

Abstract

GPR55 is an orphan G-protein coupled receptor involved in various pathophysiological conditions. However, there are only a few noncannabinoid GPR55 ligands reported so far. The lack of potent and selective GPR55 ligands precludes a deep exploration of this receptor. The studies presented here focused on a thienopyrimidine scaffold based on the GPR55 antagonist ML192, previously discovered by high-throughput screening. The GPR55 activities of the new synthesized compounds were assessed using β-arrestin recruitment assays in Chinese hamster ovary cells overexpressing human GPR55. Some derivatives were identified as GPR55 antagonists with functional efficacy and selectivity versus CB1 and CB2 cannabinoid receptors.
Keywords: GPR55, antagonist, cannabinoid, thienopyrimidine, SAR
G protein-coupled receptor 55 (GPR55) is an orphan receptor that was identified in 1999.1−3 Despite its low homology with the well-known cannabinoid receptors CB1 and CB2, being 13% and 14%, respectively, it has been suggested as a putative cannabinoid receptor. This proposed classification is due to the fact that some endogenous, plant-derived, and synthetic cannabinoid ligands are able to modulate GPR55 activity.4,5 Complex pharmacology and species-dependent activation of GPR55 have complicated the discovery of potent and selective ligands.6−8 There are only a few noncannabinoid GPR55 ligands reported so far.8−16 However, the evidence reported for therapeutic applications of GPR55 modulators has generally been encouraging.7,8,16,17 GPR55 expression has been described in diverse tissues6 such as breast, adipose tissue, testes, and spleen and in several areas of the brain,18 including the hippocampus, putamen, caudate, and thalamic nuclei. Therefore, it has been shown that GPR55 is implicated in different pathophysiological conditions such as cancer,19−22 pain inflammation,23 neurodegeneration,24−26 or energy homeostasis.27,28 On the basis of the relevance of GPR55 in these therapeutic areas, our efforts have been dedicated to its targeting.
Lysophosphatidylinositol (LPI; Figure 1), identified as an endogenous GPR55 ligand, and ML191, ML192, and ML193 (Figure 1), discovered through a high throughput screening (HTS) using a β-arrestin recruitment assay, are representative of GPR55.1 In the present study, we propose to focus on the ML192 scaffold.
Figure 1.

Structure of the putative endogenous GPR55 ligand LPI and the GPR55 hit antagonists ML191, ML192, and ML193 identified by an HTS.
As previously mentioned, ML192 and related derivatives have been tested in the HTS developed by the National Institutes of Health (NIH).1 In these initial studies, structural diversification was mainly focused on substituents in the acylated piperazine group (see ML192 in Figure 1), while at R1 and R2 (see general structure in Figure 2) only hydrogen or methyl groups were explored. From this SAR,1 a clear preference for the furan heterocycle versus other aromatic or alkyl groups was observed at the acyl piperazine. However, no further conclusion was extracted from the thienopyrimidine substitution pattern. Taking this previous structural knowledge into account, we decided to mainly focus on the exploration of diverse substituents on the thienopyrimidine moiety as well as replacement of this tricyclic core with other heterocycles (Figure 2).
Figure 2.

Proposed structural modifications of the thienopyrimidine scaffold.
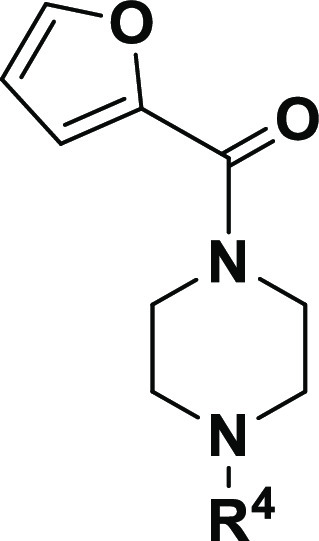
In an attempt to increase bulk and the presence of electron donor groups, hydroxyalkyl and morpholino substituents were tested at R1 (Figure 2, red). R2 was limited to hydrogen and dimethyl groups (Figure 2, green), while at R3 the furan-2-carbonylpiperazinyl group was preserved in most of the novel derivatives. Substitution of the piperazine by a diazepane was also explored in order to understand structural determinants of activity (Figure 2; blue, n = 2). The N-(3,4-dimethylisoxazol-5-yl)-4-methylbenzenesulfonamide present in ML193 (Figure 1) was also explored at position R3 (Figure 2, blue).
Moreover, a computational ligand-based scaffold hopping strategy (see the scaffold hopping workflow chart in Figure S1) was pursued to identify potential core replacement of the thienopyrimidine tricycle. Core hopping as implemented in the Maestro software (Schrödinger, LLC, New York, NY, 2019), was used for this purpose. The tricyclic moiety was set up to be replaced while preserving specific pharmacophoric requirements during the fragment-based screening of a library of over 100 000 cores. Synthesizability, lack of promiscuous structural alerts, the so-called PAINS (Pan Assay Interference Compounds), and in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties were used to filter the obtained molecules. Selected cores are shown in Figure 2 (black).
The first series of compounds (20–31) was synthesized as described in Scheme 1. A divergent synthetic route was followed that started with a Gewald multicomponent reaction by using ethyl cyanoacetate and S8 to obtain aminothiophene carboxylates.29 Intermediates 1 and 2 reacted with the corresponding nitrile under acidic conditions, yielding thienopyrimidinones (3–9).30 Dehydration using phosphoryl chloride31 provided the chlorinated compounds 12–19 that were then coupled with the appropriate piperazine or diazepine achieving the desired thienopyrimidines 20, 21, 24–26, 28, and 29. The synthesis of compounds 23 and 27 had the morpholinylalkyl group introduced through the intermediate chloroalkyl 10 and 22. As proposed in the design, the incorporation of hydroxymethyl and hydroxyethyl as R1 residues (30, 31) was obtained from the methoxy derivatives 28 and 29 using BBr3.
Scheme 1. Reagents and Conditions: (i) Gewald Reaction: Ethyl Cyanoacetate, S8, H2O, Et3N, rt, Overnight, 70–87%; (ii; a) Corresponding Nitrile, HCl (4M in dioxane), 4 h Ultrasonic Bath, (b) 100 °C, Overnight, 87–96% [For 7 (85%) and 8 (94%) only procedure A was Applied]; (iii) Morpholine, Dioxane, Et3N, Reflux, Overnight, 53%; (iv) POCl3, 90 °C, 3 h, 60–98%; (v) Corresponding Acylpiperazine, Et3N, Dioxane, Reflux, 24 h, 63–85%; (vi) Morpholine, Dioxane, MW, 120 °C, 3 h, 80% %; (vii) BBr3, CH2Cl2, −78 °C to rt, Overnight, 61–71%.

Substitution of the hexahydrobenzothienopyrimidine scaffold by N-(3,4-dimethylisoxazol-5-yl)-4-methylbenzenesulfonamide present in ML193 was realized following the procedure illustrated in Scheme 2 to afford compounds 32 and 33.
Scheme 2. Reagents and Conditions: (i) 2-Propanol, 100 °C, Overnight, 51 and 74%.

In order to expand the SAR on the 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine scaffold, an aromatic scaffold version was explored. Thus, as shown in Scheme 3, the benzothienopyrimidines 41 and 42 were prepared from the tetrahydrobenzothiophenes 1 and 34 that were oxidized in dimethyl sulfoxide to give the corresponding 2-aminobenzo[b]thiophene-3-carboxylates 35 and 36.32 Then, procedures described for 21–29 were used in the next steps to give the formation of the final aromatic compounds 41 and 42.
Scheme 3. Reagents and Conditions: (i) DMSO, 190 °C, 18 h, 5–31%; (ii; a) Acetonitrile, Hydrochloric Acid (12.1 M), 100 °C, 75 min, 24–37%; (iii) POCl3, 150 °C, 3.5 h, 61–94%; (iv; a) Furoylpiperazine, Et3N, Dry MeOH, rt for 24 h, (b) 50 °C for 24 h, 10–18%.

Based on the core hopping strategy described in Figure S1, substitutions of the thienopyrimidine tricycle by 6,8-dimethyl-2-(pyridin-2-yl)quinolone (44),33 6-phenylthieno[2,3-d]pyrimidine (45 and 46), 2,5-dimethylpyrimidin-4-ol (50), 3,5-dimethylpyridin-4-one (51), pyrazinone (52), 1-methyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidine (53), and pyrrolo[2,1-f][1,2,4]triazine (54) have been intended. Their syntheses are described in Scheme 4 where the incorporation of furan-2-yl(piperazin-1-yl)methanone on the new scaffolds has been done following the corresponding conditions.
Scheme 4. Reagents and Conditions: (i) 2-Acetylpirydine, KOH (85%), EtOH, Reflux, Overnight, 80%; (ii; a) SOCl2, Reflux, 3 h, (b) 1-(2-Furoyl)piperazine, Et3N, THF, rt, Overnight, 98%; (iii) 1-(2-Furoyl)piperazine, Et3N, Dioxane, Reflux, 24 h, 3–99%; (iii) 1-(2-Furoyl)piperazine, Et3N, Dioxane, Reflux, 24 h, 3–53%; (iv) CH3I, K2CO3, DMF, rt, Overnight, 72%;34 (v) PyBOP (Benzotriazol-1-yloxytripyrrolidinophosphonium Hexafluorophosphate), DBU (1,8-Diazabicyclo[5.4.0]undec-7-ene), Corresponding Heterocycle, ACN, Overnight, rt, 37–48%.
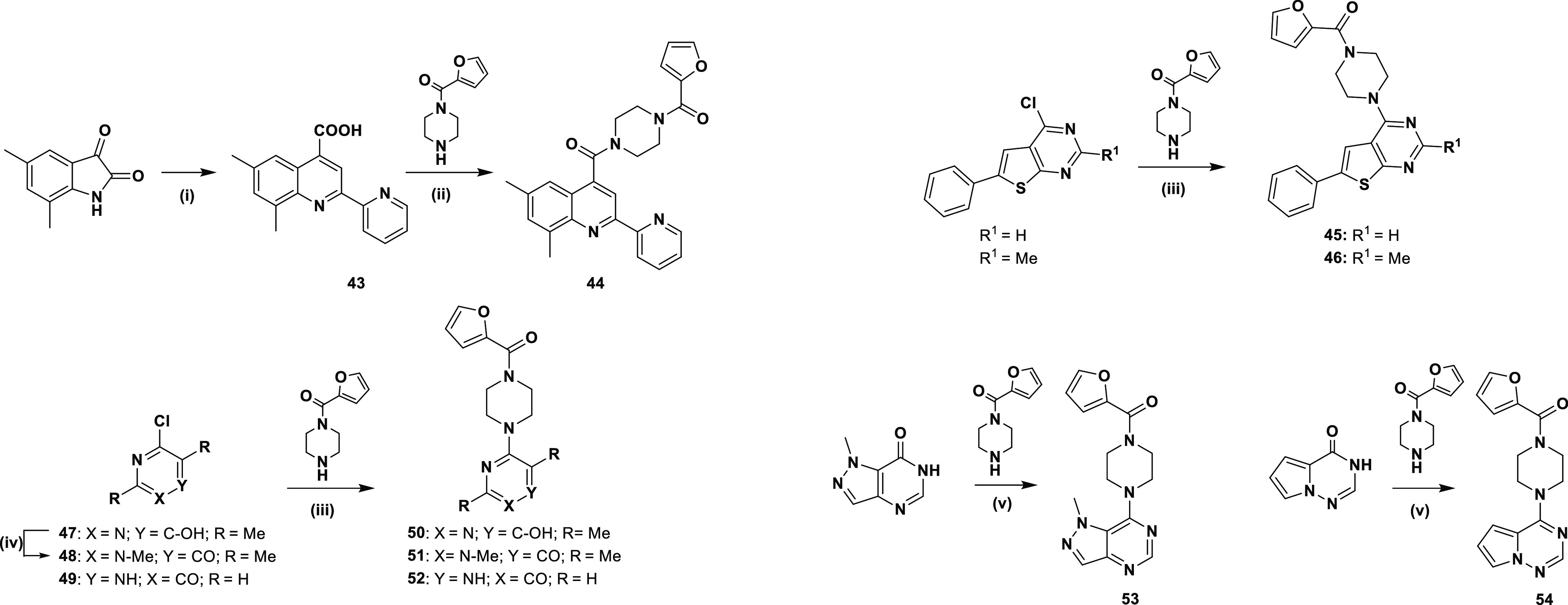
Novel ML192 derivatives were tested using functional GPR55 assays. The activity of compounds 20, 21, 23–33, 41, 42, 44–46, and 50–54 was first assessed using β-arrestin recruitment assays in CHO (Chinese hamster ovary) cells overexpressing human GPR55. Their ability to activate GPR55 by themselves was discarded by the assays. Then, antagonist activity was evaluated by ligand-mediated inhibition of LPI-induced receptor activation using coexposure of LPI with the tested compound. Dose–response experiments upon cotreatment with the agonist LPI (6 μM; EC50) were used to determine their IC50’s (Tables 1 and 2).
Table 1. Activity of ML192; CID3193014; CID655864; and Derivatives 20, 21, and 23–33 Using β-Arrestin Recruitment Assay in CHO Cells Overexpressing Human GPR55.

Data represent the mean of at least three experiments, and 95% confidence intervals (CI) for the IC50 values are given in parentheses.
IC50 (μM)/LPI (5 μM).
https://pubchem.ncbi.nlm.nih.gov/bioassay/2820#section=Protocol. n.e.: No effect up to 10 μM.
Table 2. Activity of ML192 Derivatives 41, 42, 44–46, and 50–54 Using β-Arrestin Recruitment Assay in CHO Cells Overexpressing Human GPR55.

Data represent the mean of at least three experiments, and 95% confidence intervals (CI) for the IC50 values are given in parentheses. n.e.: No effect up to 10 μM.
Table 1 refers to GPR55 activity following structural modifications around the substituents (R1, R2, and R3) of the thienopyrimidine scaffold. The first observation concerns the unsubstituted thienopyrimidine scaffold at R1 and R2 (20) that surprisingly showed a GPR55 antagonist activity similar to the reference GPR55 ligand ML192. Interestingly, R2 accommodates a dimethyl group as illustrated by compound 24 that exhibits significantly improved GPR55 antagonistic activity (submicromolar IC50) compared to the hit ML192. This tendency is not only observed when R3 is a furancarbonylpiperazinyl (24 vs ML192) but also for furancarbonyldiazepinyl (21 vs 25) and dimethylisoxazolylsulfonylanilinyl (32 vs 33) derivatives.
Despite the interesting antagonist activity of the reference GPR55 antagonist CID655864 bearing a cyclopropyl at R1, new substitutions at this position did not show any clear improvement in activity. Hydroxyalkyl derivatives 30 and 31 showed activity in the same range as that of ML192, whereas morpholinylalkyl or methoxyalkyl (26–29) resulted in a loss of activity at GPR55 except when R2 was not substituted (23).
As shown in Table 2, structural modifications of the thienopyrimidine core were able to preserve GPR55 potency in the case of the aromatic counterparts 41 and 42. The replacement with other mono- and bicyclic scaffolds (44–46, 50–54) was not able to show relevant activity.
GPR55 and the cannabinoid receptors CB1 and CB2 share natural and synthetic ligands. Thus, the new compounds described here have been evaluated in competitive binding assays for cannabinoid receptors. None of them showed significant affinity for the cannabinoid receptors (>4 μM; Table S1) showing selectivity for GPR55.
The most promising GPR55 antagonist, 24, was further selected for G protein-dependent functional assays. For this purpose, Serum Response Element (SRE) experiments were performed as previously reported.35 The SRE assay assesses the contribution of the MAPK/ERK signaling pathway for GPR55 activity. As shown in Figure 3, 24 behaves as a weak GPR55 inverse agonist by itself (EC50 = 0.9 (0.3–2.2) μM). Upon cotreatment of the tested compound with LPI, 24 was shown to be a better antagonist of LPI than the hit compound ML192 (24, IC50 = 1.5 (0.9–2.5) μM; ML192, IC50 = 7.5 (3.4–16.5) μM).
Figure 3.

SRE dose response assays of compound 24 (the agonist LPI and the antagonist ML192 were used as reference).
Measure of cell viability is essential for a possible future development of the best GPR55 antagonist 24. Thus, cell viability determined for 24 by using the Biotium XTT cell viability kit showed no cytotoxicity up to 10 μM (Figure S2). In an effort to further understand structural determinants of activity, docking studies of ML192 and 24 (KC52) were performed in our previously published GPR55 inactive state model.35 ML192 docking studies suggest that the nitrogen 1 from the pyrimidine core of the hit compound establishes a hydrogen bond with K(263), while the carbonyl oxygen interacts with Q7.36 (Figure S3).
In addition, the furoyl ring hydrogen bonds with K2.60, and it establishes aromatic stacking interactions with F6.55 and Y3.32 (Figure S3). As previously demonstrated by us, the GPR55 toggle switch residues are Y3.32, M3.36, and F6.48.35 Since ML192 sits above Y3.32, it blocks its ability to change conformation, therefore stabilizing the GPR55 inactive state. Interestingly, when R2 becomes a bulkier substituent, such as a dimethyl group (compound 24), the ligand could fill the empty space (Figure 4A), and the dimethyl may help lock the ligand into place by increasing hydrophobic interactions (with residues D13, G14, Y166, and M167) in the previously unoccupied space. Figure 4B illustrates the GPR55 R/24 complex highlighting the residues involved in the extra hydrophobic interactions in the binding site (Table S2).
Figure 4.

Docking studies of thienopyrimidine derivatives ML192 and 24 in the GPR55 R state (inactive receptor). (A) hGPR55 R/ML192 complex in which the ligand is shown as van der Waals; green tubes highlight residues that establish hydrophobic interactions; purple tubes represent the disulfide bridge formed between the extracellular loops EC3 and EC4. (B) hGPR55 R/compound 24 complex showing the residues establishing hydrophobic interactions with the dimethyl R3 in green tubes.
In this study, we have designed and synthesized a series of novel thienopyrimidine derivatives on the basis of the structural features previously reported for ML192. These compounds were synthesized and evaluated using a β-arrestin recruitment assay in CHO cells overexpressing human GPR55 and βarr2-GFP. Interestingly, compound 24 revealed increased potency and efficacy as a GPR55 antagonist (submicromolar IC50) when compared with the hit ML192. Docking studies in the GPR55 inactive state model allowed us to rationalize key molecular features leading to this activity. In summary, we show here that the combination of structure–activity relationship development and molecular modeling studies has permitted the identification of novel potent GPR55 antagonists that may serve as new tools for studying GPR55.
Acknowledgments
M.E.A., P.H.R., and N.J. are supported by National Institutes of Health grant R01 DA0455698. M.E.A. and P.Z. thank the financial support NIH P30 DA013429. P.M. and N.J. are supported by the Ministry of Science, Innovation, and Universities, Spain (MCIU)/FEDER grant RTI2018-095544-B-I00 and the Spanish National Research Council (CSIC) grant PIE-201580E033. P.M. acknowledges the Comunidad de Madrid (CM) programme “Atraccion de Talento” number 2018-T2/BMD-10819 and “Juan de la Cierva Incorporación Programme-MICIU” (IJC 2019-042182-I).
Glossary
Abbreviations
- LPI
lysophosphatidylinositol
- HTS
high throughput screening (HTS)
- NIH
National Institutes of Health
- SAR
structure–activity relationship
- PAINS
pan assay interference compounds
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- rt
room temperature
- CHO
chinese hamster ovary
- IC50
half maximal inhibitory concentration
- βarr2-GFP
β-arrestin2/green fluorescent protein conjugate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00325.
Synthetic procedures, compound structural characterizations and purity, viability assays for 24 and ML192, experimental procedures for GPR55 β-arrestin recruitment assays, Serum Response Element (SRE) assays, competitive radioligand binding assays in CB1 and CB2 cannabinoid receptors, a table with binding affinity of ML192 derivatives and the reference cannabinoid WIN55, 212–2 for hCB1 and hCB2 cannabinoid receptors, the experimental procedures for scaffold hopping including a workflow scheme, conformational analysis, docking studies, assessment of pairwise interaction energies, and a table with pairwise hydrophobic interaction energies involved in the explored unoccupied region for ML192 and 24 (PDF)
Author Contributions
⊥ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. These authors contributed equally.
National Institutes of Health grants R01 DA0455698 and P30 DA013429.
The authors declare no competing financial interest.
Supplementary Material
References
- Heynen-Genel S.; Dahl R.; Shi S.; Milan L.; Sergienko E.; Hedrick M.; Dad S.; Stonich D.; Su Y.; Chung T. D. Y.; Sharir H.; Caron M. G.; Barak L. S.; Abood M. E.. Screening for Selective Ligands for GPR55-Antagonists. Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (U.S.): Bethesda, MD, 2010. https://www.ncbi.nlm.nih.gov/books/NBK66153/ (accessed Oct 9, 2022). [PubMed] [Google Scholar]
- Saliba S. W.; Gläser F.; Deckers A.; Keil A.; Hurrle T.; Apweiler M.; Ferver F.; Volz N.; Endres D.; Bräse S.; Fiebich B. L. Effects of a Novel Gpr55 Antagonist on the Arachidonic Acid Cascade in LPS-activated Primary Microglial Cells. Int. J. Mol. Sci. 2021, 22 (5), 2503. 10.3390/ijms22052503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawzdargo M.; Nguyen T.; Lee D. K.; Lynch K. R.; Cheng R.; Heng H. H.; George S. R.; O’Dowd B. F. Identification and Cloning of Three Novel Human G Protein-Coupled Receptor Genes GPR52, PsiGPR53 and GPR55: GPR55 Is Extensively Expressed in Human Brain. Mol. brain Res. 1999, 64 (2), 193–198. 10.1016/S0169-328X(98)00277-0. [DOI] [PubMed] [Google Scholar]
- Wise A.; Brown A. J.. Identification of Modulators of GPR55. WO200186305, 2001.
- Drmota T.; Greasley P.; Groblewski T.; Drmota P.; Greasley P.; Groblewski T.. Screening Assays for Cannabinoid- Ligand Type Modulators. WO2004074844, 2004.
- Henstridge C. M.; Balenga N. a B.; Kargl J.; Andradas C.; Brown A. J.; Irving A.; Sanchez C.; Waldhoer M. Minireview: Recent Developments in the Physiology and Pathology of the Lysophosphatidylinositol-Sensitive Receptor GPR55. Mol. Endocrinol. 2011, 25 (11), 1835–1848. 10.1210/me.2011-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. A. The Enigmatic Pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30 (3), 156–163. 10.1016/j.tips.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Morales P.; Jagerovic N. Advances towards the Discovery of GPR55 Ligands. Curr. Med. Chem. 2016, 23 (20), 2087–2100. 10.2174/0929867323666160425113836. [DOI] [PubMed] [Google Scholar]
- Fakhouri L.; Cook C. D.; Al-Huniti M. H.; Console-Bram L. M.; Hurst D. P.; Spano M. B. S.; Nasrallah D. J.; Caron M. G.; Barak L. S.; Reggio P. H.; Abood M. E.; Croatt M. P. Design, Synthesis and Biological Evaluation of GPR55 Agonists. Bioorg. Med. Chem. 2017, 25 (16), 4355–4367. 10.1016/j.bmc.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. J.; Castellano-Pellicena I.; Haslam C. P.; Nichols P. L.; Dowell S. J. Structure-Activity Relationship of the GPR55 Antagonist, CID16020046. Pharmacology 2018, 102, 324–331. 10.1159/000493490. [DOI] [PubMed] [Google Scholar]
- Badolato M.; Carullo G.; Caroleo M. C.; Cione E.; Aiello F.; Manetti F. Discovery of 1,4-Naphthoquinones as a New Class of Antiproliferative Agents Targeting GPR55. ACS Med. Chem. Lett. 2019, 10 (4), 402–406. 10.1021/acsmedchemlett.8b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeder C. T.; Meyer A.; Mahardhika A. B.; Thimm D.; Blaschke T.; Funke M.; Müller C. E. Development of Chromen-4-One Derivatives as (Ant)Agonists for the Lipid-Activated G Protein-Coupled Receptor GPR55 with Tunable Efficacy. ACS Omega 2019, 4 (2), 4276–4295. 10.1021/acsomega.8b03695. [DOI] [Google Scholar]
- Abe J.; Guy A. T.; Ding F.; Greimel P.; Hirabayashi Y.; Kamiguchi H.; Ito Y. Systematic Synthesis of Novel Phosphoglycolipid Analogues as Potential Agonists of GPR55. Org. Biomol. Chem. 2020, 18 (41), 8467–8473. 10.1039/D0OB01756F. [DOI] [PubMed] [Google Scholar]
- Morales P.; Whyte L. S.; Chicharro R.; Gómez-Cañas M.; Pazos M. R.; Goya P.; Irving A. J.; Fernandez-Ruiz J.; Ross R. A.; Jagerovic N. Identification of Novel GPR55 Modulators Using Cell-Impedance-Based Label-Free Technology. J. Med. Chem. 2016, 59, 1840–1853. 10.1021/acs.jmedchem.5b01331. [DOI] [PubMed] [Google Scholar]
- Harada N.; Okuyama M.; Teraoka Y.; Arahori Y.; Shinmori Y.; Horiuchi H.; Luis P. B.; Joseph A. I.; Kitakaze T.; Matsumura S.; Hira T.; Yamamoto N.; Iuni T.; Goshima N.; Schneider C.; Inui H.; Yamaji R. Identification of G Protein-Coupled Receptor 55 (GPR55) as a Target of Curcumin. npj Sci. Food 2022, 6 (1), 4. 10.1038/s41538-021-00119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharir H.; Abood M. E. Pharmacological Characterization of GPR55, A Putative Cannabinoid Receptor. Pharmacol. Ther. 2010, 126 (3), 301–313. 10.1016/j.pharmthera.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M.; Masquelier J.; Muccioli G. G. Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharmacol. Sci. 2018, 39 (6), 586–604. 10.1016/j.tips.2018.02.011. [DOI] [PubMed] [Google Scholar]
- A. Marichal-Cancino B.; Fajardo-Valdez A.; E. Ruiz-Contreras A.; Mendez-Díaz M.; Prospero-García O. Advances in the Physiology of GPR55 in the Central Nervous System. Curr. Neuropharmacol. 2017, 15 (5), 771–778. 10.2174/1570159X14666160729155441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andradas C.; Caffarel M. M.; Pérez-Gómez E.; Salazar M.; Lorente M.; Velasco G.; Guzmán M.; Sánchez C. The Orphan G Protein-Coupled Receptor GPR55 Promotes Cancer Cell Proliferation via ERK. Oncogene 2011, 30 (2), 245–252. 10.1038/onc.2010.402. [DOI] [PubMed] [Google Scholar]
- Sutphen R.; Xu Y.; Wilbanks G. D.; Fiorica J.; Grendys E. C.; LaPolla J. P.; Arango H.; Hoffman M. S.; Martino M.; Wakeley K.; Griffin D.; Blanco R. W.; Cantor A. B.; Xiao Y.; Krischer J. P. Lysophospholipids Are Potential Biomarkers of Ovarian Cancer. Cancer Epidemiol. Biomarkers Prev. 2004, 13 (7), 1185–1191. 10.1158/1055-9965.1185.13.7. [DOI] [PubMed] [Google Scholar]
- Ford L. A.; Roelofs A. J.; Anavi-Goffer S.; Mowat L.; Simpson D. G.; Irving A. J.; Rogers M. J.; Rajnicek A. M.; Ross R. a. A Role for L-Alpha-Lysophosphatidylinositol and GPR55 in the Modulation of Migration, Orientation and Polarization of Human Breast Cancer Cells. Br. J. Pharmacol. 2010, 160 (3), 762–771. 10.1111/j.1476-5381.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñeiro R.; Maffucci T.; Falasca M. The Putative Cannabinoid Receptor GPR55 Defines a Novel Autocrine Loop in Cancer Cell Proliferation. Oncogene 2011, 30 (2), 142–152. 10.1038/onc.2010.417. [DOI] [PubMed] [Google Scholar]
- Staton P. C.; Hatcher J. P.; Walker D. J.; Morrison A. D.; Shapland E. M.; Hughes J. P.; Chong E.; Mander P. K.; Green P. J.; Billinton A.; Fulleylove M.; Lancaster H. C.; Smith J. C.; Bailey L. T.; Wise A.; Brown A. J.; Richardson J. C.; Chessell I. P. The Putative Cannabinoid Receptor GPR55 Plays a Role in Mechanical Hyperalgesia Associated with Inflammatory and Neuropathic Pain. Pain 2008, 139 (1), 225–236. 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Akimov M. G.; Gamisonia A. M.; Dudina P. V.; Gretskaya N. M.; Gaydaryova A. A.; Kuznetsov A. S.; Zinchenko G. N.; Bezuglov V. V. GPR55 Receptor Activation by the N-Acyl Dopamine Family Lipids Induces Apoptosis in Cancer Cells via the Nitric Oxide Synthase (NNOS) over-Stimulation. Int. J. Mol. Sci. 2021, 22 (2), 622. 10.3390/ijms22020622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba S. W.; Jauch H.; Gargouri B.; Keil A.; Hurrle T.; Volz N.; Mohr F.; van der Stelt M.; Bräse S.; Fiebich B. L. Anti-Neuroinflammatory Effects of GPR55 Antagonists in LPS-Activated Primary Microglial Cells. J. Neuroinflammation 2018, 15 (1), 322. 10.1186/s12974-018-1362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgaz S.; García C.; Gonzalo-Consuegra C.; Gómez-Almería M.; Ruiz-Pino F.; Unciti J. D.; Gómez-Cañas M.; Alcalde J.; Morales P.; Jagerovic N.; Rodríguez-Cueto C.; de Lago E.; Muñoz E.; Fernández-Ruiz J. Preclinical Investigation in Neuroprotective Effects of the GPR55 Ligand VCE-006.1 in Experimental Models of Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Molecules 2021, 26 (24), 7643. 10.3390/molecules26247643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudurí E.; López M.; Diéguez C.; Nadal A.; Nogueiras R. GPR55 and the Regulation of Glucose Homeostasis. Int. J. Biochem. Cell Biol. 2017, 88, 204–207. 10.1016/j.biocel.2017.04.010. [DOI] [PubMed] [Google Scholar]
- Fondevila M. F.; Fernandez U.; Gonzalez-Rellan M. J.; Da N.; Lima S.; Buque X.; Gonzalez-Rodriguez A.; Alonso C.; Iruarrizaga-Lejarreta M.; Delgado T. C.; Varela-Rey M.; Senra A.; Garcia-Outeiral V.; Novoa E.; Iglesias C.; Porteiro B.; Beiroa D.; Folgueira C.; Tojo M.; Torres J. L.; Hernández-Cosido L.; Blanco Ó.; Arab J. P.; Barrera F.; Guallar D.; Fidalgo M.; López M.; Dieguez C.; Marcos M.; Martinez-Chantar M. L.; Arrese M.; Garcia-Monzon C.; Mato J. M.; Aspichueta P.; Nogueiras R. The L-α-Lysophosphatidylinositol/G Protein-Coupled Receptor 55 System Induces the Development of Nonalcoholic Steatosis and Steatohepatitis. Hepatology 2021, 73 (2), 606–624. 10.1002/hep.31290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abaee M. S.; Cheraghi S. Efficient Three-Component Gewald Reactions under Et3N/H2O Conditions. J. Sulfur Chem. 2014, 35 (3), 261–269. 10.1080/17415993.2013.860141. [DOI] [Google Scholar]
- Bogolubsky A. V.; Ryabukhin S. V.; Plaskon A. S.; Stetsenko S. V.; Volochnyuk D. M.; Tolmachev A. A. Dry HCl in Parallel Synthesis of Fused Pyrimidin-4-Ones. J. Comb. Chem. 2008, 10 (6), 858–862. 10.1021/cc800074t. [DOI] [PubMed] [Google Scholar]
- Sleebs B. E.; Nikolakopoulos G.; Street I. P.; Falk H.; Baell J. B. Identification of 5,6-Substituted 4-Aminothieno[2,3-d]Pyrimidines as LIMK1 Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21 (19), 5992–5994. 10.1016/j.bmcl.2011.07.050. [DOI] [PubMed] [Google Scholar]
- Adib M.; Soheilizad M.; Rajai-Daryasaraei S.; Mirzaei P. An Efficient Aromatization of 2-Amino-4,5,6,7-Tetrahydrobenzo-[b]Thiophene-3-Carboxylates in Dimethyl Sulfoxide Catalyzed by p-Toluenesulfonic Acid. Synlett 2015, 26 (08), 1101–1105. 10.1055/s-0034-1379998. [DOI] [Google Scholar]
- Li J.; Chen J.; Gui C.; Zhang L.; Qin Y.; Xu Q.; Zhang J.; Liu H.; Shen X.; Jiang H. Discovering Novel Chemical Inhibitors of Human Cyclophilin A: Virtual Screening, Synthesis, and Bioassay. Bioorg. Med. Chem. 2006, 14 (7), 2209–2224. 10.1016/j.bmc.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claremon D. A.; Zhuang L.; Leftheris K.; Ye Y.; Singh S. B.; Himmelsbach F.. Cyclic Inhibitors of 11beta-Hydroxysteroid Dehydrogenase. WO2009134387, 2009.
- Lingerfelt M. A.; Zhao P.; Sharir H. P.; Hurst D. P.; Reggio P. H.; Abood M. E. Identification of Crucial Amino Acid Residues Involved in Agonist Signaling at the GPR55 Receptor. Biochemistry 2017, 56 (3), 473–486. 10.1021/acs.biochem.6b01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.