

Abstract
π-Stacking is a common descriptor for face-to-face attractive forces between aromatic hydrocarbons. However, the physical origin of this interaction remains debatable. Here we examined π-stacking in a model homodimer formed by two thiol-substituted naphthalene rings. Two isomers of the 2-naphthalenethiol dimer were discovered using rotational spectroscopy, sharing a parallel-displaced crossed orientation and absence of thiol–thiol hydrogen bonds. One of the isomers presents C2 symmetry, structurally analogous to the global minimum of the naphthalene dimer. The experimental data were rationalized with molecular orbital calculations, revealing a shallow potential energy surface. Noncovalent interactions are dominated by dispersion forces according to SAPT energy decomposition. In addition, the reduced electronic density shows a diffuse and extended region of inter-ring interactions, compatible with the description of π-stacking as a competition between dispersion and Pauli repulsion forces.
π-Stacking interactions are ubiquitous in chemical1 and biological systems2,3 and constitute a valuable tool in the engineering of supramolecular assemblies,4,5 crystal designs,6 host–guest compounds,7 catalysis,8 and materials.9 At the same time, π-stacking is considered one of the most controversial supramolecular interactions,10,11 and most of present information is still essentially descriptive. Electronic computations can establish the main features of the potential energy surface (PES), but they miss a clear understanding of the physical origin of stacking.12 Moreover, nearly all empirical data arise from crystal diffraction,4,13 and there is a lack of gas-phase experiments which could provide benchmarking structural evidence unbiased from matrix effects.
The ideas on π-stacking have evolved considerably. The Hunter–Sanders model attributed the observed geometries to a balance between (classical) quadrupolar electrostatics and London dispersion interactions.14 This model has largely influenced supramolecular chemistry5 but fails for some simple systems like benzene-hexafluorobenzene.12 Sherrill15−17 and Wheeler18,19 have criticized several aspects of this model, in particular the need of quantum electrostatics and the importance of penetration effects at the shorter distances of π–π or CH···π interactions. Grimme analyzed larger arene dimers, emphasizing the prevailing role of dispersion forces.10 More recently, Carter-Fenk and Herbert12 offered a radically different paradigm, in which electrostatics is sidelined and dispersion and Pauli repulsion constitute the main contributors to π-stacking. As computational studies become more complex, there is a recurrent need for contrasting experiments on π-bonded systems. Among gas-phase techniques, jet-cooled vibrational20−24 and rotational25−31 spectroscopies provide cluster generation and characterization of weakly bound dimers, offering high-resolution structural information directly comparable to theory. The number of gas-phase studies of π-stacking polycyclic aromatic hydrocarbon (PAH) dimers is nevertheless quite small.
In the flat and deceptively simple PES of the prototype benzene dimer, both the perpendicular (T-shape) and parallel (cofacial) canonical forms are saddle points,32−34 and the lowest-lying minima correspond to the parallel-displaced and the tilted-T-shape geometry, which was experimentally detected in the gas phase using rotational spectroscopy.26−28 Unsurprisingly, the spectrum confirmed that the weakly bound benzene dimer exhibits notorious internal dynamical effects,28 quite difficult to reproduce theoretically and impossible to observe in the crystal. We recently observed also large-amplitude motions in the rotational spectrum of the thiophenol dimer, which, unlike the hinged phenol dimer,35 combine a parallel-displaced geometry with a weak S–H···S hydrogen bond.30 A parallel-displaced geometry and torsional tunnellings were similarly observed for the o-difluorobenzene dimer.25 For bicyclic aromatic hydrocarbons, the naphthalene dimer may adopt four possible geometries, the global minimum being a stacked geometry with the two rings in a crossed V-shape geometry.36,37 However, this dimer could only be detected vibrationally because of its small dipole moment,24 and rotational observations have been limited to the dimer of 1-naphthol29 and the tricyclics of fluorene and dibenzofuran.31 In order to understand why parallel-displaced geometries are the dominant structures for larger PAHs,38 including biological compounds2 and heterocycles,39,40 other arene dimers should be studied in the gas phase.
Here we report a rotational investigation on the dimer of 2-naphthalenethiol using broadband microwave spectroscopy,41 density-functional theory (B3LYP, ωB97XD, and B2PLYP), and ab initio (SCS-MP2 and DLPNO-CCSD(T)) calculations. The rationale for this study was the introduction of a dipole moment in the molecule through the thiol polar bond, simultaneously offering comparison with the dimers of naphthalene36 and 1-naphthol,29 gauging substituent effects15,18,42,43 and comparing plausible hydrogen bonds30,44 originated by the thiol group. The results constitute the first rotational detection of π-stacking isomerism in bicyclic aromatic hydrocarbons, offering insight on the PES and the structural and electronic properties of these weak noncovalent interactions.
The monomer of 2-naphthalenethiol adopts two planar cis or trans conformations, depending on the orientation of the thiol group. cis-2-Naphthalenethiol is the global minimum but quite similar in energy to the trans conformer (B3LYP-D3(BJ): 0.6 kJ mol–1 in Figure S1). For the 2-naphthalenethiol dimer, 47 cis/cis (CC), cis/trans (CT), or trans/trans (TT) structures were investigated computationally (see Supporting Information). A priori, the dimer structure may use sandwich, parallel-displaced, or T-shaped geometries similar to the naphthalene,36 benzene,32−34 or the thiophenol30 dimers, but hydrogen-bonded hinged geometries similar to the phenol dimer35 were also considered. However, neither T-shaped nor hinged geometries converged to stable dimer structures, and all predicted isomers displayed parallel-displaced stacked geometries. The calculated PES was quite flat, and the 19 most stable isomers of Figure S2 are distributed within a small energy window of only 4.3 kJ mol–1 (B3LYP). The five lowest-lying isomers in Figures 1 and S3 differ by less than 0.9 kJ mol–1 (B3LYP) and are characterized by the absence of any S–H···S hydrogen bond (predicted at electronic energies above 1.1 kJ mol–1). The parallel-displaced forms may present different geometries depending on the orientation between the two naphthalene subunits, occasionally adopting symmetric structures like the crossed or slipped geometries, as in the C2 global minimum (CC-1). The results of the model calculations are compared in Tables S1–S3 (B3LYP), S4 (ωB97XD), S5 (B2PLYP), S6 (RI-SCS-MP2), and S7 (DLPNO-CCSD(T)). B3LYP and B2PLYP predict CC-1 as global minimum, but the prediction is reversed for ωB97XD and the ab initio methods, which favor CC-2.
Figure 1.

Two detected species and partial PES of the 2-naphthalenethiol dimer (see also Tables S1–S7 and Figure S3). The two cis-cis (CC-1 and CC-2) isomers observed experimentally are highlighted on the left. The conformational search used initially a DFT method (B3LYP-D3(BJ), blue trace) and the most stable isomers were later reoptimized at different calculation levels (the RI-SCS-MP2 and DLPNO-CCSD(T) results in green and red do not include zero-point corrections). The most stable isomers lack any S–H···S hydrogen bond. In all cases, stacked structures are preferred.
The molecular-jet microwave spectrum of Figure S4 was then surveyed for the signals of the 2-naphthalenethiol dimer, expected to peak in the 2–8 GHz region at the effective rotational temperature of ca. 2 K. The spectral analysis is discussed in the Supporting Information. Two weak spectral signatures were detected, confirming the competition between two isomers of the dimer. The experimental data set, comprising more than 250 transitions, is presented in Tables S8 and S9. The derived experimental parameters are collected in Table 1. Isotopic species in natural abundance were undetectable. No other dimer species were identified.
Table 1. Rotational Parameters for the 2-Naphthalenethiol Dimer and Computational Predictions.
| Experiment |
Theory |
|||||||
|---|---|---|---|---|---|---|---|---|
| Isomer 1 | Isomer 2 | CC-1 | CC-2 | CT-1 | CT-2 | TT-1 | CT-3 | |
| A/MHza | 308.38853(21)e | 299.45856(51) | 318.10 | 304.23 | 294.60 | 307.2 | 300.3 | 390.1 |
| B/MHz | 231.75029(16) | 246.9652(12) | 233.40 | 250.65 | 245.68 | 249.6 | 242.1 | 228.2 |
| C/MHz | 226.78483(18) | 221.5793(15) | 231.34 | 225.72 | 240.89 | 225.6 | 239.4 | 175.3 |
| DJ/kHz | 0.01761(44) | 0.1328(56) | 0.016 | 0.012 | 0.024 | 0.012 | 0.022 | 0.010 |
| DJK/kHz | [0.0]f | –0.242(14) | –0.003 | 0.028 | –0.037 | 0.032 | –0.024 | 0.005 |
| DK/kHz | 0.0234(12) | 0.164(10) | 0.023 | 0.005 | 0.055 | 0.001 | 0.044 | 0.065 |
| d1/kHz | [0.0] | 0.0434(28) | –0.002 | 0.000 | –0.007 | 0.000 | –0.009 | –0.001 |
| d2/kHz | [0.0] | [0.0] | 0.001 | –0.001 | 0.002 | –0.001 | 0.001 | –0.001 |
| |μa|/D | - | - | 0.0 | 0.2 | 1.0 | –0.7 | 0.0 | –1.1 |
| |μb|/D | - | ++ | 0.0 | 0.6 | 1.1 | 1.3 | 0.0 | 1.2 |
| |μc|/D | +++g | ++ | 1.3 | 0.6 | 0.6 | 1.2 | –2.1 | 0.0 |
| Nb | 173 | 86 | ||||||
| σ/kHz | 10.6 | 10.6 | ||||||
| ΔEZPE/kJ mol–1c | 0.0 | 0.2 | 0.5 | 0.7 | 0.9 | 1.1 | ||
| ΔG/kJ mol–1 | 1.4 | 1.8 | 0.0 | 0.2 | –1.1 | 1.3 | ||
| EC/kJ mol–1 | –48.7 | –47.4 | –47.2 | –47.2 | –46.9 | –46.9 | ||
| ΔESCS-MP2/kJ mol–1d | 1.1 | 0.0 | 0.4 | 0.4 | 1.1 | 1.3 | ||
| ΔEDLPNO-CCSD(T)/kJ mol–1 | 0.7 | 0.0 | 0.6 | 0.8 | 1.1 | 2.4 | ||
Rotational constants (A, B, C), centrifugal distortion constants (DJ, DJK, DK, d1, d2) according to Watson’s S-reduction (Ir-representation) and electric dipole moments (μα, α = a, b, c).
Number of measured transitions (N) and standard deviation of the fit (σ).
Relative energy with zero-point corrections (ΔE), Gibbs energy (ΔG, 298 K, 1 atm), and complexation energy (ΔEC including BSSE corrections) using B3LYP-D3(BJ)/def2-TZVP.
Electronic energy using SCS-MP2 and DLPNO-CCSD(T), uncorrected for zero-point vibrational energy.
Standard errors in parentheses in units of the last digit.
Parameters in square brackets fixed to zero.
The plus signs denote qualitatively the observation of the corresponding rotational transitions.
The spectroscopic parameters allowed for an unequivocal isomer identification. In particular, the presence of a symmetry axis matches the predictions for CC-1. Rotatable 3D figures and coordinates for both isomers are shown in Figures S5–S6 and Tables S10–S11. The experiment–theory comparison in Table 1 gives a good structural agreement for the B3LYP-D3(BJ) model, with relative differences in the rotational constants of 0.7–3.1% for isomer CC-1 and 1.5–1.9% for isomer CC-2. A comparison with the alternative theoretical models is shown in Table S12 (SI). Interestingly, neither ωB97XD nor the double-hybrid B2PLYP offered significant improvements over B3LYP in structural terms. The basis set dependence on B3LYP is shown in Table S13. The size of the system prevented the calculation of vibrational frequencies and zero-point energies for the B2PLYP, RI-SCS-MP2, and DLPNO-CCSD(T) models.
The noncovalent interactions (NCIs in the 2-naphthalenethiol dimers have been analyzed using structural, energetic, and electronic information. Despite only a few bicyclic aromatic hydrocarbon dimers having been detected so far in the gas phase, some structural patterns now become apparent. Noticeably, hydrogen-bonded structures are not dominant after insertion of an alcohol or thiol group in the naphthalene dimer, preserving the stacked geometry. In the naphthalene dimer, the global minimum shows the parallel-displaced crossed C2-symmetric structure of Figure 2.36,37 In this structure, the largest ring overlap occurs between two terminal rings, favoring the torsioned symmetric arrangement. This C2 structure was now observed for the 2-naphthalenethiol dimer CC-1, but not in 1-naphthol,29 where it is predicted higher in energy (1.0–1.6 kJ mol–1). However, the weak balance of intermolecular forces results in different inter-ring torsion angles, much larger in naphthalene36,37 (MP2: 135–136°) than for the CC-1 structure of 2-naphthalenethiol (B3LYP: 81°, Figure S7). A second type of limit dimer structure corresponds to ring overlapping between one of the aromatic rings and the central region of the second naphthalene molecule, rotated ca. 66° (D2d in the naphthalene dimer, Figure S8). This geometry is the global minimum of the 1-naphthol dimer, where it was denoted as V-shape.29 We also observed this second structure in the CC-2 dimer, nearly isoenergetic to (B3LYP: +0.2 kJ mol–1) or more stable than CC-1 for the ωB97XD (−0.7 kJ mol–1) and the ab initio models (SCS-MP2: −1.1 kJ mol–1; DLPNO-CCSD(T): −0.7 kJ mol–1).
Figure 2.

Comparison of the two observed structures of the 2-naphthalenethiol dimer (left) with the dimers of 1-naphthol (center) and naphthalene (right).
In both the 2-naphthalenethiol and 1-naphthol dimers, the polar bond preferentially adopts the monomer conformation (i.e., CC in the first case, TT in the second). However, CT and TT isomers generate competing isomers in the thiol. As an example, isomer TT-1 (B3LYP: 0.9 kJ mol–1) is structurally analogous to CC-1. The eventual presence of an S–H···S hydrogen bond is slightly destabilized (B3LYP: >1.1 kJ mol–1) but does not affect the global stacking arrangements, with thiol–thiol contacts of r(S–H···S) = 2.86–2.94 Å in Figure S9. Hinged structures are energetically excluded for the thiol or alcohol dimers, but they are much lower in energy for the stronger O–H···O hydrogen bond (>1.8 kJ mol–1). The weaker interaction associated to the thiol group is also reflected in the flatter and more corrugated PES of this dimer. The inter-ring distances of the most stable dimers (3.26–3.29 Å), also in Figure S9, are similar to those predicted for the naphthalene dimer, exposing the common origin of the attractive stacking interaction.
Despite the observed structures suggesting apparently small substituent effects, the complexation energies for the two 2-naphthalenethiol dimers in Tables 1 and S1–S5 range between −48.7 and 47.3 kJ mol–1 using B3LYP or B2PLYP. This value is much larger than the energies of −42.2 and −31.0 kJ mol–1 (B3LYP) for the 1-naphthol and naphthalene dimers, exposing the important stabilizer role associated to the presence of the polar bonds. The ωB97XD complexation energies of Table S4 are ca. 3 kJ mol–1 smaller. The experimental detection of two isomers suggests interconversion barriers exceeding the collisional relaxation thresholds in the jet (5–12 kJ mol–1).45 However, the weak character of the 2-napthalenethiol dimer and the multiplicity of conversion paths made the barrier calculation difficult and very sensitive to the calculation method. The GRRM-IRC calculations in Figure S10 estimated a barrier height above 14 kJ mol–1 for isomer interconversion, involving a complex multistep route of three different intermediates at B3LYP-D3(BJ)/def2-TZVP level. However, considering the tiny conformational differences, we do not exclude other classes of barriers due to sliding/reorientation, SH internal rotation, and naphthalene face-to-face flipping.
Noncovalent interactions were mapped using Johnson–Contreras’s
reduced gradient  of the electronic density (ρ).46 The NCI plot in Figure 3 reveals a wide region of weak interactions
between the two rings with small pockets of more attractive forces,
characteristic of the extended spatial distribution of stacking interactions.
The uneven distribution of attractive forces was interpreted in the
1-naphthol dimer as a competition between attractive interactions
and Pauli repulsion.29 Comparison of the
plot of the reduced density gradient vs the signed density for the
naphthalene or 1-naphthol dimers also in Figure 3 shows similar patterns. In particular, the
negative minimum associated to the most attractive interaction is
quite diffuse, and the signatures of the S–H···H
or O–H···O hydrogen bonds, observed in the phenol
and thiophenol dimers of Figure S11, are
absent here. This representation confirms the weak nature of the π-stacking
interactions in these dimers. Other interactions are difficult to
ascertain. A small tilt of the terminal thiol hydrogens may be indicative
of weak cooperative S–H···π interactions,
as suggested for the O–H···π contacts
in the 1-naphthol dimer, previously categorized as van der Waals forces.29
of the electronic density (ρ).46 The NCI plot in Figure 3 reveals a wide region of weak interactions
between the two rings with small pockets of more attractive forces,
characteristic of the extended spatial distribution of stacking interactions.
The uneven distribution of attractive forces was interpreted in the
1-naphthol dimer as a competition between attractive interactions
and Pauli repulsion.29 Comparison of the
plot of the reduced density gradient vs the signed density for the
naphthalene or 1-naphthol dimers also in Figure 3 shows similar patterns. In particular, the
negative minimum associated to the most attractive interaction is
quite diffuse, and the signatures of the S–H···H
or O–H···O hydrogen bonds, observed in the phenol
and thiophenol dimers of Figure S11, are
absent here. This representation confirms the weak nature of the π-stacking
interactions in these dimers. Other interactions are difficult to
ascertain. A small tilt of the terminal thiol hydrogens may be indicative
of weak cooperative S–H···π interactions,
as suggested for the O–H···π contacts
in the 1-naphthol dimer, previously categorized as van der Waals forces.29
Figure 3.

NCI plots (upper panel, isovalue s = 0.5) mapping the weak attractive interactions for the CC-1 isomer of the 2-naphthalenethiol dimer, together with a representation of the reduced gradient vs the signed electronic density in the dimers of 2-naphthalenethiol, 1-naphthol, and naphthalene (lower panel, see also Figure S11).
We examined in Table 2 the balance of electrostatic and dispersion forces in the dimer stability, using energy decomposition at SAPT2+(3) level. As expected, the 2-naphthalenethiol is dominated by dispersion forces, which account for 66% of the total attractive interactions compared to a 27% of electrostatic contributions. This energetic composition is quite similar to the naphthalene dimer and other previously observed mono and bicyclic homodimers displaying π-stacking structures, like thiophenol or 1-naphthol. Actually, the representations in Figures 4 and S12 show nearly disjoint regions when compared to prototype hydrogen-bonded dimers using either O–H···O or S–H···H contacts. This dichotomy is especially evident for the dimers of thiophenol and phenol, which differ only in the heteroatom. The different nature of the π-stacking regime and hydrogen-bonded interactions is thus well captured by the combination of NCI plots and SAPT calculation.
Table 2. Results from a Binding Energy Decomposition Using (Second-Order Intramonomer/Third-Order Intermonomer) Symmetry-Adapted Perturbation Theory (SAPT2+(3)/aug-cc-pVDZ) for the π-Stacking Homodimers of 2-Naphthalenethiol, Naphthalene, 1-Naphthol, and Thiophenol and Several Hydrogen-Bonded Dimers, Comparing the Magnitude of the Attractive Contributions and Stabilization Energiesa.
| ΔEElectrostatic | ΔEInduction | ΔEDispersion | ΔEExchange | ΔETotal | |
|---|---|---|---|---|---|
| (H2S)2b | –12.1 [49.0%]c | –4.8 [19.4%] | –7.8 [31.6%] | 19.3 | –5.4 |
| (H2O)2 | –35.7 [63.4%] | –11.1 [19.7%] | –9.5 [16.9%] | 37.7 | –18.6 |
| (Phenol)2 | –41.8 [48.3%] | –15.9 [18.4%] | –28.8 [33.3%] | 58.9 | –27.6 |
| (1-Naphthol)2 | –33.4 [27.5%] | –11.0 [9.1%] | –77.1 [63.5%] | 80.2 | –41.3 |
| (Naphthalene)2 | –20.5 [21.4%] | –7.0 [7.3%] | –68.1 [71.2%] | 65.2 | –30.3 |
| (2-Naphthalenethiol)2 | –34.1 [26.7%] | –8.8 [6.9%] | –84.7 [66.4%] | 80.1 | –47.5 |
| (Thiophenol)2 | –26.2 [29.8%] | –8.4 [9.6%] | –53.3 [60.6%] | 61.0 | –26.9 |
All values in kJ mol–1.
The dimer structures were optimized at B3LYP-D3(BJ)/def2-TZVP level.
The values in square brackets represent the relative percentage with respect to the total attractive interactions.
Figure 4.
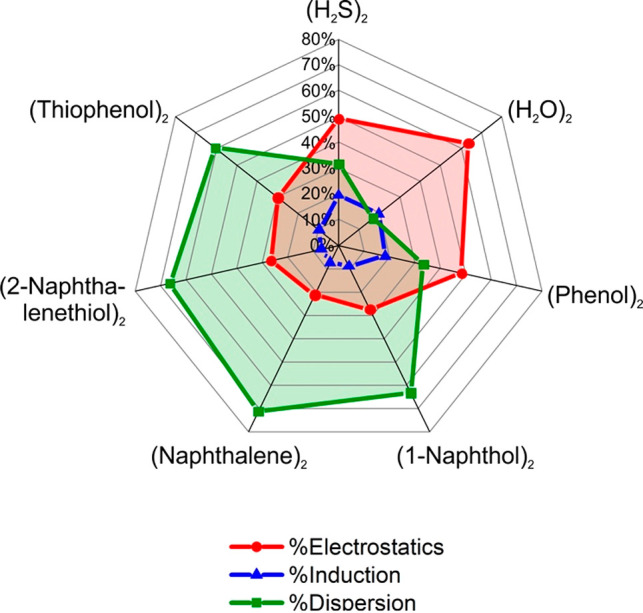
SAPT energy decomposition for the 2-naphthalenethiol dimer and related complexes. The radar chart shows the percentage of electrostatic, induction, and dispersion energy compared to the total stabilization energy in each complex. See Table 2 and Figure S12 for the total energy values.
In conclusion, gas-phase intermolecular clusters constitute chemically specific models of molecular aggregation. However, most studies have focused on hydrogen-bonding, and the observation of π-stacking aggregates has remained elusive. The detection of the 2-naphthalenethiol dimer significantly expands our understanding of π-stacking on substituted bicyclic aromatic hydrocarbons, offering valuable insight about their electronic properties, energetics, noncovalent interactions, and internal dynamics. Several conclusions are worth noticing. The 2-naphthalenethiol dimer maintains the π-stacking arrangement predicted for naphthalene,36 but its shallow PES increases in complexity and difficulty, offering a large number of low-lying isomers in the sub-kJ mol–1 window, difficult to model computationally. The experimental detection of two isomers of the dimer constitutes the first rotational observation of π-stacking isomerism and evidences the important internal dynamics observable in the gas phase, previously noticed in the dimers of benzene,27,28 difluorobenzene,25 and thiophenol.30 The observed dimers of 2-naphthalenethiol have structural resemblances with those of naphthalene and naphthol, one of them sharing the C2-symmetric arrangement of the naphthalene dimer global minimum. However, the substituent effect of the thiol group is mostly noticed in the increased dimer stabilization compared to the naphthalene or naphthol dimers (increase of +53% or +12%, respectively, in complexation energy). The energy decomposition analysis confirms the dominant character of dispersion forces, common to the three π-stacking structures and distinctive element with respect to hydrogen-bonded clusters. π-Stacking interactions are described through NCI plots as extended diffuse interactions, with localized attractive pockets compatible with the recent descriptions balancing dispersion forces and Pauli repulsion.12 Finally, the introduction of polar groups with associated bond dipoles proves an interesting chemical tool to gauge the molecular balance between the increased electrostatic forces, dispersion, and Pauli repulsion, thus offering different scenarios to test π-stacking forces. Considering that both repulsion and dispersion have size-dependent magnitudes, new experiments could now be devised examining the impact of substituents and heteroatoms in other polycyclic aromatic hydrocarbons. In this effort, the collaboration with adequate dispersion-corrected quantum mechanical models will be critical, emphasizing the synergistic role of gas-phase high-resolution rotational experiments.
Acknowledgments
Funding from the Spanish Ministerio de Ciencia e Innovación MICINN/FEDER (grants PGC2018-098561-B-C22 and PID2021-125015NB-I00) and FEDER-Junta de Castilla y León (grant INFRARED IR2020-1-UVa02) is gratefully acknowledged. C.P. thanks the Ministerio de Universidades for a BG20/00160 “Beatriz Galindo” Senior Researcher position. M.J. thanks the Ministerio de Universidades and Universidad de Valladolid for a “Margarita Salas” postdoctoral contract.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.2c03299.
Experimental and computational methods, analysis of the rotational spectrum, supplementary Figures (stable structures, microwave spectrum, rotatable drawings, interconversion barriers), and supplementary Tables (calculated energetic and rotational parameters, list of the observed rotational transitions, theoretical Cartesian coordinates) (PDF)
Author Present Address
⊥ Institut für Ionenphysik and Angewandte Chemie, Universität Innsbruck, Technikerstr. 25/4.OG, 6020 Innsbruck, Austria
The authors declare no competing financial interest.
Supplementary Material
References
- Non-Covalent Interactions in the Synthesis and Design of New Compounds; Maharramov A. M., Mahmudov K. T., Kopylovich M. N., Pombeiro A. J. L., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, 2016. [Google Scholar]
- McGaughey G. B.; Gagné M.; Rappé A. K. π-Stacking Interactions, Alive and Well in Proteins. J. Biol. Chem. 1998, 273 (25), 15458–15463. 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- Kool E. T. Hydrogen Bonding, Base Stacking, and Steric Effects in DNA Replication. Annu. Rev. Biophys. Biomol. Struct. 2001, 30 (1), 1–22. 10.1146/annurev.biophys.30.1.1. [DOI] [PubMed] [Google Scholar]
- Thakuria R.; Nath N. K.; Saha B. K. The Nature and Applications of π–π Interactions: A Perspective. Cryst. Growth Des. 2019, 19 (2), 523–528. 10.1021/acs.cgd.8b01630. [DOI] [Google Scholar]
- Fagnani D. E.; Sotuyo A.; Castellano R. K. π–π Interactions. Comprehensive Supramolecular Chemistry II 2017, 121–148. 10.1016/B978-0-12-409547-2.12485-0. [DOI] [Google Scholar]
- Stornaiuolo M.; De Kloe G. E.; Rucktooa P.; Fish A.; Van Elk R.; Edink E. S.; Bertrand D.; Smit A. B.; De Esch I. J. P.; Sixma T. K. Assembly of a π-π Stack of Ligands in the Binding Site of an Acetylcholine-Binding Protein. Nat. Commun. 2013, 4 (1), 1875. 10.1038/ncomms2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen L. M.; Ellermann M.; Diederich F. Aromatic Rings in Chemical and Biological Recognition: Energetics and Structures. Angew. Chemie - Int. Ed. 2011, 50 (21), 4808–4842. 10.1002/anie.201007560. [DOI] [PubMed] [Google Scholar]
- Neel A. J.; Hilton M. J.; Sigman M. S.; Toste F. D. Exploiting Non-Covalent π Interactions for Catalyst Design. Nature 2017, 543 (7647), 637–646. 10.1038/nature21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosser F.; Moos M.; Lambert C.; Würthner F. Redox-Switchable Intramolecular π-π-Stacking of Perylene Bisimide Dyes in a Cyclophane. Adv. Mater. 2013, 25 (3), 410–414. 10.1002/adma.201201266. [DOI] [PubMed] [Google Scholar]
- Grimme S. Do Special Noncovalent π-π Stacking Interactions Really Exist?. Angew. Chemie - Int. Ed. 2008, 47 (18), 3430–3434. 10.1002/anie.200705157. [DOI] [PubMed] [Google Scholar]
- Martinez C. R.; Iverson B. L. Rethinking the Term “π-Stacking”. Chem. Sci. 2012, 3 (7), 2191–2201. 10.1039/c2sc20045g. [DOI] [Google Scholar]
- Carter-Fenk K.; Herbert J. M. Electrostatics Does Not Dictate the Slip-Stacked Arrangement of Aromatic π-π Interactions. Chem. Sci. 2020, 11 (26), 6758–6765. 10.1039/D0SC02667K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryno S. M.; Risko C.; Brédas J. L. Noncovalent Interactions and Impact of Charge Penetration Effects in Linear Oligoacene Dimers and Single Crystals. Chem. Mater. 2016, 28 (11), 3990–4000. 10.1021/acs.chemmater.6b01340. [DOI] [Google Scholar]
- Hunter C. A.; Sanders J. K. M. The Nature of π-π Interactions. J. Am. Chem. Soc. 1990, 112 (14), 5525–5534. 10.1021/ja00170a016. [DOI] [Google Scholar]
- Hohenstein E. G.; Duan J.; Sherrill C. D. Origin of the Surprising Enhancement of Electrostatic Energies by Electron-Donating Substituents in Substituted Sandwich Benzene Dimers. J. Am. Chem. Soc. 2011, 133 (34), 13244–13247. 10.1021/ja204294q. [DOI] [PubMed] [Google Scholar]
- Sherrill C. D. Energy Component Analysis of π Interactions. Acc. Chem. Res. 2013, 46 (4), 1020–1028. 10.1021/ar3001124. [DOI] [PubMed] [Google Scholar]
- Parrish R. M.; Sherrill C. D. Quantum-Mechanical Evaluation of π–π versus Substituent−π Interactions in π Stacking: Direct Evidence for the Wheeler–Houk Picture. J. Am. Chem. Soc. 2014, 136 (50), 17386–17389. 10.1021/ja5101245. [DOI] [PubMed] [Google Scholar]
- Wheeler S. E. Understanding Substituent Effects in Noncovalent Interactions Involving Aromatic Rings. Acc. Chem. Res. 2013, 46 (4), 1029–1038. 10.1021/ar300109n. [DOI] [PubMed] [Google Scholar]
- Wheeler S. E.; Bloom J. W. G. Toward a More Complete Understanding of Noncovalent Interactions Involving Aromatic Rings. J. Phys. Chem. A 2014, 118 (32), 6133–6147. 10.1021/jp504415p. [DOI] [PubMed] [Google Scholar]
- Busker M.; Svartsov Y. N.; Häber T.; Kleinermanns K. IR-UV Double Resonance Spectra of Pyrazine Dimers: Competition between CH ··· π, π ··· π and CH ··· N Interactions. Chem. Phys. Lett. 2009, 467 (4–6), 255–259. 10.1016/j.cplett.2008.10.091. [DOI] [Google Scholar]
- Maity S.; Patwari G. N.; Sedlak R.; Hobza P. A π-Stacked Phenylacetylene Dimer. Phys. Chem. Chem. Phys. 2011, 13 (37), 16706–16712. 10.1039/c1cp20677j. [DOI] [PubMed] [Google Scholar]
- Kundu A.; Sen S.; Patwari G. N. The Propargylbenzene Dimer: C-H···π Assisted π-π Stacking. Phys. Chem. Chem. Phys. 2015, 17 (14), 9090–9097. 10.1039/C5CP00162E. [DOI] [PubMed] [Google Scholar]
- Kundu A.; Sen S.; Patwari G. N. π-Stacking in Heterodimers of Propargylbenzene with (Fluoro)Phenylacetylenes. ACS Omega 2021, 6 (27), 17720–17725. 10.1021/acsomega.1c02385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens A. K.; Chopra P.; Garg D.; Steber A. L.; Schnell M.; Buma W. J.; Rijs A. M. High-Resolution Infrared Spectroscopy of Naphthalene and Acenaphthene Dimers. Mol. Phys. 2021, 119 (1–2), e1811908. 10.1080/00268976.2020.1811908. [DOI] [Google Scholar]
- Goly T.; Spoerel U.; Stahl W. The Microwave Spectrum of the 1,2-Difluorobenzene Dimer. Chem. Phys. 2002, 283 (1–2), 289–296. 10.1016/S0301-0104(02)00500-1. [DOI] [Google Scholar]
- Arunan E.; Gutowsky H. S. The Rotational Spectrum, Structure and Dynamics of a Benzene Dimer. J. Chem. Phys. 1993, 98 (5), 4294–4296. 10.1063/1.465035. [DOI] [Google Scholar]
- Schnell M.; Erlekam U.; Bunker P. R.; Von Helden G.; Grabow J.-U.; Meijer G.; Van Der Avoird A. Unraveling the Internal Dynamics of the Benzene Dimer: A Combined Theoretical and Microwave Spectroscopy Study. Phys. Chem. Chem. Phys. 2013, 15 (25), 10207–10223. 10.1039/C3CP51181B. [DOI] [PubMed] [Google Scholar]
- Schnell M.; Erlekam U.; Bunker P. R.; Vonhelden G.; Grabow J.-U.; Meijer G.; Vanderavoird A. Structure of the Benzene Dimer - Governed by Dynamics. Angew. Chemie - Int. Ed. 2013, 52 (19), 5180–5183. 10.1002/anie.201300653. [DOI] [PubMed] [Google Scholar]
- Seifert N. A.; Hazrah A. S.; Jäger W. The 1-Naphthol Dimer and Its Surprising Preference for π-π Stacking over Hydrogen Bonding. J. Phys. Chem. Lett. 2019, 10 (11), 2836–2841. 10.1021/acs.jpclett.9b00646. [DOI] [PubMed] [Google Scholar]
- Saragi R. T.; Juanes M.; Pérez C.; Pinacho P.; Tikhonov D. S.; Caminati W.; Schnell M.; Lesarri A. Switching Hydrogen Bonding to π-Stacking: The Thiophenol Dimer and Trimer. J. Phys. Chem. Lett. 2021, 12 (5), 1367–1373. 10.1021/acs.jpclett.0c03797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima M.; Steber A. L.; Poblotzki A.; Pérez C.; Zinn S.; Schnell M. Rotational Signatures of Dispersive Stacking in the Formation of Aromatic Dimers. Angew. Chemie - Int. Ed. 2019, 58 (10), 3108–3113. 10.1002/anie.201812556. [DOI] [PubMed] [Google Scholar]
- Sinnokrot M. O.; Valeev E. F.; Sherrill C. D. Estimates of the Ab Initio Limit for π–π Interactions: The Benzene Dimer. J. Am. Chem. Soc. 2002, 124 (36), 10887–10893. 10.1021/ja025896h. [DOI] [PubMed] [Google Scholar]
- Sinnokrot M. O.; Sherrill C. D. Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer: Sandwich, T-Shaped, and Parallel-Displaced Configurations. J. Phys. Chem. A 2004, 108 (46), 10200–10207. 10.1021/jp0469517. [DOI] [Google Scholar]
- Bludský O.; Rubeš M.; Soldán P.; Nachtigall P. Investigation of the Benzene-Dimer Potential Energy Surface: DFT/CCSD(T) Correction Scheme. J. Chem. Phys. 2008, 128 (11), 114102. 10.1063/1.2890968. [DOI] [PubMed] [Google Scholar]
- Seifert N. A.; Steber A. L.; Neill J. L.; Pérez C.; Zaleski D. P.; Pate B. H.; Lesarri A. The Interplay of Hydrogen Bonding and Dispersion in Phenol Dimer and Trimer: Structures from Broadband Rotational Spectroscopy. Phys. Chem. Chem. Phys. 2013, 15 (27), 11468–11477. 10.1039/c3cp51725j. [DOI] [PubMed] [Google Scholar]
- Saeki M.; Akagi H.; Fujii M. Theoretical Study on the Structure and the Frequency of Isomers of the Naphthalene Dimer. J. Chem. Theory Comput. 2006, 2 (4), 1176–1183. 10.1021/ct050278n. [DOI] [PubMed] [Google Scholar]
- Dubinets N. O.; Safonov A. A.; Bagaturyants A. A. Structures and Binding Energies of the Naphthalene Dimer in Its Ground and Excited States. J. Phys. Chem. A 2016, 120 (17), 2779–2782. 10.1021/acs.jpca.6b03761. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Lawson K. R.; Perkins J.; Urch C. J. Aromatic Interactions. J. Chem. Soc. Perkin Trans. 2 2001, (5), 651–669. 10.1039/b008495f. [DOI] [Google Scholar]
- Hohenstein E. G.; Sherrill C. D. Effects of Heteroatoms on Aromatic π–π Interactions: Benzene–Pyridine and Pyridine Dimer. J. Phys. Chem. A 2009, 113 (5), 878–886. 10.1021/jp809062x. [DOI] [PubMed] [Google Scholar]
- Geng Y.; Takatani T.; Hohenstein E. G.; Sherrill C. D. Accurately Characterizing the π–π Interaction Energies of Indole–Benzene Complexes. J. Phys. Chem. A 2010, 114 (10), 3576–3582. 10.1021/jp9099495. [DOI] [PubMed] [Google Scholar]
- Park G. B.; Field R. W. Perspective: The First Ten Years of Broadband Chirped Pulse Fourier Transform Microwave Spectroscopy. J. Chem. Phys. 2016, 144 (20), 200901. 10.1063/1.4952762. [DOI] [PubMed] [Google Scholar]
- Sinnokrot M. O.; Sherrill C. D. Substituent Effects in π–π Interactions: Sandwich and T-Shaped Configurations. J. Am. Chem. Soc. 2004, 126 (24), 7690–7697. 10.1021/ja049434a. [DOI] [PubMed] [Google Scholar]
- Wheeler S. E. Local Nature of Substituent Effects in Stacking Interactions. J. Am. Chem. Soc. 2011, 133 (26), 10262–10274. 10.1021/ja202932e. [DOI] [PubMed] [Google Scholar]
- Saragi R. T.; Juanes M.; Pinacho R.; Rubio J. E.; Fernández J. A.; Lesarri A. Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer. Symmetry (Basel) 2021, 13 (11), 2022. 10.3390/sym13112022. [DOI] [Google Scholar]
- Florio G. M.; Christie R. A.; Jordan K. D.; Zwier T. S. Conformational Preferences of Jet-Cooled Melatonin: Probing Trans- and Cis-Amide Regions of the Potential Energy Surface. J. Am. Chem. Soc. 2002, 124 (34), 10236–10247. 10.1021/ja0265916. [DOI] [PubMed] [Google Scholar]
- Contreras-García J.; Johnson E. R.; Keinan S.; Chaudret R.; Piquemal J. P.; Beratan D. N.; Yang W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7 (3), 625–632. 10.1021/ct100641a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.