

Abstract
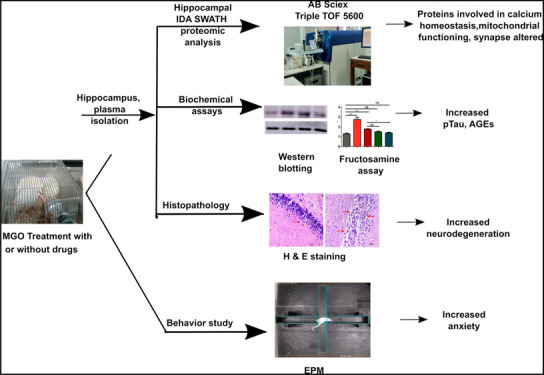
Diabetes is one of the major risk factors for Alzheimer’s disease (AD) development. The role of elevated levels of glucose, methylglyoxal (MGO), and advanced glycation end products (AGEs) in the pathogenesis of AD is not well understood. In this pursuit, we studied the role of methylglyoxal in the pathogenesis of AD in rat models. The elevated plus-maze (EPM) behavioral study indicated that MGO induces anxiety. Treatment of telmisartan (RAGE expression inhibitor) and aminoguanidine (MGO quencher) attenuated MGO induced anxiety. Further, hippocampal proteomics demonstrated that MGO treated rats differentially regulate proteins involved in calcium homeostasis, mitochondrial functioning, and apoptosis, which may affect neurotransmission and neuronal plasticity. The hippocampal tau phosphorylation level was increased in MGO treated rats, which was reduced in the presence of aminoguanidine and telmisartan. The plasma fructosamine level was increased upon MGO treatment. Hippocampal histochemistry showed vascular degeneration and neuronal loss upon MGO treatment. This study provides mechanistic insight into the role of MGO in the diabetes-associated development of AD.
Keywords: Alzheimer’s disease, diabetes, methylglyoxal, tau, proteomics, mass spectrometry
Alzheimer’s disease (AD) is one of the major neurodegenerative diseases among the old-aged population, which is characterized by dementia, progressive memory loss, a decline in thinking ability, mental confusion, and disorientation, ultimately leading to death. Extracellular amyloid-β (Aβ) senile plaques (SPs) and the intracellular neurofibrillary tangles (NFTs) are two major hallmarks of AD. SPs and NFTs are majorly formed in the hippocampus and cortical areas of the brain, which leads to neurodegeneration and loss of synaptic plasticity. The etiology of AD is multifactorial, and diabetes is one of the major risk factors for the development of AD. Population-based studies have shown a strong association between diabetes and AD, with diabetes as a risk factor for AD development. Diabetes and AD share a few common pathological symptoms, such as brain atrophy, insulin resistance, increased inflammation, etc.1 It acts as a potential predisposal factor for AD development, suggesting that studying pathological changes at the cellular level in diabetes can help to understand AD development.2 Insulin resistance can increase neuritic plaque formation and cause cognitive impairment.3 Treatment with moderate insulin levels has been shown to benefit the brain, whereas excess insulin reduces Aβ clearance from the brain.4 Insulin deficiency is associated with cognitive impairment with a concomitant increase in neurotoxic Aβ peptide and tau hyperphosphorylation, along with a decrease in p-Akt and phosphorylated glycogen synthase kinase-3β (GSK-3β) in a transgenic mouse model.5 Tau hyperphosphorylation has also been observed in streptozotocin (STZ) induced diabetes characterized by insulin deficiency along with protein phosphatase (PP2A) inhibition.6 Type 1 diabetes-induced rats have been shown to cause cognitive impairment along with an increase in Aβ and tau hyperphosphorylation.7 The result of insulin deficiency and insulin resistance is hyperglycemia and an increase in methylglyoxal (MGO), both of which increase protein glycation by forming advanced glycation end products (AGEs). The ratio of glycated albumin to HbA1c has been found to be associated with AD development in a population-based study.8
MGO is a toxic byproduct of glycolysis whose levels are elevated in diabetes. It is a potent glycating agent and forms AGEs. AGEs interact with the cell surface receptor for advanced glycation end products (RAGE) and initiate the pathological cascade. The effect of MGO on neuronal cells and AD pathophysiology has been studied earlier. MGO has also been found to increase the Aβ aggregation rate and size of aggregates.9 MGO is detrimental to neurons via AGEs formation and the generation of reactive oxygen species (ROS).10 MGO has been found to decrease mitochondrial membrane potential and increase intracellular ROS production in SHSY-5Y cells.11 In neuro-2a cells, MGO activates the proapoptotic mitogen-activated protein kinase (MAPK) signaling pathway involving c-Jun N-terminal kinases (JNK) and p38.12 A higher MGO level has also been found to be associated with poor memory and cerebral atrophy in older people, and it induces cognitive deficit in the rat model.13,14 Also, the concentration of MGO in cerebrospinal fluid (CSF) of AD patients is found to be higher than in the control.15 Proteomic analysis of neuronal cells indicated that MGO differentially regulates the expression of proteins involved in AD, calcium signaling, and neurotransmission. Alterations in these important pathways can make diabetes a risk factor for AD development. Small molecule intervention to reduce AGEs has been found beneficial in several previous studies. Aminoguanidine (AMG) is known to reduce MGO toxicity by quenching. It is a selective inhibitor of inducible nitric oxide synthase (iNOS)1.16 AMG has been found to reduce anxiety in various conditions. Telmisartan (TELMI) is an angiotensin II (Ang II) receptor blocker (ARB), which is used for hypertension treatment. TELMI also reduces RAGE expression by stimulating peroxisome proliferator-activated receptor-γ (PPAR-γ) activity.17 TELMI has a partial PPAR-γ agonistic activity in addition to its role in controlling hypertension through the renin-angiotensin system.18 PPARs are a group of nuclear receptors that controls various transcription factors upon ligand binding. These transcription factors regulate diverse cellular processes such as inflammation, adipogenesis, and maintenance of metabolic homeostasis.19 PPAR-γ agonists have been found to be helpful in restoring learning and memory deficits in AD rodent models.20 To this end, we have assessed the effect of AMG and TELMI drugs in MGO treated rats by performing behavioral and proteomics studies. We have used a label-free, untargeted DIA (data-independent acquisition) SWATH (sequential window acquisition of all theoretical fragments) method for understanding differential protein expression in the rat hippocampus region.
Results
Diabetes Induction
To understand the molecular mechanisms involved in MGO-associated AD development, behavioral experiments, proteomic analysis, and histopathological studies were conducted in MGO treated rats. STZ-induced diabetic rats were used as a positive control, as diabetes is associated with cognitive decline. Bodyweight, blood glucose, and HbA1c were measured before and after treatments. Diabetes was confirmed by measuring blood glucose and HbA1c. Two-tailed t tests were performed to calculate the significance of body weight, blood glucose, and HbA1c between the control and diabetic group. Figure 1 represents the workflow of the animal experiment performed.
Figure 1.
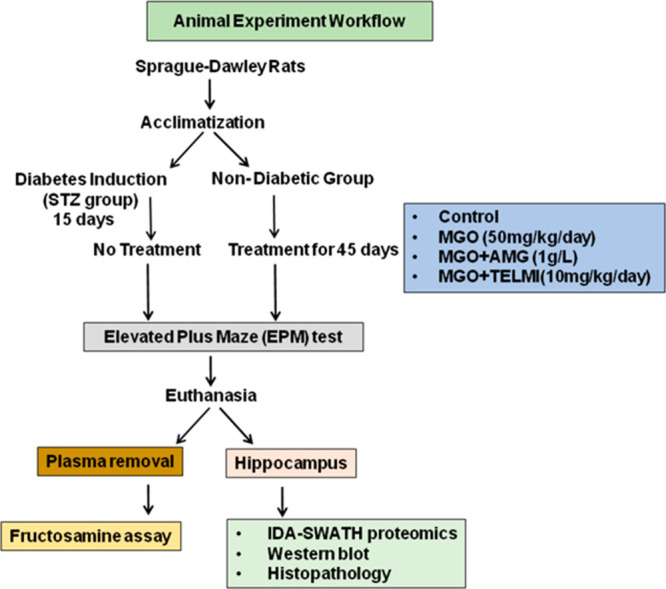
Overall view of the animal experiment indicating the dosage and duration of treatment. It also represents further experiments performed with plasma and hippocampus at the end of the treatment.
The average body weight of nondiabetic rats was 339.84 ± 39.55 g, while the diabetic rats were 279.50 ± 64.86 g*, data expressed as mean (±SD). A significant decrease in the body weight of diabetic rats was observed. Nondiabetic rats had a blood glucose of 87.71 ± 12.80 mg/dL as compared to diabetic rats with 537.20 ± 72.04 mg/dL***. Along with increased blood glucose levels, diabetic rats showed an increase in HbA1c value 7.03 ± 0.55%*** as compared to nondiabetic rats 4.1 ± 0.12% (Supporting Information, Sheet 1). After 45 days of MGO treatment, all the body parameters were measured again. The STZ group showed a reduction in body weight as compared to the control group. Blood glucose and HbA1c were significantly higher in the STZ group as compared to the control group. There was no difference in body weight, blood glucose, and HbA1c between the control and MGO treated groups (Figure S1). HbA1c reflects the glycemic status of the preceding three months, and 45 days may not be enough to see a large a difference in HbA1c. Therefore, we have estimated plasma fructosamine, which reflects total plasma protein glycation.
Plasma Fructosamine Assay
Fructosamine is one of the intermediate products during AGEs formation. To understand the effect of MGO treatments on protein glycation, we measured plasma fructosamine. Plasma fructosamine was normalized with total protein content. MGO treatment showed a significant increase in fructosamine content w.r.t. the control. MGO cotreatment with AMG and TELMI reduced fructosamine levels similar to that of the control. In the STZ treatment, due to a higher glucose level in the blood, increased fructosamine content was observed w.r.t. the control (Figure 2).
Figure 2.

Fructosamine assay of the rat plasma after 45 days of treatment. It depicts the glycation of all plasma proteins. (n = 3, One way ANOVA, **p < 0.01, ***p < 0.001, data expressed as mean ± SD).
Behavior Study
The effect of MGO treatment (with or without AMG or TELMI) on rat behavior was studied by the elevated plus-maze test (EPM). EPM detects the development of anxiety if present in the early stages of AD development. A reduced number of entries and reduced distance traveled in open arms compared to closed arms indicated anxiety development. A representative trajectory of rats in all groups is depicted in Figure 3A. Control rats were found to explore open as well as closed arms, indicating healthy condition; MGO rats displayed a significantly reduced number of entries along with reduced distance traveled in open arms, confirming anxiety development (Figure 3B). Cotreatment of MGO with AMG has been found to reduce anxiety as compared to MGO alone. AMG treatment significantly increased the number of entries in open arms with respect to MGO (Figure 3B). Like AMG, cotreatment with TELMI also increased the total number of entries and distance traveled in open arms, indicating a reduction in anxiety (Figure 3B,C). STZ-treated rats also showed a significantly reduced number of entries w.r.t. the control. The number of entries into open arms in the STZ group was significantly decreased w.r.t. the control, indicating increased anxiety in the diabetic rats (Figure 3B).
Figure 3.
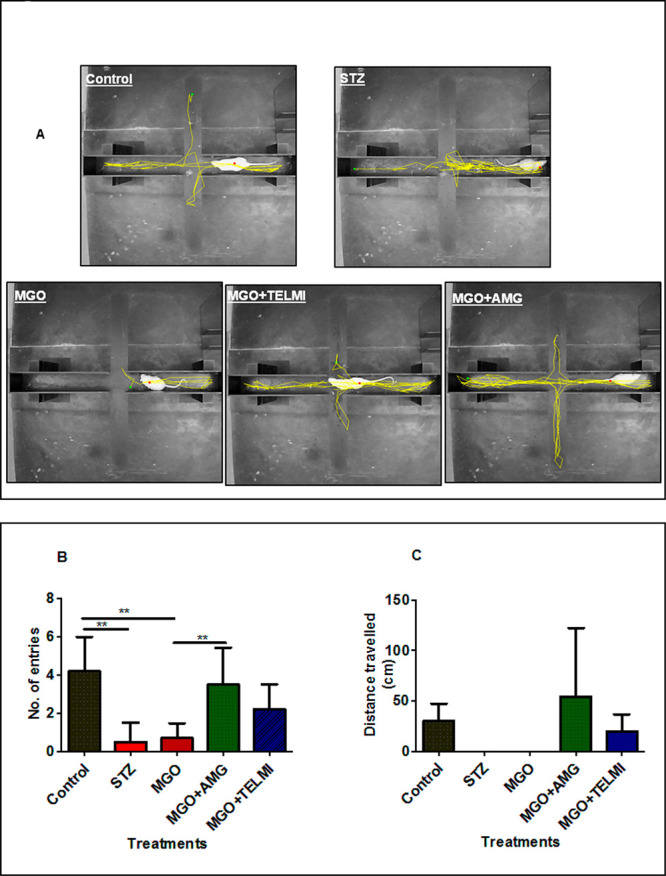
Elevated plus maze test (EPM) to detect the development of anxiety in rats after 45 days of treatment. (A) Representative trajectories of rats in all 5 groups during EPM study. (B) Number of entries made by the rats from all 5 groups in open arms during the study. (C) Average distance traveled by rats from all 5 groups in open arms during the study (n = 5, One way ANOVA, **p < 0.01, data expressed as mean ± SD).
Proteomics Analysis of Rat Brain Hippocampus
Proteomic analysis of rat hippocampal tissue was performed to establish a molecular link between diabetes, MGO, and AD. In order to analyze the reproducibility of the mass spectrometric analysis, a principal component analysis was done. The PCA plot shows consistency between biological and technical triplicates of mass spectrometric acquisition of hippocampal tissue (Figure 4A). The methodology of IDA-SWATH has been depicted in Figure S2. A total of 1692 proteins were identified with at least two unique peptides (Supplementary table, Sheet 2). A list of identified peptides and distinct peptide summary is given in Supporting Information Sheets 3 and 4, respectively. Figure 4B indicates the number of proteins common between MGO and STZ treatment. A total of 60 proteins were shared between STZ and MGO. Out of 60 proteins, 31 were upregulated, and 29 were downregulated w.r.t. the control (Figure 4C,D). Supporting Information Sheet 5 represents a list of commonly differentially expressed proteins between STZ and MGO w.r.t. the control. TELMI cotreatment restored the expression levels of 34 proteins, and AMG cotreatment restored that of 33 proteins compared to MGO treatment alone (Supporting Information Sheets 6 and 7, respectively).
Figure 4.

PCA and Venn diagram of rat hippocampal proteomic acquisition. (A) PCA plot showing consistency between biological and technical proteomic acquisitions (biological and technical triplicates). (B) Venn diagram representing total number of differentially expressed proteins in MGO (289 proteins) and STZ (210 proteins) treatment w.r.t. the control and common proteins differentially expressed upon both the treatments (60 proteins). (C) Common upregulated proteins between MGO and STZ treatment w.r.t. the control (31 proteins out of 60 proteins). (D) Common downregulated proteins between MGO and STZ treatment w.r.t. the control (29 proteins out of 60 proteins).
Functional analysis was performed by the online DAVID software.21,22 All the differentially expressed proteins in STZ and MGO w.r.t. the control were used for DAVID analysis. All the 210 proteins differentially expressed proteins upon STZ treatment were considered for DAVID analysis. Proteins upregulated in STZ treated rat brain tissues were involved in AD, glycolysis, glutathione metabolism, oxidative phosphorylation, and the citrate cycle (Figure 5A). While downregulated proteins were involved in long-term potentiation, dopaminergic synapse, GABAergic synapse, and the synaptic vesicle cycle (Figure 5B). MGO altered expression of 289 proteins was considered for DAVID analysis. Functional analysis of differentially expressed proteins in response to MGO treatment shared pathways similar to those that are activated in response to STZ treatment except glycolysis, however, with few additional pathways (Figure 5C,D). Proteins altered after MGO treatment were involved in the same pathways observed in STZ altered pathways except for glycolysis. Along with the same pathways observed after STZ treatment, downregulated proteins were found to be involved in insulin signaling, insulin secretion, regulation of actin cytoskeleton, the MAPK signaling pathway, the calcium signaling pathway, proteasomes, and synapses (serotonergic, glutamatergic, cholinergic).
Figure 5.

DAVID analysis of differentially expressed proteins in STZ (A, B) and MGO (C, D) treatment. (A,B) Involvement of differentially expressed proteins upon STZ treatment into different cellular pathways: (A) pathways involving STZ upregulated proteins w.r.t. the control; (B) pathways involving STZ downregulated proteins w.r.t. the control. (C,D) Different pathways involving proteins affected by MGO treatment: (C) pathways involving MGO upregulated proteins w.r.t. the control; (D) pathways involved in proteins downregulated in MGO w.r.t. the control.
MGO+TELMI and MGO+AMG Cotreatment Restored MGO Altered Proteins
Cotreatment of TELMI and AMG along with MGO restored the expression of 34 proteins w.r.t. MGO treatment. TELMI restored some of the dysregulated proteins such as mitochondrial proteins (cytochrome c oxidase subunit 5B, [pyruvate dehydrogenase [{acetyl-transferring}ss]-phosphatase 1, Frataxin) (Figure 6A). Similarly, AMG cotreatment restored 33 proteins w.r.t. MGO treatment, such as septin-5, and neuronal-specific septin-3 (Figure 6B).
Figure 6.

Heat maps of the fold change of MGO altered proteins upon TELMI and AMG cotreatment. (A) Heat map of MGO treatment altered proteins w.r.t. the control and its restoration by MGO-TELMI cotreatment. (B) Heat map of MGO treatment altered proteins w.r.t. the control and its restoration by MGO-AMG cotreatment.
Effect of STZ and MGO Treatments on RAGE Expression
To understand the role of increased AGEs upon MGO treatment, we further studied RAGE expression in the hippocampus by Western blotting. RAGE expression was normalized with GAPDH expression. RAGE expression was elevated in MGO and STZ treatment w.r.t. the control (Figure S3A). MGO cotreatment with AMG and TELMI reduced the RAGE expression (Figure S3B).
Effect of STZ and MGO Treatments on Tau Phosphorylation
The effect of MGO treatment on tau phosphorylation was studied, as it is one of the essential pathological hallmarks of AD. Tau phosphorylation was increased in MGO and STZ treatment w.r.t. the control (Figure 7A,C). Total tau expression was normalized with GAPDH (Figure 7B). This normalized factor was then used for pTau normalization (Figure 7C).
Figure 7.

Western blot analysis for pTau in hippocampus (n = 3, One way ANOVA, data expressed as mean ± SD). (A) Western blotting ECL image of the pTau, Tau, and GAPDH in rat brain tissue proteins. (B) Densitometric analysis of fold change Total Tau/GAPDH in all 5 groups. (C) Densitometric analysis of fold change pTau/Total Tau in all 5 groups.
Hippocampal Histochemistry
The effect of MGO treatment on the structure CA1 region of the hippocampus was studied by H&E staining (Figure 8). MGO and STZ treatments have been found to cause a neuronal loss in the CA1 region as compared to the control.
Figure 8.

H&E staining of the hippocampus (n = 3). The top panel shows neuronal health in the control (A), STZ (B), and MGO (C) group. The lower panel indicates the effect of treatment on the neuronal cells in MGO+AMG cotreatment (D) and MGO+TELMI cotreatment (E).
Discussion
Diabetes has been a major risk factor for AD development, yet the exact mechanism is not fully understood. Herein we have tried to understand the role of the toxic, highly active glycating agent MGO whose level is elevated in diabetes in AD development. In our study, after chronic treatment of MGO for 45 days, the behavioral study showed that MGO treatment causes anxiety development, as evident from reduced entries and reduced distance traveled in open arms. This observation supports increased anxiety in diabetic patients having elevated MGO levels. Also, AD CSF has been found to contain more MGO than control CSF samples.15 One study has found that diabetes is an important risk factor for AD, which promotes depression and anxiety in patients.23 Cotreatment of MGO with AMG and TELMI reduced anxiety. AMG has shown its antianxiety effect in nondiabetic stressed mice via activation of the inducible nitric oxide synthase cyclic guanosine monophosphate (iNOS-cGMP) pathway. iNOS leads to nitric oxide production (NO), increasing cyclic guanosine monophosphate (cGMP), a secondary messenger that carries neuronal communications and increases anxiety. AMG acts by reducing NO synthesis, thereby reducing cGMP and anxiety. AMG has also been shown to inhibit the activity of guanylyl cyclase, an essential enzyme for cGMP formation.24 Current study also found that MGO cotreatment with TELMI reduced anxiety. Earlier studies support our observation where TELMI has been shown to reduce anxiety in diabetic rats by reducing levels of serum cortisol, NO, IL-6, IL-1β. Also, TELMI has been found to increase the expression of peroxisome proliferator-activated receptors δ (PPARδ) and serotonin transporter (5 HTT), thereby reducing anxiety. PPARδ expression has been found to have anti-inflammatory and neuroprotective activity, reducing neuronal degeneration in AD and reducing Aβ levels.25−27 A recent clinical study has found that TELMI is reducing the risk of AD in Africans Americans.28
Development of anxiety in STZ diabetic rats is well established in many research papers. STZ treatment has led to increased anxiety in rats as detected by the EPM test.29 A previous study has demonstrated that a deficiency of serotonin, adenylyl cyclase type VIII, and tuberoinfundibular peptide of 39 residues can develop anxiety in diabetic rats.29 In our study along with MGO, diabetic rats also showed anxiety development.
Functional annotation of the differentially expressed proteins of diabetic rat hippocampal tissues showed that the proteins found to be involved in AD were primarily mitochondrial (cytochrome b-c1 complex subunit 1, NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5, NADH dehydrogenase [ubiquinone] flavoprotein 2, cytochrome b-c1 complex subunit 2, ATP synthase subunit gamma, cytochrome c oxidase subunit 5B, cytochrome c oxidase subunit 5A, cytochrome b-c1 complex subunit Rieske, succinate dehydrogenase [ubiquinone] iron–sulfur subunit, cytochrome b-c1 complex subunit 6, ATP synthase subunit beta, succinate dehydrogenase [ubiquinone] flavoprotein subunit) and calcineurin subunit B type 1. Homeostasis of these proteins plays a crucial role in maintaining mitochondrial functioning. The role of mitochondrial dysfunction has been well studied in AD.30 Disturbances in mitochondrial functioning have also been reported to initiate apoptosis.31 MGO induces ROS production, which might trigger mitochondrial dysfunction.32 Conversely, mitochondrial dysfunction can produce more ROS, such as H2O2, OH–, and O2, through electron leakage during oxidative phosphorylation.31 Dysfunctional mitochondria have also been found to contribute to calcium homeostasis disturbances, which is crucial for proper neurotransmission.32 Diabetes is characterized by oxidative stress, triggering cellular response via the glutathione mechanism. The upregulation of proteins involved in glutathione metabolism indicates its role as an antioxidant. The brain is one of the highest energy-requiring organs in the body. Upregulation of proteins involved in oxidative phosphorylation and citrate cycle indicates higher energy demand by the brain in diabetic conditions. Downregulated proteins in STZ were involved in the synaptic vesicle cycle, GABAergic synapse, dopaminergic synapse, and long-term potentiation (LTP). Synapses and synaptic vesicle cycles play an important role in neurotransmission. A study in the mouse model has shown that microglia and complementary systems are involved in synapse loss in the early stages of AD.33 Also, the synaptic vesicle cycle is essential, as they carry neurotransmitters that release it by exocytosis into the synaptic cleft upon fusion with the cell membrane. It is then taken up by specific receptors present on the neighboring dendrite.34 AD brains have been found to show dysregulation in LTP.35 Proteins involved in these pathways were downregulated upon STZ treatment indicating improper functioning of the neurotransmission system in diabetes.
Functional analysis suggested that differentially expressed proteins shared common pathways in response to MGO and STZ treatment w.r.t. the control (Figure 5), albeit with few additional pathways.
Also, both MGO and STZ treatments shared common downregulated proteins involved in insulin signaling, insulin secretion, regulation of actin cytoskeleton, MAPK signaling pathway, calcium signaling pathway, proteasome, and synapses (serotonergic, glutamatergic, cholinergic). The actin cytoskeleton helps neurons hold their normal structure and carry out functions. Alteration in the actin cytoskeleton has been reported in AD.36
Cotreatment of AMG with MGO, restored the expression of proteins involved in neurotransmission, such as synaptosomal-associated protein 25, which has been reported to be decreased in AD.37 This protein helps in the exocytosis of neurotransmitters.38 Glutamate decarboxylase 2 plays an essential role in producing the principal inhibitory neurotransmitter γ aminobutyric acid (GABA). This enzyme carries out the decarboxylation of glutamate, which leads to GABA formation. Expression of this enzyme has been found to be downregulated in AD.39 In our study, this enzyme was downregulated upon MGO treatment, suggesting perhaps lesser GABA production. This may lead to anxiety development, as seen in the EPM study. A lower level of GABA and glutamate has been observed in AD.40 Another important enzyme that plays a significant role in GABAergic synapse is Septin-11, which was downregulated upon MGO treatment. This protein has been found to play a role in neuronal architecture and GABAergic synaptic connectivity.41 TELMI treatment restored septin-11 expression. TELMI also was found to restore the expression of tau protein, which plays an essential role in maintaining neuronal architecture. One of the major lysosomal proteins involved in AD is acid ceramidase, which was upregulated upon MGO treatment and downregulated by TELMI.42 TELMI also restored proteins involved in maintaining cell homeostasis by regulating reactive oxygen species, apoptosis, chaperones, and degradation of ubiquitinated proteins such as glutathione S-transferase, complement C3, 10 kDa heat shock protein, and proteasome subunit beta type-5. The heat map of TELMI restored proteins is shown in Figure 6A.
Cotreatment of AMG with MGO restored some of the proteins that were restored by TELMI, such as acid ceramidase, proteasome subunit beta type-5, and glutamate decarboxylase 2. In addition, AMG exclusively restored a few proteins. Important synaptic protein Septin-5 downregulated by MGO reported to be involved in AD was restored by AMG.43 Neuronal-specific septin-3 plays a role in neurite outgrowth and synaptic vesicle fusion, and recycling was also found to be restored by AMG.44 Likewise, protein phosphatase 1 regulatory subunit 1B was also restored by AMG. Its role in tau dephosphorylation is debatable, but it has been found to play an essential role in neuronal processes.45 The heat map of AMG restored proteins is shown in Figure 6B.
MGO is a potent precursor for AGEs formation. MGO can exert its toxic effect by direct methods or by forming AGEs. To confirm this observation, we measured plasma fructosamine levels. It was found to be increased upon MGO and STZ treatments. MGO has been found to elevate fructosamine formation.46 Hyperglycemic condition in diabetes promotes AGEs formation via fructosamine formation.47 Cotreatment of MGO along with either AMG or TELMI reduced plasma fructosamine. AMG is known to reduce fructosamine formation by quenching MGO.48 TELMI is known to improve insulin sensitivity via the PPAR γ pathway; perhaps the decreased fructosamine in rat plasma upon TELMI treatment could be due to an increase in glucose uptake, thus leading to a reduction in fructosamine (Figure 2). Increased plasma AGEs indicated upregulation of its receptor RAGE. Hippocampal RAGE expression was found to be increased upon MGO and STZ treatments. Both the drugs downregulated RAGE expression. TELMI is known to reduce RAGE expression by activating the PPAR γ pathway.49 AMG being an MGO quencher, has reduced AGEs content, thereby reducing RAGE upregulation via AGE-RAGE axis (Figure S3A,B).50
Further, we have looked at phosphorylated tau protein, which is one of the important pathological hallmarks of AD. Tau phosphorylation was increased upon MGO and STZ treatment, and its phosphorylation was decreased in the presence of both drugs. We found an increase in tau phosphorylation upon MGO treatment (Figure 7A,C). MGO activates the AGE-RAGE axis, thereby activating kinases GSK-3β and p38, leading to tau phosphorylation.51 MGO has been found to induce tau phosphorylation by activating kinase activities and reducing PP2A activity. The MGO induced tau phosphorylation was reduced upon AMG treatment, due to MGO quenching effect of AMG (Figure 7A,C). TELMI has been reported to show a neuroprotective effect by reducing Aβ accumulation and phosphorylated tau, and reduced tau phosphorylation with its cotreatment was evident in our study as well.52 TELMI mediated reduction in tau phosphorylation could be due to reduced RAGE expression and thus decreased AGE-RAGE signaling. Two major kinases responsible for tau phosphorylation, Akt and GSK-3β, have been found to alter their activities in response to blood glucose levels. It has been postulated that hyperglycemia in diabetes might alter the balance between these kinases and phosphatase (PP2A), leading to tau hyperphosphorylation.53 Hyperglycemia in STZ diabetic rats can cause upregulation of the AGE-RAGE axis, causing increased expression of asparagine endopeptidase (AEP). AEP has been found to increase tau phosphorylation by inhibiting PP2A activity.45,54 It has been postulated that hyperglycemia in diabetes might alter the balance between these kinases and phosphatase (PP2A), leading to tau hyperphosphorylation.53
The hippocampus is the first region of the brain to be affected in the early stages of AD. It is responsible for memory as well as the regulation of anxiety. The Hippocampus region regulates anxiety.55 This region has shown a neuronal loss in AD, specifically in the region called cornu ammonis (CA1).56 MGO and STZ have been found to cause neuronal loss in the CA1 region as compared to the control. H&E staining of tissue sections of the CA1 region of the brain showed that MGO causes minimal to mild neuronal degeneration with a reduced number of cells in the layer and the presence of dark-stained neurons. Focal congestion with minimal to mild neuronal degeneration with a decreased density of neuronal tissue was observed in the MGO treated group. It was observed that MGO cotreatment with AMG and TELMI treated groups showed comparatively fewer degenerative changes in neuronal tissue compared to MGO. Neurodegenerative changes have been reported in the brain’s CA1 region in STZ treated rats. In the STZ treated rats, mild vascular congestion in the brain tissue was observed along with mild neuronal degeneration with focal neuronal swelling. Focal loss of neuronal tissue with irregular arrangement, along with a decreased thickness of small pyramidal cell layers at multiple foci was observed. Microscopically, the CA1 region of the hippocampus showed normal histomorphological features of neuronal cells with uniform homogeneous layers of neuronal cells with oval to round shaped neurons, intact nuclei, and uniform cellular morphology. STZ and MGO treatments have been found to cause a neuronal loss in the CA1 region with mild neurodegenerative changes characterized by shrinkage, focal neuronal swelling with vacuolar changes, and a decreased number of layers of cells and pyramidal cells as compared to the control. These cellular histomorphological features of neuronal tissue in the CA1 region were significantly decreased in MGO cotreatment with AMG and TELMI and most of the neuronal changes were focal and minimal degree as compared to the STZ group and MGO group, suggestive of restoration of cellular morphology with an intact nucleus and uniform cellular layer arrangement in the CA1 region (Figure 8).
In conclusion, MGO treatment leads to altered expression of proteins involved in key pathways of AD and diabetes in the rat model. Proteins involved in the synapse, glutathione metabolism, and calcium signaling are found to be altered. MGO treatment also caused an increase in fructosamine with a concomitant increase in RAGE expression. We observed an increase in tau phosphorylation upon MGO treatment, which was reduced upon AMG and TELMI treatments. We also found that MGO caused anxiety, which is contradictory to the reported role of MGO in showing anxiolytic effects. MGO caused neurodegenerative changes in the brain, which were reduced in AMG and TELMI treated groups. This study has found that MGO causes alterations in protein expression involved in various biological processes that increase the risk of AD development in diabetes. We have also found that AGEs formed due to MGO activating the AGE-RAGE signaling leading to increased tau phosphorylation. MGO also caused neurodegenerative changes in the hippocampus. Perhaps these changes are responsible for increased anxiety in MGO-treated rats, and these effects were reduced by AMG and TELMI.
The current animal study has a few limitations, such as we could not estimate the actual uptake of MGO. Since MGO is highly active, it is possible that it might get metabolized in the blood before reaching to the brain. It is uncertain to determine if the observed changes in rat behavior and protein are due to MGO alone or due to MGO induced AGEs. Since MGO is detoxified by the glyoxalase system involving GLO1 and GLO2 enzymes, there is scope to perform these experiments in GLO1 and GLO2 deficient rat models. The deficiency of these enzymes will help in the accumulation of MGO in vivo, which will aid in a better understanding of the observed effect of MGO. Also, by using transgenic AD mice, the effect of AMG and TELMI can be studied. Such studies will help in understanding the role of these drugs in reducing AD symptoms.
Methods
Chemicals
MGO (Sigma, #M0252) and aminoguanidine (Sigma, #396494), fructosamine kit (Abbexa, #abx098427), HbA1c measurement kit (NycoCard, #1042184), anti-Th 205/AT8 pTau antibody (AT8) (Thermo scientific, #MN1020), anti-Tau antibody (Dako, #A0024), anti-RAGE (Sigma, #R5278), anti-GAPDH (Sigma, #G8795), protease inhibitor cocktail (Sigma, #P8340), phosphatase inhibitor cocktail (Sigma, #P0044), Amersham ECL prime (GE Healthcare, #RPN2232), and polyvinylidene difluoride (Millipore, #ISEQ10100) membranes were used. Telmisartan (Commercially available as Micardis tablet) was used. RapiGest SF surfactant (Waters Corporation, MA, #186001860), LC–MS grade water, and acetonitrile (J T. Baker, PA) were used.
Animal Experiment
All the animal experiments were carried out at the Experimental Animal Facility, Symbiosis School of Biological Sciences, Pune, India. Experiments were approved by the Institutional Animal Ethics committee (IAEC proposal no.-SSBS/AIEC/03-2017). Male Sprague–Dawley rats were obtained from National Biosciences, Pune. A behavior study was performed at Sinhgad Institute of Pharmacy (SIOP), Pune, India. Rats were supplied with a standard rodent chow diet and had access to water. These animals were housed in a room at an ambient temperature of 25 ± 2 °C. A light and dark cycle of 12 ± 1 h was maintained. Diabetes was induced by injecting a single intraperitoneal injection of 55 mg/kg streptozotocin in 10 mM sodium citrate buffer pH (4.0) to 4 h starved rats, whereas nondiabetic rats received saline as the vehicle control. After 15 days, blood glucose and HbA1c were measured to confirm the establishment of diabetes. Diabetic rats were used as a positive control and nondiabetic rats were given oral MGO treatment or in combination with TELMI and AMG. MGO (50 mg/kg/day) was given with or without TELMI (10 mg/kg/day) and AMG (1 g/L/day) for 45 days.57−59 MGO and TELMI were given orally, and AMG was given through drinking water. Body weight, blood glucose, and HbA1c were measured before and at the end of the treatment. After the behavior study, laboratory animals under experimentation were sacrificed under anesthesia with chloroform, and brain dissection was performed. The hippocampus region of the brain and blood samples was collected for further analysis.
Behavior Study
Rats were acclimatized to the environment 2 days prior to behavior studies. The elevated plus maze (EPM) platform consisted of 2 opposing open arms (zone 1, 2) (10 × 50 cm) and 2 opposing closed arms (zone 3, 4) (50 × 20 × 50 cm) with side walls (15 cm). The platform is at the height of 40 cm from the ground. Distraction due to the manual presence was avoided by placing a wall between the experimental area and the observer. Rats were placed on the central square platform (zone 5) (10 × 10 cm) and allowed to explore the maze freely for 5 min. Rat’s behavior was videographically recorded for 5 min by Maze Master (VJ instruments, India) software. Behavioral parameters such as a number of entries, distance traveled were counted by the software.
Brain Tissue Processing
After sacrifice, the hippocampus was selectively isolated and washed with sterile chilled PBS. The tissue was frozen immediately in liquid nitrogen and stored at −80 °C until further use. Part of the hippocampal tissue was stored in formalin for histochemical analysis. Frozen tissue was washed once with 0.9% chilled saline to remove blood traces for proteomic analysis. It was followed by ice-cold milli-Q washes. Tissue was homogenized in fine powder using mortar pestle in liquid nitrogen. Powdered tissue was weighed, and aliquots were kept at −80 °C until further use.
Hippocampal Brain Sample Preparation for Mass Spectrometric Analysis
For mass spectrometric analysis, proteins were extracted by using 0.1% RapiGest in 50 mM ammonium bicarbonate buffer. Tissue was homogenized for 30 min on ice with brief vortexing every 5 min. Homogenate was cleared by centrifugation at 17,000 rpm at 4 °C for 1 h. The clear supernatant was used for protein estimation by Bradford assay (Bio-Rad). A total of 100 μg of protein was processed by in-solution digestion. Proteins were denatured at 80 °C for 20 min. It was followed by reduction with dithiothreitol (100 mM) at 60 °C for 15 min and alkylation with iodoacetamide (200 mM) at ambient temperature in the dark for 30 min. Proteomic grade porcine trypsin (4 μg) was added and kept at 37 °C for 18 h with shaking (200 rpm). The trypsin reaction was stopped by adding 2 μL of formic acid. The digest was briefly vortexed and kept on ice for 5 min to precipitate acid-labile RapiGest. The digest was centrifuged at 4 °C, 20,000 rpm for 30 min, and the supernatant was collected. Tryptic peptides were desalted by using ZipTip and concentrated by using a Speed-Vac vacuum concentrator. Peptides were reconstituted in 3% ACN with 0.1% formic acid before mass spectrometric analysis.
Liquid Chromatography–Mass Spectrometry Analysis (SWATH-MS)
All samples were analyzed on an AB Sciex Triple-TOF 5600 mass spectrometer coupled with micro LC 200 (Eksigent) in high-sensitivity mode. To generate the SWATH spectral library, peptide digests of each treatment were analyzed by LC–MS/MS in an Information Dependent Acquisition (IDA) mode. A spectral library was created by combining the files of all the treatments. The accumulation time for MS and MS/MS was set to 0.25 and 0.01 ms, respectively, and fragmentation was undertaken using rolling collision energy. MS scans were performed in the mass range 350–1800 m/z, with a charge state 2 to 5, and MS/MS was triggered for ions exceeding 120 cps. SWATH-MS data sets were acquired in biological triplicates n = 3 each with technical triplicates n = 3 total (n = 9/group) on micro LC-Triple TOF 5600. The desalted tryptic peptides were injected onto an Eksigent C18-RP HPLC column (100 × 0.3 mm, 3 μm, 120 Å) at the flow rate of 8 μL/min over 120 min gradient conditions, solvent A (water with 0.1% formic acid) and solvent B (ACN with 0.1% formic acid): held at 97% A for 5 min, 97–90% A over 20 min, 90–70% A over 70 min, 70–50% A over 5 min, 50–10% A over 1 min, at 10% A for 7 min, 10–97% A over 1 min and held at 97% A for 11 min. For SWATH-MS data acquisition, the instrument was tuned to optimize the quadrupole settings for the precursor ion selection window of 25 Da wide using 34 windows of 25 Da effective isolation width (with an additional 1 Da overlap) and with a dwell time of 70 ms to cover the mass range 350–1200 m/z in 3.4 s. Before each cycle, an MS1 scan was acquired, and then the MS2 scan cycle started (350–375 m/z precursor isolation window for the first scan, 474–500 m/z for the second, and 1174–1200 m/z for the last scan). The collision energy for each window was set using the collision energy of a 2+ ion centered in the middle of the window with a spread of 15 eV. To obtain a spectral library from IDA runs, data were analyzed by ProteinPilot v5.0 software. The Rattus norvegicus database (UniProt release 2019_11) was used from UniProt. Trypsin was set as an enzyme used for digestion, cysteine alkylation was set to iodoacetamide, and rapid ID was performed. A false discovery rate (FDR) was set to 1% for protein identification. IDA analysis result was used as a spectral library for PeakView v 2.2 software, and SWATH runs were processed using FDR 1%, mass error 50 ppm, retention time window 5 min, and 95% confidence interval. The number of peptides per protein was set to 10, and 6 was the number of transitions per peptide. SWATH data were exported to MarkerView v1.2.1 for quantitative and statistical analysis. Proteins with a fold change >1.3 and a p value <0.05 were considered for further analysis. Mass spectrometric raw files data are available via ProteomeXchange with the identifier PXD037673.
Statistical Considerations for Proteomic Analysis
A total of three biological replicates, each with three technical replicates, were considered for rat hippocampal proteomic analysis. Proteins were normalized with the total area sum. Proteins with fold change >1.3 and p value <0.05 were considered for further analysis.60
Plasma Fructosamine Assay
A total of 0.5 mL of blood was collected before sacrifice and mixed with EDTA to prevent coagulation. The blood sample was centrifuged at 13,000 rpm, and plasma was collected and stored in aliquots at −80 °C to avoid repeated freeze–thaw cycles. Plasma fructosamine was performed per the manufacturer’s instructions.
Western Blotting
Hippocampal proteins were extracted in RIPA buffer (50 mM Tris HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton 100, 1 mM sodium orthovanadate) containing protease inhibitor and phosphatase inhibitor for 30 min on ice with brief vortexing after every 5 min. Tissue homogenate was cleared by centrifugation at 17,000 rpm, 4 °C for 1 h. Supernatant protein estimation was performed by Bradford’s method. 40 μg of hippocampal proteins was separated on 10% SDS-PAGE gel. Proteins were then transferred onto polyvinylidene difluoride membrane. Ponceau S solution was used to monitor equal loading and uniform transfer of proteins and incubated for 1 h at room temperature in a blocking buffer containing 3% BSA. It was then further incubated with the primary antibody in a blocking buffer. Following antibody dilutions were used, Th205/AT8 (1:1000), Tau (1:8000), RAGE (1:3000), GAPDH (1:1000). The membranes were incubated either with antimouse or antirabbit peroxidise conjugated secondary antibody for 1 h at RT at a dilution of 1:5000 in PBST. Detection was performed using Amersham ECL prime per the manufacturer’s instructions. Harsh stripping buffer followed by blocking was used before probing with another primary antibody.
Hippocampal Histochemistry
The brain tissue samples were collected from the respective experimental groups in 10% neutral buffer formalin as a standard fixative and tissue preservative for histology. After 72 h of fixation, the tissue sections of brain were grossed and processed on an automated tissue processor for the histopathological protocol using ascending grades of alcohol and cleared in xylene. The tissue was embedded in paraffin blocks for microsectioning on an automated tissue microtome (Leica, Germany), and tissue sections of 5 μm were taken on glass slides. The tissue sections were stained by routine Hematoxylin and Eosin protocol and were observed under a binocular microscope with a microphotography unit (Nikon, Japan). The histopathological examination was performed for observation of any pathological and cellular changes in the cerebrum and in the C-region of the brain.
Acknowledgments
G.P. would like to acknowledge Lady Tata Memorial Trust, Mumbai, India, for fellowship. We would also like to thank Babasaheb Sonawane, Shakuntala Kolewar, and Arvind Chaurasiya for help in animal experiments.
Glossary
Abbreviations
- AGEs
advanced glycation end products
- AD
Alzheimer’s disease
- ARB
angiotensin II receptor blocker
- AMG
aminoguanidine
- Aβ
amyloid beta
- CSF
cerebrospinal fluid
- JNK
c-Jun N-terminal kinases
- EPM
elevated plus maze
- GABA
γ aminobutyric acid
- GSK-3β
glycogen synthase kinase-3β
- iNOS
inducible nitric oxide synthase
- MGO
methylglyoxal
- MAPK
mitogen-activated protein kinase
- NFTs
neurofibrillary tangles
- PPAR-γ
peroxisome proliferator-activated receptor-γ
- PP2A
protein phosphatase
- pTau
phosphorylated Tau
- ROS
reactive oxygen species
- RAGE
receptor for advanced glycation end products
- SPs
senile plaques
- STZ
Streptozotocin
- TELMI
Telmisartan
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00143.
Figure S1, parameters of the rats under study ((A) body weight, (B) blood glucose, (C) HbA1c); Figure S2, overall view of the IDA-SWATH methodology used; Figure S3, RAGE Western blot (A) ECL image, (B) Densitometric analysis) (PDF)
Sheet 1, parameters of the rats under study; Sheet 2, list of proteins identified in IDA SWATH analysis with at least 2 unique peptides; Sheet 3, list of peptides identified for each protein; Sheet 4, list of distinct peptides identified for each protein; Sheet 5, list of common differentially expressed proteins between STZ and MGO w.r.t control; Sheet 6, list of MGO+TELMI cotreatment regulated proteins; Sheet 7, list of MGO+AMG cotreatment regulated proteins (XLS)
Author Contributions
$ S. Kulsange and R. Kazi contributed equally.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “Autism and Neurodevelopmental Disorders”.
Supplementary Material
References
- Watson G. S.; Craft S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs 2003, 17 (1), 27–45. 10.2165/00023210-200317010-00003. [DOI] [PubMed] [Google Scholar]
- Profenno L. A.; Porsteinsson A. P.; Faraone S. V. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 2010, 67 (6), 505–512. 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Matsuzaki T.; Sasaki K.; Tanizaki Y.; Hata J.; Fujimi K.; Matsui Y.; Sekita A.; Suzuki S. O.; Kanba S.; Kiyohara Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 2010, 75 (9), 764–770. 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- S. Roriz-Filho J.; Sa-Roriz T. M.; Rosset I.; Camozzato A. L.; Santos A. C.; Chaves M. L.F.; Moriguti J. C.; Roriz-Cruz M. (Pre)diabetes, brain aging, and cognition. Biochim. Biophys. Acta 2009, 1792 (5), 432–443. 10.1016/j.bbadis.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Imamura T.; Yanagihara Y. T.; Ohyagi Y.; Nakamura N.; Iinuma K. M.; Yamasaki R.; Asai H.; Maeda M.; Murakami K.; Irie K.; et al. Insulin deficiency promotes formation of toxic amyloid-β42 conformer co-aggregating with hyper-phosphorylated tau oligomer in an Alzheimer’s disease model. Neurobiol Dis 2020, 137, 104739. 10.1016/j.nbd.2020.104739. [DOI] [PubMed] [Google Scholar]
- Gratuze M.; Julien J.; Petry F. R.; Morin F.; Planel E. Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology. Sci. Rep 2017, 7, 46359. 10.1038/srep46359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsrijai U.; Wongchitrat P.; Nopparat C.; Satayavivad J.; Govitrapong P. Melatonin attenuates streptozotocin-induced Alzheimer-like features in hyperglycemic rats. Neurochem. Int. 2020, 132, 104601. 10.1016/j.neuint.2019.104601. [DOI] [PubMed] [Google Scholar]
- Mukai N.; Ohara T.; Hata J.; Hirakawa Y.; Yoshida D.; Kishimoto H.; Koga M.; Nakamura U.; Kitazono T.; Kiyohara Y.; et al. Alternative Measures of Hyperglycemia and Risk of Alzheimer’s Disease in the Community: The Hisayama Study. Journal of clinical endocrinology and metabolism 2017, 102 (8), 3002–3010. 10.1210/jc.2017-00439. [DOI] [PubMed] [Google Scholar]
- Angeloni C.; Zambonin L.; Hrelia S. Role of methylglyoxal in Alzheimer’s disease. BioMed. research international 2014, 2014, 238485. 10.1155/2014/238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I.; Bélanger M.; Magistretti P. J. Methylglyoxal, the dark side of glycolysis. Frontiers in neuroscience 2015, 9, 23. 10.3389/fnins.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Arriba S. G.; Stuchbury G.; Yarin J.; Burnell J.; Loske C.; Munch G. Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells-protection by carbonyl scavengers. Neurobiology of aging 2007, 28 (7), 1044–1050. 10.1016/j.neurobiolaging.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Huang S.-M.; Chuang H.-C.; Wu C.-H.; Yen G.-C. Cytoprotective effects of phenolic acids on methylglyoxal-induced apoptosis in Neuro-2A cells. Molecular nutrition & food research 2008, 52 (8), 940–949. 10.1002/mnfr.200700360. [DOI] [PubMed] [Google Scholar]
- Hansen F.; Pandolfo P.; Galland F.; Torres F. V.; Dutra M. F.; Batassini C.; Guerra M. C.; Leite M. C.; Gonçalves C.-A. Methylglyoxal can mediate behavioral and neurochemical alterations in rat brain. Physiology & behavior 2016, 164 (Pt A), 93–101. 10.1016/j.physbeh.2016.05.046. [DOI] [PubMed] [Google Scholar]
- Srikanth V.; Westcott B.; Forbes J.; Phan T. G.; Beare R.; Venn A.; Pearson S.; Greenaway T.; Parameswaran V.; Münch G. Methylglyoxal, cognitive function and cerebral atrophy in older people. journals of gerontology. Series A, Biological sciences and medical sciences 2013, 68 (1), 68–73. 10.1093/gerona/gls100. [DOI] [PubMed] [Google Scholar]
- KUHLA B.; LUTH H.-J.; HAFERBURG D.; BOECK K.; ARENDT T.; MUNCH G. Methylglyoxal, glyoxal, and their detoxification in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 2005, 1043, 211–216. 10.1196/annals.1333.026. [DOI] [PubMed] [Google Scholar]
- Farhad A. R.; Razavi S.; Jahadi S.; Saatchi M. Use of aminoguanidine, a selective inducible nitric oxide synthase inhibitor, to evaluate the role of nitric oxide in periapical inflammation. Journal of oral science 2011, 53 (2), 225–230. 10.2334/josnusd.53.225. [DOI] [PubMed] [Google Scholar]
- Tsukuda K.; Mogi M.; Iwanami J.; Min L.-J.; Sakata A.; Jing F.; Iwai M.; Horiuchi M. Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension (Dallas, Tex.: 1979) 2009, 54 (4), 782–787. 10.1161/HYPERTENSIONAHA.109.136879. [DOI] [PubMed] [Google Scholar]
- Benson S. C.; Pershadsingh H. A.; Ho C. I.; Chittiboyina A.; Desai P.; Pravenec M.; Qi N.; Wang J.; Avery M. A.; Kurtz T. W. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension (Dallas, Tex.: 1979) 2004, 43 (5), 993–1002. 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- Ahmadian M.; Suh J. M.; Hah N.; Liddle C.; Atkins A. R.; Downes M.; Evans R. M. PPARγ signaling and metabolism: the good, the bad and the future. Nature medicine 2013, 19 (5), 557–566. 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano L.; Simon A.-M.; Perez-Mediavilla A.; Salazar-Colocho P.; Rio J. D.; Frechilla D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer’s disease mouse model. Biochemical and biophysical research communications 2009, 379 (2), 406–410. 10.1016/j.bbrc.2008.12.071. [DOI] [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists.. Nucleic Acids Res. 2009, 37, 1–13. 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4 (1), 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Zhou F.; Mei J.; Yang H.; Li H. The Effect of Type 2 Diabetes Mellitus on Neuropsychological Symptoms in Chinese Early Alzheimer’s Disease Population. Neuropsychiatr Dis Treat 2020, 16, 829–836. 10.2147/NDT.S240529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhotra N.; Dhingra D. Involvement of NO-cGMP pathway in anti-anxiety effect of aminoguanidine in stressed mice. Progress in neuro-psychopharmacology & biological psychiatry 2009, 33 (8), 1502–1507. 10.1016/j.pnpbp.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Aswar U.; Chepurwar S.; Shintre S.; Aswar M. Telmisartan attenuates diabetes induced depression in rats. Pharmacological reports: PR 2017, 69 (2), 358–364. 10.1016/j.pharep.2016.12.004. [DOI] [PubMed] [Google Scholar]
- Li Y.; Cheng K.-C.; Liu K.-F.; Peng W.-H.; Cheng J.-T.; Niu H.-S. Telmisartan Activates PPARδ to Improve Symptoms of Unpredictable Chronic Mild Stress-Induced Depression in Mice. Sci. Rep. 2017, 7 (1), 14021. 10.1038/s41598-017-14265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm T.; Mariani M.; Donovan L. J.; Neilson L.; Landreth G. E. Activation of the nuclear receptor PPARδ is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. Journal of neuroinflammation 2015, 12, 7. 10.1186/s12974-014-0229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Hou Y.; Tu W.; Campbell N.; Pieper A. A.; Leverenz J. B.; Gao S.; Cummings J.; Cheng F. Population-based discovery and Mendelian randomization analysis identify telmisartan as a candidate medicine for Alzheimer’s disease in African Americans. Alzheimer’s Dementia 2022, 10.1002/alz.12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajashree R.; Kholkute S. D.; Goudar S. S. Effects of duration of diabetes on behavioural and cognitive parameters in streptozotocin-induced juvenile diabetic rats. Malays. J. Med. Sci. 2011, 18 (4), 26–31. [PMC free article] [PubMed] [Google Scholar]
- Serý O.; Povová J.; Míšek I.; Pešák L.; Janout V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: a review. Folia neuropathologica 2013, 51 (1), 1–9. 10.5114/fn.2013.34190. [DOI] [PubMed] [Google Scholar]
- Bose A.; Beal M. F. Mitochondrial dysfunction in Parkinson’s disease. Journal of neurochemistry 2016, 139, 216–231. 10.1111/jnc.13731. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang W.; Li L.; Perry G.; Lee H.-g.; Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochimica et biophysica acta 2014, 1842 (8), 1240–1247. 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.; Beja-Glasser V. F.; Nfonoyim B. M.; Frouin A.; Li S.; Ramakrishnan S.; Merry K. M.; Shi Q.; Rosenthal A.; Barres B. A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (New York, N.Y.) 2016, 352 (6286), 712–716. 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T. C. The synaptic vesicle cycle. Annual review of neuroscience 2004, 27, 509–547. 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Auffret A.; Gautheron V.; Mattson M. P.; Mariani J.; Rovira C. Progressive age-related impairment of the late long-term potentiation in Alzheimer’s disease presenilin-1 mutant knock-in mice. J. Alzheimers Dis. 2010, 19 (3), 1021–1033. 10.3233/JAD-2010-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg J. R.; Bloom G. S. Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskeleton 2009, 66 (8), 635–649. 10.1002/cm.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber S.; Lubec G.; Cairns N.; Fountoulakis M. Decreased levels of synaptosomal associated protein 25 in the brain of patients with Down syndrome and Alzheimer’s disease. Electrophoresis 1999, 20 (4–5), 928–934. 10.1002/(SICI)1522-2683(19990101)20:4/5<928::AID-ELPS928>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Noor A.; Zahid S. A review of the role of synaptosomal-associated protein 25 (SNAP-25) in neurological disorders. International journal of neuroscience 2017, 127 (9), 805–811. 10.1080/00207454.2016.1248240. [DOI] [PubMed] [Google Scholar]
- Burbaeva G. S.; Boksha I. S.; Tereshkina E. B.; Starodubtseva L. I.; Savushkina O. K.; Vorob’eva E. A.; Prokhorova T. A. [A role of glutamate decarboxylase in Alzheimer’s disease]. Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova 2014, 114 (4), 68–72. [PubMed] [Google Scholar]
- Gueli M. C.; Taibi G. Alzheimer’s disease: amino acid levels and brain metabolic status. Neurological sciences: official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 2013, 34 (9), 1575–1579. 10.1007/s10072-013-1289-9. [DOI] [PubMed] [Google Scholar]
- Li X.; Serwanski D. R.; Miralles C. P.; Nagata K.-i.; De Blas A. L. Septin 11 is present in GABAergic synapses and plays a functional role in the cytoarchitecture of neurons and GABAergic synaptic connectivity. J. Biol. Chem. 2009, 284 (25), 17253–17265. 10.1074/jbc.M109.008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Tanimukai H.; Liu F.; Iqbal K.; Grundke-Iqbal I.; Gong C.-X. Elevation of the level and activity of acid ceramidase in Alzheimer’s disease brain. European journal of neuroscience 2004, 20 (12), 3489–3497. 10.1111/j.1460-9568.2004.03852.x. [DOI] [PubMed] [Google Scholar]
- Musunuri S.; Wetterhall M.; Ingelsson M.; Lannfelt L.; Artemenko K.; Bergquist J.; Kultima K.; Shevchenko G. Quantification of the brain proteome in Alzheimer’s disease using multiplexed mass spectrometry. J. Proteome Res. 2014, 13 (4), 2056–2068. 10.1021/pr401202d. [DOI] [PubMed] [Google Scholar]
- Takehashi M.; Alioto T.; Stedeford T.; Persad A. S.; Banasik M.; Masliah E.; Tanaka S.; Ueda K. Septin 3 gene polymorphism in Alzheimer’s disease. Gene Expr. 2004, 11 (5–6), 263–270. 10.3727/000000003783992243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite S. P.; Stock J. B.; Lombroso P. J.; Nairn A. C. Protein phosphatases and Alzheimer’s disease. Progress in molecular biology and translational science 2012, 106, 343–379. 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. A.; Arif Z.; Khan M. A.; Moinuddin; Alam K. Methylglyoxal produces more changes in biochemical and biophysical properties of human IgG under high glucose compared to normal glucose level. PLoS One 2018, 13 (1), e0191014 10.1371/journal.pone.0191014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese E.; Montagnana M.; Nouvenne A.; Lippi G. Advantages and pitfalls of fructosamine and glycated albumin in the diagnosis and treatment of diabetes. Journal of diabetes science and technology 2015, 9 (2), 169–176. 10.1177/1932296814567227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley P. J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Archives of biochemistry and biophysics 2003, 419 (1), 31–40. 10.1016/j.abb.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Yamagishi S.; Nakamura K.; Matsui T.; Imaizumi T.; Takeuchi M.; Koga H.; Ueno T.; Sata M. Telmisartan inhibits AGE-induced C-reactive protein production through downregulation of the receptor for AGE via peroxisome proliferator-activated receptor-gamma activation. Diabetologia 2006, 49 (12), 3094–3099. 10.1007/s00125-006-0437-7. [DOI] [PubMed] [Google Scholar]
- Lohwasser C.; Neureiter D.; Weigle B.; Kirchner T.; Schuppan D. The receptor for advanced glycation end products is highly expressed in the skin and upregulated by advanced glycation end products and tumor necrosis factor-alpha. Journal of investigative dermatology 2006, 126 (2), 291–299. 10.1038/sj.jid.5700070. [DOI] [PubMed] [Google Scholar]
- Li X.-H.; Xie J.-Z.; Jiang X.; Lv B.-L.; Cheng X.-S.; Du L.-L.; Zhang J.-Y.; Wang J.-Z.; Zhou X.-W. Methylglyoxal induces tau hyperphosphorylation via promoting AGEs formation. Neuromolecular medicine 2012, 14 (4), 338–348. 10.1007/s12017-012-8191-0. [DOI] [PubMed] [Google Scholar]
- Kurata T.; Lukic V.; Kozuki M.; Wada D.; Miyazaki K.; Morimoto N.; Ohta Y.; Deguchi K.; Ikeda Y.; Kamiya T.; et al. Telmisartan reduces progressive accumulation of cellular amyloid beta and phosphorylated tau with inflammatory responses in aged spontaneously hypertensive stroke resistant rat. J. Stroke Cerebrovasc Dis 2014, 23 (10), 2580–2590. 10.1016/j.jstrokecerebrovasdis.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Qu Z.; Jiao Z.; Sun X.; Zhao Y.; Ren J.; Xu G. Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res. 2011, 1383, 300–306. 10.1016/j.brainres.2011.01.084. [DOI] [PubMed] [Google Scholar]
- Basurto-Islas G.; Grundke-Iqbal I.; Tung Y. C.; Liu F.; Iqbal K. Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J. Biol. Chem. 2013, 288 (24), 17495–17507. 10.1074/jbc.M112.446070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman D. M.; Grubb M.; Deacon R. M. J.; Yee B. K.; Feldon J.; Rawlins J. N. P. Ventral hippocampal lesions affect anxiety but not spatial learning. Behavioural brain research 2003, 139 (1–2), 197–213. 10.1016/S0166-4328(02)00268-1. [DOI] [PubMed] [Google Scholar]
- Kerchner G. A.; Hess C. P.; Hammond-Rosenbluth K. E.; Xu D.; Rabinovici G. D.; Kelley D. A. C.; Vigneron D. B.; Nelson S. J.; Miller B. L. Hippocampal CA1 apical neuropil atrophy in mild Alzheimer disease visualized with 7-T MRI. Neurology 2010, 75 (15), 1381–1387. 10.1212/WNL.0b013e3181f736a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso S.; Carvalho C.; Marinho R.; Simões A.; Sena C. M.; Matafome P.; Santos M. S.; Seiça R. M.; Moreira P. I. Effects of methylglyoxal and pyridoxamine in rat brain mitochondria bioenergetics and oxidative status. Journal of bioenergetics and biomembranes 2014, 46 (5), 347–355. 10.1007/s10863-014-9551-2. [DOI] [PubMed] [Google Scholar]
- Bhat S.; Jagadeeshaprasad M. G.; Patil Y. R.; Shaikh M. L.; Regin B. S.; Mohan V.; Giri A. P.; Balasubramanyam M.; Boppana R.; Kulkarni M. J. Proteomic Insight Reveals Elevated Levels of Albumin in Circulating Immune Complexes in Diabetic Plasma. Molecular & cellular proteomics: MCP 2016, 15 (6), 2011–2020. 10.1074/mcp.M116.058008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salum E.; Butlin M.; Kals J.; Zilmer M.; Eha J.; Avolio A. P.; Arend A.; Aunapuu M.; Kampus P. Angiotensin II receptor blocker telmisartan attenuates aortic stiffening and remodelling in STZ-diabetic rats. Diabetol Metab Syndr 2014, 6, 57. 10.1186/1758-5996-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary S.; Kulkarni M. J.; Malakar D.; Joshi S. R.; Mehendale S. S.; Giri A. P. Placental Proteomics Provides Insights into Pathophysiology of Pre-Eclampsia and Predicts Possible Markers in Plasma. J. Proteome Res. 2017, 16 (2), 1050–1060. 10.1021/acs.jproteome.6b00955. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.