

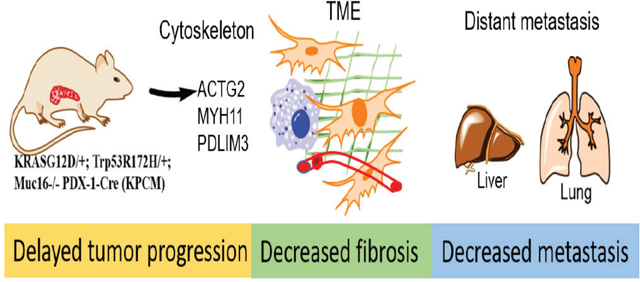
Abstract
MUC16, membrane-bound mucin, plays an oncogenic role in pancreatic ductal adenocarcinoma (PDAC). However, the pathological role of MUC16 in the PDAC progression, tumor microenvironment, and metastasis in cooperation with KrasG12D and Trp53R172H mutations remains unknown. Deletion of Muc16 with activating mutations KrasG12D/+ and Trp53R172H/+ in mice significantly decreased progression and prolonged overall survival in KrasG12D/+; Trp53R172H/+; Pdx-1-Cre; Muc16−/− (KPCM) and KrasG12D/+; Pdx-1-Cre; Muc16−/− (KCM), as compared to KrasG12D/+; Trp53R172H/+; Pdx-1-Cre (KPC) and KrasG12D/+; Pdx-1-Cre (KC) mice, respectively. Muc16 knockout pancreatic tumor (KPCM) displays decreased tumor microenvironment factors and significantly reduced incidence of liver and lung metastasis compared to KPC. Furthermore, in silico data analysis showed a positive correlation of MUC16 with activated stroma and metastasis- associated genes. KPCM mouse syngeneic cells had significantly lower metastatic and endothelial cell binding abilities than KPC cells. Similarly, KPCM organoids significantly decreased the growth rate than KPC organoids. Interestingly, RNA-seq data revealed that the cytoskeletal proteins Actg2, Myh11, and Pdlim3 were downregulated in KPCM tumors. Further knockdown of these genes showed reduced metastatic potential. Overall, our results demonstrate that Muc16 alters the tumor microenvironment factors during pancreatic cancer progression and metastasis by changing the expression of Actg2, Myh11, and Pdlim3 genes.
Keywords: MUC16, pancreatic ductal adenocarcinoma, metastasis, tumor microenvironment, mouse models
Graphical Abstract
Introduction
Pancreatic adenocarcinoma (PDAC) is one of the most aggressive and metastatic diseases, with a 5-year survival rate of ~10% (1). Approximately 80% of patients with PDAC have locally advanced or metastatic disease at initial diagnosis, making it a challenging condition (2, 3). This poor prognosis is due to the lack of early biomarkers and intense therapeutic resistance (4). Several studies have demonstrated that KRAS mutation occurs early in the development of PDAC, whereas deletion or inactivation of TP53 occurs during the late stages and metastasis (5, 6). Emerging studies suggest that additional genetic events and tumor microenvironment factors contribute to PDAC development and metastasis (7, 8). However, the detailed understanding and mechanistic events of PDAC metastasis are unknown.
Various oncogenes play a critical role in pancreatic cancer initiation, progression, and metastasis (5, 9, 10). Several studies have shown that mucins expression is drastically altered in various cancers, including PDAC tumor growth and metastasis (10–13). Cancer antigen 125/MUC16 is a high molecular weight glycoprotein, highly expressed in PDAC, and associated with a poor prognosis (14–16). MUC16 has also been associated with enhanced tumorigenesis and metastasis (14, 16–18). In addition, MUC16 is involved in cancer stem cell enrichment and chemoresistance in pancreatic cancer by regulating JAK2 and Lmo2 (19). Mechanistically, MUC16 interacts with multiple proteins such as mesothelin (20), JAK2 (17), Src (21), FAK (22) and Siglec 9 (23), facilitating tumor growth and metastasis in various cancers, suggesting that MUC16 could play a critical role in determining the therapy response and metastasis.
Genetically engineered mouse (GEM) models are essential for identifying the molecular mechanisms associated with disease progression and metastasis (24). Various mice models have been developed to understand the biology of pancreatic cancer by activating KrasG12D and Trp53R172H mutations under the influence of pancreas-specific recombinase (Cre) (Pdx-1 Cre or Ptf1a-Cre) (24–28). The KPC mouse model is well-established as it recapitulates the biology of human pancreatic cancer (29). We previously reported that the KrasG12D; Ptf1a-CreERTM (iKC) pancreatic mouse model showed increased expression of Muc16 and was associated with disease progression (30). In this study, we have developed the Muc16 knockout (Muc16−/−) pancreatic cancer mouse models by crossing with well-established KPC (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre) and KC (KrasG12D/+; Pdx-1-Cre) to generate composite KPCM (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre; Muc16−/−) and KCM (KrasG12D/+; Pdx-1-Cre; Muc16−/−) mouse models. We investigated the difference in the development of precancerous lesions, survival, tumor stage, and metastatic phenotype between KPCM and KCM mice lacking Muc16. We also explored the impact of Muc16 loss on the modulation of tumor microenvironment factors associated with metastasis.
Materials and Methods
Muc16 mouse model generation and tissue collection
To generate the Muc16 knockout model, KPC-LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1Cre mice were crossed with Muc16−/− mice. The LSL- KrasG12D/+, LSL-Trp53R172H/+(B6;129S4- Trp53tm2Tyj/J), Muc16−/−, and Pdx-1 Cre mouse strains (C57BL/6J) were used (27). These strains were first bred to generate the genotypes LSL-KrasG12D/+; Trp53R172H/+(KP) and Muc16−/−; Pdx-1- cre (MC). The KP mice were intercrossed with MC mice to produce the experimental cohorts, KPC-LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1 Cre, and KPCM-LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1 Cre/+; Muc16−/−), KC-LSL-KrasG12D/+; Pdx-1 Cre, and KCM-LSL-KrasG12D/+; Pdx-1Cre; Muc16−/−).The Muc16 whole-body knockout model (C57BL/6J) mice (Gifted by Richard R. Behringer) (31). Mice were supplied with food and water and subjected to 12 hrs light/dark cycle. The pancreatic tumor and metastatic tissue harvested from each mouse were grossly macro-dissected from surrounding normal tissues and divided into three equal regions at the experimental endpoint. The first part was immediately snap-frozen in liquid nitrogen and used for RNA isolation. The other two parts were used for IHC and protein studies, respectively. We performed RNA sequence analysis from KPCM and KPC pancreatic tumor tissues and respective controls (KPC and KC). The mouse studies were conducted by the US Public Health Service Guidelines for the Care and Use of Laboratory Animals under an approved protocol by the Institutional Animal Care and Use Committee, University of Nebraska Medical Center.
Gene silencing by Small-Interfering RNA
Transient knockdown (KD) of Actg2 siRNA (cat# SR412703), Pdlim3 siRNA (cat# SR410257), and Myh11 siRNA (cat# SR423332, OriGene, Rockville, MD, USA) were used to silence Actg2, Pdlim3, and Myh11 in KPC and KPCM mice tumor-derived syngeneic cells. Experiments or cell lysate were collected after 48 hrs of post-transfections.
DNA extraction and genotype
Using a DNA extraction machine (Maxwell RSC48, Promega, Madison, WI, USA), genomic DNA was extracted (cat# AS1610) from the tail of various strains. The genotype of each gene was analyzed by PCR technique using specific primers (Supplementary Table 1) and confirmed by gel electrophoresis. Further, genotype was established for KPCM, KCM, KPC, and KC mice.
Cell and organoid culture
KPCM and KPC cell lines or organoids were established from tumor tissues of pancreatic adenocarcinoma derived from KPCM and KPC mice (32–34). In brief, tumor samples were chopped and digested enzymatically using digestion media containing 100 mg collagenase II (cat# C7657, Millipore Sigma, Burlington, MA, USA), 20 mg Dispase II (cat# 17105041, ThermoFisher, Carlsbad. CA, USA), and 1% FBS. After digestion, the cells were washed with wash buffer containing 1 M Hepes, 100X Glutamine, 1X primocin, and 10 % FBS and centrifuged at 200 RCF for 5 mins. After digestion and washing, the cell pellet was suspended in 50 ul of matrigel (cat# 356255, Corning, Kennebunk, ME, USA) and plated as the dome in 48 well plates. Once the matrigel domed solidified after 15 to 20 mins of incubation, the organoids were supplemented with 200 ul of pre-warmed organoids specific media [Wnt3A (1X), R-Spondin (1X), B27 Supplement (1X), Nicotinamide (10 mM), N-N-acetylcysteine (1.25 mM), Primocin (100 ug/ml), Noggin (100 ng/ml), EGF (50 ng/ml), FGF (100 ng/ml), Gastrin I (10 nM), A83–01 (500 nM)]. Real-time images of the organoids were captured every 3 hours for 48 hours using the IncuCyte-S5 live-cell imaging system (Sartorius). The kinetic data (organoid average growth and darkness) were analyzed and graphically represented using IncuCyte software (Sartorius). Mouse CAFs were isolated from KPC mice pancreas by enzymatic digestion and further enriched by differential trypsinization method for subsequent ten passages to get rid of epithelial and cancer cells. Isolated cells were seeded in high dilution in 96 well plate single cells/well. Single colonies were expanded, characterized for expression of various CAF-specific markers such as α-SMA, Fibronectin, fibroblast activation protein (FAP), and Vimentin, and the absence of epithelial cell marker CK19 was also confirmed (35). CAFs were maintained in 10% FBS + RPMI media at 37°C and 5% CO2. KPCM, KPC, and human (Capan1 and SW1990) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were then incubated at 37°C in a humidified atmosphere with 5% CO2.
3D Spheroid culture
Tumor cells (KPCM and KPC cells) and CAFs cells were cultured as monolayers. Then cells were counted, resuspended in a 1:2 ratio (tumor cells & CAFs), and seeded in a low attachment plate in DMEM high glucose, supplemented with 10% FBS. These 3D spheroid cultures were allowed for ten days, and then the spheroid pellet was collected for the paraffin-embedded process.
Endothelial and p-selectin binding assays
Mouse bone marrow endothelial cell-1 (BMEC1) cells were cultured in a 48-well plate to form a uniform monolayer. Recombinant mouse p-selectin protein (10 μg/mL) was coated in 96 well plates, the protocol followed by Li et al. (36). Tumor cells (KPCM and KPC) were then pre-labeled with a fluorescent dye (Cat# CBA215, Cell Biolabs, Inc. San Diego, CA, USA) and added to endothelial cells or p-selectin coated plates and incubated for 90 minutes. After incubation, cells were gently washed and photographed using a fluorescence microscope, and 96 well plates were read in the fluorescence plate reader at 480/520nm.
Bioinformatic analysis
RNAseq processed data from tumor-stromal subtypes (also known as Moffitt data; n = 357) was obtained from the NCBI GEO repository (GSE71729). Only primary PDAC samples (n = 145) with annotation of stromal subtype were considered. The sample sizes with stromal subtype annotations of Low, Normal, and Activated are 22, 39, and 84. The heatmap of collagen/SMC markers (COL11A1, COL10A1, COL3A1, COL5A1, FN1, PDPN & SPARC) was made with respect to the stromal subtypes using the heatmap R package, and hierarchical clustering using R cluster function was used for intragroup clustering independently. The pairwise t-test with FDR correction evaluated the statistical significance between gene expression and the stromal subtype. Similarly, we used human scRNA data (NCBI accession number: GSE155698) comprising 14698 cells (of 41178 cells) from 15 PDAC patients were preprocessed and were subsequently normalized using standard (logarithmic) normalization with scale factor of 10000. The top 2000 highly variable features identified using Variance Stabilizing Transformation (VST) were used for principal component analysis (PCA). We used the first 35 principal components for cell type clustering, including fibroblast and epithelial cells.
Data analysis
Statistical significance was evaluated with the Student t-test using GraphPad Prism 8.1.2 software. Differences between groups were statistically significant when the P-value was less than 0.05. All experiments were performed in triplicate.
Additional material and methods are available in the supplementary information.
Results
Loss of Muc16 exhibits reduced PDAC initiation, progression, and the increased survival of mice
To understand the role of Muc16 in PDAC initiation, progression, and metastasis, Muc16−/− mice were crossed with KPC (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre) and KC (KrasG12D/+; Pdx-1-Cre) mice. Intercrossing of their F1 progeny resulted in Muc16 (Muc16−/−) knockout mice with KPC (KPCM) (Supplementary Figure 1A). Muc16 knockout was confirmed by the RNA scope method (Supplementary Figure 1B). We have generated mice with various genotypes such as KPCM (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre; Muc16−/−) and KCM (KrasG12D/+; Pdx-1-Cre; Muc16−/−) and respected control models such as KPC (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre) and KC (KrasG12D/+; Pdx-1-Cre). KPCM, KCM, and age-matched controls (KPC and KC) were euthanized at various time-point (15–50 weeks). Pancreas tissues and vital organs (liver, peritoneum, intestine, spleen, and lung, were collected and analyzed for tumor incidence.
In these models, we observed that the KPCM and KCM mice showed relatively low pancreatic intraepithelial neoplasia (PanIN) lesions than the KPC and KC mice. For instance, KC mice appear to show precancerous lesions at 25 weeks and fully developed PDAC at 50 weeks, while KCM mice developed precancerous lesions at 40 weeks and later developed PDAC (Figure 1A). Similarly, KPC mice showed precancerous lesions (PaNIII) at 15 weeks and tumors developed at 25 weeks, whereas KPCM mice showed precancerous lesions (PaNIII) at 25 weeks (Figure 1B). This finding suggests that the loss of Muc16 delays the initiation and progression of PDAC.
Figure 1. Establishment of Muc16 mediated pancreatic cancer mouse models.
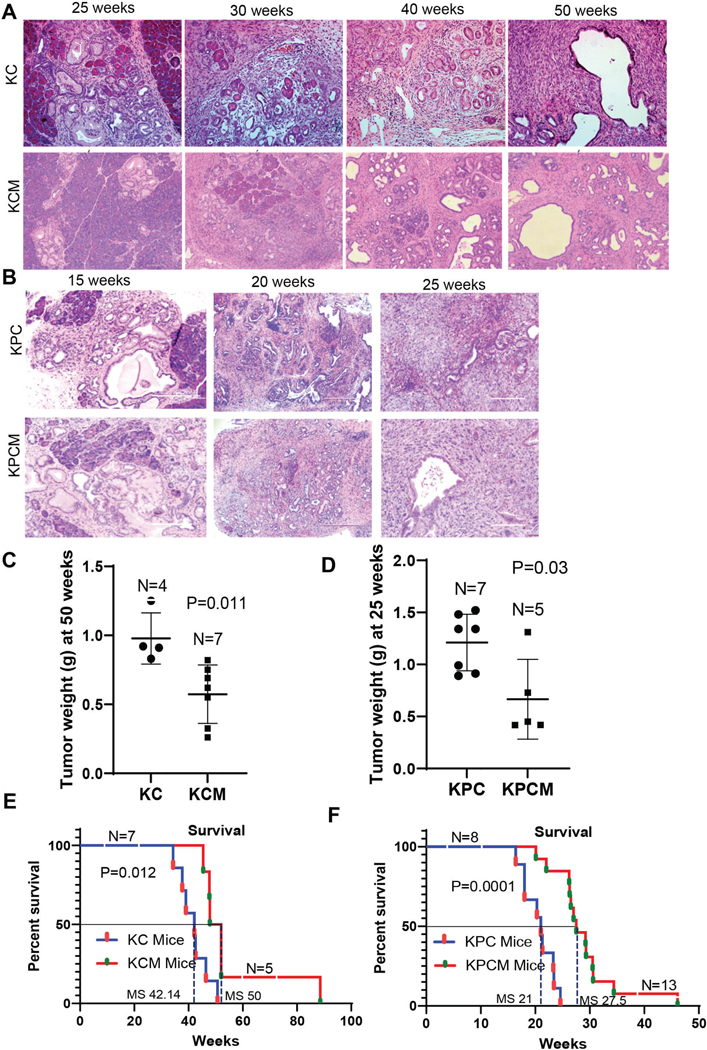
(A) Representative HSE images of Muc16 knockout in Kras background KCM (KrasG12D/+; Pdx-1-Cre; Muc16−/−) mice displayed delayed precancerous formation compared to KC (KrasG12D/+; Pdx-1-Cre), 50 weeks KC mice showing PDAC, whereas 50 weeks KCM showing only PaNIII. (B) Similarly, KPCM (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre; Muc16−/−) also showed delayed precancerous lesions and PDAC development compared to KPC mice (KrasG12D/+; Trp53R172H/+; Pdx-1-Cre). (C & D) Pancreatic tumor weight of KCM (n=7) and KPCM (n=5) as compared to KC (n=4) and KPC (n=7), respectively. (E & F) Kaplan-Meier survival curves display the overall survival of KCM (n=5) and KPCM (n= 13) as compared to KC (n=7) and KPC (n=8), respectively. Median survival (MS) was observed in KC 42.14, KPC 21, KCM 50, and KPCM 27.5 weeks.
Muc16 deficient mice display increased survival in the PDAC model
Next, we evaluated pancreatic tumor weight between Muc16 knockout KC and KPC composite mice. The results indicate that the overall weight of pancreatic tumors in KCM & KPCM mice was significantly (P=0.011, P=0.03) lesser than KC and KPC tumors, respectively (Figure 1C & D). Further, the overall survival analysis revealed that KCM (N=5) tumor-bearing mice had significantly longer overall survival (P=0.012) than KC (N=7) mice (Figure 1E), with a median survival (MS) of KC 42.14 and KCM 50 weeks. Similarly, tumor-bearing KPCM (N=13) mice (P=0.0001) showed significantly longer survival compared to KPC (N=8) mice (Figure 1F), with median survival, KPC 21, and KPCM 27.5 weeks, suggesting that Muc16 is associated with PDAC pathogenesis. These findings indicate that Muc16 plays a critical role in pancreatic cancer development and pathogenesis.
Muc16 loss diminishes pancreatic cancer metastasis
Next, we analyzed the metastatic potential of Muc16 in pancreatic cancer mouse models; the results indicate that Muc16 knockout mice with KC background (KCM) (N=6) did not develop any metastasis, but KC (N=6) mice showed liver metastasis (Figure 2A). Similarly, tumors of KPCM mice had significantly less propensity toward liver (0/7), lung (0/7), peritoneum (0/7), intestinal (1/7, 14.28%) metastasis compared to KPC mice with liver (4/7, 57.14%), lung (3/7, 42.86%), peritoneum (3/7, 42.86%), and intestinal (2/7, 28.57%) metastases (Figure 2B & C). We have also analyzed the metastatic incidence of the individual mouse, where we noticed that liver and lung metastasis is more common in KPC mice than in other models (Figure 2D). The representative histology of pancreatic liver metastasis in KPC mice and no liver metastasis was observed in KPCM mice, as shown in Figure 2E. Since Muc16 significantly enhanced the incidence of PDAC metastasis, we analyzed the pancreatic cancer patients’ dataset (GSE42952) for MUC16 expression. GSE dataset revealed that the significantly (P=0.0162) elevated expression of MUC16 was associated with pancreatic cancer liver metastasis (Figure 2F). Furthermore, our results of QT-PCR analysis showed that Muc16 expression is significantly (***P<0.001) high in liver and lung metastasis tissues than in primary PDAC tumors (Supplementary Figure 2A). In addition, our western blot results revealed that expression of mesenchymal markers N-cadherin and Twist were down-regulated in KPCM compared to KPC. The expression of the epithelial marker cytokeratin18 did not affect both KPC and KPCM tumor tissues. These results suggest that MUC16 mediated epithelial to mesenchymal transition (EMT) in pancreatic cancer (Supplementary Figure 2B).
Figure 2. Muc16 knockout mice displayed reduced PDAC metastasis.
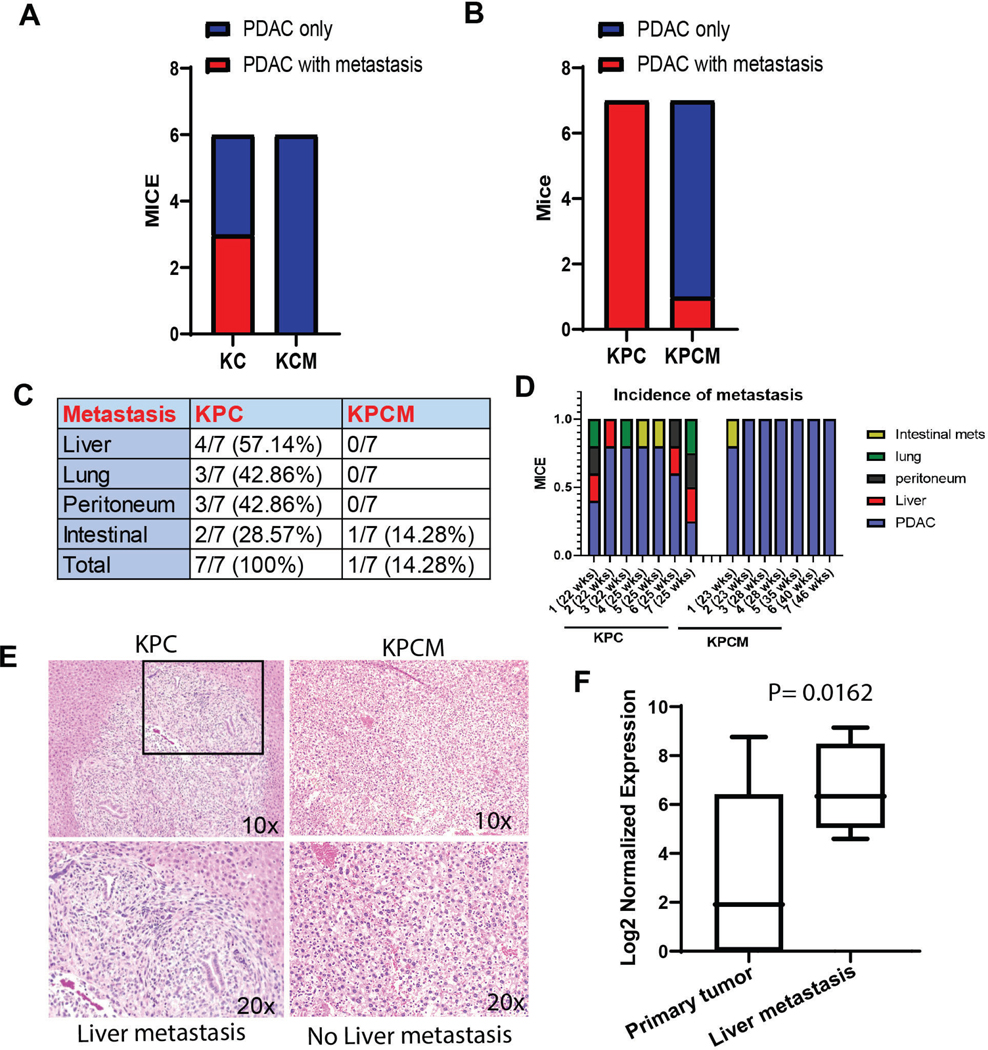
(A & B) Muc16 knockout mice (KPCM and KCM) showed significantly reduced pancreatic cancer metastatic incidence compared to the control mice (KPC and KC). (C) The table shows the percentage of metastasis in KPCM and KPC mice. (D) The bar diagram displays the individual mouse with various metastatic incidences. (E) Histology of liver metastasis of KPC control and KPCM mice. (F) The human patient’s GSE42952 dataset shows that MUC16 is significantly elevated in liver metastatic (number of samples 7) tissues than in PDAC (number of samples 23).
Muc16 contributes to fibrosis formation in pancreatic tumor
Fibrosis and extracellular matrix deposition are high in pancreatic tumors, critical in disease aggressiveness, and resistant to therapy. This study aimed to analyze whether Muc16 affects extracellular matrix (ECM) deposition or stromal levels in the pancreatic tumor microenvironment. We have performed Masson’s trichrome staining method to identify the fibrosis and collagen content in pancreatic tumor tissues (37). The pathological evaluation of trichrome-stained tissues indicates that KPCM tumor tissues displayed significantly (P=0.002) decreased fibrosis and collagen content compared to KPC tumor tissues (Figure 3A). Furthermore, the confocal experiment shows that αSMA, a fibroblast marker, and Ki67, a proliferation marker, were significantly (P<0.011; P<0.0004) decreased in KPCM tumor tissues compared to the KPC tumors (Figure 3B).
Figure 3. Muc16 decreased the fibrosis and ECM content in pancreatic tumors.
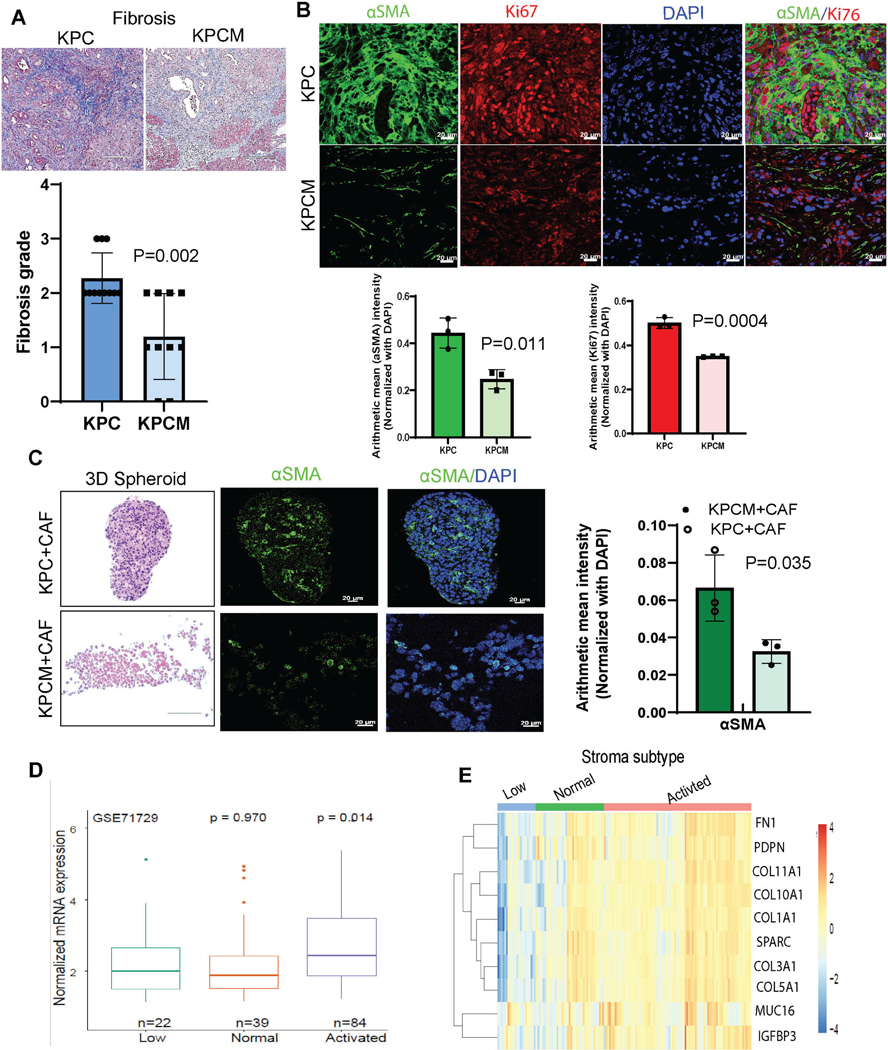
(A) Representative image of Masson’s trichrome staining demonstrates the content of connective tissues or fibrosis in Muc16 knockout pancreatic tumors (KPCM) (number of samples 10) and control (KPC) (number of samples 11), values in bar graphs indicate a mean ± S.D. of experimental replicates. (B) Representative co-immunofluorescence confocal images show the stromal marker, αSMA, and proliferation marker Ki67 in KPCM (number of tumor samples 3) pancreatic tumor tissues compared to KPC (number of tumor samples 3) tumors. Values in bar graphs indicate a mean ± SD of three experimental replicates (C) Co-culture of KPCM with cancer-associated fibroblasts (CAFs) 3D spheroid was stained with H&E and immunofluorescence staining of the stromal marker αSMA and their quantification. (D) The patients’ data set (GSE71729) shows the boxplot representing MUC16 expression across different stromal subtypes of primary PDAC samples. (E) Heatmap illustrates the expression of MUC16 and associated collagen/smooth muscle markers across different stromal subtypes in human primary PDAC samples. The annotation bar indicates the human PDAC tumor with different stromal subtypes denoted as follows low or no stroma (top-left; blue color), Normal stroma (top-middle; green color), and Activated stroma (top-right; pink color).
In addition, we have performed 3D spheroid co-culture assays by culturing cancer cells (KPC or KPCM) with mouse cancer-associated fibroblasts (CAFs) (Supplementary Figure 3A & B). The hematoxylin and eosin analysis indicate Muc16 knockout (KPCM) and KPC cells with CAFs 3D spheroid co-culture (Figure 3C). Next, our confocal images represent the expression of αSMA is significantly (P=0.035) less in KPCM/CAFs spheroids than in KPC/CAFs (Figure 3C). Next, we wanted to analyze the clinical correlation of MUC16 with pancreatic cancer fibrosis. The publicly available data set (GSE71729) showed that MUC16 levels were significantly (P=0.014) higher in the activated stroma containing human pancreatic tumor tissues (Figure 3D). In primary PDAC samples, the expression of MUC16 was associated with collagen/smooth muscle cell markers such as FN1, PDPN, COL11A1, COL10A1, COL1A1, SPARC, COL3A1, COL5A1, and IGFBP3 in different stromal subtypes (Figure 3E). Our in silico analysis (GSE155698) indicated that MUC16 is primarily expressed in pancreatic cancer cells but not fibroblasts (Supplementary Figure 3C). However, the data indicate that the activated stromal area has higher MUC16 expression in tumor cells, suggesting crosstalk between MUC16 expressing tumor cells and stromal factors during pancreatic cancer development.
Muc16 enhances endothelial cell and p-selectin binding in PDAC
We have developed mouse pancreatic syngeneic cell lines (Supplementary Figure 4A) and organoids from KPC and KPCM models. KPC and KPCM cell lines were used to identify the metastatic properties and other functional assays. Here, we have investigated whether loss of Muc16 affects the binding ability of cancer cells with endothelial cells (ECs). Our results showed that KPCM cells had significantly (***P<0.001) reduced endothelial cell binding affinity than KPC tumor cells (Figure 4A). In addition, we also studied the binding properties of KPCM tumor cells and p-selectin. Our results showed a significantly (*P<0.05) reduced p-selectin binding effect of KPCM cells when compared to control KPC cells (Figure 4B). Overall, these results suggested that the Muc16 promotes the interaction of PDAC cells with endothelial cells and selectins during the metastatic spread of PDAC cells.
Figure 4. Muc16 in endothelial cell binding, p-selectin, colony formation, and cell migration abilities in pancreatic cancer.

(A) The bar diagram represents that KPCM cells showed a less binding affinity with mouse-specific endothelial cells. (B) Muc16 knockout (KPCM) cells had significantly reduced binding affinity to p-selectin. (C&D) Muc16 knockout (KPCM) cells showed substantially less colony formation and migration abilities than KPC cell lines. (E) KPCM organoids had significantly less proliferation capacity compared to KPC organoids. (F) Confocal microscopy shows the decreases in the proliferation marker Ki67 in organoids derived from KPCM pancreatic tumors compared to KPC tumors. Values in bar graphs indicate a mean ± SD of three replicates (***P<0.001, **P<0.01, *P<0.05) obtained through the student’s t-test.
Muc16 promotes colony formation and migration abilities
Next, we wanted to examine the metastatic properties of KPCM cells. Our results showed that the KPCM pancreatic syngeneic cells showed a significantly (**P<0.01) decreased colony formation ability compared to KPC cells (Figure 4C). Our results have shown that migratory potential was significantly (***P<0.001) reduced in KPCM cells when compared to KPC cells (Figure 4D). We further tested the colony formation ability of KPCM cells using a transwell migration assay. These results confirmed that Muc16 contributes to the clonogenic and migratory potential, which helps PDAC cells metastasize to distant organs.
Effect of Muc16 on pancreatic cancer organoid growth
We have also developed organoids from KPC and KPCM mouse models. Organoids are excellent in vitro models for studying the tumor microenvironment, including tumor cells, tumor-associated fibroblasts, and endothelial cells. Here, we performed the proliferation assay using the Incucyte® live imaging system at various time points. The results showed that the KPCM organoid shows significantly (***P<0.001) reduced growth than the KPC organoid (Figure 4E). Confocal analysis indicates that Ki67, a proliferation marker, was drastically (**P<0.01) decreased in KPCM tumor-derived organoids from KPCM compared to the organoids from KPC (Figure 4F). These results strongly suggest that the KPCM models showed significantly reduced organoid growth activity compared to the KPC models.
Muc16-associated gene signature in pancreatic cancer
To determine the Muc16-associated molecular mechanism in PDAC, we performed RNA sequence analysis using Muc16 knockout pancreatic tumor tissues (KPCM) and control pancreatic cancer tissues (KPC). The result showed that the expression of Clca1, Eno1b, Actg2, Myh11, Pdlim3, Ppargc1a, Zswim5, Des, Synpo2, Vil1, Chdh, Atp8a2, Rragd, Synm, Gprin3, Scg2, and Rnf144b were significantly down-regulated in KPCM tumors as compared to KPC tumors (Figure 5A). Furthermore, we have analyzed the expression of these genes in pancreatic cancer patients’ samples using the TCGA dataset. We observed that the expression of genes ACTG2, MYH11, PDLIM3, RNF144B, SYNPO2 and VIL1 were significantly (*P<0.05) high in PDAC patients’ samples (N=179) as compared to healthy individuals (N=171) (Figure 5B-G).
Figure 5. Transcriptome analysis, correlation of MUC16 and its gene signature in PDAC.

(A) Global transcriptome (RNA-seq) analysis indicated that expression of Clca1, Eno1b, Actg2, Myh11, Pdlim33, Ppargc1a, Zswim5, Des, Synpo2, Vil1, Chdh, Atp8a2, Rragd, Synm, Gprin3, Scg2, and Rnf144b were significantly downregulated in KPCM tumors as compared to KPC as indicated by heatmap. (B-G) TCGA patient data set showed that the ACTG2, MYH11, PDLIM3, RNF144B, SYNPO2, and VIL1 were significantly higher in PDAC patients than in healthy individuals. (H-J) The patients’ data set of pancreatic cancer indicates that MUC16 is correlated substantially with ACTG2, MYH11, and PDLIM3, suggesting that these genes may associate with MUC16 during the development of pancreatic cancer. (K) Gene ontology pathway analysis indicated that MUC16 and its gene signature are associated with the cytoskeleton, cell motility, endothelial cell proliferation, and migration.
Correlation of MUC16 and its gene signature in PDAC
Next, we performed the correlation analysis using pancreatic cancer patients’ samples (TCGA). The results showed that the MUC16 significantly correlated with ACTG2 (R=0.25, P=0.0008), MYH11 (R=0.16, P=0.033), and PDLIM3 (R=0.31, P=2e-05) in PDAC patients’ samples (Figure 5H-J). Furthermore, the analysis of the pathway of down-regulated genes of KPCM tumors showed that actin-based cell motility, cell migration, and endothelial cell migration pathways were significantly altered (Figure 5K). These findings suggested that the MUC16 may regulate pancreatic cell motility and endothelial cell migration through the Actg2, Myh11, and Pdlim3 genes.
Expression of Muc16-associated genes (Actg2, Myh11, Pdlim3) in pancreatic tumors
We validated the top 10 altered genes in KPCM and KPC tumors using quantitative real-time PCR. We identified a significant variation of Actg2, Myh11, and Pdlim3 expression of mRNA and protein in KPCM knockout pancreatic tissue than in KPC tumors (Figure 6A and Supplementary Figure 4B). Furthermore, the immunofluorescence analysis of cytoskeleton protein Actg2, Myh11, and Pdlim3 expression was decreased in KPCM tumor tissues compared to the KPC tumors (Figure 6B-D). Furthermore, ACTG2 and PDLIM3 protein expression were analyzed in the human Capan1 CRISPR/Cas9 MUC16 knockout (KO) and SW1990 MUC16 Knockdown (KD) PC cell lines. The results demonstrated that ACTG2, and PDLIM3 were downregulated in MUC16 KO/KD cells compared to control cells (Supplementary Figure 4C). These findings strongly indicate that MUC16 regulates cytoskeleton rearrangement proteins, essential for PDAC development.
Figure 6. Muc16 associated gene signatures in pancreatic cancer and their role in metastasis.
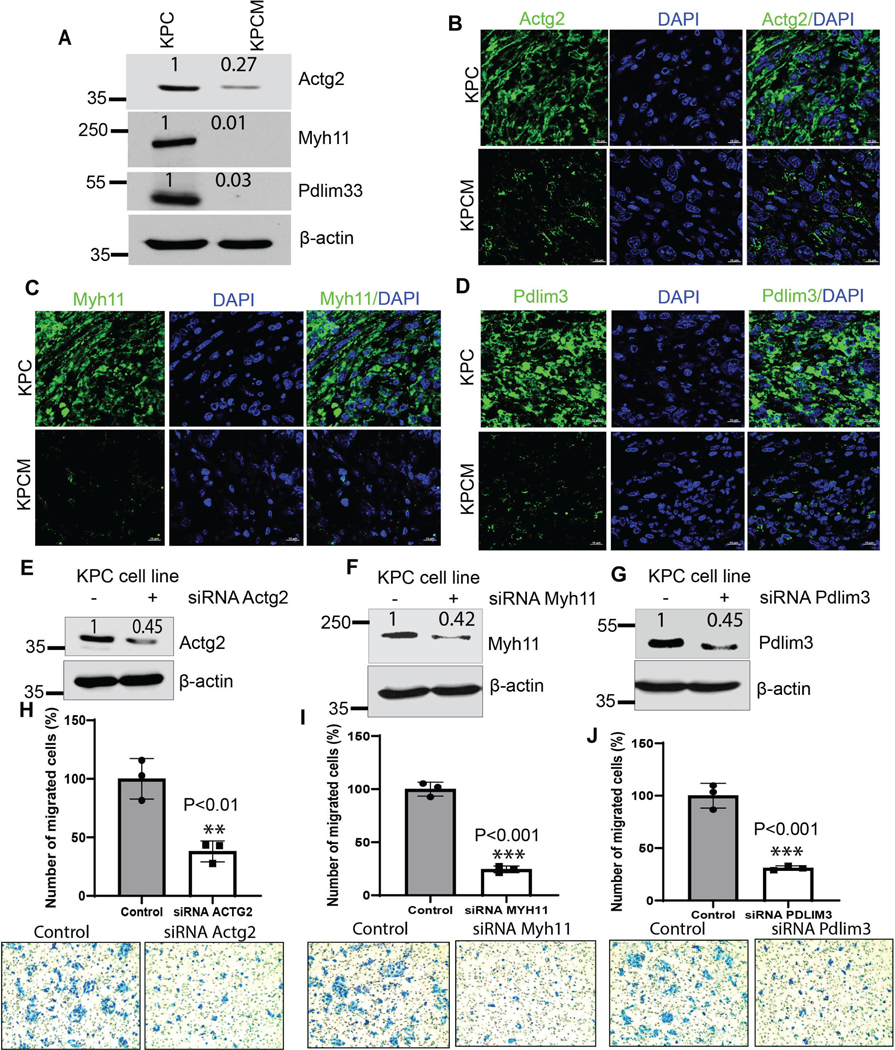
(A) Muc16 knockout (KPCM) cells drastically reduced the expression of Actg2, Myh11, and Pdlim3 KPCM tumor tissues compared to KPC tumors. (B-D) Immunofluorescence analysis of cytoskeletal proteins Actg2, Myh11, and Pdlim3 decreased in KPCM (Sample size-3 individual mice) pancreatic tumor tissues compared to KPC tumors (Sample size-3 individual mice). (E-G) siRNA-mediated knockdown of Actg2, Myh11, and Pdlim3 in KPC cells. (H-J) Actg2, Myh11, and Pdlim3 knockdown cells showed significantly reduced migration potential compared to control cells. ß-actin was used as an internal control. All western blot data were quantified by relative expression. The values in the bar graphs indicate a mean ± SD of three replicates (***P<0.001, **P<0.01) obtained through Student’s t-test.
Silencing of Actg2, Myh11, and Pdlim3 in PDAC cells and their impact on metastasis properties
We have performed the mouse-specific siRNA knockdown and functional studies to identify the functional effect of Muc16-associated genes (Actg2, Myh11, and Pdlim3) in PDAC. Immunoblot analysis validates the silencing of Actg2, Myh11, and Pdlim3 in syngeneic KPC cells (Figure 6E-G). Furthermore, the motility assays demonstrated that knockdown of Actg2, Myh11, and Pdlim3 cells significantly (**P<0.01, **P<0.01, ***P<0.001) decreased motility properties compared to control siRNA cells (Figure 6H-J). These findings suggest that Muc16 and its associated genes (Actg2, Myh11, and Pdlim3) are required for pancreatic cancer metastasis. Our study demonstrated that Muc16 alters the tumor microenvironment and promotes PDAC cell metastasis by regulating Actg2, Myh11, and Pdlim3 genes (Figure 7).
Figure 7. Schematic diagram of Muc16 mediated tumor progression, altered tumor microenvironment, and metastasis in PDAC.

This study revealed that Muc16 promotes PDAC progression and metastasis through altering cytoskeletal rearrangement protein and tumor microenvironment.
Discussion
PDAC is the leading cause of cancer-related mortality worldwide, with limited therapeutic options and poor outcomes (38). Hence, understanding the biology of pancreatic tumor progression and metastasis and the associated molecular mechanism is essential to improving PDAC patient survival outcomes. We previously showed the impact of MUC16 on PDAC cell growth and metastasis (14, 16, 22, 39, 40). Studies have also indicated the functional role of the MUC16 carboxyl-terminal (Cter) in tumorigenesis and metastasis of PDAC (14, 18, 21). Furthermore, it has been shown that KPC-driven tumors are highly aggressive, and we observed a high level of Muc16, suggesting that Muc16 might be involved in disease progression and development (41). In this study, we showed the pathobiological role of Muc16 in PDAC development using the Muc16 knockout mice model in the background of KrasG12D/+ and Trp53R172H/+ mutations.
We observed that Muc16 knockout mice (KPCM and KCM) showed significantly longer overall survival than KPC and KC mice. Initiation and development of pancreatic intraepithelial neoplasia (PanIN) lesions were relatively delayed in the pancreas of Muc16 knockout mice (KPCM and KCM) compared to KPC and KC, respectively. Similarly, Muc16 knockout (KPCM and KCM) mice exhibited significantly decreased tumor weight than KC and KPC models. These results demonstrate that Muc16 plays a potential role in the progression of PDAC. Further, the TME factors and stroma predominantly contribute to the aggressiveness of PDAC tumors (8, 41). The primary characteristic features of pancreatic tumors are high fibrotic and extracellular matrix (ECM) associated with disease progression and drug resistance (2–4, 6). Therefore, we analyzed the ECM and fibrosis formation in the pancreas of Muc16 knockout mice. The Muc16 gene deletion in the background of KPC models shows decreased cancer-associated fibrosis during PDAC development. A recent study demonstrated that CAFs co-cultured with pancreatic cancer cells in a 3D model displayed increased stemness features. The interplay between CAF and the increased stemness population happens via the osteopontin/secreted phosphoprotein 1-CD44 axis in pancreatic cancer (42). In support of these findings, MUC16 is significantly correlated with high fibrosis and ECM molecules in PDAC tumors. MUC16 is not present in normal pancreatic ducts but is overexpressed on the surface of pancreatic cancer cells. A recent study shows that direct cell-cell interaction influences the activation of CAFs (43). Our 3D spheroid data showed that Muc16 expressing tumor cells and CAFs co-culture increased the αSMA expression in CAFs. The αSMA is the marker for the activated CAFs. These findings reveal that Muc16 could directly communicate with CAFs and activates the stroma. The complex process of crosstalk between pancreatic cancer cells and the surrounding stroma needs to be explored in the future. Our functional studies also demonstrated that KPCM tumor-derived syngeneic cells had fewer associations with mouse pancreatic cancer-associated fibroblast cells. Similarly, KPCM cells had a significantly lower binding affinity to endothelial cells. These findings suggest that Muc16 is associated with tumor microenvironment factors during the development of pancreatic cancer. Muc16 knockout with KPC (KPCM) tumor-derived syngeneic cells showed reduced migration colony formation ability compared to KPC cells. Furthermore, KPCM-derived organoid also shows decreased growth compared to KPC organoids, suggesting that Muc16 plays a critical role in the tumor microenvironment during PDAC development. This result indicates that deletion of Muc16 in Kras and p53 mutated animals significantly alters the TME factors and disease aggressiveness. Pancreatic cancer patients often develop metastasis in the liver, lung, and peritoneum and are strongly associated with poor outcomes (44–46).
We have demonstrated that KPC mice develop distant metastasis such as the liver, peritoneum, lung, and intestine (25, 27, 30). Muc16 knockout (KPCM and KCM) mice did not develop liver, lung, or peritoneal metastasis in this study. Interestingly, the patient’s dataset also indicated an elevated level of MUC16 in pancreatic liver metastasis patients. Our functional studies have demonstrated that KPCM syngeneic cells had significantly less migration and colony formation properties. These findings indicated that Muc16 promotes metastasis of pancreatic cancer cells.
Next, we analyzed the molecular mechanism of Muc16-mediated pancreatic cancer cell metastasis. Muc16-associated gene (Actg2, Myh11, and Pdlim3) signature was identified using KPCM mouse models. The ACTG2, MYH11, and PDLIM3 genes were significantly overexpressed in PDAC patients and positively correlated with MUC16. Further silencing of these (Actg2, Myh11, and Pdlim3) also decreased migratory properties of pancreatic cancer syngeneic cells. These results suggest that Muc16 is associated with these genes (Actg2, Myh11, and Pdlim3) required for the growth and metastasis of pancreatic cancer cells.
Overall, our findings revealed that Muc16 knockout pancreatic cancer mouse models exhibit delayed cancer progression and metastasis. Muc16 also affects pancreatic cancer-associated fibrosis during pancreatic cancer development. We also identified MUC16-associated gene signatures required for pancreatic cancer metastasis. Therefore, targeting MUC16 and its gene signature could prevent pancreatic cancer development and metastasis. Our future study explores how MUC16 expressed tumor cells alter the tumor microenvironment, stromal cell-tumor cell interaction mechanisms, and how direct physical interaction between MUC16 expressing tumor cells and CAFs promotes PDAC progression.
Supplementary Material
Acknowledgments
We thank Corinn Grabow, UNMC core facilities, Advanced Microscopy Core Facility, Genomics Core Facility, and Tissue Science Facility, University of Nebraska Medical Center.
Funding information
This work was supported by the support from the National Institutes of Health P01 CA217798, R01 CA210637, R01 CA183459, R01 CA195586, R01 CA201444, R01 CA228524, F99 CA234962, U01 CA200466, and U01 CA210240, and the Nebraska Department of Health and Human Services LB595, US Department of Veterans Affairs (I01 BX004676).
Abbreviations
- IHC
Immunohistochemistry
- KC
KrasG12D/+
- Pdx-1 Cre
- KPC
KrasG12D/+
- Trp53R172H/+
- Pdx-1 Cre
- KCM
KrasG12D/+
- Pdx-1 Cre
Muc16−/−
- KPCM,
KrasG12D/+
- Trp53R172H/+
- Pdx-1 Cre
Muc16−/−
- KD
Knockdown
- KO
Knockout
- MUC
Mucins
- PDAC
pancreatic ductal adenocarcinoma
- PCR
Polymerase chain reaction
- TCGA
The cancer genome atlas
- TP53
Tumor protein 53
- TPM
Transcripts per million
- Trp53
Transformation related protein 53
- UNMC
University of Nebraska Medical Center
Footnotes
Competing Interests: Surinder K Batra (S.K.B) is one of the co-founders of Sanguine Diagnostics and Therapeutics, Inc. The other authors declare no potential conflicts of interest.
Data Accessibility
The RNA sequence data and materials associated with the current study are available from the corresponding author upon reasonable request.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: a cancer journal for clinicians. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2.Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17(8):487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Encarnación-Rosado J, Kimmelman AC. Harnessing metabolic dependencies in pancreatic cancers. Nat Rev Gastroenterol Hepatol. 2021. [DOI] [PMC free article] [PubMed]
- 4.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17(3):153–68. [DOI] [PubMed] [Google Scholar]
- 5.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32(2):185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian C, Huang Y, Clauser KR, Rickelt S, Lau AN, Carr SA, et al. Suppression of pancreatic ductal adenocarcinoma growth and metastasis by fibrillar collagens produced selectively by tumor cells. Nat Commun. 2021;12(1):2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(39):19609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bafna S, Kaur S, Momi N, Batra SK. Pancreatic cancer cells resistance to gemcitabine: the role of MUC4 mucin. British journal of cancer. 2009;101(7):1155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol. 2013;10(10):607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caffrey T, Sagar S, Thomas D, Lewallen ME, Hollingsworth MA, Radhakrishnan P. The glycoprotein mucin-1 negatively regulates GalNAc transferase 5 expression in pancreatic cancer. FEBS letters. 2019;593(19):2751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen SH, Dallas MR, Balzer EM, Konstantopoulos K. Mucin 16 is a functional selectin ligand on pancreatic cancer cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26(3):1349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen SH, Hung WC, Wang P, Paul C, Konstantopoulos K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci Rep. 2013;3:1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das S, Rachagani S, Torres-Gonzalez MP, Lakshmanan I, Majhi PD, Smith LM, et al. Carboxyl-terminal domain of MUC 16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget. 2015;6(8):5772–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felder M, Kapur A, Gonzalez-Bosquet J, Horibata S, Heintz J, Albrecht R, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haridas D, Chakraborty S, Ponnusamy MP, Lakshmanan I, Rachagani S, Cruz E, et al. Pathobiological implications of MUC16 expression in pancreatic cancer. PLoS One. 2011;6(10):e26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lakshmanan I, Ponnusamy MP, Das S, Chakraborty S, Haridas D, Mukhopadhyay P, et al. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene. 2012;31(7):805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakshmanan I, Salfity S, Seshacharyulu P, Rachagani S, Thomas A, Das S, et al. MUC16 Regulates TSPYL5 for Lung Cancer Cell Growth and Chemoresistance by Suppressing p53. Clin Cancer Res. 2017;23(14):3906–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das S, Batra SK. Understanding the Unique Attributes of MUC16 (CA125): Potential Implications in Targeted Therapy. Cancer research. 2015;75(22):4669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gubbels JA, Belisle J, Onda M, Rancourt C, Migneault M, Ho M, et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 2006;5(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akita K, Tanaka M, Tanida S, Mori Y, Toda M, Nakada H. CA125/MUC16 interacts with Src family kinases, and over-expression of its C-terminal fragment in human epithelial cancer cells reduces cell-cell adhesion. Eur J Cell Biol. 2013;92(8–9):257–63. [DOI] [PubMed] [Google Scholar]
- 22.Muniyan S, Haridas D, Chugh S, Rachagani S, Lakshmanan I, Gupta S, et al. MUC16 contributes to the metastasis of pancreatic ductal adenocarcinoma through focal adhesion mediated signaling mechanism. Genes & cancer. 2016;7(3–4):110–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belisle JA, Horibata S, Jennifer GA, Petrie S, Kapur A, Andre S, et al. Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Molecular cancer. 2010;9:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+); LSL-Trp53(R172H/+); Pdx-1- Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr Protoc Pharmacol. 2016;73:14.39.1–14.39.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nimmakayala RK, Leon F, Rachagani S, Rauth S, Nallasamy P, Marimuthu S, et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene. 2021;40(1):215–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chugh S, Barkeer S, Rachagani S, Nimmakayala RK, Perumal N, Pothuraju R, et al. Disruption of C1galt1 Gene Promotes Development and Metastasis of Pancreatic Adenocarcinomas in Mice. Gastroenterology. 2018;155(5):1608–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. [DOI] [PubMed] [Google Scholar]
- 28.Lakshmanan I, Seshacharyulu P, Haridas D, Rachagani S, Gupta S, Joshi S, et al. Novel HER3/MUC4 oncogenic signaling aggravates the tumorigenic phenotypes of pancreatic cancer cells. Oncotarget. 2015;6(25):21085–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boj SF, Hwang CI, Baker LA, Chio II, n DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallya K, Haridas D, Seshacharyulu P, Pothuraju R, Junker WM, Krishn SR, et al. Acinar transformed ductal cells exhibit differential mucin expression in a tamoxifen-induced pancreatic ductal adenocarcinoma mouse model. Biol Open. 2020;9(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheon DJ, Wang Y, Deng JM, Lu Z, Xiao L, Chen Cm, et al. CA125/MUC16 is dispensable for mouse development and reproduction. PLoS One. 2009;4(3):e4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaushik G, Seshacharyulu P, Rauth S, Nallasamy P, Rachagani S, Nimmakayala RK, et al. Selective inhibition of stemness through EGFR/FOXA2/SOX9 axis reduces pancreatic cancer metastasis. Oncogene. 2021;40(4):848–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rauth S, Karmakar S, Batra SK, Ponnusamy MP. Recent advances in organoid development and applications in disease modeling. Biochim Biophys Acta Rev Cancer. 2021;1875(2):188527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakshmanan I, Rachagani S, Hauke R, Krishn SR, Paknikar S, Seshacharyulu P, et al. MUC5AC interactions with integrin β4 enhances the migration of lung cancer cells through FAK signaling. Oncogene. 2016;35(31):4112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G, Kim YJ, Mantel C, Broxmeyer HE. P-selectin enhances generation of CD14+CD16+ dendritic-like cells and inhibits macrophage maturation from human peripheral blood monocytes. J Immunol. 2003;171(2):669–77. [DOI] [PubMed] [Google Scholar]
- 37.Lennon S, Oweida A, Milner D, Phan AV, Bhatia S, Van Court B, et al. Pancreatic Tumor Microenvironment Modulation by EphB4-ephrinB2 Inhibition and Radiation Combination. Clin Cancer Res. 2019;25(11):3352–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet. 2020;395(10242):2008–20. [DOI] [PubMed] [Google Scholar]
- 39.Das S, Majhi PD, Al-Mugotir MH, Rachagani S, Sorgen P, Batra SK. Membrane proximal ectodomain cleavage of MUC16 occurs in the acidifying Golgi/post-Golgi compartments. Sci Rep. 2015;5:9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marimuthu S, Lakshmanan I, Muniyan S, Gautam SK, Nimmakayala RK, Rauth S, et al. MUC16 Promotes Liver Metastasis of Pancreatic Ductal Adenocarcinoma by Upregulating NRP2-Associated Cell Adhesion. Mol Cancer Res. 2022;20(8):1208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chugh S, Barkeer S, Rachagani S, Nimmakayala RK, Perumal N, Pothuraju R, et al. Disruption of C1galt1 Gene Promotes Development and Metastasis of Pancreatic Adenocarcinomas in Mice. Gastroenterology. 2018. [DOI] [PMC free article] [PubMed]
- 42.Nallasamy P, Nimmakayala RK, Karmakar S, Leon F, Seshacharyulu P, Lakshmanan I, et al. Pancreatic Tumor Microenvironment Factor Promotes Cancer Stemness via SPP1-CD44 Axis. Gastroenterology. 2021;161(6):1998–2013.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sperb N, Tsesmelis M, Wirth T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. International journal of molecular sciences. 2020;21(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connor AA, Denroche RE, Jang GH, Lemire M, Zhang A, Chan-Seng-Yue M, et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell. 2019;35(2):267–82.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRInOx in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017;18(6):770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab- Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol. 2018;36(4):359–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequence data and materials associated with the current study are available from the corresponding author upon reasonable request.