

Abstract
Microtubule (MT)-stabilizing 1,2,4-triazolo[1,5-a]pyrimidines (TPDs) hold promise as candidate therapeutics for Alzheimer’s disease (AD) and other neurodegenerative conditions. However, depending on the choice of substituents around the TPD core, these compounds can elicit markedly different cellular phenotypes that likely arise from the interaction of TPD congeners with either one or two spatially distinct binding sites within tubulin heterodimers (i.e., the seventh site and the vinca site). In the present study, we report the design, synthesis, and evaluation of a series of new TPD congeners, as well as matched molecular pair analyses and computational studies, that further elucidate the structure–activity relationships of MT-active TPDs. These studies led to the identification of novel MT-normalizing TPD candidates that exhibit favorable ADME-PK, including brain penetration and oral bioavailability, as well as brain pharmacodynamic activity.
Introduction
Neurodegenerative tauopathies are characterized by the hyperphosphorylation and aggregation of the microtubule (MT)-associated protein tau, with evidence of dysregulation of MTs in the axons of neurons that could result in axonal transport deficits and axonal dystrophy that may ultimately contribute to neuronal loss.1 Relatively low doses of brain-penetrant MT-stabilizing compounds have been shown to provide therapeutic benefits in mouse models of tauopathy by normalizing axonal MTs and, consequently, by restoring axonal transport.2−6 Similarly, MT-stabilizing molecules have been shown to provide benefits in other CNS disease models where MT deficits are thought to occur, including models of amyloid plaque formation,7 traumatic brain injury,8 and spinal cord injury.9 Among the different MT-stabilizing agents tested to date in preclinical animal models,3−7 selected members of the 1,2,4-triazolo[1,5-a]pyrimidine (TPD) class (e.g., 1, Figure 1) have been identified as potentially promising candidates due to a generally favorable combination of MT-stabilizing activity and brain penetration. MT-stabilizing TPDs are the first and thus far the only known examples of MT-directed molecules that stabilize MTs through interactions with the vinca site on β-tubulin,10 a binding site that is normally targeted by MT-depolymerizing compounds, such as vinblastine. However, cell-based studies from our laboratories demonstrated that different TPD congeners produce significantly different effects on cellular tubulin and MTs.11 In particular, short-term (4 h) incubation of QBI293 cells with certain TPD compounds, such as 1 and 2 (Figure 1), produced a dose-dependent increase in markers of stable MTs, such as acetylated α-tubulin (AcTub) and de-tyrosinated α-tubulin (GluTub), without alterations in total tubulin levels. In contrast, other TPD congeners, typified by 3 (Figure 1), caused a bell-shaped dose–response curve with respect to AcTub and GluTub, as well as a proteasome-dependent degradation of tubulin. Considering that MT-normalizing therapies to treat neurodegenerative disease are expected to provide benefits to the extent that they can effectively preserve/restore the normal function of MTs, the different phenotypic responses elicited by TPD congeners are likely to have important ramifications. In this context, preferred candidate compounds would clearly be those that do not cause tubulin degradation. To distinguish the different compounds based on the phenotypic response, MT-active TPDs have been classified as “Class I” if they cause a dose-dependent increase in markers of stable MTs without causing loss of total tubulin, or “Class II” if they cause bell-shaped dose–response curve with respect to AcTub and GluTub and/or a reduction of total tubulin levels. The fragments linked at C6 and C7 of the TPD core were found to play an important role in determining both the phenotypic response and the ADME-PK properties of TPDs.3,11 Structure–activity relationship (SAR) studies12 revealed that Class I activity generally requires (a) the presence of fluoro substituents at either one or both of the ortho positions of the phenyl ring at C6; (b) an electron-withdrawing group in the para position; and (c) a relatively lipophilic, aliphatic amine at C7. Meanwhile, X-ray co-crystal structures10,13 of selected TPD congeners from both classes bound to a tubulin assembly revealed that although the MT-stabilizing Class I compound, 4 (Figure 1), interacts exclusively within the vinca binding site on β-tubulin, the Class II TPD, cevipabulin14 (5, Figure 1) binds with similar affinity to both the vinca site as well as a related, but spatially distinct, intradimer binding site known as the gatorbulin15 or seventh site. Moreover, these studies showed that the proteasome-dependent degradation of tubulin caused by cevipabulin may be a direct consequence of this compound binding to the seventh site, which possibly leads to the release of the nonexchangeable GTP from α-tubulin with resulting unfolding and degradation of tubulin.13 Although these SAR and structural biology data provide important insights into the mechanism(s) underlying the different phenotypic responses elicited by TPD congeners in cells, further studies are necessary to better elucidate the role played by the fragments at C6 and C7 in determining the relative affinity for the two binding sites and the resulting effects on MT stability. For example, although our prior SAR studies highlighted the importance of the stereo-electronic properties of the substituent in the para position of the phenyl ring at C6, which is exemplified by electron-donating (e.g., alkoxy) and electron-withdrawing (e.g., nitrile) substituents producing opposite overall effects on the cellular phenotype (Class II and Class I phenotype, respectively), the X-ray structure of 5 bound within the seventh site indicates that the fluorinated amine fragment at C7 may also play a prominent role in triggering tubulin degradation as this fragment establishes direct interactions with the deoxyribose of the nonexchangeable GTP.13
Figure 1.

Structure of selected examples of MT-binding triazolopyrimidines.
To further assess the SAR of TPDs, we have designed and synthesized a series of congeners and conducted matched molecular pair (MMP) analyses. Test compounds were evaluated for MT-stabilizing activity in QBI293 cells, and for their calculated binding energies at the vinca and the seventh site. These analyses further elucidate the interplay between the C6 and C7 fragments in determining the differential affinity of TPDs for the two binding sites and, consequently, the cellular MT phenotype. Moreover, these studies illustrate how docking data can be utilized to predict Class I activity of TPDs. As a result, new Class I TPD congeners were identified that are characterized by the presence of a relatively rigid alkyne-containing side chain at the para position of the aromatic ring at C6 and that are endowed with favorable pharmacokinetic (PK) properties and brain pharmacodynamic (PD) effects. Moreover, this study also identified selected TPD congeners that elicit an unusual cellular phenotype that appears to be hybrid between the Class I and II phenotypes, characterized by a dose-response where higher concentrations show a reduction in activity with respect to MT AcTub modification (i.e., Class II-like) without evidence of a significant decrease in total tubulin levels (i.e., Class I-like). An evaluation of representative Class I, II and hybrid TPDs in a neuronal assay with tau hyperphosphorylation and MT deficits indicates that selected examples of these hybrid compounds act like Class I molecules in the context of neurons by effectively normalizing MTs, suggesting that these compounds may also be considered, in addition to Class I molecules, for further development as candidates for neurodegenerative disease.
Chemistry
The synthesis of TPD congeners of general structure A and B (Scheme 1) started with the TPD dichloride, 6.16 Chemoselective displacement of the chloro-substituent at C7 upon treatment with the appropriate amine fragment furnished TPDs of general structure A, which include previously described 1,112,14 and 7–12.12 Further treatment of 1–17 with the appropriate alcohol in the presence of NaH led to the formation of TPD derivatives of general structure B (18–32, Scheme 1).
Scheme 1.


Reduction of the nitrile of TPD 33(12) to the corresponding aldehyde and conversion of the latter to the alkyne upon treatment with the Bestmann–Ohira reagent17 provided acetylene derivative 34 (Scheme 2).
Scheme 2.

For the synthesis of the disubstituted alkyne derivatives of general structure C (Scheme 3), the 1,2,3-trifluoro-5-nitrobenzene (35)12 was treated in an SNAr reaction with diethylmalonate, followed by a palladium-catalyzed reduction of the nitro group to obtain the corresponding aniline, 36. A Sandmayer reaction was then used to convert 36 into the corresponding aryl iodide, 37. Next, condensation of 37 with the aminotriazole, followed by treatment with POCl3, led to the expected TPD dichloride, 38, which was then reacted with the appropriate aliphatic amines to furnish aryl iodides 39–41. Finally, Sonogashira coupling reactions involving the aryl iodides 39–41 and the appropriately substituted alkynes led to derivatives 42–48, 50, 52–56, 58, and 60. The structure of 53 was confirmed by X-ray crystallography (Scheme 3 and Supporting Information). Deprotection of the Boc-protecting group of 48, 50, 56, and 58 led, respectively, to final products 49, 51, 57, and 59 (Scheme 3).
Scheme 3.

Results
All test compounds were initially screened in the same cell-based (QBI293) assay of MT stabilization used in our previous SAR studies11,12 so as to allow direct comparison with literature data. Changes in levels of acetylated α-tubulin (AcTub) and total α-tubulin (α-Tub), relative to vehicle-treated negative control cells, were determined after 4 h of incubation with test compounds at 1 and 10 μM. To account for possible day-to-day differences in the responsiveness of cells to MT-stabilizing treatment, compound-dependent changes in AcTub levels relative to DMSO-treated cells were also normalized to the corresponding changes caused by 100 nM of 5 that was used as a positive control. In addition, docking studies, binding free energy calculations, and in selected cases, molecular dynamic simulations were conducted using an X-ray structure of the vinca and seventh sites (PDB: 5NJH,107CLD13). The results of this screening, alongside the observed cellular phenotype and the calculated binding energy of each of the compounds for the vinca site and the seventh site, are summarized in Table 1. For selected compounds, additional studies included testing in the QBI293 cell AcTub assay at additional compound concentrations, and/or evaluation of test compounds for their ability to prevent MT collapse in primary neuron cultures treated with the phosphatase inhibitor, okadaic assay (OA). Finally, cytotoxicity studies in QBI293 and HeLa cells were also conducted for representative examples (Supporting Information).
Table 1. MT-Stabilizing Activity and Calculated Binding Energies for the Vinca Site and the Seventh Site of Test Compoundsa.
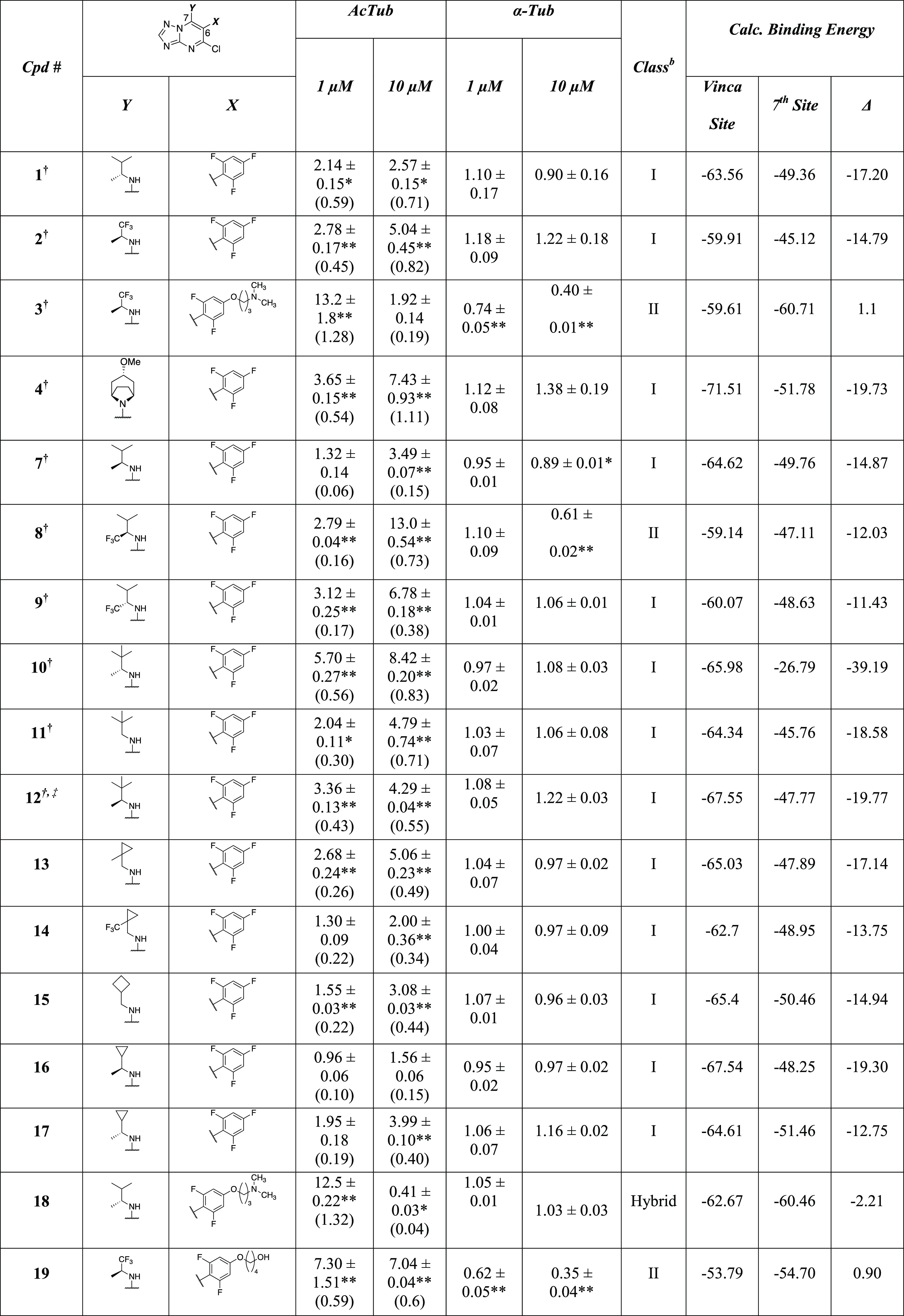
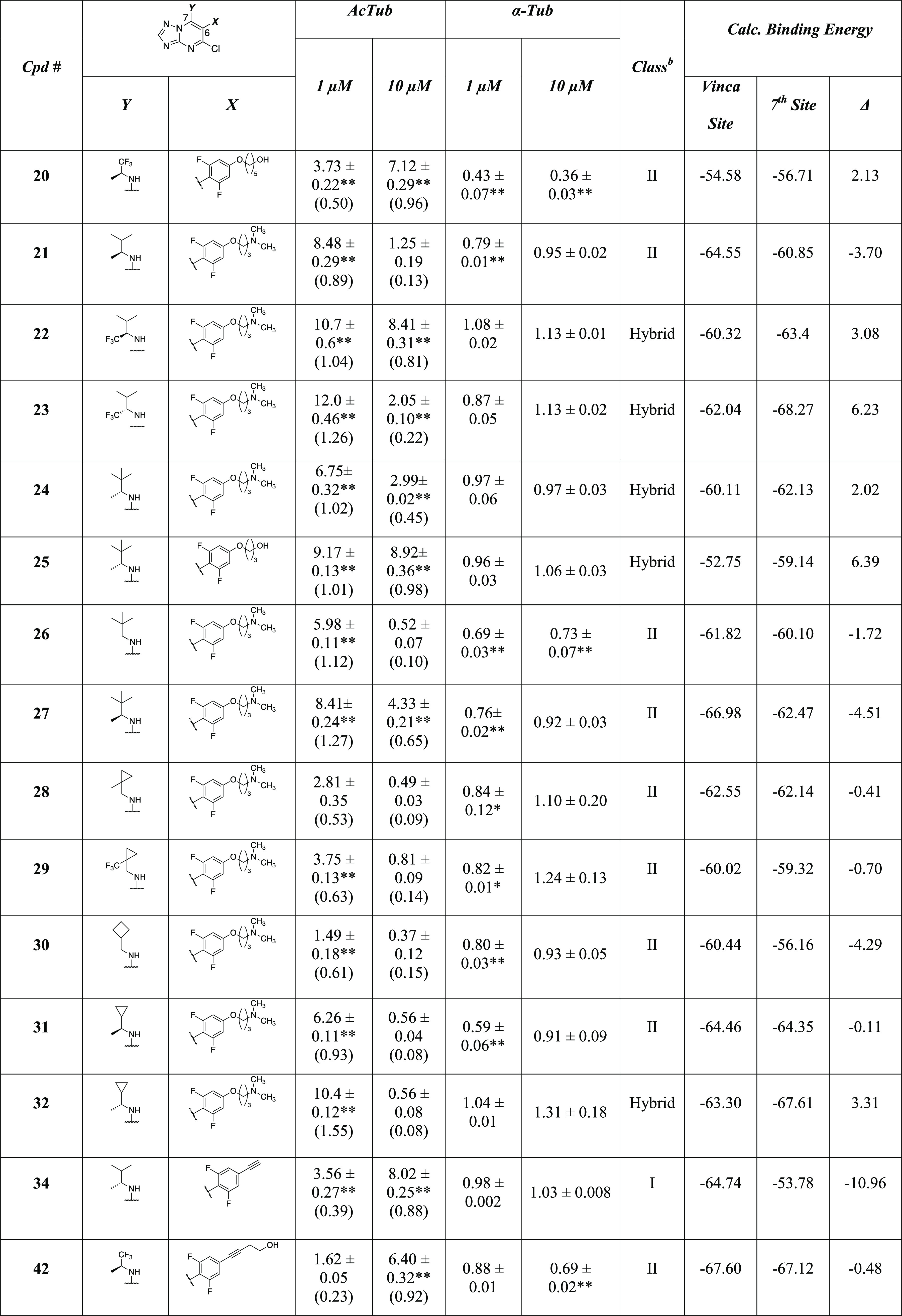
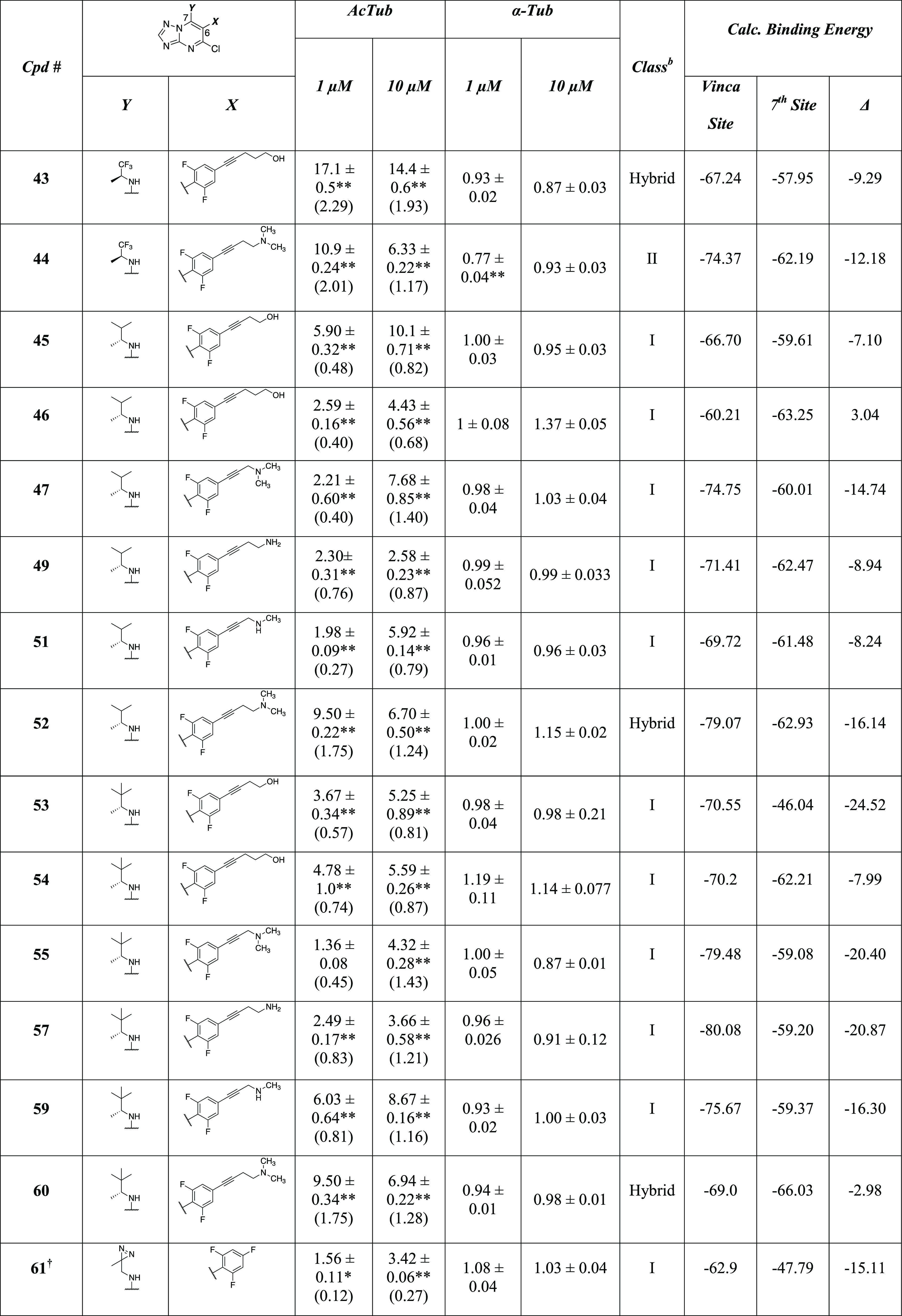
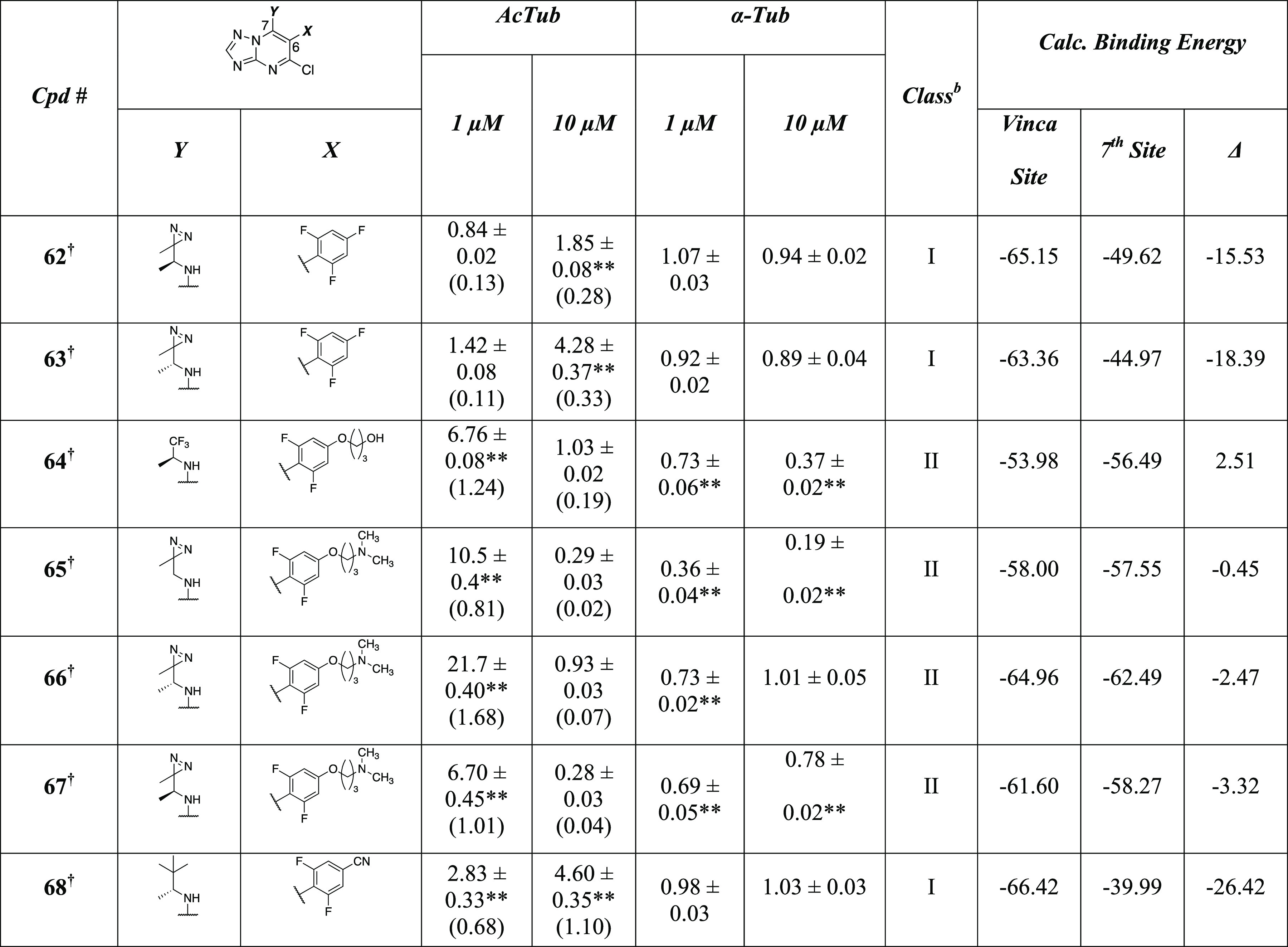
Unless otherwise noted, fold-changes in acetylated α-tubulin (AcTub) and total α-tubulin (α-Tub) levels in QBI293 cells were determined after 4 h incubation with test compounds at either 1 or 10 μM. Reported values for AcTub and α-Tub represent the fold-change compared to vehicle (DMSO)–treated cells (*p < 0.05 and **p < 0.01 by one-way analysis of variance (ANOVA)); numbers in parentheses represent the fold-change of AcTub compared to cells treated with the positive control compound 5 (100 nM).
Class I compounds are those producing a concentration-dependent increase in AcTub levels and that do not cause >15% reduction in α-tubulin at either concentration; Class II compounds are those that cause >15% decrease in α-tubulin at either 1 or 10 μM compound concentration. Hybrid compounds are those producing elevations in AcTub levels at 10 μM that are lower than the corresponding levels obtained at 1 μM but that do not cause >15% reduction in α-tubulin at either concentration. †The activity in the AcTub and α-tubulin assays have been reported in previous studies11,12 and are shown here for comparison. ‡Compound was tested at 10 and 30 μM.
The data summarized in Table 1 allow for several comparisons between series of MMPs that differ only by a single chemical transformation. First, a comparison of 3 with 15 match paired congeners (18, 21–24, 26–32, 65–67) bearing different amine fragments at C7, including examples of fluorinated (22, 23, and 29) and nonfluorinated amines, reveals that none of the 15 analogues exhibit Class I activity. However, while 10 of these analogues (21, 26–31, 65–67), like 3, produce a significant reduction (i.e., >15%) of total α-Tub levels when tested at 1 and/or 10 μM (i.e., Class II phenotype), in other cases (18, 22–24, and 32), the replacement of the fluorinated amine of 3 resulted in derivatives that do not produce a meaningful reduction in total tubulin levels at either concentration. However, similar to the parent TPD 3, these latter matched pair compounds produce an unusual dose–response in the AcTub assay as evidenced by relative increases in this marker of stable MTs that are more pronounced at the low compound concentration (1 μM) than at the high concentration (10 μM). To exemplify and confirm this phenomenon, Figure 2 illustrates a representative example (24) that was also tested at a higher (30 μM) concentration, showing a further concentration-dependent decrease in cellular AcTub without a reduction of total α-Tub.
Figure 2.

ELISA measuring acetylated AcTub levels (A) and total α-Tub levels (B) in response to 4-h treatment with TPD 5 (100 nM, used as positive control) or 24 at different concentrations. Error bars represent SEM, with *, p < 0.05 and **, p < 0.01 relative to vehicle as assessed by one-way ANOVA.
We refer to these compounds as “hybrids” since in QBI293 cells they elicit an unusual dose-dependence in the AcTub assay, which is often seen in Class II compounds (e.g., 3, 21, 26–31, 65–67), without reduction in total tubulin levels, which is a defining feature of Class I compounds (e.g., 1 and 68).
The observation that the replacement of the amine fragment of 3 can result in analogues that no longer trigger degradation of tubulin appears to be in general agreement with Yang et al.13 and suggests that the fluorinated amine fragment of 3 plays an important role in contributing to the Class II activity of this compound and likely other related congeners (e.g., 64). However, our data also show that the presence of a fluorinated amine at C7 per se may not be necessary nor sufficient for Class II activity. This is exemplified by the fact that TPDs bearing fluorinated amines at C7, such as 22 and 23, exhibit similar hybrid behavior as 24, and that different TPDs bearing nonfluorinated amines at C7 (e.g., 21, 26, 27, 28, 30, 31, 65–67) produce the Class II defining >15% reduction in total tubulin at either 1 or 10 μM as observed with 3.
Further comparison of 3 and the above-mentioned analogues (18, 21–24, 26–32, 65–67) with the corresponding matched pair compounds in which the alkoxide side chain is replaced by a fluorine atom showed that in all cases but one (MMP 22–8), substitution of the alkoxide side chain with a fluorine results in compounds with Class I activity (see MMPs: 3–2; 21–7; 23–9; 24–10; 26–11; 27–12; 28–13; 29–14; 30–15; 31–16; 32–17; 18–1; 65–61; 66–63; 67–62). These results agree with our prior SAR studies12 and confirm that in the majority of cases the presence of the alkoxide side chain of 3 and related congeners (e.g., 19, 20, and 64) is conducive to eliciting the Class II phenotype. Moreover, although 2 was found to produce a reduction in total tubulin levels in Yang et al.,13 evaluation of this compound in the QBI293 AcTub assay at higher concentrations did not produce evidence of significant reduction of total tubulin levels (see the Supporting Information, Figure S1A). The reason for this discrepancy is not clear, although it may be due to the different cell-type used for the studies (HeLa cells were used in Yang et al.) and/or the different incubation time used in the experiments (16 h incubation time was used in Yang et al.). However, the observation that selected derivatives of 3 (18, 22–24, and 32) and 64 (25) show hybrid behavior and do not trigger reductions in total tubulin levels clearly highlights the importance that specific aliphatic amines at C7 can have and suggests a more complex interplay between the nature of the amine fragment at C7 and the substituent in the para position of the aromatic ring at C6 in determining the phenotype of TPDs. This situation is also highlighted by comparing MMPs that differ only by the chiral configurations of the amine fragment. In the case of TPDs equipped with an alkoxide side chain, the chiral configuration of the amine was found to have an impact on the phenotype in the pairs 18–21, 24–27, and 31–32, but not in the corresponding compounds bearing a fluorine atom in the para position (cf., 1–7, 10–12, and 16–17).
To further investigate the role of the side chain in the para position of the fluorinated ring, we evaluated a series of 16 alkyne derivatives. In particular, since prior SAR studies12 suggested that electron-withdrawing groups in para are generally preferred for Class I activity, we evaluated the effect of incorporating an alkyne linkage within the aliphatic side chain off of the para position of TPDs. Consistent with prior SAR data, terminal alkyne TPD derivative, 34, was found to exhibit Class I activity. Furthermore, among the 15 disubstituted alkyne derivatives examined in this study (42–47, 49, 51–55, 57, 59, 60), ten examples (45–47, 49, 51, 53–55, 57, 59) were found to exhibit Class I activity, while two compounds (42 and 44) were deemed to be Class II, and three (43, 52, and 60) were hybrids. Thus, in the majority of cases, the replacement of the alkoxy linkage with a rigid and electron-withdrawing alkyne was found to result in relatively potent compounds that do not cause loss of total tubulin (Table 1 and Figure 3). The only derivatives found to be Class II within our set of alkyne derivatives (42 and 44) were those equipped with the same (S)-1,1,1-trifluoropropan-2-amine at C7 as in 3, which again suggests that this particular amine fragment is likely to play a role in the tubulin degradation properties of Class II TPDs. However, even in these cases, the incorporation of the alkyne moiety resulted in derivatives that produced a more moderate reduction in tubulin levels compared to the corresponding alkoxy congeners (cf., 3–44, 64–42, Table 1 and Figure 3). In the particular case of TPD alkyne derivative, 43, which also features an (S)-1,1,1-trifluoropropan-2-amine at C7 like its alkoxy MMP compound (19), a reduction in total tubulin levels could be detected only at higher concentrations (i.e., 30 μM, see Supporting Information, Figure S1B).
Figure 3.

Plot showing the activity of test compounds normalized to positive control (i.e., 100 nM of 5) in the AcTub assay at 1 μM (Y-axis) and 10 μM (X-axis). Class I compounds are shown as blue circles; Class II compounds as red squares; and hybrid compounds as green triangles. The average loss of total α-Tub after compound treatment at 1 and 10 μM relative to vehicle-treated controls is plotted as background red color based on the heatmap shown. Note that 12 is not plotted as the activity of this compound was determined at 10 and 30 μM.
To further evaluate the activity of Class I alkyne derivatives and hybrid compounds, several compounds were also assessed for their ability to compensate for MT deficits in primary neuron cultures that were treated with the phosphatase inhibitor, okadaic acid (OA). As previously described,11 treatment of rodent neurons with OA results in tau hyperphosphorylation and MT collapse that presumably results, at least in part, from tau disengagement from MTs. Co-treatment of neurons with Class I TPDs, such as 1, along with OA prevents this MT collapse, with a resulting normalization of MT structure and neuronal processes which can be observed upon visual inspection of the neurons or through an assessment of neuronal AcTub levels, while Class II TPDs are generally unable to provide meaningful compensation of MT deficits, especially when tested at higher (i.e., 10 μM) compound concentration.11,12 This phenomenon is illustrated in Figure 4A, in which Class I TPD, 1, is evaluated for its ability to normalize AcTub levels in cortical neurons in the OA assay in comparison with TPD enantiomers 8 (S) and 9 (R) that exhibit, respectively, Class II and Class I phenotype in QBI293 cells (Table 1). Interestingly, evaluation of Class I alkyne derivatives 45, 46, 53, and 54 in the OA assay confirmed that these compounds can prevent the OA-induced MT collapse in cortical neurons in a manner similar to 1 (Figure 4B). Notably, in the case of the hybrid compounds, evaluation in the OA assay revealed that some compounds (e.g., 24 and 60) provided a similar dose-dependent correction of MT deficits in OA-treated neurons as seen for Class I TPDs, while other compounds (e.g., 25) caused a lesser recovery of OA-induced MT collapse (see Supporting Information, Figure S2).
Figure 4.

ELISA determination of AcTub levels in homogenates from primary rat cortical neurons treated with either vehicle, or OA (15 nM) in the presence of vehicle or TPD 1, 8, 9 (A); and primary mouse cortical neurons treated with vehicle, or OA (15 nM) in the presence of vehicle or TPD 1, 24, 45, 46, 53, 54 (B). Error bars represent SEM, with ***, p < 0.001 relative to vehicle treatment in the absence of OA as determined by one-way ANOVA.
Although the SAR data emerging from these studies highlight an apparently complex interplay between the fragments at C6 and C7, comparison of biological activity data for each of the listed 53 TPD congeners with the corresponding calculated binding energies for the vinca and the seventh sites revealed general trends. In particular, as shown in Figure 5, Class I compounds appear to be generally associated with a greater calculated affinity for the vinca site, whereas Class II and hybrid compounds exhibit a smaller differential of calculated binding energies for the two sites and are thus expected to show little or no selectivity for either one of the two sites. Moreover, a plot of the differential binding affinity for the vinca and seventh site compared to the level of tubulin degradation induced by the compounds reveals a distinct clustering of the Class I, II and hybrid TPDs (Figure 5B).
Figure 5.

(A) Whisker plot showing the distribution of differential calculated binding energies (i.e., ΔGvinca – ΔG7th site) within each class of compounds. The more negative the differential binding energy, the greater the relative binding to the vinca site compared to the seventh site. Mean values are indicated as thick horizontal red lines. (B) Scatter plot showing the differential calculated binding energies for the two sites (X-axis) and the average fold-change in total α-tubulin levels caused by compound treatment at 10 and 1 μM in QBI293 cells [i.e., (α-tub10μM + α-tub1μM)/2] relative to vehicle-treated control cells (Y-axis). Although Class I compounds are largely characterized by a higher calculated affinity for the vinca than seventh site with little decrease in α-tubulin levels, most of the Class II and hybrid compounds group together with a smaller differential of calculated binding energies with Class II compounds also exhibiting an average fold-change in α-tubulin value that generally is <0.90.
Evaluation of the relative contribution of van der Waals, electrostatic, hydrogen bond, and π–π interactions showed that hydrophobic and electrostatic effects account for the majority of the ΔG difference observed between the two pockets (Supporting Information, Table S1), with Class I compounds forming more hydrophobic interactions in the vinca site compared to the Class II and hybrid compounds. For example, a two-dimensional (2D) interaction plot of a Class II compound (64), a hybrid compound (25), and a Class I compound (53) in both the vinca and seventh site (Figure 6) shows that the Class I congener is expected to establish stronger hydrophobic interactions with β-tubulin residues in the vinca binding site (i.e., Val353, Val328, and Leu248). Moreover, docking studies clearly indicate that the side chain in the para position of the fluorinated phenyl ring at C6 plays a critical role in determining the relative binding affinity for the two pockets (e.g., Figure 6C,E).
Figure 6.

2D interaction plot of Class II compound 64 (A, B), hybrid compound 25 (C, D), and Class I compound 53 (E, F) in the vinca site (left, from PDB: 5NJH) and seventh site (right, from PDB: 7CLD), respectively. Turquoise dashed lines indicate weak H-bonds, green dashed lines indicate strong H-bonds, gray dashed lines indicate hydrophobic interactions, and purple dashed lines indicate electrostatic and π–π interactions.
To further investigate the role of the side chains in the binding selectivity, a series of 100 ns molecular dynamic simulations (MD) were conducted using two MMPs (25–53 and 64–42) that produce differing effects on the cellular phenotype. All of the simulations showed an initial rearrangement of the ligand complex and a consecutive stabilization after 50 ns (Supporting Information, Figure S3). The dynamic results showed that all of the compounds could maintain a stable binding pose during the entire MD simulation in both pockets; however, the alkyne derivatives were found to adjust their orientation to establish interactions with the β-tubulin residues in the vinca site (Figure 7A vs B). Comparison of TPD derivatives 53 and 25 that feature the same amine fragment at C7 revealed that the alkyne 53 can establish multiple hydrophobic contacts in the vinca site throughout the MD simulation (i.e., α-tubulin residue Val177, as well as β-tubulin residues Pro325 and Val353) and that the presence of the rigid alkyne linkage can also facilitate the H-bonding interaction between the terminal hydroxyl and the α-tubulin residue, Asp211, in the vinca site, improving the overall affinity for this binding region (Figure 7A vs C). In contrast, the replacement of the t-butyl group with the smaller CF3 in the amine fragment at C7, combined with the presence of a more flexible alkoxide side chain (64), resulted in a similar affinity for both pockets (Figures 6A,B and 7D).
Figure 7.

Binding mode for 53 in both pockets (A, vinca site, from PDB: 5NJH; B, seventh site, from PDB: 7CLD) and 25 and 64 in the vinca site (C and D, respectively) after MD simulation, and the corresponding protein interactions throughout the simulation after reaching stability. Protein–ligand interactions are categorized into two groups: hydrogen bond (green), and hydrophobic (purple-gray) interactions. The interaction fraction indicates the normalized percentage of the simulation time for which the specific interaction is maintained.
To evaluate the potential of the newly identified Class I and hybrid alkyne TPD derivatives as potential candidates for tauopathy treatment, selected compounds (53 and 60) underwent PK analysis in mice. Determination of brain-to-plasma ratios (B/P) for these two compounds 1 h post-administration (i.p. injections of 1.5 mg/kg) indicated that both derivatives are brain penetrant with a B/P of approximately 0.5 (data not shown). Furthermore, a more comprehensive plasma PK study with compound 60 after i.v. and oral (p.o.) dosing revealed favorable properties (Figure 8A), including a relatively long half-life of >6 h and an oral bioavailability of >50% (see also Supporting Information, Figure S4, for plasma PK of 53). Finally, to evaluate target engagement in the CNS, PD studies were conducted to determine compound-dependent elevations in the stable MT marker, de-tyrosinated α-tubulin (GluTub), in brain homogenates from compound-treated WT mice. As summarized in Figure 8B, 53 and 60, as well as the previously identified Class I TPD 68, produced significant elevations in brain GluTub when administered at the low dose of 1 mg/kg (i.p.) once daily for two consecutive days.
Figure 8.

(A) Plasma PK of 60 after administration of 5 mg/kg (i.v. injection; black circles) or 20 mg/kg (p.o.; black squares). Error bars represent standard deviation (SD). (B) Increases in the GluTub-to-total α-Tub ratio in the brains of WT mice after i.p. dosing of 68, 53, and 60 at 1 mg/kg once daily for two consecutive days, relative to vehicle-treated mice. The data represent a summary of independent experiments with each compound in which n = 4 CD-1 mice received a test compound or vehicle. Error bars represent SEM, with *, p < 0.05 and **, p < 0.01 as determined by a two-tailed t-test.
Discussion
MT-normalizing TPDs hold promise as candidate therapeutics for the treatment of tauopathies and potentially other neurodegenerative diseases. However, this class of MT-active compounds presents an apparently complex SAR that arises from the fact that TPDs can interact with varying affinities with at least two spatially distinct binding sites, the vinca site and the seventh site, producing substantially different cellular phenotypes. The availability of the co-crystal structures of 4(10) and 5(13) with assembled tubulin, combined with our findings from prior SAR studies,12 provided the basis for further exploration of SAR of the fragments at C6 and C7 of the TPD core and the possible link between the calculated relative affinity for the two binding sites and the cellular phenotype associated with different TPD congeners.
Structural analysis of the two binding sites revealed a high similarity between the two pockets (Figure 9). However, the vinca site, located at the interface between β1 and α2 tubulin, exhibits different spatial arrangements of several amino acid residues relative to the seventh site, which is located at the α2-β2 interface. These amino acid residues include the β1 residues Val324, Pro325, Lys326, Val328, Asn329, Ala330, Ile332, Val353, and Ile355 that may potentially play a role in the binding of TPDs.
Figure 9.

Overview of the vinca binding site between β1 and α2-tubulin, and the sevenths site between α2 and β2-tubulin (from co-crystal structure with compound 5; PDB: 7CLD(13)).
Prior SAR studies already highlighted that with few exceptions, compounds of general structure A (Scheme 1) in which the aliphatic amine fragment is a relatively bulky and lipophilic exhibit Class I activity. However, an assessment of the same amine fragments in the context of TPDs of general structure B (Scheme 1) was not conducted. Thus, one of the objectives of the present study was to evaluate/compare more systematically the effect of different amine fragments at C7 in the context of compounds of general structure A and B. Moreover, considering that our prior SAR studies indicated that the presence of an electron-withdrawing group in the para position of the fluorinated ring at C6 (i.e., substituents characterized by a positive Hammett σp value) is generally conducive to Class I activity, we expanded the exploration of TPDs to a series of novel congeners that are characterized by the presence of a side chain connected to the para position via an intervening alkyne moiety (general structure C, Scheme 3).
Comparison of biological activity in the QBI293 assay of a series of MMPs of general structure A, B, and C illustrates an apparently complex interplay between the nature of the amine fragment at C7 and the substituent in the para position of the aromatic ring at C6. Furthermore, in addition to the previously described Class I and II phenotypes, these studies led to the identification of hybrid TPD congeners that do not cause reduction in total tubulin levels yet show an unusual dose–response relationship in the QBI293 AcTub assay. Interestingly, the biological results within our set of compounds correlate generally well with calculated binding energies for both the vinca site and the seventh site (Table 1 and Figures 3 and 5). Out of 27 Class I TPDs examined in this study, 25 examples exhibit a ΔG of binding for the vinca site that is at least −60 kcal/mol or lower, as well as a differential of binding energies for the two sites (i.e., ΔGvinca – ΔGseventh) that is at least −5 kcal/mol or lower. However, in the case of Class II and hybrid compounds, the calculated binding affinities for the two sites indicate a smaller differential of binding affinity, with 15/18 Class II compounds and 7/8 hybrid compounds exhibiting a differential of binding energies for the two sites that is above −5 kcal/mol. These observations are consistent with the notion that the MT-stabilizing activity of TPDs is mediated by interactions at the vinca binding site on β-tubulin and suggest that relatively simple estimations of the binding energies of TPDs in the two sites may be used to predict whether TPDs will exhibit Class I activity. Moreover, the fact that no major differences in the calculated relative binding affinities for the two binding sites were observed when comparing Class II and hybrid compounds may suggest that the hybrid TPDs interact, like Class II compounds, both at the vinca and the seventh site but possibly via different binding modes. For example, prior studies13 found that the degradation of cellular tubulin observed after addition of Class II compound, 5, may be linked to the ability of this compound to promote the dissociation of the nonexchangeable GTP in the seventh site. Therefore, it is possible that relatively small differences in binding modes of Class II and hybrid TPDs in the seventh site may result in differing abilities of these compounds to trigger, directly or indirectly, GTP release and subsequent tubulin degradation. In this context, although the AcTub and total α-Tub assays in QBI293 cells provide a generally effective method to sort compounds between different groups based on the magnitude and characteristics of the cellular response, our studies show that further assessment/confirmation of compound activity in the neuronal OA assay with MT deficits is desirable when prioritizing/selecting candidate compounds for further development.
With respect to the alkyne derivatives of general structure C, a comparison with the corresponding derivatives of general structure B that bear an alkoxide side chain (Figure 10) indicates that, in general, the presence of the alkyne linkage results in a relative increase in binding affinity for the vinca site (Figure 10A) and enlargement of the differential of binding affinity between the vinca and the seventh site. This effect is likely due at least in part to the rigidification of the side chain provided by the presence of the alkyne moiety, which ultimately promotes interactions within the vinca site (Figures 6 and 7). Eleven of the 16 alkyne derivatives synthesized and tested were found to exhibit Class I activity. Moreover, even in those cases in which incorporation of the alkyne linkage was not sufficient to impart Class I activity (e.g., see Class II derivatives 42 and 44, and hybrid derivatives 43, 52, and 60), the presence of the alkyne moiety in these molecules generally led to a notable attenuation of the Class II (cf., 3–44; 19–43; and 64–42) or hybrid (cf., 24–60; 18–52) character, as evidenced by a reduced loss of total tubulin and/or a reduced tendency to show a bell-shaped dose–response curve in the AcTub assay compared to the corresponding alkoxy side-chain derivatives (Table 1 and Figure 3).
Figure 10.
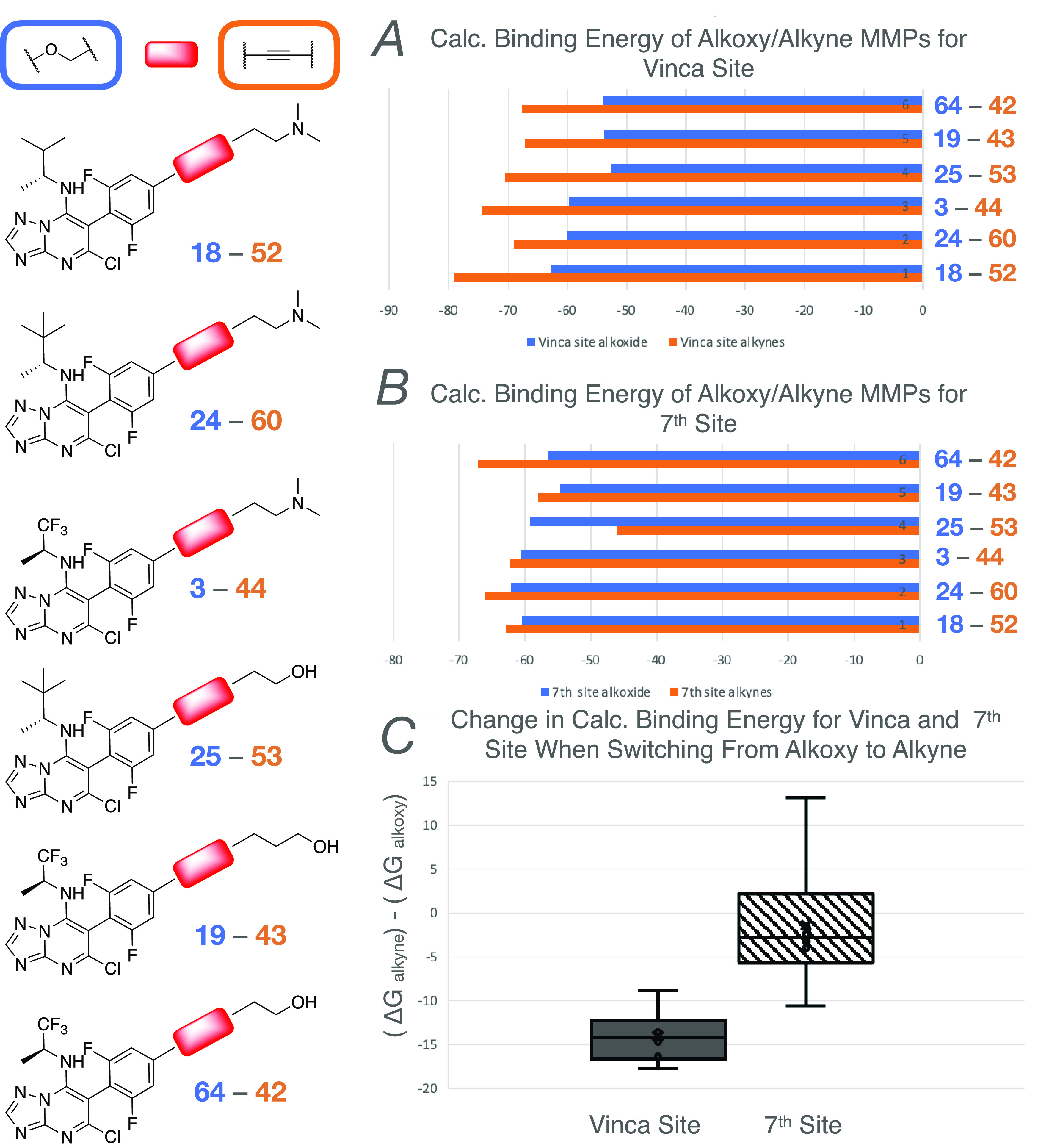
Structure of six pairs of compounds differing only by the nature of the linkage in the para position (left). (A) Comparison of calculated binding energy for the vinca site of the alkoxy (blue) and alkyne (orange) derivatives within each MMP. (B) Comparison of calculated binding energy for the seventh site of the alkoxy (blue) and alkyne (orange) derivatives within each MMP. (C) Average change in calculated binding energy for the vinca and the seventh site when switching from alkoxy to alkyne linkage in the six MMPs (average values are indicated with a cross and median values with a solid line).
With respect to the amine fragment at C7, comparison of MMPs indicates that these fragments generally do not significantly alter the calculated binding energy for the vinca site. However, depending on the nature of the fragment at C6, the relatively bulkier and more lipophilic amines can reduce the binding affinity for the seventh site. For example, direct comparison of six MMPs bearing either the (S)-1,1,1-trifluoropropan-2-amine or the (R)-3,3-dimethylbutan-2-amine (Figure 11) reveals that in all cases, the change in calculated binding energy for the vinca site between the two series of compounds is relatively small (Figure 11A,C). However, when examining the calculated binding energies for the seventh site, in selected cases (cf., 2–10 and 42–53) the presence of the bulkier and more lipophilic (R)-3,3-dimethylbutan-2-amine results in a drastic lowering of the binding affinity for this site and a consequent increase in the differential of binding energy between the vinca and seventh sites.
Figure 11.

The structure of six pairs of compounds differing only by the nature of the amine fragment at C7 (left). (A) Comparison of calculated binding energy for the vinca site of MMPs bearing either the (S)-1,1,1-trifluoropropan-2-amine (blue) or the (R)-3,3-dimethylbutan-2-amine (orange) at C7. (B) Comparison of calculated binding energy for the seventh site of MMPs bearing either the (S)-1,1,1-trifluoropropan-2-amine (blue) or the (R)-3,3-dimethylbutan-2-amine (orange) at C7. (C) Average change in calculated binding energy for the vinca and the seventh site when switching from (S)-1,1,1-trifluoropropan-2-amine to (R)-3,3-dimethylbutan-2-amine at C7 (average values are indicated with a cross and mean values with a solid line).
Importantly, further evaluation of representative Class I and hybrid alkyne derivatives (respectively, 53 and 60) in mice revealed that both compounds are brain penetrant (i.e., B/P ∼ 0.5). Moreover, plasma PK studies of 53 and 60 demonstrated a very favorable PK profile, including relatively prolonged half-life and, in the case of 60, good oral bioavailability (Figure 8A). Equally important, PD studies with 53, 60, and the previously described Class I TPD, 68,12 showed that these alkyne derivatives can produce a significant elevation in markers of stable MTs (i.e., GluTub) in the brain of WT mice at a low dose (Figure 8B). Thus, these results indicate that these novel acetylene derivatives may be considered promising alternatives to 68 as candidate therapeutics for neurodegenerative tauopathies and possibly other neurodegenerative indications.
Conclusions
Previous studies have shown that, depending on the substitution pattern, MT-targeting TPDs can interact with either one or two spatially distinct binding regions within tubulin heterodimers (i.e., the seventh site and the vinca site) producing different effects on tubulin and MT function. Our structure–activity relationship studies, based on matched molecular pair analyses, provide additional insight into the complex interplay between the fragments at C6 and C7 in determining the biological activity and the relative affinity of TPDs for the two binding sites. Calculated binding energies of TPDs for the vinca and the seventh site were found to largely correlate with biological data and indicate that compounds with preferential affinity for the vinca site exhibit dose-dependent MT-stabilizing activity without causing reduction in total tubulin levels (i.e., Class I activity). The Class II TPDs that have an atypical dose–response profile in assays of MT stabilization and trigger tubulin degradation interact with both binding sites with similar affinity, and our studies have now identified an additional hybrid series of congeners that bind to both sites with comparable affinity but do not decrease total tubulin levels. However, like the Class II compounds, these hybrid molecules are characterized by a nonlinear dose–response relationship in the QBI293 AcTub assay. Finally, our findings that the incorporation of an acetylene linkage in the aliphatic side chain at the para position of the fluorinated ring at C6 can provide potent Class I MT-stabilizing derivatives that exhibit excellent PK/PD profiles allows for a significant expansion of the possible structures that may be considered as potential candidates for CNS-directed MT-stabilizing therapies. In this context, the acetylene derivatives, 53 and 60, identified in these studies represent promising alternatives to 68 as candidate therapeutics for neurodegenerative tauopathies.
Experimental Section
Materials and Methods
All solvents were of reagent grade. All reagents were purchased from Aldrich or Acros Organics and used as received. Thin-layer chromatography (TLC) was performed with 0.25 mm E. Merck precoated silica gel plates. Silica gel column chromatography was performed with silica gel 60 (particle size 0.040–0.062 mm) supplied by Silicycle and Sorbent Technologies. TLC spots were detected by viewing under a UV light. Proton (1H) and carbon (13C) NMR spectra were recorded on a 500 MHz Bruker AMX-500 spectrometer and on a 600 MHz Bruker AVANCE III. Fluorine (19F) NMR spectra were recorded on a 400 MHz Varian Mercury Plus spectrometer using trifluorotoluene as an internal standard. Chemical shifts were reported relative to solvents. Data for 1H, 13C, and 19F NMR spectra are reported as follows: chemical shift [ppm, referenced to protium; s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, dd = doublet of doublets, td = triplet of doublets, ddd = doublet of doublet of doublets, bs = broad singlet, m = multiplet, coupling constant (Hz), and integration]. Not all 13C NMR signals were detected for compounds 42–49, 51, 52, 54, 57, and 58 despite multiple attempts with exhaustive signal averaging. Some signals are expected to be difficult to detect due to 19F coupling reducing their intensity and the longer relaxation times found in a system where an aromatic ring is conjugated with an alkyne. 13C NMR spectra of compounds 44, 50, 55, 56, and 59 were not featured for the same reasons of low signal/noise ratio. High-resolution mass spectra (HRMS) were measured using an Agilent 6230 time-of-flight mass spectrometer with a Jet stream electrospray ionization source. Single-crystal X-ray structure determinations were performed Bruker MicroStar with an APEX II detector, double-bounce micro-focus optics, and a Cu rotating anode source. Analytical reversed-phase (Sunfire C18; 4.6 mm × 50 mm, 5 mL) high-performance liquid chromatography (HPLC) was performed with a Gilson HPLC equipped with UV and a mass detector. All samples were analyzed employing a linear gradient from 10 to 90% of MeCN in H2O over 6 min and a flow rate of 2 mL/min. Preparative reversed-phase HPLC purifications were performed on a Gilson instrument employing Waters SunFire preparative C18 OBD columns (5 μm 19 mm × 50 mm or 19 mm × 100 mm). Purifications were carried out employing a linear gradient from 10 to 90% of MeCN in water using formic acid or trifluoroacetic acid as a 0.1% modifier. All final compounds were found to be >95% pure by HPLC analysis.
General Procedure A (Addition of the Amine)
According to a reported procedure,14 to 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (1.0 equiv) in N,N-dimethylformamide (DMF) (0.1 M) at rt was added the appropriate amine or amine hydrochloride (1.5 to 3.0 equiv) followed by Et3N (3.0 equiv) if necessary. The reaction mixture was stirred for 0.5–2 h at rt and diluted with H2O. The aqueous phase was extracted with EtOAc (×3), and the combined organic layers were washed with brine (×2), dried (Na2SO4), filtered, and concentrated. The products were purified by flash chromatography.
General Procedure B (Addition of the Alkoxy Side Chain)
According to a reported procedure,14 to a suspension of NaH (4.0 equiv) in DMSO (0.35 M) was added the appropriate amino alcohol (4.0 equiv), and the mixture was heated to 60 °C for 1 h. The resulting solution was treated with a solution of trifluoroarene (1.0 equiv) in DMSO (0.5 M). The reaction mixture was stirred at 60 °C for 3 h and monitored by liquid chromatography–mass spectrometry (LCMS). Following complete consumption of the starting material, the reaction mixture was cooled to rt and diluted with H2O and EtOAc. The aqueous layer was extracted with EtOAc (×2). The combined organic layers were washed with brine (×2), dried (Na2SO4), filtered, and concentrated. Purification by reversed-phase HPLC yielded the desired product.
General Procedure C (Sonogashira Coupling)
To a solution of the appropriate Iodo-TPD (1 equiv) in degassed DMF (0.2 M) were added successively CuI (0.15 equiv), Et3N (3 equiv), and the desired alkyne (3 equiv). The mixture was degassed and backfilled with nitrogen before Tetrakis palladium (0.1 equiv) was added. The mixture was degassed and backfilled with nitrogen three times and then stirred at rt for 8 h before it was diluted with water. The aqueous layer was extracted with EtOAc (×2). The combined organic fractions were washed with brine, dried, and concentrated under reduced pressure. Finally, purification via silica gel flash chromatography (Hexanes/EtOAc) or by reversed-phase HPLC (water/MeCN + 0.1% FA or trifluoroacetic acid (TFA), (10 to 90%), 20 mL/min, 20 min ramp time) yielded the desired compound.
General Procedure D (Boc Deprotection)
To a solution of the appropriate Boc-protected alkynylated TPD (1 equiv) in MeOH (0.1 M) was added a 4 M solution of HCl in 1,4-dioxane (18 equiv). After 3 h at rt, the mixture was evaporated under reduced pressure to furnish the title compound as a HCl salt as a yellow or brown solid.
5-Chloro-N-((1-methylcyclopropyl)methyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (13)
General procedure A was followed using dichloride 6 (50 mg, 0.16 mmol), (1-methylcyclopropyl)methanamine hydrochloride (29 mg, 0.24 mmol), and Et3N (66 μL, 0.47 mmol), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 80/20) provided compound 13 as a white powder (46 mg, 0.13 mmol, 80%). 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 6.84 (dt, J = 6.0, 6.0 Hz, 2H), 6.43 (bs, 1H), 2.81 (d, J = 4.5 Hz, 2H), 1.09 (s, 3H), 0.44–0.42 (m, 2H), 0.40–0.38 (m, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ 164.09 (dt, J = 253.7, 15.1 Hz), 161.75 (dd, J = 250.7, 7.5 Hz), 161.67 (dd, J = 250.7, 7.5 Hz), 158.12, 155.12, 153.65, 146.47, 107.33 (dt, J = 19.6, 4.5 Hz), 101.09–100.76 (m), 88.93, 52.68, 20.99, 16.20, 11.93 ppm. IR (film) ν 1620, 1606, 1596, 1566, 1555, 1459, 1435, 1371, 1260, 1250, 1234, 1139, 1120, 1035, 1022, 997, 836, 763 cm–1. HRMS (ES+) calcd for C16H14ClF6N5 [M + H]+, 368.0884; found 368.0881.
5-Chloro-N-((1-(trifluoromethyl)cyclopropyl)methyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (14)
General A procedure was followed using dichloride 6 (50 mg, 0.16 mmol), (1-(trifluoromethyl)cyclopropyl)methanamine hydrochloride (41 mg, 0.24 mmol), and Et3N (66 μL, 0.47 mmol), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 80/20) provided compound 14 as a white powder (64 mg, 0.15 mmol, 97%). 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 6.89–6.86 (m, 2H), 6.32 (bs, 1H), 3.51 (s, 2H), 1.11 (d, J = 6.0 Hz, 2H), 0.74 (d, J = 6.0 Hz, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ 164.33 (dt, J = 255.2, 15.1 Hz), 161.71 (dd, J = 252.4, 9.0 Hz), 161.56 (dd, J = 251.4, 9.0 Hz), 157.89, 155.28, 154.07, 146.32, 126.48 (q, J = 274.8 Hz), 101.42–101.05 (m), 89.84, 45.60, 23.56 (q, J = 33.2 Hz), 8.54 ppm. IR (film) ν 1624, 1613, 1571, 1560, 1469, 1440, 1383, 1359, 1246, 1146, 1121, 1056, 1034, 997, 909, 844, 766 cm–1. HRMS (ES+) calculated for C16H10ClF6N5Na [M + Na]+, 444.0421; found 444.0419.
5-Chloro-N-(cyclobutylmethyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (15)
General procedure A was followed using dichloride 6 (50 mg, 0.16 mmol), cyclobutylmethanamine hydrochloride (29 mg, 0.24 mmol), and Et3N (66 μL, 0.47 mmol), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 80/20) provided compound 15 as a white powder (39 mg, 0.11 mmol, 68%). 1H NMR (600 MHz, CDCl3) δ 8.33 (s, 1H), 6.85 (dd, J = 6.0, 6.0 Hz, 2H), 6.31 (bs, 1H), 3.00 (t, J = 6.0 Hz, 2H), 2.47 (hept, J = 6.0 Hz, 1H), 2.90–2.03 (m, 2H), 1.96–1.89 (m, 1H), 1.86–1.80 (m, 1H), 1.62–1.55 (m, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ 164.06 (dt, J = 253.7, 15.1 Hz), 161.82 (dd, J = 250.7, 9.1 Hz), 161.72 (dd, J = 250.7, 9.1 Hz), 158.06, 155.04, 153.68, 146.62, 107.28 (dt, J = 21.1, 6.0 Hz), 101.03–100.66 (m), 89.00, 48.75, 34.91, 25.44, 18.21 ppm. IR (film) ν 1579, 1571, 1562, 1545, 1492, 1478, 1335, 1252, 1204, 1168, 1121, 1034, 999, 844, 764, 513 cm–1. HRMS (ES+) calcd for C16H14ClF3N5 [M + H]+, 368.0884; found 368.0886.
(S)-5-Chloro-N-(1-cyclopropylethyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (16)
General procedure A was followed using dichloride 6 (150 mg, 0.47 mmol) and (S)-1-cyclopropylethan-1-amine (105 μL, 0.99 mmol, 2.1 equiv), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 60/40) provided compound 16 as a white powder (136 mg, 0.37 mmol, 79%). 1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1H), 7.11–6.65 (m, 2H), 6.40 (d, J = 9.2 Hz, 1H), 2.98–2.89 (m, 1H), 1.11 (d, J = 6.6 Hz, 3H), 0.89 (dq, J = 11.2, 4.0 Hz, 1H), 0.50 (ddt, J = 22.1, 10.1, 4.4 Hz, 2H), 0.16 (dq, J = 10.3, 5.1 Hz, 1H), 0.03 (dq, J = 9.5, 4.9 Hz, 1H) ppm. 13C NMR (151 MHz, CDCl3) δ 164.08 (dt, J = 30.9, 16.2 Hz), 162.84–160.35 (m), 157.91, 155.06, 154.81, 153.88, 145.89, 107.23 (t, J = 21.4 Hz), 101.81–100.32 (m), 88.90, 54.32, 20.92, 17.66, 3.27 ppm. HRMS (ES+) calcd for C16H14ClF3N5 [M + H]+, 368.0884; found 368.0882.
(R)-5-Chloro-N-(1-cyclopropylethyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (17)
General procedure A was followed using dichloride 6 (100 mg, 0.31 mmol) and (R)-1-cyclopropylethan-1-amine (67 μL, 0.63 mmol, 2.1 equiv), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 60/40) provided compound 17 as a white powder (82 mg, 0.22 mmol, 71%). 1H NMR (600 MHz, CDCl3) δ 8.30 (s, 1H), 6.86–6.77 (m, 2H), 6.39 (d, J = 9.1 Hz, 1H), 2.94 (q, J = 7.1 Hz, 1H), 1.11 (d, J = 6.5 Hz, 3H), 0.94–0.82 (m, 1H), 0.56–0.44 (m, 2H), 0.17 (dq, J = 10.3, 5.1 Hz, 1H), 0.03 (dq, J = 9.5, 4.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 164.13 (dt, J = 30.3, 15.0 Hz), 162.85–160.25 (m), 157.95, 154.98, 153.93, 145.93, 107.36 (dd, J = 20.7, 4.5 Hz), 101.78–100.28 (m), 88.94, 54.44, 20.97, 17.73, 3.39 ppm. HRMS (ES+) calcd for C16H14ClF3N5 [M + H]+, 368.0884; found 368.0883.
(R)-3-(4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (18)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (89 μL, 0.76 mmol), NaH (60% in oil, 30 mg, 0.76 mmol), and 1 (70 mg, 0.19 mmol). Purification via preparative reversed-phase chromatography provided the title compound as a white solid (15 mg, 16% yield). 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), 8.31 (s, 1H), 6.59 (d, J = 9.1 Hz, 2H), 6.31 (s, 1H), 4.11 (t, J = 5.2 Hz, 2H), 3.29 (s, 1H), 3.06–3.01 (m, 2H), 2.66 (s, 6H), 2.24 (dt, J = 12.3, 6.0 Hz, 2H), 1.64 (dq, J = 13.1, 6.6 Hz, 1H), 1.06 (d, J = 6.6 Hz, 3H), 0.80 (t, J = 6.1 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.81, 161.78 (dd, J = 247.9, 8.6 Hz), 161.73 (dd, J = 247.8, 8.7 Hz), 161.52 (t, J = 13.8 Hz), 158.62, 154.83, 153.63, 146.28, 103.18 (t, J = 20.7 Hz), 98.80 (d, J = 25.9 Hz), 89.77, 66.27, 55.07, 54.71, 43.30, 33.67, 25.03, 18.21, 18.12, 18.01 ppm. HRMS (ES+) calcd for C21H28ClF2N6O [M + H]+, 453.1976; found 453.1979.
(S)-4-(4-(5-Chloro-7-((1,1,1-trifluoropropan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)butan-1-ol (19)
General procedure B was followed using butane-1,4-diol (36 mg, 0.40 mmol), NaH (60% in oil, 16 mg, 0.40 mmol), and 2 (40 mg, 0.10 mmol). Purification via preparative reversed-phase chromatography provided compound 19 as a white solid (38 mg, 82% yield). 1H NMR (500 MHz; CDCl3): δ 8.40 (s, 1H), 6.65–6.63 (m, 2H), 5.92 (d, J = 10.2 Hz, 1H), 4.77 (br s, 1H), 4.08 (t, J = 6.3 Hz, 2H), 3.78 (t, J = 6.3 Hz, 2H), 1.99–1.93 (m, 2H), 1.82–1.76 (m, 2H), 1.41 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (126 MHz; CDCl3): δ 162.87 (t, J = 13.8 Hz), 161.77 (dd, J = 249.8, 9.0 Hz), 161.51 (dd, J = 248.2, 9.0 Hz), 158.87, 155.06, 153.93, 146.11, 127.99, 125.75, 123.51, 121.27, 100.44 (t, J = 21.1 Hz), 99.31 (ddd, J = 30.4, 25.6, 3.4 Hz), 92.82, 69.08, 62.48, 50.87 (q, J = 32.1 Hz), 29.17, 25.66, 15.31 ppm. IR: ν 3344, 2945, 2882, 1616, 1575, 1558, 1184, 1156, cm–1. HRMS (ES+) calcd for C18H18ClF5N5O2 [M + H]+, 466.1069; found 466.1069.
(S)-5-(4-(5-Chloro-7-((1,1,1-trifluoropropan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)pentan-1-ol (20)
General procedure B was followed using pentane-1,5-diol (31 mg, 0.32 mmol), NaH (60% in oil, 13 mg, 0.32 mmol), and 2 (30 mg, 0.08 mmol). Purification via preparative reversed-phase chromatography provided compound 20 as a white solid (25 mg, 68% yield). 1H NMR (500 MHz; CDCl3): δ 8.38 (s, 1H), 6.63 (dd, J = 9.1, 2.3 Hz, 2H), 5.93 (d, J = 9.8 Hz, 1H), 4.77–4.76 (br s, 1H), 4.03 (t, J = 6.3 Hz, 2H), 3.71 (t, J = 6.3 Hz, 2H), 1.88 (dt, J = 14.2, 6.9 Hz, 2H), 1.67 (dt, J = 14.2, 6.9 Hz, 2H), 1.62–1.56 (m, 2H), 1.40 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (126 MHz; CDCl3): δ 162.90 (t, J = 14.0 Hz), 161.76 (dd, J = 249.9, 9.1 Hz), 161.50 (dd, J = 248.1, 9.0 Hz), 158.63, 155.33, 154.12, 146.08, 124.65 (q, J = 282.0 Hz), 100.45 (t, J = 21.0 Hz), 99.25 (ddd, J = 29.3, 25.7, 3.4 Hz), 92.60, 69.15, 62.74, 50.82 (q, J = 32.1 Hz), 32.42, 28.84, 22.48, 15.30 ppm. IR (film): ν 3336, 2949, 2873, 1617, 1575, 1553, 1184, 1146, 1036 cm–1. HRMS (ES+) calcd for C19H20ClF5N5O2 [M + H]+, 480.1226; found 480.1232.
(S)-3-(4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (21)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (63 μL, 0.54 mmol), NaH (60% in oil, 22 mg, 0.54 mmol), and 7 (50 mg, 0.14 mmol). Purification via preparative reversed-phase chromatography provided compound 21 as a white solid (26 mg, 39% yield). 1H NMR (600 MHz, CDCl3) δ 9.21 (s, 1H), 8.41 (s, 1H), 8.30 (s, 1H), 6.59 (d, J = 9.1 Hz, 2H), 6.31 (s, 1H), 4.11 (t, J = 5.2 Hz, 2H), 3.29 (s, 1H), 3.20–2.96 (m, 2H), 2.70 (s, 6H), 2.26 (dq, J = 11.9, 5.9 Hz, 1H), 1.64 (dq, J = 13.0, 6.6 Hz, 1H), 1.05 (d, J = 6.6 Hz, 3H), 0.79 (t, J = 6.2 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.60, 161.76 (dd, J = 247.9, 8.7 Hz), 161.71 (dd, J = 247.7, 8.8 Hz), 161.47 (t, J = 13.9 Hz), 158.60, 154.80, 153.61, 146.27, 103.18 (t, J = 20.8 Hz), 98.80 (dd, J = 25.9, 2.4 Hz), 89.75, 66.17, 54.99, 54.72, 43.13, 33.65, 24.84, 18.20, 18.10, 18.00 ppm. HRMS (ES+) calcd for C21H28ClF2N6O [M + H]+, 453.1976; found 453.1979.
(R)-3-(4-(5-Chloro-7-((1,1,1-trifluoro-3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (22)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (66 μL, 0.57 mmol), NaH (60% in oil, 23 mg, 0.57 mmol), and 8 (60 mg, 0.14 mmol). Purification via preparative reversed-phase chromatography provided compound 21 as a white solid (43 mg, 55% yield). 1H NMR (500 MHz, CDCl3) δ 8.38 (s, 1H), 6.65 (t, J = 10.0 Hz, 2H), 6.42 (s, 1H), 4.09 (t, J = 6.1 Hz, 2H), 2.48 (t, J = 6.9 Hz, 2H), 2.28 (s, 6H), 2.20–2.10 (m, 1H), 2.01 (p, J = 6.5 Hz, 2H), 1.01 (d, J = 6.8 Hz, 3H), 0.96 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.49, 162.83 (t, J = 13.8 Hz), 161.62 (dd, J = 250.0, 8.9 Hz), 161.44 (dd, J = 248.0, 8.9 Hz), 158.87, 155.31, 153.88, 146.62, 124.59 (q, J = 283.6 Hz), 100.10, 99.34 (dd, J = 25.6, 3.5 Hz), 99.22 (t, J = 25.2 Hz)*, 67.38, 58.62 (q, J = 29.4 Hz), 56.05, 45.57, 28.25, 27.20, 19.71, 16.67 ppm. * two C–H of aryl ring in the same signal. IR (KBr) ν 3423, 2970, 1617, 1574, 1574, 1494, 1445, 1357, 1263, 1157, 1090, 1037, 936, 842, 768, 731, 652 cm–1 HRMS (ES+) calcd for C21H25ClF5N6O [M + H]+, 507.1693; found 507.1706.
(S)-3-(4-(5-Chloro-7-((1,1,1-trifluoro-3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (23)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (63 μL, 0.54 mmol), NaH (60% in oil, 22 mg, 0.54 mmol), and 9 (57 mg, 0.14 mmol). Purification via preparative reversed-phase chromatography provided compound 21 as a white solid (49 mg, 66% yield). 1H NMR (500 MHz, CDCl3) δ 8.38 (s, 1H), 6.66 (t, J = 9.8 Hz, 2H), 4.09 (t, J = 6.4 Hz, 2H), 2.48 (t, J = 7.0 Hz, 2H), 2.28 (s, 6H), 2.15 (dq, J = 13.2, 6.6 Hz, 1H), 2.01 (p, J = 6.7 Hz, 2H), 1.01 (d, J = 6.7 Hz, 3H), 0.96 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (126 MHz, CDCl3) δ 162.91 (t, J = 13.8 Hz), 161.63 (dd, J = 250.0, 9.0 Hz), 161.45 (dd, J = 247.9, 8.9 Hz), 155.32, 146.64, 124.59 (q, J = 283.6 Hz), 99.53–98.89 (m), 67.38, 58.62 (q, J = 29.0 Hz), 56.04, 45.57, 28.25, 27.19, 19.71, 16.66 ppm. HRMS (ES+) calcd for C21H25ClF5N6O [M + H]+, 507.1693; found 507.1706.
(R)-3-(4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (24)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (61 μL, 0.52 mmol), NaH (60% in oil, 21 mg, 0.52 mmol), and 10 (50 mg, 0.13 mmol). Purification via preparative reversed-phase chromatography provided compound 24 as a formic salt adduct as a white powder (62 mg, 0.12 mmol, 93%). 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), 8.28 (s, 1H), 8.02 (s, 1H), 6.58 (d, J = 6.0 Hz, 2H), 6.37 (bs, 1H), 4.09 (t, J = 6.0 Hz, 2 H), 3.03 (t, J = 6.0 Hz, 2 H), 2.66 (s, 6H), 2.22 (quint, J = 6.0 Hz, 2H), 0.98 (t, J = 6.0 Hz, 3H) 0.80 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.94, 161.27 (dd, J = 247.6, 9.1 Hz), 161.56 (t, J = 13.6 Hz), 161.45 (dd, J = 247.6, 9.1 Hz), 158.56, 154.72, 153.54, 146.42, 103.09 (t, J = 21.1 Hz), 98.83 (dd, J = 26.4, 1.5 Hz), 98.74 (dd, J = 25.7, 1.5 Hz), 89.57, 66.26, 57.85, 54.94, 43.20, 34.68, 25.79, 24.93, 16.61 ppm. 19F NMR (376 MHz, DMSO-D6) δ −112.70, −114.09 (d, J = 11.3 Hz) ppm. IR (film) ν 1607, 1571, 1496, 1443, 1349, 1259, 1152, 1029, 765 cm–1. HRMS (ES+) calcd for C22H30ClF2N6O [M + H]+, 467.2132; found 467.2133.
(R)-3-(4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)propan-1-ol (25)
General procedure B was followed using propan-1,3-diol (29 μL, 0.40 mmol), NaH (60% in oil, 16 mg, 0.40 mmol) and 10 (39 mg, 0.10 mmol). Purification via preparative reversed-phase chromatography provided compound 25 as a white powder (9 mg, 0.02 mmol, 20%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 6.63 (d, J = 9.7 Hz, 2H), 6.37 (bs, 1H), 4.19 (t, J = 6.2 Hz, 2H), 3.90 (t, J = 5.9 Hz, 2H), 3.28 (bs, 1H), 2.22–1.94 (m, 2H), 1.02 (d, J = 6.7 Hz, 3H), 0.84 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) δ 162.15 (t, J = 13.6 Hz),161.80 (dd, J = 248.6, 8.6 Hz), 161.57 (dd, J = 247.6, 8.6 Hz), 158.78, 154.78, 146.59, 100.86–96.50 (m), 89.88, 66.28, 59.43, 57.91, 34.75, 31.88, 25.87, 16.70 ppm. HRMS (ES+) calcd for C20H24ClF2N5O2 [M + H]+, 439.1587; found 439.1588.
5-Chloro-6-(4-(3-(dimethylamino)propoxy)-2,6-difluorophenyl)-N-neopentyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (26)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (51 μL, 0.43 mmol), NaH (60% in oil, 17 mg, 0.43 mmol), and 11 (40 mg, 0.11 mmol). Purification by reversed-phase HPLC provided compound 26 as a white powder (24 mg, 0.053 mmol, 49%). 1H NMR (600 MHz, CDCl3) δ 8.33 (s, 1H), 6.61 (d, J = 6.0 Hz, 2H), 6.36 (bs, 1H), 4.07 (t, J = 6.0 Hz, 2 H), 2.89 (d, J = 6.0 Hz, 2 H), 2.48 (t, J = 6.0 Hz, 2H), 2.28 (s, 6H), 2.01 (quint, J = 6.0 Hz, 2H), 0.89 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) δ 162.19 (t, J = 15.1 Hz), 161.86 (dd, J = 247.6, 9.1 Hz), 158.68, 154.92, 153.67, 146.94, 102.51 (t, J = 22.7 Hz), 98.82–98.63 (m), 90.13, 67.27, 56.08, 54.47, 45.58, 31.87, 27.22, 26.91 ppm. IR (film) ν 1610, 1571, 1561, 1462, 1441, 1429, 1352, 1247, 1152, 1138, 1027, 765 cm–1. HRMS (ES+) calcd for C21H28ClF2N6O [M + H]+, 453.1976; found 453.1974.
(S)-5-Chloro-6-(4-(3-(dimethylamino)propoxy)-2,6-difluorophenyl)-N-(3,3-dimethylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-aminium Formate (27)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (61 μL, 0.52 mmol), NaH (60%, 21 mg, 0.52 mmol), and 12 (50 mg, 0.13 mmol). Purification by reversed-phase HPLC provided compound 27 as a formic salt adduct as a white powder (66 mg, 0.12 mmol, 98%). 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), 8.27 (s, 1H), 7.95 (s, 1H), 6.57 (d, J = 6.0 Hz, 2H), 6.36 (bs, 1H), 4.0 (t, J = 6.0 Hz, 2 H), 3.04 (t, J = 6.0 Hz, 2 H), 2.66 (s, 6H), 2.22 (quint, J = 6.0 Hz, 2H), 0.97 (t, J = 6.0 Hz, 3H) 0.79 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.93, 161.27 (dd, J = 247.6, 9.1 Hz), 161.56 (t, J = 13.6 Hz), 161.45 (dd, J = 247.6, 9.1 Hz), 158.53, 154.68, 153.51, 146.41, 103.09 (t, J = 21.1 Hz), 98.83 (dd, J = 26.4, 1.5 Hz), 98.74 (dd, J = 25.7, 1.5 Hz), 89.56, 67.99, 66.23, 57.83, 54.89, 43.15, 34.65, 25.77, 24.88, 16.58 ppm. IR (film) ν 1607, 1571, 1496, 1443, 1349, 1259, 1152, 1029, 954, 765 cm–1. HRMS (ES+) calcd for C22H30ClF2N6O [M + H]+, 467.2132; found 467.2132.
5-Chloro-6-(4-(3-(dimethylamino)propoxy)-2,6-difluorophenyl)-N-((1-methylcyclopropyl)methyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (28)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (44 μL, 0.37 mmol), NaH (60%, 15 mg, 0.37 mmol), and 13 (34 mg, 0.092 mmol). Purification by reversed-phase HPLC provided compound 28 as a white powder (25 mg, 0.055 mmol, 60%). 1H NMR (600 MHz, CDCl3) δ 8.33 (s, 1H), 6.59 (d, J = 12.0 Hz, 2H), 6.37 (bs, 1H), 4.07 (t, J = 6.0 Hz, 2 H), 2.50 (t, J = 6.0 Hz, 2 H), 2.29 (s, 6H), 2.01 (quint, J = 6.0 Hz, 2H), 1.08 (s, 3H), 0.42–0.38 (m, 4H) ppm. 13C NMR (151 MHz, CDCl3) δ 162.13 (t, J = 13.6 Hz), 161.93 (dd, J = 246.1, 9.1 Hz), 158.68, 154.95, 153.62, 146.66, 102.47(t, J = 21.1 Hz), 98.77–98.58 (m), 90.05, 67.19, 56.08, 52.57, 45.54, 27.18, 21.01, 16.26, 11.91 ppm. IR (film) ν 1608, 1571, 1561, 1495, 1455, 1441, 1426, 1364, 1342, 1260, 1246, 1205, 1153, 1139, 1119, 1057, 1030, 1017, 927, 912, 820, 765, 559, 540, 515 cm–1. HRMS (ES+) calcd for C21H25ClF2N6O [M + H]+, 451.1819; found 451.1816.
5-Chloro-6-(4-(3-(dimethylamino)propoxy)-2,6-difluorophenyl)-N-((1-(trifluoromethyl)cyclopropyl)methyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (29)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (40 μL, 0.34 mmol), NaH (60%, 14 mg, 0.34 mmol), and 14 (36 mg, 0.085 mmol). Purification by reversed-phase HPLC provided compound 29 as a white powder (25 mg, 0.050 mmol, 58%). 1H NMR (600 MHz, CD3CN) δ 8.33 (s, 1H), 6.76 (d, J = 6.0 Hz, 2H), 6.59 (bs, 1H), 4.11 (t, J = 6.0 Hz, 2 H), 3.85 (bs, 2H), 2.48 (t, J = 6.0 Hz, 2 H), 2.24 (s, 6H), 1.97–1.94 (m, 2H), 0.97 (t, J = 6 Hz, 2H), 0.82 (bs, 2H) ppm. 13C NMR (151 MHz, CD3CN): selected representative peaks δ 163.56, 161.93, 155.67, 148.32, 100.06–99.87 (m), 68.04, 56.10, 45.21, 44.52, 27.31, 7.88 ppm. HRMS (ES+) calculated for C21H23ClF5N6O [M + H]+, 505.1537; found 505.1533.
5-Chloro-N-(cyclobutylmethyl)-6-(4-(3-(dimethylamino)propoxy)-2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (30)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (55 μL, 0.47 mmol), NaH (60%, 19 mg, 0.47 mmol), and 15 (43 mg, 0.12 mmol). Purification by reversed-phase HPLC provided compound 30 as a white powder (30 mg, 0.067 mmol, 57%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 6.60 (d, J = 6.0 Hz, 2H), 6.22 (bs, 1H), 4.07 (t, J = 6.0 Hz, 2 H), 3.06 (t, J = 6.0 Hz, 2 H), 2.49–2.45 (m, 3H), 2.27 (s, 6H), 2.06–1.99 (m, 4H), 1.95–1.79 (m, 2H), 1.59 (quint, J = 6.0 Hz, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ 162.14 (t, J = 13.6 Hz), 161.98 (dd, J = 246.89, 9.1 Hz), 158.68, 154.91, 153.62, 146.58, 102.40 (t, J = 22.7 Hz), 98.75–98.55 (m), 90.17, 67.22, 56.08, 48.66, 45.56, 34.98, 27.20, 25.47, 18.24 ppm. IR (film) ν 1611, 1572, 1562, 1461, 1441, 1353, 1336, 1248, 1206, 1151, 1137, 1027, 838 cm–1. HRMS (ES+) calcd for C21H25ClF2N6O [M + H]+, 451.1819; found 451.1816.
(S)-3-(4-(5-Chloro-7-((1-cyclopropylethyl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (31)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (64 μL, 0.54 mmol), NaH (60%, 21 mg, 0.54 mmol), and 16 (50 mg, 0.14 mmol). Purification by reversed-phase HPLC provided compound 31 as a white powder (22 mg, 0.04 mmol, 33%). 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 8.30 (s, 1H), 6.56 (t, J = 10.9 Hz, 2H), 6.37 (d, J = 8.6 Hz, 1H), 4.10 (t, J = 4.9 Hz, 2H), 3.32–3.01 (m, 2H), 3.02 (s, 1H), 2.72 (s, 6H), 2.25 (s, 2H), 1.11 (d, J = 6.5 Hz, 3H), 0.88 (dt, J = 8.5, 4.7 Hz, 1H), 0.49 (ddt, J = 18.6, 9.0, 5.0 Hz, 2H), 0.18 (dq, J = 9.9, 4.8 Hz, 1H), 0.05 (dt, J = 9.3, 4.8 Hz, 1H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.60, 161.76 (dd, J = 247.5, 8.7 Hz), 161.58 (dd, J = 248.0, 9.1 Hz), 161.47 (t, J = 13.9 Hz), 158.41, 154.77, 153.77, 145.98, 102.89 (t, J = 20.9 Hz), 98.78 (ddd, J = 50.7, 25.8, 2.9 Hz), 89.84, 66.10, 54.93, 43.07, 24.75, 21.10, 17.75, 3.52, 3.46 ppm. HRMS (ES+) calcd for C21H26ClF2N6O [M + H]+, 451.1819; found 451.1817.
(R)-3-(4-(5-Chloro-7-((1-cyclopropylethyl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenoxy)-N,N-dimethylpropan-1-aminium Formate (32)
General procedure B was followed using 3-(dimethylamino)propan-1-ol (64 μL, 0.54 mmol), NaH (60%, 21 mg, 0.54 mmol), and 17 (50 mg, 0.14 mmol). Purification by reversed-phase HPLC provided compound 31 as a formic acid salt as a white powder (23 mg, 0.04 mmol, 34%). 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 8.30 (s, 1H), 6.56 (t, J = 10.9 Hz, 2H), 6.37 (d, J = 8.6 Hz, 1H), 4.10 (t, J = 4.9 Hz, 2H), 3.32–3.01 (m, 2H), 3.02 (s, 1H), 2.72 (s, 6H), 2.25 (s, 2H), 1.11 (d, J = 6.5 Hz, 3H), 0.88 (dt, J = 8.5, 4.7 Hz, 1H), 0.49 (ddt, J = 18.6, 9.0, 5.0 Hz, 2H), 0.18 (dq, J = 9.9, 4.8 Hz, 1H), 0.05 (dt, J = 9.3, 4.8 Hz, 1H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.60, 161.76 (dd, J = 247.5, 8.7 Hz), 161.58 (dd, J = 248.0, 9.1 Hz), 161.47 (t, J = 13.9 Hz), 158.41, 154.77, 153.77, 145.98, 102.89 (t, J = 20.9 Hz), 98.78 (ddd, J = 50.7, 25.8, 2.9 Hz), 89.84, 66.10, 54.93, 43.07, 24.75, 21.10, 17.75, 3.52, 3.46 ppm. HRMS (ES+) calcd for C21H26ClF2N6O [M + H]+, 451.1819; found 451.1814.
(R)-5-Chloro-6-(4-ethynyl-2,6-difluorophenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (34)
To a solution of 33 (0.047 g, 0.125 mmol, 1 equiv) in dry toluene (12 mL) at 0 °C was added DiBAl-H (0.170 mL, 1.1 M, 0.027 g, 0.182 mmol, 1.5 equiv) dropwise, and the mixture was stirred for 1 h at this temperature before it was quenched with a 1 M solution of HCl. The aqueous layer was extracted twice with EtOAc, and then the combined organic fractions were washed with brine, dried, and concentrated under reduced pressure to furnish the aldehyde intermediate, which was directly dissolved into MeOH (4 mL). K2CO3 (0.025 g, 0.182 mmol, 1.5 equiv) and dimethyl (1-diazo-2-oxopropyl)phosphonate (0.035 g, 0.182 mmol, 1.5 equiv) were successively added, and the mixture was stirred at rt for 3 h. The reaction was then filtered over sintered glass, and the filtrate was evaporated under reduced pressure. Purification via silica gel flash chromatography (Hexanes/EtOAc: 90/10 to 75/25) provided compound 34 as an off-white solid (0.005 g, 0.012 mmol, 10% over two steps). 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 1H), 7.20 (d, J = 8.3 Hz, 2H), 6.36 (d, J = 9.8 Hz, 1H), 3.31 (s, 1H), 3.16 (bs, 1H), 1.75–1.49 (m, 1H), 1.06 (d, J = 6.7 Hz, 3H), 0.81 (d, J = 6.7 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (151 MHz, CDCl3) δ 160.89 (dd, J = 249.9, 7.7 Hz), 160.82 (dd, J = 250.0, 6.9 Hz), 157.85, 154.94, 145.99, 126.40 (t, J = 12.0 Hz), 115.66 (ddd, J = 24.0, 7.7, 3.6 Hz), 112.16 (t, J = 20.4 Hz), 89.18, 81.37, 80.79, 54.95, 33.69, 29.83, 18.11, 18.08, 17.92 ppm. HRMS (ES+) calcd for C18H17N5F2Cl [M + H]+, 376.1135; found 376.1139.
Diethyl 2-(4-Amino-2,6-difluorophenyl)malonate (36)
To a cooled solution of NaH (1.887 g, 47.44 mmol, 2.1 equiv) in tetrahydrofuran (THF, 60 mL) was added diethylmalonate (6.891 mL, 7.236 g, 45.18 mmol, 2 equiv) and stirred for 1 h at rt before adding 35 (2.63 mL, 4.000 g, 22.59 mmol, 1 equiv) dropwise and stirred for 2 h at rt. The reaction was quenched with a NH4ClSat. solution and extracted with EtOAc twice. The combined organic fractions were washed with water and brine, dried over Na2SO4, and concentrated under vacuum. Filtration over a silica pad (Hexanes/EtOAc: 95/5) provided the nitro malonate intermediate + diethylmalonate (ratio 1/1), which will be used in the next step without further purification. To a solution of the intermediate nitro malonate in MeOH (150 mL) backfilled with N2 was added Pd/C (2.404 g, 10% weight, 2.259 mmol, 0.1 equiv). The mixture was put under atmospheric hydrogen pressure for 4 h. The flask was backfilled three times with N2 before the solution was filtered over Celite and rinsed with MeOH. The filtrate was concentrated and purified over silica gel flash chromatography (Hexanes/EtOAc: 98/02 to 85/15) to provide compound 36 (2.590 g, 9.02 mmol, 39% over 2 steps) as a pale yellow oil. 1H NMR (599 MHz, CDCl3) δ 6.18 (d, J = 9.9 Hz, 2H), 4.80 (s, 1H), 4.23 (q, J = 7.2 Hz, 4H), 3.82 (bs, 2H), 1.26 (t, J = 7.2 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) δ 167.72, 162.81 (d, J = 10.5 Hz), 161.17 (d, J = 10.4 Hz), 148.66 (t, J = 14.1 Hz), 99.62 (t, J = 19.5 Hz), 98.34–97.10 (m), 62.08, 46.92, 14.05 ppm. LCMS: [M + H]+, 288.
Diethyl 2-(2,6-Difluoro-4-iodophenyl)malonate (37)
To a solution of 36 (2.050 g, 7.136 mmol, 1 equiv) in 6 N HCl (12 mL, 71.36 mmol, 10 equiv) cooled to 0 °C, a solution of NaNO2 (0.492 g, 7.136 mmol, 1 equiv) in water (2.7 mL) was added dropwise. The resulting solution was added dropwise to a solution of KI (4.916 g, 29.62 mmol, 4.15 equiv) in water (5 mL) keeping the temperature at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 3 h, then it was stopped and extracted twice with EtOAc. The combined layers were washed in sequence with 10% Na2S2O3 and brine, then dried over Na2SO4 and concentrated under vacuum. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 75/25) provided compound 37 (2.054 g, 5.159 mmol, 72%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.31 (d, J = 7.0 Hz, 1H), 4.89 (s, 1H), 4.25 (q, J = 7.1 Hz, 4H), 1.27 (t, J = 7.2 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) δ 166.36, 160.77 (dd, J = 255.2, 7.8 Hz), 121.41 (dd, J = 23.6, 4.8 Hz), 101.61 (dt, J = 20.3, 14.7 Hz), 62.45, 47.22, 14.05 ppm. LCMS: [M + H]+, 339.
5,7-Dichloro-6-(2,6-difluoro-4-iodophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (38)
A pressure flask flushed with nitrogen was charged with 37 (1.920 g, 6.459 mmol, 1 equiv), 1H-1,2,4-triazol-5-amine (0.570 g, 6.782 mmol, 1.05 equiv), and tributylamine (1.620 mL, 6.782 mmol, 1.05 equiv). The mixture was heated to 170 °C for 2 h. Toluene (10 mL) was then added at 110 °C followed by a 50% NaOH solution (1.023 mL, 19.38 mmol, 3 equiv) at 50 °C. Once at rt, the mixture was filtered over sintered glass and rinsed twice with toluene to furnish the bis-phenolate intermediate (2.068 g, 6.621 mmol, 97%) that is engaged in the next step without further purification. In a pressure flask charged with the intermediate bis-phenolate (2.000 g, 4.608 mmol, 1 equiv) was added phosphoryl chloride (7.670 mL, 82.020 mmol, 17.8 equiv) and the mixture was heated to 130 °C for 6 h. The reaction was poured over ice, the aqueous phase was extracted twice with CH2Cl2, and the combined organic fractions were washed with brine, dried, and concentrated under reduced pressure. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 70/30) provided compound 38 as an off-white solid (1.520 g, 3.56 mmol, 77%). 1H NMR (600 MHz, CDCl3) δ 8.61 (s, 1H), 7.52 (d, J = 6.4 Hz, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ 160.45 (d, J = 6.2 Hz), 158.75 (d, J = 6.2 Hz), 157.28, 156.13, 153.85, 141.28, 122.05 (dd, J = 23.0, 4.2 Hz), 112.35, 109.18 (t, J = 19.8 Hz), 95.68 (t, J = 10.2 Hz) ppm. HRMS (ES+) calcd for C11H4N4F2ICl2 [M + H]+, 426.8820; found 426.8812.
(S)-5-Chloro-6-(2,6-difluoro-4-iodophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (39)
General procedure A was followed: To a solution of 38 (0.200 g, 0.468 mmol, 1 equiv) in DMF (2 mL) was added (S)-1,1,1-trifluoropropan-2-amine (0.097 mL, 0.111 g, 0.984 mmol, 2.1 equiv), and the mixture was stirred for 24 h at 40 °C. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 70/30) provided compound 39 as an off-white solid (0.121 g, 0.240 mmol, 51%). 1H NMR (600 MHz, CDCl3) δ 8.38 (s, 1H), 7.49 (d, J = 6.6 Hz, 2H), 5.97 (d, J = 9.6 Hz, 1H), 4.84 (bs, 1H), 1.43 (d, J = 6.8 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 160.61 (dd, J = 257.4, 5.9 Hz), 160.29 (dd, J = 255.8, 6.0 Hz), 157.41, 155.41, 154.26, 145.87, 124.59 (q, J = 282.1 Hz), 122.32 (dd, J = 63.0, 3.8 Hz), 122.16 (dd, J = 62.9, 3.7 Hz), 109.10 (t, J = 20.2 Hz), 95.55 (t, J = 10.1 Hz), 91.74, 51.08 (q, J = 32.1 Hz), 15.16 ppm. HRMS (ES+) calcd for C14H9N5F5ICl [M + H]+, 503.9506; found 503.9502.
(R)-5-Chloro-6-(2,6-difluoro-4-iodophenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (40)
General procedure A was followed: To a solution of 38 (0.156 g, 0.365 mmol, 1 equiv) in DMF (1 mL) were added (R)-3-methylbutan-2-amine (0.064 mL, 0.048 g, 0.548 mmol, 1.5 equiv) and triethylamine (0.153 mL, 0.111 g, 1.100 mmol, 3 equiv), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 70/30) provided compound 40 as an off-white solid (0.161 g, 0.337 mmol, 92%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.46 (d, J = 6.5 Hz, 2H), 6.35 (d, J = 7.8 Hz, 1H), 3.18 (s, 1H), 1.64 (dq, J = 13.2, 6.7 Hz, 1H), 1.06 (d, J = 6.6 Hz, 3H), 0.79 (dd, J = 11.6, 6.8 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) δ 159.60 (dd, J = 254.9, 8.4 Hz), 159.56 (dd, J = 254.8, 8.4 Hz), 156.51, 153.82, 152.51, 144.76, 120.65 (dt, J = 24.4, 3.7 Hz), 110.04 (t, J = 21.1 Hz), 93.34 (t, J = 10.2 Hz), 87.81, 53.81, 32.50, 16.95, 16.94, 16.77 ppm. HRMS (ES+) calculated for C16H16N5F2ICl [M + H]+, 478.0101; found 478.0098.
(R)-5-Chloro-6-(2,6-difluoro-4-iodophenyl)-N-(3,3-dimethylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (41)
General procedure A was followed: To a solution of 38 (0.156 g, 0.365 mmol, 1 equiv) in DMF (1 mL) were added (R)-3,3-dimethylbutan-2-amine hydrochloride (0.075 g, 0.548 mmol, 1.5 equiv) and triethylamine (0.153 mL, 0.111 g, 1.100 mmol, 3 equiv), and the mixture was stirred for 1 h at rt. Purification via silica gel flash chromatography (Hexanes/EtOAc: 100/0 to 70/30) provided compound 41 as an off-white solid (0.158 g, 0.321 mmol, 88%). 1H NMR (600 MHz, CDCl3) δ 8.31 (s, 1H), 7.46 (d, J = 6.4 Hz, 2H), 6.42 (s, 1H), 3.10 (s, 1H), 1.00 (d, J = 6.7 Hz, 4H), 0.82 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) δ 160.67 (dd, J = 254.9, 6.2 Hz), 160.40 (dd, J = 254.8, 6.0 Hz), 157.59, 154.89, 153.58, 146.03, 121.82 (dd, J = 21.7, 3.9 Hz), 121.67 (dd, J = 21.7, 3.9 Hz), 111.23 (t, J = 18.2 Hz), 94.41 (t, J = 10.1 Hz), 88.71, 58.10, 34.74, 25.77, 16.56, 16.55 ppm. HRMS (ES+) calcd for C17H18N5F2ICl [M + H]+, 492.0258; found 492.0256.
(S)-4-(4-(5-Chloro-7-((1,1,1-trifluoropropan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)but-3-yn-1-ol (42)
General procedure C was followed using 39 (0.032 g, 0.064 mmol) and but-3-yn-1-ol (0.014 mL, 0.013 g, 0.190 mmol). Purification by reversed-phase HPLC provided compound 42 as an off-white solid (0.011 g, 0.025 mmol, 39%). 1H NMR (600 MHz, CD3OD) δ 8.50 (s, 1H), 7.24 (d, J = 9.0 Hz, 2H), 5.77 (bs, 1H), 3.76 (t, J = 5.9 Hz, 2H), 3.32 (bs, 1H), 2.68 (t, J = 5.9 Hz, 2H), 1.46 (d, J = 6.5 Hz, 3H) ppm. 13C NMR (151 MHz, CDCl3) (selected representative peaks) δ 157.85, 155.52, 145.75, 143.53, 115.82, 115.65, 115.31, 115.16, 94.99, 61.61, 31.16, 16.12, 15.26 ppm. HRMS (ES+) calcd for C18H14N5F5ClO [M + H]+, 446.0802; found 446.0804.
(S)-5-(4-(5-Chloro-7-((1,1,1-trifluoropropan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)pent-4-yn-1-ol (43)
General procedure C was followed using 39 (0.033 g, 0.066 mmol) and pent-4-yn-1-ol (0.020 mL, 0.017 g, 0.200 mmol). Purification by reversed-phase HPLC provided compound 43 as an off-white solid (0.010 g, 0.022 mmol, 33%). 1H NMR (600 MHz, CD3OD) δ 8.50 (s, 1H), 7.21 (d, J = 8.8 Hz, 2H), 5.80 (bs, 1H), 3.71 (t, J = 6.0 Hz, 2H), 3.32 (bs, 1H), 2.57 (t, J = 7.0 Hz, 2H), 1.94–1.71 (m, 2H), 1.46 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (151 MHz, CD3OD) δ 163.05 (dd, J = 115.0, 7.0 Hz), 161.40 (dd, J = 114.6, 7.0 Hz), 158.58, 155.77, 149.07, 130.24 (t, J = 12.4 Hz), 126.50 (q, J = 281.6 Hz), 116.38 (dd, J = 23.5, 3.3 Hz), 116.08 (dd, J = 23.6, 3.3 Hz), 109.98 (t, J = 20.8 Hz), 95.25, 94.42, 79.42, 61.46, 52.43 (q, J = 31.7 Hz) ppm. HRMS (ES+) calcd for C19H16N5F5ClO [M + H]+, 460.0958; found 460.0961.
(S)-5-Chloro-6-(4-(4-(dimethylamino)but-1-yn-1-yl)-2,6-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (44)
General procedure A was followed using 39 (0.033 g, 0.066 mmol) and N,N-dimethylprop-2-yn-1-amine (0.023 mL, 0.019 g, 0.197 mmol). Purification by reversed-phase HPLC provided compound 44 as an off-white solid (0.012 g, 0.025 mmol, 39%). 1H NMR (600 MHz, CDCl3) δ 8.40 (s, 1H), 7.14 (dd, J = 8.4, 5.6 Hz, 2H), 5.94 (s, 1H), 4.67 (s, 1H), 2.84 (d, J = 7.1 Hz, 2H), 2.76 (d, J = 7.2 Hz, 2H), 2.48 (s, 6H), 1.40 (d, J = 6.8 Hz, 3H) ppm. HRMS (ES+) calcd for C20H19N5F6Cl [M + H]+, 473.1274; found 473.1277.
(R)-4-(4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)but-3-yn-1-ol (45)
General procedure C was followed using 40 (0.040 g, 0.084 mmol) and but-3-yn-1-ol (0.019 mL, 0.018 g, 0.250 mmol). Purification by reversed-phase HPLC provided compound 45 as an off-white solid (0.012 g, 0.029 mmol, 34%). 1H NMR (600 MHz, CD3OD) δ 8.44 (s, 1H), 7.26 (d, J = 8.4 Hz, 2H), 3.77 (t, J = 6.5 Hz, 2H), 3.31 (bs, 1H), 2.68 (t, J = 6.5 Hz, 2H), 1.80–1.66 (m, 1H), 1.11 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.7 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) (selected representative peaks) δ 161.58, 160.00, 157.65, 127.89, 115.19 (dd, J = 23.2, 8.8 Hz), 91.50, 89.36, 79.89, 60.95, 55.04, 33.69, 23.91, 18.11, 18.10, 17.97 ppm. 19F NMR (376 MHz, MeCN-D3) δ −110.61, −111.02 to −111.11 (m) ppm. HRMS (ES+) calcd for C20H21N5F2ClO [M + H]+, 420.1397; found 420.1398.
(R)-5-(4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)pent-4-yn-1-ol (46)
General procedure C was followed using 40 (0.027 g, 0.057 mmol) and pent-4-yn-1-ol (0.016 mL, 0.014 g, 0.170 mmol). Purification by reversed-phase HPLC provided compound 46 as an off-white solid (0.012 g, 0.028 mmol, 49%). 1H NMR (600 MHz, CD3OD) δ 8.43 (s, 1H), 7.22 (d, J = 8.4 Hz, 2H), 3.71 (t, J = 6.2 Hz, 2H), 3.31 (bs, 1H), 2.58 (t, J = 7.1 Hz, 2H), 1.84 (p, J = 6.7 Hz, 2H), 1.73 (dq, J = 13.6, 6.8 Hz, 2H), 1.11 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.7 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) (selected representative peaks) δ 161.67, 158.02, 154.94, 146.02, 128.27, 115.06 (d, J = 20.3 Hz), 94.29, 89.46, 61.63, 54.86, 33.70, 31.20, 18.11, 17.95, 16.12 ppm. HRMS (ES+) calcd for C21H23N5F2ClO [M + H]+, 434.1554; found 434.1553.
(R)-5-Chloro-6-(4-(3-(dimethylamino)prop-1-yn-1-yl)-2,6-difluorophenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (47)
General procedure C was followed using 40 (0.070 g, 0.147 mmol) and N,N-dimethylprop-2-yn-1-amine (0.047 mL, 0.037 g, 0.440 mmol). Purification by reversed-phase HPLC provided compound 47 as an off-white solid (0.043 g, 0.099 mmol, 68%). 1H NMR (599 MHz, CD3OD) δ 8.53 (bs, 1H), 8.44 (s, 2H), 7.32 (d, J = 8.3 Hz, 2H), 3.61 (s, 2H), 2.44 (s, 6H), 1.73 (h, J = 6.8 Hz, 1H), 1.11 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.8 Hz, 6H) ppm. 13C NMR (151 MHz, CD3OD) δ 162.96 (dd, J = 18.0, 6.8 Hz), 161.31 (dd, J = 18.1, 6.8 Hz), 158.80, 155.44, 155.02, 147.99, 128.64 (t, J = 12.3 Hz), 116.28 (dt, J = 24.2, 3.4 Hz), 112.93–112.48 (m), 90.42, 89.00, 84.17, 57.10, 44.24, 34.73, 19.27, 18.69, 18.18 ppm. HRMS (ES+) calcd for C21H24N6F2Cl [M + H]+, 433.1714; found 433.1715.
tert-Butyl (R)-(4-(4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)but-3-yn-1-yl)carbamate (48)
General procedure C was followed using 40 (0.040 g, 0.084 mmol) and tert-butyl but-3-yn-1-ylcarbamate (0.043 g, 0.250 mmol). Purification via silica gel flash chromatography (Hexanes/EtOAc: 90/10 to 60/40) provided compound 48 as a yellow solid (0.043 g, 0.083 mmol, 99%). 1H NMR (599 MHz, CDCl3) δ 8.34 (s, 1H), 7.10 (d, J = 7.9 Hz, 2H), 6.34 (s, 1H), 4.85 (s, 1H), 3.40 (t, J = 5.9 Hz, 2H), 3.19 (s, 1H), 2.66 (t, J = 6.5 Hz, 2H), 1.66–1.62 (m, 1H), 1.47 (s, 9H), 1.06 (d, J = 6.6 Hz, 3H), 0.80 (t, J = 7.4 Hz, 6H) ppm. 13C NMR (151 MHz, CDCl3) (selected representative peaks) δ 154.98, 146.00, 54.87, 33.71, 28.54, 21.29, 18.12 ppm. HRMS (ES+) calcd for C25H29N6F2ClO2 [M + Na]+, 519.2081; found 519.2083.
(R)-6-(4-(4-Aminobut-1-yn-1-yl)-2,6-difluorophenyl)-5-chloro-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine Hydrochloride 49
General procedure D was followed using 48 (0.040 g, 0.077 mmol) to provide compound 49 (0.034 g, 0.075 mmol, 97%) as a yellow solid. 1H NMR (600 MHz, CD3OD) δ 8.87 (s, 1H), 7.37 (d, J = 8.3 Hz, 2H), 3.33 (bs, 1H), 3.23 (t, J = 6.7 Hz, 2H), 2.92 (t, J = 6.8 Hz, 2H), 1.77 (dq, J = 13.6, 6.8 Hz, 1H), 1.15 (d, J = 6.6 Hz, 3H), 0.81 (t, J = 7.3 Hz, 6H) ppm. 13C NMR (151 MHz, CD3OD) (selected representative peaks) δ 158.87, 155.47, 148.02, 128.96, 116.35 (d, J = 24.0 Hz), 90.13, 81.36, 57.10, 34.79, 19.30, 18.72, 18.17 ppm. HRMS (ES+) calcd for C20H22N6F2Cl [M + H]+, 419.1557; found 419.1560.
tert-Butyl (R)-(3-(4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)prop-2-yn-1-yl)(methyl)carbamate (50)
General procedure D was followed using 40 (0.050 g, 0.105 mmol) and tert-butyl methyl(prop-2-yn-1-yl)carbamate (0.053 g, 0.314 mmol). Purification via silica gel flash chromatography (Hexanes/EtOAc: 90/10 to 50/50) provided compound 50 as a yellow solid (0.020 g, 0.039 mmol, 37%). 1H NMR (600 MHz, CDCl3) δ 8.33 (s, 1H), 7.12 (d, J = 8.0 Hz, 2H), 6.35 (d, J = 8.1 Hz, 1H), 4.32 (bs, 2H), 3.16 (bs, 1H), 2.99 (s, 3H), 1.65–1.60 (m, 1H), 1.49 (s, 9H), 1.05 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.9 Hz, 3H), 0.78 (d, J = 6.9 Hz, 3H) ppm. HRMS (ES+) calcd for C25H30N6F2ClO2 [M + H]+, 519.2081; found 519.2078.
(R)-5-Chloro-6-(2,6-difluoro-4-(3-(methylamino)prop-1-yn-1-yl)phenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine Hydrochloride (51)
General procedure D was followed using 50 (0.015 g, 0.029 mmol) to provide compound 51 (0.011 g, 0.024 mmol, 84%) as a brown solid. 1H NMR (600 MHz, CD3OD) δ 8.59 (s, 1H), 7.42 (d, J = 8.2 Hz, 2H), 4.23 (s, 2H), 3.31 (bs, 1H), 2.85 (s, 3H), 1.74 (dq, J = 13.4, 6.7 Hz, 1H), 1.11 (d, J = 6.5 Hz, 3H), 0.79 (d, J = 4.4 Hz, 6H) ppm. HRMS (ES+) calcd for C20H22N6F2Cl [M + H]+, 419.1557; found 419.1554.
(R)-5-Chloro-6-(4-(4-(dimethylamino)but-1-yn-1-yl)-2,6-difluorophenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine 2,2,2-Trifluoroacetate (52)
General procedure C was followed using 40 (0.029 g, 0.0612 mmol) and N,N-dimethylprop-2-yn-1-amine (0.022 mL, 0.018 g, 0.182 mmol). Purification by reversed-phase HPLC provided compound furnish compound 52 as a TFA salt as an off-white solid (0.010 g, 0.022 mmol, 37%). 1H NMR (600 MHz, CDCl3) δ 8.31 (s, 1H), 7.08 (d, J = 7.7 Hz, 2H), 6.35 (d, J = 8.3 Hz, 1H), 4.95 (bs, 2H), 3.16 (bs, 1H), 2.89 (t, J = 7.3 Hz, 2H), 2.78 (t, J = 7.3 Hz, 2H), 1.75–1.48 (m, 1H), 1.04 (d, J = 6.6 Hz, 3H), 0.89–0.68 (m, 6H) ppm. 13C NMR (151 MHz, CD3OD) (selected representative peaks) δ 153.54, 129.18, 67.28, 51.18, 32.83, 26.48, 17.43, 16.74, 16.18 ppm. HRMS (ES+) calcd for C22H26ClF2N6 [M + H]+, 447.1870; found 447.1873.
(R)-4-(4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)but-3-yn-1-ol (53)
General procedure C was followed using 41 (0.040 g, 0.081 mmol) and but-3-yn-1-ol (0.018 mL, 0.017 g, 0.240 mmol). Purification by reversed-phase HPLC provided compound 53 as an off-white solid (0.014 g, 0.032 mmol, 40%). 1H NMR (600 MHz, CD3OD) δ 8.46 (s, 1H), 7.28 (dd, J = 8.1, 1.5 Hz, 2H), 3.77 (t, J = 6.5 Hz, 2H), 3.31 (bs, 1H), 2.68 (t, J = 6.5 Hz, 2H), 1.07 (d, J = 6.7 Hz, 3H), 0.84 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) δ 159.85 (dd, J = 249.8, 7.0 Hz), 159.60 (dd, J = 249.8, 6.9 Hz), 157.03, 153.87, 152.67, 145.22, 126.93, 114.20 (td, J = 23.5, 3.2 Hz), 110.06, 90.55, 88.27, 78.86, 59.92, 57.09, 33.81, 24.85, 22.91, 15.60 ppm. 19F NMR (376 MHz, MeCN-D3) δ −108.51 (dd, J = 6.9 Hz, 6.9 Hz), −109.14 (dd, J = 6.8 Hz, 6.8 Hz) ppm. HRMS (ES+) calcd for C21H23N5F2ClO [M + H]+, 434.1554; found 434.1556.
(R)-5-(4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)pent-4-yn-1-ol (54)
General procedure C was followed using 41 (0.040 g, 0.081 mmol) and pent-4-yn-1-ol (0.023 mL, 0.021 g, 0.240 mmol). Purification by reversed-phase HPLC provided compound 54 as an off-white solid (0.036 g, 0.031 mmol, 38%). 1H NMR (600 MHz, CD3OD) δ 8.46 (s, 1H), 7.25 (dd, J = 8.0, 1.5 Hz, 2H), 3.72 (t, J = 6.2 Hz, 2H), 3.31 (bs, 1H), 2.58 (t, J = 7.1 Hz, 2H), 1.85 (p, J = 6.7 Hz, 2H), 1.08 (d, J = 6.7 Hz, 3H), 0.84 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) (selected representative peaks) δ 160.86 (dd, J = 250.1, 7.1 Hz), 160.61 (dd, J = 249.0, 7.1 Hz), 158.04, 153.67, 146.23, 134.55–130.43 (m), 128.51 (dd, J = 46.6, 12.1 Hz), 115.07, 94.36, 89.31, 78.80, 61.58, 58.07, 34.80, 31.20, 25.85, 16.61, 16.12 ppm. 19F NMR (376 MHz, MeCN-D3) δ −112.20 (dd, J = 8.1 Hz, 8.1 Hz), −112.83 (dd, J = 8.7 Hz, 8.1 Hz) ppm. HRMS (ES+) calcd for C22H25N5F2ClO [M + H]+, 448.1710; found 448.1711.
(R)-5-Chloro-6-(4-(3-(dimethylamino)prop-1-yn-1-yl)-2,6-difluorophenyl)-N-(3,3-dimethylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (55)
General procedure C was followed using 41 (0.070 g, 0.142 mmol) and N,N-dimethylprop-2-yn-1-amine (0.046 mL, 0.036 g, 0.427 mmol). Purification by reversed-phase HPLC provided compound 55 as an off-white solid (0.043 g, 0.096 mmol, 68%). 1H NMR (600 MHz, CD3OD) δ 8.50 (s, 1H), 7.33 (d, J = 7.3 Hz, 2H), 3.58 (bs, 2H), 3.31 (bs, 1H), 2.42 (s, 6H), 1.08 (d, J = 6.6 Hz, 3H), 0.83 (s, 9H) ppm. HRMS (ES+) calcd for C22H26N6F2Cl [M + H]+, 447.1870; found 447.1872.
tert-Butyl (R)-(4-(4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)but-3-yn-1-yl)carbamate (56)
General procedure C was followed, using 41 (0.040 g, 0.081 mmol) and tert-butyl but-3-yn-1-ylcarbamate (0.041 g, 0.240 mmol). Purification via silica gel flash chromatography (Hexanes/EtOAc: 90/10 to 60/40) provided compound 56 as a yellow solid (0.042 g, 0.079 mmol, 97%). 1H NMR (600 MHz, CDCl3) δ 8.40 (s, 1H), 7.11 (d, J = 7.7 Hz, 2H), 6.43 (bs, 1H), 4.85 (bs, 1H), 3.41 (d, J = 5.1 Hz, 2H), 3.14 (bs, 1H), 2.66 (t, J = 6.4 Hz, 2H), 1.47 (s, 9H), 1.02 (d, J = 6.6 Hz, 3H), 0.84 (s, 9H) ppm. HRMS (ES+) calculated for C26H32N6F2ClO2 [M + H]+, 533.2238; found 533.2238.
(R)-6-(4-(4-Aminobut-1-yn-1-yl)-2,6-difluorophenyl)-5-chloro-N-(3,3-dimethylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine Hydrochloride (57)
General procedure D was followed using 41 (0.040 g, 0.075 mmol) to provide compound 57 (0.034 g, 0.072 mmol, 97%) as a yellow solid. 1H NMR (600 MHz, CD3OD) δ 1H NMR (599 MHz, CD3OD) δ 8.55 (s, 1H), 8.46 (s, 1H), 7.34 (d, J = 8.9 Hz, 2H), 3.21–3.17 (m, 2H), 2.89 (s, 2H), 1.06 (d, J = 6.8 Hz, 3H), 0.82 (s, 9H) ppm. 13C NMR (151 MHz, CD3OD) (selected representative peaks) δ 170.30, 162.82 (d, J = 51.9 Hz), 161.88–160.69 (m), 158.96, 155.57, 154.88, 148.10, 129.12, 116.40 (d, J = 23.9 Hz), 90.42, 81.26, 59.54, 39.47, 36.06, 26.07, 19.64, 16.17 ppm. 19F NMR (376 MHz, CD3OD) δ −110.82 (bs), −111.85 to −112.24 (m) ppm. HRMS (ES+) calcd for C21H24N6F2Cl [M + H]+, 433.1714; found 433.1716.
tert-Butyl (R)-(3-(4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorophenyl)prop-2-yn-1-yl)(methyl)carbamate (58)
General procedure C was followed using 41 (0.050 g, 0.102 mmol) and tert-butyl methyl(prop-2-yn-1-yl)carbamate (0.052 g, 0.305 mmol). Purification via silica gel flash chromatography (Hexanes/EtOAc: 90/10 to 50/50) provided compound 58 as a yellow solid (0.043 g, 0.081 mmol, 79%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.12 (d, J = 7.5 Hz, 2H), 6.43 (bs, 1H), 4.31 (bs, 2H), 2.98 (s, 3H), 1.49 (s, 9H), 1.47–1.45 (m, 1H), 1.01 (d, J = 6.7 Hz, 3H), 0.83 (s, 9H) ppm. 13C NMR (151 MHz, CDCl3) (selected representative peaks) δ 160.83 (dd, J = 250.5, 6.9 Hz), 160.58 (dd, J = 250.0, 6.1 Hz), 157.88, 154.90, 153.60, 146.16, 132.20 (d, J = 9.9 Hz), 128.63 (d, J = 12.2 Hz), 115.19 (t, J = 23.2 Hz), 89.29, 89.07, 80.92, 80.55, 58.09, 34.78, 33.94, 28.48, 25.80, 16.56 (d, J = 1.6 Hz) ppm. HRMS (ES+) calcd for C26H32N6F2ClO2 [M + H]+, 533.2238; found 533.2234.
(R)-5-Chloro-6-(2,6-difluoro-4-(3-(methylamino)prop-1-yn-1-yl)phenyl)-N-(3,3-dimethylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine Hydrochloride (59)
General procedure D was followed using 58 (0.035 g, 0.066 mmol) to provide compound 59 (0.028 g, 0.060 mmol, 91%) as a HCl salt as a light brown solid. 1H NMR (600 MHz, CD3OD) δ 8.74 (s, 1H), 7.47 (dd, J = 8.4, 3.1 Hz, 2H), 4.24 (s, 2H), 3.33 (bs, 1H), 2.86 (s, 3H), 1.10 (d, J = 6.7 Hz, 3H), 0.84 (s, 9H) ppm. HRMS (ES+) calcd for C21H24N6F2Cl [M + H]+, 433.1714; found 433.1716.
(R)-5-Chloro-6-(4-(4-(dimethylamino)but-1-yn-1-yl)-2,6-difluorophenyl)-N-(3,3-dimethylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (60)
General procedure C was followed using 41 (0.040 g, 0.081 mmol) and N,N-dimethylbut-3-yn-1-amine (0.029 mL, 0.024 g, 0.224 mmol). Purification by reversed-phase HPLC provided compound furnish compound 60 as an off-white solid (0.023 g, 0.050 mmol, 61%). 1H NMR (599 MHz, CD3OD) δ 8.48 (s, 1H), 7.48–7.19 (m, 2H), 3.47 (t, J = 6.9 Hz, 2H), 3.05 (t, J = 6.9 Hz, 2H), 3.00 (s, 5H), 1.07 (d, J = 6.7 Hz, 3H), 0.83 (s, 9H) ppm. 13C NMR (151 MHz, CD3OD) δ 160.68, 159.03, 156.92, 153.63, 114.52, 114.36, 87.26, 57.57, 54.66, 41.66, 34.06, 24.07, 14.63, 14.16 ppm. 19F NMR (376 MHz, MeCN-D3) δ −75.26, −110.00 to −110.13 (m), −110.69 (dd, J = 11.0, 7.5 Hz) ppm. HRMS (ES+) calcd for C23H28ClF2N6 [M + H]+, 461.2027; found 461.2031.
In Silico Studies
All molecular modeling experiments were performed using Maestro software package (Schrodinger Release 2021–1).18 Molecular Operating Environment (MOE)19 and Flare (Cresset)20 were used to visualize the structures and protein–ligand interaction, and acquire the images.
The X-ray crystal structures of αβ-tubulin in complex with 4 and 5 were downloaded from the Protein Data Bank (PDB ID: 5NJH and 7CLD, respectively). Both proteins were prepared with Protein Preparation Wizard. Waters and other co-crystallized molecules were removed, except for the ligand and GDP. Predicting protonation states of protein residues were calculated considering a temperature of 300 K and a pH of 7. A 15 Å docking grid (inner box, 10 Å; outer box, 20 Å) was prepared using as centroid the co-crystallized ligands. The TPD compounds were prepared considering the ionization states at pH 7 ± 2. The docking studies were performed using Glide SP precision keeping the default parameters and setting, and it was combined with molecular mechanics generalized Born surface area (MMGBSA), implemented in the Prime module from Maestro to re-score, the three output docking poses of each compound.
Molecular dynamics simulations were performed using Desmond module, employing OPLS4 force field in the explicit solvent and the TIP3 water model. The initial coordinates for the MD simulation were taken from the best docking poses obtained for each compound. A cubic water box was used for the solvation of the system, ensuring a buffer distance of 12 Å between each box side and the protein–ligand complex. The system was minimized and preequilibrated using the default parameters. A 100 ns MD simulation was performed. The protein–ligand during the MD simulations was analyzed using the Simulation Interaction Diagram tool implemented in the Desmond package.
QBI293 Cell and Neuronal Culture Acetyl-Tubulin and α-Tubulin Determinations
Compound-induced changes in acetylated-tubulin and α-tubulin in QBI293 cells or primary mouse or rat neurons were as previously described.11,12 For compound testing, QBI293 cells were plated at a density of 2 × 105 cells/well in 12-well plates. The plated cells were incubated overnight, after which the medium was aspirated and fresh medium containing vehicle (0.25–0.3% DMSO) or test compound was added. Test compounds were prepared as 4–20 mM stock solutions in 100% DMSO, and these were diluted in culture medium prior to addition to the QBI293 cells. After 4 h incubation, whole-cell extracts were prepared from the QBI293 cells as described11 and the supernatant fraction from each sample was assessed for protein content by bicinchoninic acid (BCA) assay. The amount of acetyl-tubulin (AcTub) and α-tubulin (αTub) within the cell extract samples was quantified using specific enzyme-linked immunosorbent assays (ELISA), as previously described.11 Acetyl-tubulin levels were determined in a similar manner in rat or mouse cortical neuron cultures in which embryonic dissociated cortical neurons were plated at 3 × 105 cells/well in poly-d-lysine-coated 12-well plates, essentially as previously described.11 The neurons were allowed to grow for 10 days before they were treated for 8 h with 15 nM okadaic acid in the presence or absence of test compound prepared as described above, or vehicle (0.25% DMSO). Culture homogenates were prepared for the determination of AcTub levels by ELISA as previously described.11
Determination of Plasma Pharmacokinetics of Compound 60
Studies to determine the pharmacokinetic properties of compounds 53 and 60 were conducted at Touchstone Biosciences (Plymouth Meeting, PA). For the determination of plasma pharmacokinetics, three male CD-1 mice (2–3 months of age) were injected i.v. (tail vein) with test compound at a dose of 5 mg/kg in a solution of 10% DMSO, 40% polyethylene glycol, and 50% water, and another three male CD-1 mice received oral gavage dosing of 60 formulated as above at a dose of 20 mg/kg. Blood samples were collected from the saphenous or submandibular veins at multiple time points after dosing and collected in Greiner MiniCollect K2EDTA tubes. Compound levels in plasma were determined by LC-MS/MS, using protocols similar to those previously described.16,22
Assessment of Glu-Tubulin in Brain Tissue after Compound Treatment
All animal protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). Groups of four CD-1 mice received two i.p. injections of compounds 53, 60, or 68 at 1 mg/kg or vehicle only spaced approximately 24 h apart. Compounds were diluted from 20 or 40 mM DMSO stock solutions into 30% Kolliphor EL (Sigma-Aldrich) phosphate-buffered saline, pH 7.4. After 4 h following the second injection, mice were euthanized and cortices and hippocampi were isolated from each brain and placed in 0.2 mL of ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), pH 8.0) containing protease inhibitor (PI) cocktail (Sigma-Aldrich), 1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich), and 3 μM trichostatin A (TSA) (Sigma-Aldrich). The brain samples were homogenized with a handheld battery-operated mixer and subsequently sonicated. After centrifugation at 100,000g for 30 min at 4 °C, supernatant samples were transferred to a new tube and the pellets were resuspended in 0.15 mL of RIPA buffer and again homogenized, sonicated, and centrifuged. The supernatant from the second centrifugation was pooled with that from the first. The combined supernatant samples were assessed for protein concentration using a BCA assay (Thermo Fisher Scientific), and equal protein amounts from each brain sample underwent separation by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer onto nitrocellulose membranes essentially as previously described.11 Membranes were incubated overnight in primary antibodies in blocking buffer at 4 °C, utilizing antibodies to de-tyrosinated (Glu)-tubulin, α-tubulin, and glyceraldehyde-3-phosphate dehydrogenase as previously described.11 Incubation with infrared dye-conjugated secondary antibodies and subsequent imaging and quantification of the blots on the Odyssey imaging system were as described.11
Acknowledgments
Financial support for this work was provided by the NIH/NIA (AG061173, AG044332).
Glossary
Abbreviations Used
- AcTub
acetylated α-tubulin
- α-Tub
α-tubulin
- MMGBSA
molecular mechanics generalized Born surface area
- MT
microtubule
- NFTs
neurofibrillary tangles
- NTs
neuropil threads
- OA
okadaic acid
- PD
pharmacodynamic
- PK
pharmacokinetic
- Tg
transgenic
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01411.
NMR spectra of test compounds and X-ray crystal structures of compound 53 (CCDC 2178417) (PDF)
The authors will release the atomic coordinates and experimental data upon article publication; PDB coordinates for Figure 6A (PDB)
PDB coordinates for Figure 6B (PDB)
PDB coordinates for Figure 6C (PDB)
PDB coordinates for Figure 6D (PDB)
PDB coordinates for Figure 6E (PDB)
PDB coordinates for Figure 6F (PDB)
PDB coordinates for Figure 7A (PDB)
PDB coordinates for Figure 7B (PDB)
PDB coordinates for Figure 7C (PDB)
PDB coordinates for Figure 7D (PDB)
SMILES string structures along the full data set in tabular form (CSV)
Author Present Address
# Vysoká Škola Chemiko-Technologiká v Praze Technická 5, 16628 Prague 6, Czech Republic
The authors declare no competing financial interest.
Dedication
The authors dedicate this article to the late Dr. John Q. Trojanowski, Professor of Geriatric Medicine and Gerontology at University of Pennsylvania, who championed the idea that microtubule-normalizing drugs may provide benefit in tauopathies and who provided continued encouragement of the studies described here.
Supplementary Material
References
- Ballatore C.; Lee V. M. Y.; Trojanowski J. Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Maiti A.; Shively S.; Lakhani F.; McDonald-Jones G.; Bruce J.; Lee E. B.; Xie S. X.; Joyce S.; Li C.; Toleikis P. M.; Lee V. M. Y.; Trojanowski J. Q. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 227–231. 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Yao Y.; Cornec A.-S.; Oukoloff K.; James M. J.; Koivula P.; Trojanowski J. Q.; Smith A. B. 3rd; Lee V. M.-Y.; Ballatore C.; Brunden K. R. A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model. Mol. Neurodegener. 2018, 13, 59 10.1186/s13024-018-0291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makani V.; Zhang B.; Han H.; Yao Y.; Lassalas P.; Lou K.; Paterson I.; Lee V. M. Y.; Trojanowski J. Q.; Ballatore C.; Smith A. B. 3rd; Brunden K. R. Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta Neuropathol. Commun. 2016, 4, 106 10.1186/s40478-016-0378-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Carroll J.; Trojanowski J. Q.; Yao Y.; Iba M.; Potuzak J. S.; Hogan A. M.; Xie S. X.; Ballatore C.; Smith A. B. 3rd; Lee V. M.; Brunden K. R. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. 10.1523/JNEUROSCI.4922-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden K. R.; Zhang B.; Carroll J.; Yao Y.; Potuzak J. S.; Hogan A. M.; Iba M.; James M. J.; Xie S. X.; Ballatore C.; Smith A. B. 3rd; Lee V. M.; Trojanowski J. Q. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.; Nzou G.; Alle T.; Tsering W.; Maimaiti S.; Trojanowski J. Q.; Lee V. M.; Ballatore C.; Brunden K. Correction of microtubule defects within Aβ plaque-associated dystrophic axons results in lowered Aβ release and plaque deposition. Alzheimer’s Dement. 2020, 16, 1345–1357. 10.1002/alz.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D. J.; Garwin G. G.; Cline M. M.; Richards T. L.; Yarnykh V.; Mourad P. D.; Ho R. J.; Minoshima S. Paclitaxel improves outcome from traumatic brain injury. Brain Res. 2015, 1618, 299–308. 10.1016/j.brainres.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschel J.; Bradke F. Systemic administration of epothilone D improves functional recovery of walking after rat spinal cord contusion injury. Exp. Neurol. 2018, 306, 243–249. 10.1016/j.expneurol.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Sáez-Calvo G.; Sharma A.; Balaguer F. D. A.; Barasoain I.; Rodríguez-Salarichs J.; Olieric N.; Muñoz-Hernández H.; Berbís M. Á.; Wendeborn S.; Peñalva M. A.; Matesanz R.; Canales Á.; Prota A. E.; Jímenez-Barbero J.; Andreu J. M.; Lamberth C.; Steinmetz M. O.; Díaz J. F. Triazolopyrimidines Are Microtubule-Stabilizing Agents that Bind the Vinca Inhibitor Site of Tubulin. Cell Chem. Biol. 2017, 24, 737–750 e6. 10.1016/j.chembiol.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Kovalevich J.; Cornec A. S.; Yao Y.; James M.; Crowe A.; Lee V. M.; Trojanowski J. Q.; Smith A. B. 3rd; Ballatore C.; Brunden K. R. Characterization of brain-penetrant pyrimidine-containing molecules with differential microtubule-stabilizing activities developed as potential therapeutic agents for Alzheimer’s disease and related tauopathies. J. Pharmacol. Exp. Ther. 2016, 357, 432–450. 10.1124/jpet.115.231175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oukoloff K.; Nzou G.; Varricchio C.; Lucero B.; Alle T.; Kovalevich J.; Monti L.; Cornec A. S.; Yao Y.; James M. J.; Trojanowski J. Q.; Lee V. M.; Smith A. B. 3rd; Brancale A.; Brunden K. R.; Ballatore C. Evaluation of the Structure-Activity Relationship of Microtubule-Targeting 1,2,4-Triazolo[1,5-a]pyrimidines Identifies New Candidates for Neurodegenerative Tauopathies. J. Med. Chem. 2021, 64, 1073–1102. 10.1021/acs.jmedchem.0c01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yu Y.; Li Y.; Yan W.; Ye H.; Niu L.; Tang M.; Wang Z.; Yang Z.; Pei H.; Wei H.; Zhao M.; Wen J.; Yang L.; Ouyang L.; Wei Y.; Chen Q.; Li W.; Chen L. Cevipabulin-tubulin complex reveals a novel agent binding site on α-tubulin with tubulin degradation effect. Sci. Adv. 2021, 7, eabg4168 10.1126/sciadv.abg4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Ayral-Kaloustian S.; Nguyen T.; Afragola J.; Hernandez R.; Lucas J.; Gibbons J.; Beyer C. Synthesis and SAR of [1,2,4]triazolo[1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition. J. Med. Chem. 2007, 50, 319–327. 10.1021/jm060717i. [DOI] [PubMed] [Google Scholar]
- Matthew S.; Chen Q. Y.; Ratnayake R.; Fermaintt C. S.; Lucena-Agell D.; Bonato F.; Prota A. E.; Lim S. T.; Wang X.; Diaz J. F.; Risinger A. L.; Paul V. J.; Oliva M. A.; Luesch H. Gatorbulin-1, a distinct cyclodepsipeptide chemotype, targets a seventh tubulin pharmacological site. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2021847118 10.1073/pnas.2021847118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou K.; Yao Y.; Hoye A. T.; James M. J.; Cornec A. S.; Hyde E.; Gay B.; Lee V. M.; Trojanowski J. Q.; Smith A. B. 3rd; Brunden K. R.; Ballatore C. Brain-penetrant, orally bioavailable microtubule-stabilizing small molecules are potential candidate therapeutics for Alzheimer’s disease and related tauopathies. J. Med. Chem. 2014, 57, 6116–6127. 10.1021/jm5005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S.; Liepold B.; Roth G. J.; Bestmann H. J. An Improved One-pot Procedure for the Synthesis of Alkynes from Aldehydes. Synlett 1996, 1996, 521–522. 10.1055/s-1996-5474. [DOI] [Google Scholar]
- Schrödinger Release 2022-1: Maestro; Schrödinger, LLC: New York, NY, 2021.
- Molecular Operating Environment (MOE), 2022.02 Chemical Computing Group ULC1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2022.
- Flare, version 6, Cresset: Litlington, Cambridgeshire, U.K.
- Cornec A. S.; James M. J.; Kovalevich J.; Trojanowski J. Q.; Lee V. M.; Smith A. B. 3rd; Ballatore C.; Brunden K. R. Pharmacokinetic, pharmacodynamic and metabolic characterization of a brain retentive microtubule (MT)-stabilizing triazolopyrimidine. Bioorg. Med. Chem. Lett. 2015, 25, 4980–4982. 10.1016/j.bmcl.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.