; Paredes-Sabja D. Clostridioides (Clostridium) difficile infection: current and alternative therapeutic strategies. Future Microbiol. 2018, 13, 469–482. 10.2217/fmb-2017-0203. [DOI] [PubMed] [Google Scholar]
- Orenstein R.; Patron R. L.; Seville M. T. Why Does Clostridium difficile Infection Recur?. J. Am. Osteopath. Med. 2019, 119, 322–326. 10.7556/jaoa.2019.054. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Saha S.; Khanna S. Therapies to modulate gut microbiota: Past, present and future. World J. Gastroenterol. 2020, 26, 777–788. 10.3748/wjg.v26.i8.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S. I.; Draper L. A.; Ross R. P.; Hill C. Bacteriophage endolysins as a potential weapon to combat Clostridioides difficile infection. Gut Microbes 2020, 12, 1813533. 10.1080/19490976.2020.1813533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll M.; et al. Prevention of Clostridioides difficile in hospitals: A position paper of the International Society for Infectious Diseases. Int. J. Infect. Dis. 2021, 102, 188–195. 10.1016/j.ijid.2020.10.039. [DOI] [PubMed] [Google Scholar]
- Turner N. A.; Smith B. A.; Lewis S. S. Novel and emerging sources of Clostridioides difficile infection. PLoS Pathog. 2019, 15, e1008125 10.1371/journal.ppat.1008125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guh A. Y.; Kutty P. K. Clostridioides difficile Infection. Ann. Intern. Med. 2018, 169, ITC49–ITC64. 10.7326/AITC201810020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits W. K.; Lyras D.; Lacy D. B.; Wilcox M. H.; Kuijper E. J. Clostridium difficile infection. Nat. Rev. Dis. Primers 2016, 2, 16020. 10.1038/nrdp.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dureja C.; Olaitan A. O.; Hurdle J. G. Mechanisms and impact of antimicrobial resistance in Clostridioides difficile. Curr. Opin. Microbiol. 2022, 66, 63–72. 10.1016/j.mib.2022.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; et al. Increasing antibiotic resistance in Clostridioides difficile: A systematic review and meta-analysis. Anaerobe 2019, 58, 35–46. 10.1016/j.anaerobe.2019.102072. [DOI] [PubMed] [Google Scholar]
- Boekhoud I. M.; et al. Haem is crucial for medium-dependent metronidazole resistance in clinical isolates of Clostridioides difficile. J. Antimicrob. Chemother. 2021, 76, 1731–1740. 10.1093/jac/dkab097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Shen W. J.; Deshpande A.; Olaitan A. O.; Palmer K. L.; Garey K. W.; Hurdle J. G. The Integrity of Heme Is Essential For Reproducible Detection of Metronidazole-Resistant <i>Clostridioides difficile</i> By Agar Dilution Susceptibility Tests. J. Clin. Microbiol. 2021, 59, e00585-21 10.1128/JCM.00585-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesniak N. A.; Schubert A. M.; Sinani H.; Schloss P. D. Clearance of Clostridioides difficile Colonization Is Associated with Antibiotic-Specific Bacterial Changes. Msphere 2021, 6, e01238–e01220. 10.1128/mSphere.01238-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; et al. Targeting Clostridioides difficile: New uses for old drugs. Drug Discovery Today 2022, 27, 1862–1873. 10.1016/j.drudis.2022.03.021. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lu Y.; Du Y.; Wang H.; Wu Q. Recent development of small-molecular inhibitors against Clostridioides difficile infection. Bioorg. Chem. 2022, 125, 105843 10.1016/j.bioorg.2022.105843. [DOI] [PubMed] [Google Scholar]
- Oliveira P. H.; et al. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat. Microbiol. 2020, 5, 166–180. 10.1038/s41564-019-0613-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert M.; O’Keeffe T. A.; Purdy D.; Elmore M.; Minton N. P. Gene transfer into Clostridium difficile CD630 and characterisation of its methylase genes. FEMS Microbiol. Lett. 2003, 229, 103–110. 10.1016/S0378-1097(03)00795-X. [DOI] [PubMed] [Google Scholar]
- van Eijk E.; et al. Complete genome sequence of the Clostridium difficile laboratory strain 630Deltaerm reveals differences from strain 630, including translocation of the mobile element CTn5. BMC Genomics 2015, 16, 1–4. 10.1186/s12864-015-1252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D.; Anwar F.; Vedantam G. Anti-virulence strategies for Clostridioides difficile infection: advances and roadblocks. Gut Microbes 2020, 12, 1802865. 10.1080/19490976.2020.1802865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Horton J. R.; Blumenthal R. M.; Zhang X.; Cheng X. Clostridioides difficile specific DNA adenine methyltransferase CamA squeezes and flips adenine out of DNA helix. Nat. Commun. 2021, 12, 3436. 10.1038/s41467-021-23693-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; et al. Repurposing epigenetic inhibitors to target the Clostridioides difficile-specific DNA adenine methyltransferase and sporulation regulator CamA. Epigenetics 2022, 17, 970–981. 10.1080/15592294.2021.1976910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusachi S.; Thompson R. D.; Yamada N.; Daly D. T.; Olsson R. A. Dog coronary artery adenosine receptor: structure of the N6-aryl subregion. J. Med. Chem. 1986, 29, 989–996. 10.1021/jm00156a016. [DOI] [PubMed] [Google Scholar]
- Trivedi B. K.; Bristol J. A.; Bruns R. F.; Haleen S. J.; Steffen R. P. N6-(arylalkyl)adenosines. Identification of N6-(9-fluorenylmethyl)adenosine as a highly potent agonist for the adenosine A2 receptor. J. Med. Chem. 1988, 31, 271–273. 10.1021/jm00396a044. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A.; et al. Synthesis and biological activity of N6-(p-sulfophenyl)alkyl and N6-sulfoalkyl derivatives of adenosine: water-soluble and peripherally selective adenosine agonists. J. Med. Chem. 1992, 35, 4143–4149. 10.1021/jm00100a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenichev M. S.; et al. Modification of the length and structure of the linker of N(6)-benzyladenosine modulates its selective antiviral activity against enterovirus 71. Eur. J. Med. Chem. 2016, 111, 84–94. 10.1016/j.ejmech.2016.01.036. [DOI] [PubMed] [Google Scholar]
- Sako M.; Ishikura H.; Hirota K.; Maki Y. A Newly Devised Method for the Debenzylation of N 6-Benzyladenosines. A Convenient Synthesis of [6-15 N]-Labeled Adenosines. Nucleosides, Nucleotides Nucleic Acids 1994, 13, 1239–1246. 10.1080/15257779408012148. [DOI] [Google Scholar]
- Bressi J. C.; et al. Adenosine analogues as inhibitors of Trypanosoma brucei phosphoglycerate kinase: elucidation of a novel binding mode for a 2-amino-N(6)-substituted adenosine. J. Med. Chem. 2000, 43, 4135–4150. 10.1021/jm000287a. [DOI] [PubMed] [Google Scholar]
- Nsumiwa S.; Kuter D.; Wittlin S.; Chibale K.; Egan T. J. Structure-activity relationships for ferriprotoporphyrin IX association and beta-hematin inhibition by 4-aminoquinolines using experimental and ab initio methods. Bioorg. Med. Chem. 2013, 21, 3738–3748. 10.1016/j.bmc.2013.04.040. [DOI] [PubMed] [Google Scholar]
- Yi J. S.; et al. Structure-guided DOT1L probe optimization by label-free ligand displacement. ACS Chem. Biol. 2015, 10, 667–674. 10.1021/cb500796d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila M. A.; Garcia-Trevijano E. R.; Lu S. C.; Corrales F. J.; Mato J. M. Methylthioadenosine. Int. J. Biochem. Cell Biol. 2004, 36, 2125–2130. 10.1016/j.biocel.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Moroz-Omori E. V.; et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem 2021, 16, 3035–3043. 10.1002/cmdc.202100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankova E.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberkampf M.; et al. c-di-AMP signaling is required for bile salt resistance, osmotolerance, and long-term host colonization by Clostridioides difficile. Sci. Signaling 2022, 15, eabn8171. 10.1126/scisignal.abn8171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw J. M.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. A.; Uchida K.; Weiss A.; Taunton J. Essential biphasic role for JAK3 catalytic activity in IL-2 receptor signaling. Nat. Chem. Biol. 2016, 12, 373–379. 10.1038/nchembio.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Molina J. B.; Tseng S. C.; Simonett S. P.; Taunton J.; Ansari A. Z. Engineered covalent inactivation of TFIIH-kinase reveals an elongation checkpoint and results in widespread mRNA stabilization. Mol. Cell 2016, 63, 433–444. 10.1016/j.molcel.2016.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V.; et al. Structure-based design of a covalent inhibitor of the SET domain-containing protein 8 (SETD8) lysine methyltransferase. J. Med. Chem. 2016, 59, 9881–9889. 10.1021/acs.jmedchem.6b01244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. S.; et al. Discovery of the First-in-Class G9a/GLP Covalent Inhibitors. J. Med. Chem. 2022, 65, 10506–10522. 10.1021/acs.jmedchem.2c00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; et al. Structure-Based Engineering of Irreversible Inhibitors against Histone Lysine Demethylase KDM5A. J. Med. Chem. 2018, 61, 10588–10601. 10.1021/acs.jmedchem.8b01219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinsmade S. R. CodY, a master integrator of metabolism and virulence in Gram-positive bacteria. Curr. Genet. 2017, 63, 417–425. 10.1007/s00294-016-0656-5. [DOI] [PubMed] [Google Scholar]
- Usenik A.; et al. The CWB2 Cell Wall-Anchoring Module Is Revealed by the Crystal Structures of the Clostridium difficile Cell Wall Proteins Cwp8 and Cwp6. Structure 2017, 25, 514–521. 10.1016/j.str.2016.12.018. [DOI] [PubMed] [Google Scholar]
- Kirk J. A.; et al. New class of precision antimicrobials redefines role of Clostridium difficile S-layer in virulence and viability. Sci. Transl. Med. 2017, 9, 406. 10.1126/scitranslmed.aah6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker D. B.; Wardle H. M.; Boote V. Phospholipid profiles of Clostridium difficile. J. Bacteriol. 1996, 178, 5844–5846. 10.1128/jb.178.19.5844-5846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferentz A. E.; Verdine G. L. Aminolysis of 2′-Deoxyinosine Aryl Ethers: Nucleoside Model Studies for the Synthesis of Functionally Tethered Oligonucleotides. Nucleosides, Nucleotides Nucleic Acids 1992, 11, 1749–1763. 10.1080/07328319208017821. [DOI] [Google Scholar]
- Pignot M.; Pljevaljcic G.; Weinhold E. Efficient Synthesis of S-Adenosyl-L-Homocysteine Natural Product Analogues and Their Use to Elucidate the Structural Determinant for Cofactor Binding of the DNA Methyltransferase M·HhaI. Eur. J. Org. Chem. 2000, 549–555. 10.1002/(SICI)1099-0690(200002)2000:3<549::AID-EJOC549>3.0.CO;2-7. [DOI] [Google Scholar]
- Horton J. R.; et al. The cell cycle-regulated DNA adenine methyltransferase CcrM opens a bubble at its DNA recognition site. Nat. Commun. 2019, 10, 4600. 10.1038/s41467-019-12498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Liebert K.; Hattman S.; Jeltsch A.; Cheng X. Transition from nonspecific to specific DNA interactions along the substrate-recognition pathway of dam methyltransferase. Cell 2005, 121, 349–361. 10.1016/j.cell.2005.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Liebert K.; Bekes M.; Jeltsch A.; Cheng X. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J. Mol. Biol. 2006, 358, 559–570. 10.1016/j.jmb.2006.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H.; et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012, 40, 4841–4849. 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; et al. Structural characterization of dicyanopyridine containing DNMT1-selective, non-nucleoside inhibitors. Structure 2022, 30, 793–802.e5. 10.1016/j.str.2022.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y.; et al. The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b-mediated DNA methylation. Genes Dev. 2020, 34, 1546–1558. 10.1101/gad.341925.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Kaur G.; Blumenthal R. M.; Zhang X.; Cheng X. Enzymatic characterization of three human RNA adenosine methyltransferases reveals diverse substrate affinities and reaction optima. J. Biol. Chem. 2021, 296, 100270 10.1016/j.jbc.2021.100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; et al. Enzymatic characterization of mRNA cap adenosine-N6 methyltransferase PCIF1 activity on uncapped RNAs. J. Biol. Chem. 2022, 298, 101751 10.1016/j.jbc.2022.101751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock C. B.; et al. Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discovery 2019, 5, 63. 10.1038/s41421-019-0136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; et al. Human MettL3-MettL14 RNA adenine methyltransferase complex is active on double-stranded DNA containing lesions. Nucleic Acids Res. 2021, 49, 11629–11642. 10.1093/nar/gkab460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q.; et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12, 1052–1058. 10.1016/S0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- Cao R.; Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Chang Y.; et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 2003, 11, 509–520. 10.1016/S0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan L. C.; et al. Cloning and expression of an A1 adenosine receptor from rat brain. Mol. Pharmacol. 1991, 40, 1–7. [PubMed] [Google Scholar]
- Brambilla R.; et al. Activation of the A3 adenosine receptor affects cell cycle progression and cell growth. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 361, 225–234. 10.1007/s002109900186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Borek D.; Majewski W.; Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr., Sect. A: Found. Crystallogr. 2003, 59, 228–234. 10.1107/S0108767303005488. [DOI] [PubMed] [Google Scholar]
- Afonine P. V.; et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A. T. Free R value: cross-validation in crystallography. Methods Enzymol. 1997, 277, 366–396. 10.1016/S0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental data that support the findings of this study are contained within the article. The authors have deposited the X-ray structure (coordinates) and the source data (structure factor file) of CamA-DNA with bound inhibitors to the PDB and will be released upon article publication under accession numbers PDB 8CXS (MTA), PDB 8CXT (1), 8CXU (2), 8CXV (3), 8CXW (4), 8CXX (6), 8CXY (8), 8CXZ (14), 8CY0 (18), 8CY1 (19), 8CY2 (9), 8CY3 (15), 8CY4 (16) and 8CY5 (39).