

Abstract
Due to improved efficiency and reduced cost of viral sequencing, molecular cluster analysis can be feasibly utilized alongside existing human immunodeficiency virus (HIV) prevention strategies. The goal of this paper is to elucidate how HIV molecular cluster and social network analyses are being integrated to implement HIV response interventions. We searched PubMed, Scopus, PsycINFO, and Cochrane Library databases for studies incorporating both HIV molecular cluster and social network data. We identified 32 articles that combined analyses of HIV molecular sequences and social or sexual networks. All studies were descriptive. Six studies described network interventions informed by molecular and social data but did not fully evaluate their efficacy. There is no current standard for incorporating molecular and social network analyses to inform interventions or data demonstrating its utility. More research must be conducted to delineate benefits and best practices for leveraging molecular data for network-based interventions.
Keywords: Cluster analysis, HIV, Social networks, Contact tracing, Systematic review, Cluster detection and response
Resumen
Debido a mejor eficiencia y costo reducido de la secuenciación viral, el análisis de complejos moleculares se puede utilizar de manera factible junto con las estrategias de prevención del virus de inmunodeficiencia humana (VIH) existentes. El objetivo de este repaso es de aclarar como integrar los análisis de las redes sociales y de los complejos moleculares del VIH para implementar intervenciones para controlar el VIH. Buscamos en las bases de datos de PubMed, Scopus, PsycINFO y Cochrane Library por estudios que incorporaran datos de redes sociales y grupos moleculares del VIH. Identificamos 32 estudios que combinaban análisis de secuencias moleculares del VIH y datos de redes sociales. Todos los estudios fueron descriptivos. Seis estudios describieron intervenciones informadas por datos moleculares y sociales, pero no evaluaron completamente su eficacia. No existe un estándar actual para incorporar análisis moleculares y sociales para informar intervenciones o datos que demuestren su eficacia. Se deben realizar más investigaciones para delinear los beneficios y las mejores prácticas de aplicar los datos moleculares y sociales para crear intervenciones del VIH.
Introduction
Network-level surveillance methods are one of the primary strategies employed by health departments to monitor the transmission of Human Immunodeficiency Virus (HIV) and reduce subsequent infections [1, 2]. Partner services, a common social network intervention, involves collecting partner information from individuals newly diagnosed with HIV and informing these sexual and drug-use contacts of possible exposure, often with the aid of health providers, disease intervention specialists (DIS) [2, 3], or internet-based referral services [4]. Due to its high yield in identifying new HIV diagnoses [5, 6] and preventing onward HIV transmission [7, 8], forms of partner services or partner notification, along with linkage-to-care, have become the gold standard for health department HIV identification and prevention services provided upon identifying new diagnoses.
With recent advances in genomic sequencing technology, public health officials have shown interest in utilizing molecular cluster analysis alongside partner notification to bolster existing HIV surveillance methods [9, 10] and ultimately inform targeted interventions [11]. Molecular cluster analysis uses viral genetic sequences to group viral samples into transmission clusters with the goal of pinpointing transmission risk factors, monitoring growing clusters, identifying individuals at risk of HIV acquisition [11–14], and reducing subsequent transmission [15, 16]. Molecular cluster analysis is well suited for HIV surveillance due to HIV’s predictable error rate, which provides an easily interpretable “molecular clock” [17]. The viral sequences required to conduct molecular cluster analyses are increasingly available to health departments and researchers due to recommendations made by the Department of Health and Human Services (DHHS) that newly diagnosed individuals receive drug resistance testing upon entry to care [18]. Molecular cluster analysis is particularly appealing because, when integrated with traditional surveillance approaches, it could potentially identify gaps in existing HIV testing and treatment programs, inform cluster-specific interventions [15, 19], direct scarce resources to transmission “hotspots” [13, 20], and conversely determine when additional resources may not be required.
While cluster detection and response utilizing molecular and social network data holds promise, the efficacy of this approach remains largely unknown. Prior systematic reviews have explored the effectiveness of partner services [21], online and phone messaging-based referral services [4], and HIV testing interventions [22]. The ethical implications of applying molecular cluster analysis to HIV have also been analyzed [23]. No review, however, systematically examines the utilization of molecular and social network data together to develop network-based interventions that interrupt ongoing transmission. We evaluated peer-reviewed publications and grey literature from major HIV conferences to assess how molecular cluster analysis has been used with partner services or other social network approaches to investigate HIV transmission and inform targeted interventions. A better understanding of these processes is important if we are to effectively use molecular data to respond to new diagnoses and move towards eliminating HIV transmission.
Methods
Article Identification
The systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) (Table S1) [24]. An exhaustive list of search terms was compiled based on prior investigator knowledge in consultation with an academic librarian and is available as part of the supplemental materials (Online Appendix A). Database searches of PubMed, Scopus, PsycINFO, and Cochrane Library were completed on March 27, 2020, and again on May 7, 2021, yielding a total of 1201 articles. In addition, online abstracts from conference proceedings for the following conferences and years were reviewed: International AIDS Conference (2001–2016), Conferences on Retroviruses and Opportunistic Infections (2014–2020), AIDS (2016, 2018), HIV Research for Prevention (2016, 2018), IDWeek (2018), National HIV Prevention Conference (2019), and the Laboratory of Biotechnology and Molecular Virology Conference (2019). The review of conference proceedings identified two eligible abstracts. Reference lists of included articles were also reviewed, yielding one additional eligible article. Articles suggested by subject experts were additionally reviewed for inclusion, identifying an additional six studies.
Article Eligibility and Screening Process
Articles were screened and selected based on two primary inclusion criteria: (1) molecular cluster analysis of HIV sequence data was described in the study, and (2) relational data between individuals (referred to as “social network data” throughout) was captured and utilized in the analysis including sexual and social contact data from partner services, contact tracing, other social network sources (i.e., Facebook friends), respondent-driven sampling (RDS), and/or connections through shared venue attendance. No restrictions were placed on article publication date or study location. We excluded articles that focused primarily on hypothetical modeling or laboratory research in order to focus specifically on how molecular analysis has been implemented in real-world settings. We also excluded studies that only presented molecular or social network connections between dyads, ignoring the larger network, including those describing transmission between one index case and multiple partners without successive rounds of transmission. The search terms were inclusive of a broad range of diseases to ensure all HIV transmission articles were identified, but all non-HIV disease conditions were ultimately excluded. If multiple articles utilized the same data source over the same time period, the article with the most robust molecular/social network data analysis or response intervention was included. Detailed reasons for article ineligibility are presented in Fig. 1.
Fig. 1.
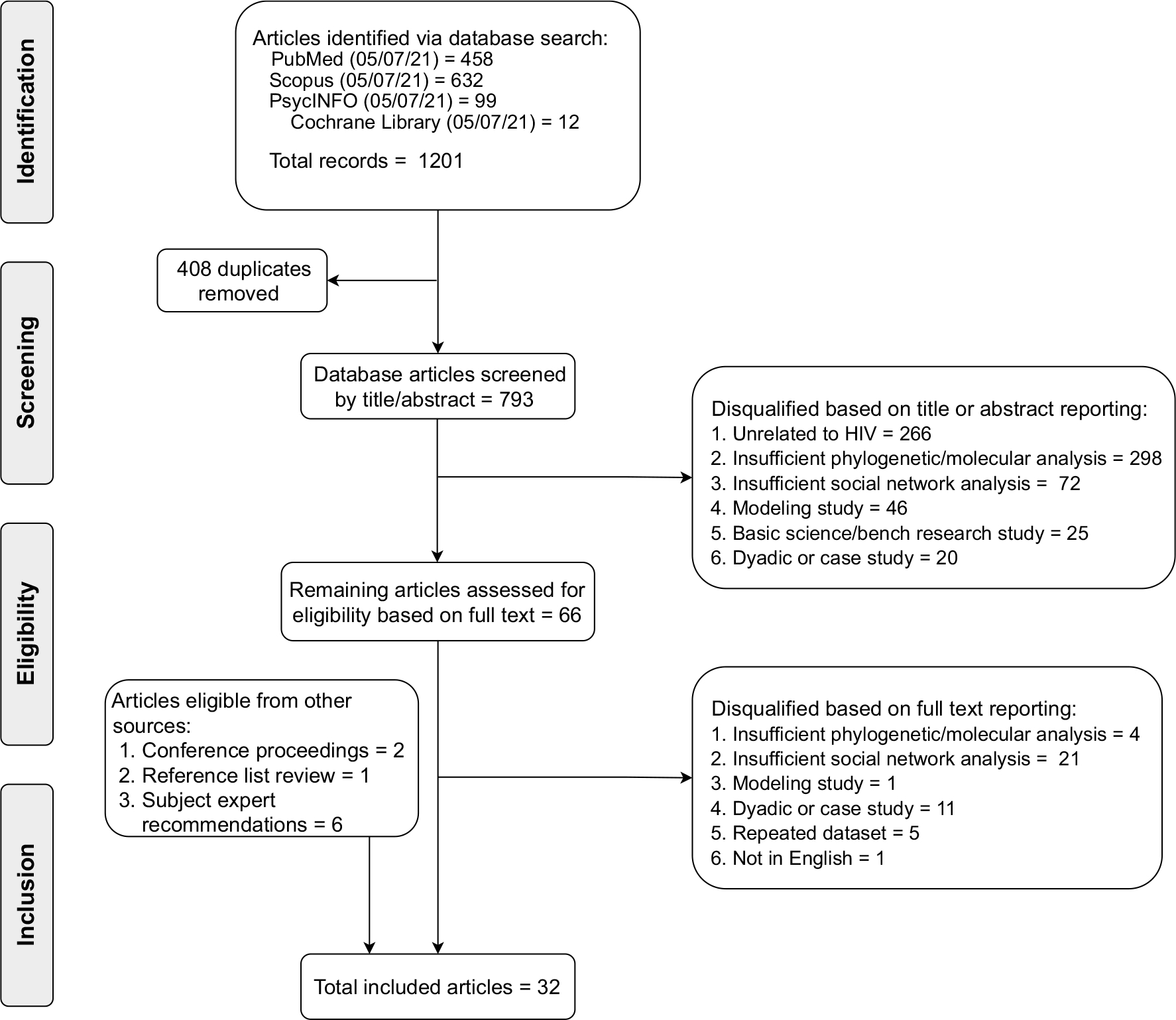
Preferred reporting items for systematic review and meta-analyses (PRISMA) flow diagram detailing systematic article screening and selection process
All 1201 identified articles were exported into an Endnote X7 library (Clarivate, Philadelphia, PA). Duplicates (n = 408) were removed, and the remaining 793 articles were assessed for eligibility. Authors DG, KS, and AU conducted preliminary screening of titles and abstracts, with conflicts adjudicated by senior author JS. To ensure reliability and consistency, DG, KS, and AU screened groups of 10 articles and sorted them into categories of qualification or disqualification for further review based on criteria outlined in Fig. 1. Authors DG, KS, AU, and JS discussed all inconsistencies in paper sorting until each categorization was consistent among reviewers (five rounds). Full-text review of 66 articles was completed by DG, KS, and SR, and all articles eligible for inclusion were reviewed by the senior author JS. Undecided studies were reviewed and adjudicated together by DG, KS, SR, and senior author JS during team meetings. A total of 727 articles were excluded, primarily because they did not involve molecular cluster analysis or because they focused on a disease other than HIV.
Data Abstraction & Charting
Data from articles were abstracted by DG, KS, and SR utilizing a standardized format and all abstractions were thoroughly audited for accuracy and consistency by a single author (DG) [25]. For each study, we collected information on the year of publication, study location, time period, study design, data source (i.e., a government agency, academic or clinical institution, or named cohort study), sample size, and characteristics of the studied population/s. We also documented detailed summaries of how both the social network data and HIV sequence data were obtained and analyzed, how they were integrated into the study, and the study’s main results. Quantitative ratings of evidence presented in articles were not possible due to the exploratory, descriptive nature of included articles and a lack of consistent outcomes reported across studies.
Role of the Funding Source
Study funders were not involved in study design, collection, analysis and interpretation of data, nor the decision to submit this work for publication.
Results
Descriptive Characteristics
Among the 32 included studies, almost all were conducted in either high-income (n = 26) or upper-middle income countries (n = 4) (Table 1) [26]. The majority (n = 20) were conducted in the United States, with the Midwest being the most represented region. No included studies were conducted before the year 2001, and the majority were conducted between 2016 and 2020 (n = 19). Major populations of interest in the studies included men who have sex with men [19, 20, 27–40], racial minorities [29–31, 34, 35, 39, 41–43], female sex workers [33, 44–46], and persons who inject drugs [39–41, 46–53]. The size of the study population varied greatly, with five studies having small sample sizes under 20 people [27, 34, 46, 52, 54] and eight studies including greater than 1000 individuals [19, 29, 32, 33, 43, 44, 48, 55], though complete molecular cluster and social network analyses were sometimes performed only on subsets. We identified three different forms of relational network data that were utilized by studies: (1) sexual or drug use contact tracing data from partner services or DIS interviews [19, 27–32, 36–44, 46–48, 50–54, 56, 57], (2) RDS data or other data capturing social contacts (i.e., reported social contacts, Facebook friends) [33–35, 38, 47, 49], and (3) information about individuals’ sex or drug-use venue attendance [20, 34, 45]. We categorized the included articles into the following groups based on the purpose of the analysis: (1) studies that characterized transmission clusters by assessing the overlap between social and molecular networks (n = 20), (2) studies documenting HIV outbreak investigations using molecular and social data (n = 6), and (3) studies depicting response interventions informed by molecular and social data (n = 6). For the purpose of this review, response interventions were defined as public health interventions implemented along the HIV surveillance, testing, care linkage, treatment, and prevention continuum beyond standard contact tracing and linkage-to-care measures typically provided by health departments during HIV outbreaks. Interventions were further required to present quantitative data on their HIV outcomes for inclusion. All studies were observational in design.
Table 1.
Summary characteristics of included articles (N = 32)
| Study region | N |
|---|---|
|
| |
| United States | 20 |
| South | 7 |
| Northeast | 2 |
| West | 5 |
| Midwest | 5 |
| Multiple regions | 1 |
| International | 12 |
| Year | |
| < 2005 | 1 |
| 2006–2010 | 6 |
| 2011–2015 | 4 |
| 2016–2020 | 19 |
| 2021 | 2 |
| Study type | |
| Cohort study | 20 |
| Outbreak investigation | 12 |
| Journal impact factora | |
| < 3 | 9 |
| 3–5 | 12 |
| 5–7 | 6 |
| 7+ | 3 |
| Source of molecular data | |
| Government-associated database | 20 |
| Research cohort | 10 |
| Unknown | 2 |
| Type of network datab | |
| Sexual and/or drug-use contact tracing (i.e., partner services) | 26 |
| Respondent-driven sampling or other social contact network | 5 |
| Venue-based | 3 |
| Populations of interest | |
| Men who have sex with men | 12 |
| Female sex workers | 3 |
| Acute HIV infection | 8 |
| Persons who inject drugs | 9 |
Total does not sum to 32, two included studies were in the form of a conference abstract
Total does not sum to 32, some studies utilized multiple types of network data
Studies Characterizing Transmission Networks
Twenty studies were identified that investigated the overlap between networks generated with social or sexual network data and networks generated from molecular cluster analysis of HIV sequences (Table 2) [20, 28–38, 43–45, 47–49, 56, 57]. Many studies were secondary analyses utilizing data from HIV research cohorts, though a portion (n = 8) utilized routine HIV surveillance data from a government source [28–31, 43, 44, 48, 57]. All 20 studies were descriptive in nature.
Table 2.
Studies combining phylogenetic and social network analysis to characterize transmission networks (N = 20)
| Authors | Title | Pub. yeara | Study design | Locationb | Time period | Data source/project | Population characteristics | Social/sexual network data | Molecular cluster analysis | Data integration | Resultsc |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Campbell et al. | Phylodynamic Analysis Complements Partner Services by Identifying Acute and Unreported HIV Transmission | 2020 | Cohort | North Carolina, New York City, San Francisco | 2011–2013 | Screening Targeted Populations to Interrupt Ongoing Chains of HIV Transmission with Enhanced Partner Notification (STOP) Project | 1326 cohort members with newly diagnosed acute HIV infection. 94.8% male, 65.0% non-white, median age of 30 years, 78% MSM | Enhanced partner notification was offered to participants who screened positive at one of 12 participating HIV testing sites | Partial HIV pol sequences from persons with available blood specimens (70 participants with AHI and 547 total participants) were analyzed with Microbe-Trace using genetic distance threshold of d< 1.5% | Genetic networks and sexual contact networks were constructed independently and then integrated into a multi-partite network | 12.6% of HIV positive participants (n = 1326) had an Acute HIV Infection. Among participants with available pol sequences, 465 high-risk contacts were reported, 65.7% of which were genetically supported as a transmission pair. Five links between individuals with AHI were reported, but none were genetically supported. Phylodynamic analysis identified an additional 102 unreported putative transmission links. Participants who were in a cluster were more likely to be younger. Black men who met partners online were most likely to be in the largest clusters |
| Deng et al. | Molecular Epidemiological Tracing of HIV-1 Outbreaks in Hainan Island of Southern China | 2009 | Cohort | Hainan Island Province, China | 1991–2006 | Chinese national HIV/AIDS surveillance system and sentinel surveillance program | 499,752 individuals from high-risk groups (IVDUs, female sex workers & their clients, STD clinic attendees, truck drivers, pregnant women, MSM, TB patients, former plasma donors, and children of HIV-infected mothers) | Participants completed standardized questionnaire administered by the Hainan Center for Disease Control & Prevention that included demographics, risk factors, mode of transmission, and sexual contacts. A second round of contact tracing was conducted on individuals found to be in the molecular clusters that had not previously completed a questionnaire | Blood samples were collected from a representative subset of all participants based on HIV prevalence in each county (n = 88). Phylogenetic relationships between viral pol sequences were estimated using neighbor-joining trees and maximum likelihood models | Phylogenetic tree was used to confirm or reject possible transmission events suggested by contact tracing data | 523 of the 499,752 participants were diagnosed with HIV infection. The 83 available patient samples segregated into one large and three small clusters. Phylogenetic analysis confirmed four purported heterosexual transmission links suggested by contact tracing and rejected two purported links |
| Dennis et al. | Social Network Based Recruitment Successfully Reveals HIV-1 Transmission Networks Among High Risk Individuals in El Salvador | 2013 | Cohort | San Salvador, San Miguel, Sonsonate; El Salvador | 2008 | Encuesta Centroamericano de Vigilancia de VIH y Comportamiento en Poblaciones Vulnerables (ECVC) | 699 MSM and 757 Female Sex Workers (FSW) | HIV-positive MSM and female sex workers were recruited via multiples waves of respondent driven sampling, which was used to construct a social network. Recruits were interviewed about their demographics, sex practices, and history of STIs | A phylogenetic tree was constructed from pol sequences from 119 HIV-positive individuals recruited via respondent driven sampling. Mean pairwise genetic distance <0.015 was used to identify transmission clusters | The phylogenetic network was compared to the network built from respondent-driven sampling | Of the available sequences, 43% of MSM and 10% of FSW had viral sequences that grouped into 14 transmission clusters. Clusters were grouped by risk factors (12 MSM clusters) and by geography. Only two transmission clusters had participants directly linked through respondent recruitment. All clusters greater than two individuals spanned more than one RDS chain. Cluster membership was associated with recent HIV infection, sex with a stable male partner, and sex with three or more partners in the past year |
| Fujimoto et al. | Lack of Support for Socially Connected HIV-1 Transmission Among Young Adult Black Men Who Have Sex with Men | 2017 | Cohort | Houston, TX | 2015 | Integrated Molecular & Affiliation Network analysis of HIV transmission (iMAN) | 10 study participants. All Black MSM aged 17–29, average age of 25 | Young black MSM were recruited via respondent driven sampling. A peer-referral network based on social and sexual connections was constructed from an adjacency matrix which was then maximally symmetrized. Two venue-based networks among the 10 individuals were also constructed | Clinical samples from subset of 10 iMAN participants were analyzed. Models of sequence evolution were chosen using Akaike’s Information Criteria. Maximum likelihood and Bayesian phylogenetic analyses were performed using AIC-chosen models. GenBank was used to identify 20 control HIV-1 sequences | Sequences from individuals found to be connected in RDS-derived network were analyzed for genetic similarity to assess for evidence of transmission pairs | No evidence was found to suggest transmission of viruses between any individuals in the socially-connected RDS chain. Support for relationships among viruses sampled from different individuals were low, thus did not conform to the structure expected for a recent transmission cluster. One viral sample was not placed in the clade with the other case samples, but instead was most closely related to a GenBank control sequence |
| Grande et al. | Transmission Patterns in a Low HIV-Morbidity State—Wisconsin, 2014–2017 | 2019 | Cohort | WI | 2014–2017 | Wisconsin Department of Public Health | 1401 individuals with molecular sequences reported to Wisconsin DPH during 2014–2017. Characteristics only provided for the 433 individuals in molecular clusters (88.4% male, 56.1% Black, 80.8% MSM, 3.5% MSM + IDU) | Data from partner services interviews were obtained from Wisconsin’s Partner Services Web | HIV-1 sequence data from persons with HIV infection in Wisconsin were analyzed using HIV-TRACE. Sequences considered related if genetic distances were <0.015. Transmission clusters defined as group of greater than two linked sequences | Named linkages between sexual contacts were compared to molecular linkages to assess whether named partners had highly genetically similar sequences | 30.9% of HIV sequences reported to Wisconsin DPH during 2014–2017 had molecular linkage to at least one other person. People were most commonly molecularly linked to persons of their own racial group, sexual orientation, and age range, except for Hispanics/Latinos and persons aged 13–19 years old. 33.8% of named partner linkages identified in public health interviews also had a molecular linkage |
| Kostaki et al. | Molecular Analysis of Human Immunodeficiency Virus Type 1 (HIV-1)–Infected Individuals in a Network-Based Intervention (Transmission Reduction Intervention Project): Phylogenetics Identify HIV-1-Infected Individuals With Social Links | 2018 | Cohort | Athens, Greece | 2013–2015 | Transmission Reduction Intervention Project (TRIP) | 356 participants 18+ years old comprised of HIV+ “seeds” (both recent and chronic infections as two discrete recruitment arms), their social network recruits, and HIV negative controls. 42.1% HIV+, 90.2% IDU | Social network data was obtained through study recruitment links, network questionnaires, and field/venue observations. 1st and 2nd degree social contacts of HIV+ seeds were recruited to the study. Contacts were defined as people who had sex with or injected with/in the presence of a seed (1st degree) or a first-degree contact of the seed (2nd degree). If a newlydiagnosed individual was identified among network recruits, two steps of contact tracing was then performed with this new seed | Samples were obtained for 118 out of 150 HIV+ study participants. Subtypes were identified using the online COMET tool and confirmed using 247 globally sampled reference sequences. Phylogenetic trees were constructed using maximum likelihood, neighbor joining, and Bayseian methods. Transmission clusters (2 or more sequences) were considered “highly supported” if they met confidence criteria for all three methods | The proportion of individuals with social network ties within their corresponding transmission clusters was assessed. These findings were compared to the expected proportion and distribution found in simulated random social mixing | 13 highly supported transmission clusters with 2–5 individuals each were generated by phylogenetic analysis. 43.8% of individuals had social network ties within their own clusters. Hypothesis testing indicated that this was significantly higher than the expected proportion with random mixing and significantly different from the distribution of expected proportions |
| Lee et al. | An Exploratory Study on the Social and Genotypic Clustering of HIV Infection in Men Having Sex with Men | 2009 | Cohort | Hong Kong | 2007–2008 | Hong Kong Integrated Treatment Centre (HIV specialist clinic) | 73 MSM receiving care at the Hong Kong Integrated Treatment Center clinic who were diagnosed with HIV in the preceding year, 18+, and of Chinese ethnicity. Mean age 33.5 years | Participants completed a questionnaire that collected sociodemographic data, estimated year of HIV infection, the type and number of sexual partners, and physical venues/internet sites used to seek sexual partners. Likert scales assessed frequency of venue usage and sexual behaviors. Venue-based clusters were constructed using Likert score strength | HIV-pol genes from 49 patients were sequenced and aligned using Clustal X. A phylogenetic tree was constructed using the maximum likelihood method with 1000 bootstrap replications. Clusters were defined as 3+ closely related sequences | Venue-based clusters constructed via social network analysis were compared to phylogenetic clusters, and genetic relatedness within venue clusters was assessed | Out of three identified phylogenetic clusters, two featured participants who mainly sourced partners from the internet (9/10 and 5/6 participants). Those in the third phylogenetic cluster mainly sourced partners from saunas and not from the internet. The sauna-based sourcing cluster also had viral sequences that were less genetically related to one another than the internet-based cluster |
| Lepej et al. | Phylogenetic Analysis of HIV Sequences Obtained in a Respondent-Driven Sampling Study of Men Who Have Sex with Men | 2009 | Cohort | Zagreb, Croatia | 2006 | Croatian Ministry of Health | 360 MSM in Zagreb recruited via respondent-driven sampling. 100% MSM | Zagreb-based MSM were recruited via multiples waves of respondent driven sampling, which was used to construct a social network. Participants were asked about their sexual behaviors and tested for various STIs | Sequences were obtained from HIV+ study participants. Reference sequences were obtained from 20 unrelated Croatians through routine clinical monitoring. Phylogenetic analysis was performed on pol sequences using the neighbor-joining method with the Kimura two-parameter distance model. Bootstrap analysis was used to assess branching pattern reliability, with clusters above 98% considered significant | HIV Molecular clusters were compared with the respondent-driven sampling recruitment chain to determine the relationship between both data sources and help characterize HIV transmission within the cohort | Out of 360 total participants, 18 (5%) were HIV+. Sequencing was successfully performed on 12 (3.3%) samples. Five of the sequences clustered together. HIV+ phylogenetically related cases were recruited in later waves: 1 in the 4th wave, 2 in the 6th, and 1 in the 8th. This phenomenon suggests the role of a sexual network in HIV transmission, though it appears unlikely that participants recruited their sexual partners |
| Lin et al. | Behavioral and molecular tracing of risky sexual contacts in a sample of Chinese HIV-infected men who have sex with men | 2013 | Cohort | Taizhou Prefecture, China | 2008–2010 | Chinese National Information System for AIDS Prevention and Control | 100 MSM index cases newly diagnosed with HIV between 2008 and 2010 in Taizhou Prefecture. Mean age 30.3 482 sexual contacts identified via contact tracing. 89% MSM | Index cases filled out egocentric contact tracing surveys and identified sexual partners were offered HIV testing. Sexual partners testing HIV+ were also asked to fill out egocentric contact tracing surveys. This process was repeated until no new HIV cases were identified | Sequences of gag and env from 72 HIV+ participants were compared with control sequences and aligned using Clustal X. Phylogenetic and molecular evolutionary analyses were conducted. Evolutionary distances were calculated and phylogenetic dendrograms were constructed using the neighbor-joining method | Identification of potential HIV transmission pairs was based on both sexual and molecular connections between the pair. A transmission cluster was defined as a group with at least 1 potential HIV transmission pair | 51 of the 100 HIV+ index cases were newly identified through contact tracing. Index cases together reported 1534 sexual contacts and provided contact information for 482 (31%), of which 115 (24%) ultimately received HIV testing. 7 out of 49 independent sexual networks were deemed HIV transmission clusters and were supported by phylogenetic analysis of HIV sequences. 16 networks included HIV transmission pairs linked by contact tracing that were not supported genetically |
| Monroe-Wise et al. | Peer-Mediated HIV Assisted Partner Services to Identify and Link to Care HIV-Positive and HCV-Positive People who Inject Drugs: a Cohort Study Protocol | 2021 | Cohort | Nairobi, Kilifi, Mambosa Counties; Kenya | 2018—Ongoing | Kenya Medical Research Institute, Kenya Ministry of Health | 1000 HIV + IDU index cases enrolled across 8 study sites and their sex/drug use partners identified via partner services | Index cases complete questionnaires identifying all of their sex and drug use partners over the last three years. Clinicians and peer educators then engage community partners for study enrollment, partner services/demographics questionnaire completion, and rapid HIV/HCV testing | HIV and HCV sequencing will be attempted for all study participants at the KwaZulunatal Research Innovation and sequencing Platform. Study HIV sequences will be combined with publicly available molecular data prior to performing molecular cluster analysis | Sexual/drug-use partner and molecular cluster data will be utilized to characterize viral transmission among high-risk populations and identify traits associated with increased infectivity | Trial currently at preresults stage |
| Morgan et al. | Sexual, Social, and Genetic Network Overlap: A Socio-Molecular Approach Toward Public Health Intervention of HIV | 2018 | Cohort | Chicago, II | 2013–2016 | uConnect Cohort | 266 HIV+ Black MSM aged 16–29 living in South Side Chicago. 69% identified as gay, 57% reported condom-less sex in the last 12 months | Participants were recruited via respondent driven sampling. All participants completed surveys that collected information on sociodemographics, substance use, close confidants, and recent sexual partners. Consenting participants (n = 12) also provided lists of their Facebook friends. Researchers used this data to construct recruitment, confidant, sexual, and Facebook networks | HIV pol sequences (n = 86) were obtained from all persons whose viral load was ≥ 2000 copies/mL through a combination of dried blood spot collection and surveillance data from the Chicago Department of Public Health. Phylogenetic tree analyses were performed by using the neighbor-joining method. Specimens were linked if the genetic distance between pol sequences was less than or equal to 0.015 substitutions/ site. Clusters were defined as 2 or more specimens linked by 1 + ties | Molecular, sexual, and confidant networks were consecutively integrated to postulate likely routes of HIV transmission. All analyses were restricted to named partners who were also study participants | 35 HIV sequences (41%) formed clusters with 55 total molecular ties. There were no significant differences in demographics, sexual activity, drug use, or confidant, sexual, and Facebook network ties between clustered and unclustered sequences. None of the molecular ties were identified by 1st, 2nd, or 3rd degree confidant or sexual networks, or 1st degree recruitment networks. There was a consistent 45–50% overlap in confidant, sexual, Facebook, and recruitment networks |
| Pasquale et al. | Leveraging Phylogenetics to Understand HIV Transmission and Partner Notification Networks | 2018 | Cohort | Wake County, NC | T2012–2013 | NC Department of Health and Human Services (NC-DHHS), North Carolina Screening and Tracing for Active HIV-1 Transmission (NC-STAT) program | 280 index cases, defined as newly diagnosed HIV cases in state of NC during 2012–2013. 83% male, 65% Black, 40% under 30 years old. 383 sexual partners or high-risk social contacts | Data on demographics, HIV testing history, HIV lab results, and sexual and social contacts were abstracted from DIS interviews performed by NC-DHHS or Wake County DHHS for all persons in NC newly diagnosed with HIV. A Partner Notification network was constructed using named partner and high-risk contact data | HIV-1 pol sequences sampled between 1997 and 2014 were obtained from the largest reference lab in NC (n = 15,246). Sequences that did not match an index case or contact were considered background sequences. Sequences were aligned using MUSCLE and phylogenetic trees were constructed using maximum-likelihood phylogenies. Putative transmission clusters were defined as clades with high branch support, maximum pairwise distance less than 3.5% between all sequences, and inclusion of at least one index or partner case | Index and partner cases were matched with HIV sequences using date of birth, gender, and lab test dates. Separate partner notification and transmission cluster networks were created and differences between the two networks were compared | 80% of index cases were interviewed, reporting a total of 854 sex partners and 34 social contacts. Of these partnerships, 383 were unique non-index partners and 335 were located during investigation. 34% of partners were found to be HIV+, while 27% had an unknown HIV status. 73% of individuals in the partner notification network who had sequences were in one of the 116 transmission clusters identified. 59% of transmission clusters contained links between sexual network components that were not apparent from the partner notification network |
| Pilon et al. | Transmission Patterns of HIV and Hepatitis C Virus among Networks of People Who Inject Drugs | 2011 | Cohort | Ottowa, Canada | 2007 | Ontario HIV Treatment Network | 407 IDUs enrolled from September to December 2007 consisting of 7 ‘seeds’ and their recruits. 100% IDUs, 80% Male, 10.1% HIV+ | 7 Initial IDU ‘seeds’ were selected based on participation in previous studies. Seeds were asked to recruit 3 IDU peers via a chain-referral method. New recruits were also instructed to recruit 3 additional peers, with this process continuing until a target of 400 participants was reached | HIV pol and HCV sequences were amplified via RTPCR. Sequences were aligned via Clustalx software, and phylogenetic trees were constructed using Saitou and Nei’s neighbor-joining method with the Kimura 2-Parameter model. Bootstrapping was used to identify significant clusters using a threshold of 80% reliability when resampling | Chain-referral social networks and phylogenetic data were compared to decipher if and how social relationships influenced transmission dynamics | Out of 407 total recruits, 41 (10.1%) were HIV+, of which 40 (9.9%) were HIV/HCV coinfected. 29 (71%) HIV samples were ultimately sequenced, 18 (62%) of which fell into one of 7 distinct clusters ranging from 2 to 4 participants. Two of the unique clusters contained participants separated by 2 or less recruitment cycles, while the remaining 5 clusters contained more |
| Pines et al. | Concurrency and HIV Transmission Network Characteristics among MSM with Recent HIV Infection | 2016 | Cohort | San Diego, CA | 1996–2015 | San Diego Primary Infection Resource Consortium (SD-PIRC) & Center for AIDS Research Network of Integrated Clinical Systems (CNICS) | 986 HIV+ individuals recruited through: (1) The UCSD HIV screening centers between 1996 and 2005, aged 16+, recently HIV diagnosed, and ART-naive (SD-PIRC cohort, n = 800), (2) The UCSD HIV clinic between July 2007 and August 2013 (CNICS cohort, n = 186). Characteristics provided for subgroup of 285 recently infected, cisgender MSM in SD-PIRC cohort used in partner concurrency analysis (White 60%, 24% Hispanic, median age 33 years) | Individuals in the SD-PIRC cohort completed interviews on socio-demographics, drug use, sexual behavior, and partner-specific data on up to three recent sexual partners. Partner concurrency was assessed based on reported overlapping timelines of sexual partnerships within 3 months prior to interview completion | HIV-1 pol sequences were collected from both cohorts (n = 986). HIV TRACE was used to infer partial transmission networks. Putative transmission links were inferred when the genetic distance between two sequences was less than 1.5% | Data on the dates of HIV diagnosis, sample collection, and estimated dates of HIV infection were examined for putative transmission links to assess direction of transmission and whether transmission occurred during a recent infection | In the subgroup of recently HIV infected MSM (n = 285), 54% phylogenetically clustered with others in the network by one or more putative transmission links. 54% reported having concurrent partners. There was a positive association between concurrency and transmission network clustering and network degree |
| Resik et al. | Limitations to Contact Tracing and Phylogenetic Analysis in Establishing HIV Type 1 Transmission Networks in Cuba | 2007 | Cohort | Cuba | Not identified | Not identified | 127 HIV-1 seropositive individuals, both male and female, identified as members of two discrete contact tracing networks. HIV seroconversion of network members occurred predominantly from 1982 to 1993 | Contact tracing was used to identify one network of 38 HIV+ individuals and another of 89, with an emphasis on partners at the time of HIV diagnosis | Blood samples were obtained from consenting study participants. RTPCR was used to amplify the gag, env1, and env2 genomic portions of viral samples. Phylogenetic trees were constructed using a maximum likelihood heuristics approach. Subtyping was performed using reference sequences from GenBank | Phylogenetic analysis was performed among samples in each contact tracing network and was used to verify or negate possible transmission links | In network 1 (N = 38), phylogenetic analysis of gag supported 4 transmission events, rejected 6, and did not reject 2. Analysis of env supported 2 transmission events, rejected 3, and did not reject 9. Combined env/gag analysis supported 3 transmission events and did not reject 2. In network 2 (N = 89), combined env/gag analysis supported 5 transmission events and did not reject 3 |
| Smith et al. | A Public Health Model for the Molecular Surveillance of HIV Transmission in San Diego, California | 2009 | Cohort | San Diego County, CA | 2006–2007 | San Diego County Resistance Testing program (SDC cohort), First Choice Program (FCP) | 268 HIV+ individuals recruited through referrals to drug resistance testing (SDC cohort). 89% male, 62% Caucasian, mean age 35 years, 80% MSM, 7% MSM + IDU. 369 recruited through the Acute Infection and Early Disease Research Program (FCP cohort) from 1996 to 2007. 96% male, 79% Caucasian, 92% MSM; 36 sexual partners recruited through contact tracing of the FCP cohort. 92% male, 62% Caucasian, 91% MSM | Contact tracing was performed with members of the FCP cohort to elicit contact information of sexual partners. Epidemiologically linked partners were defined as sexual partners with available contact information who the index cases believed to be the likely source of their HIV infections | Phylogenetic analysis was performed on HIV-1 pol sequences obtained from all SDC and FCP cohort members and listed sexual partners using BioEdit and MUSCLE. Transmission clusters were defined when pol sequences from any two people were >99% genetically similar | Phylogenetic analysis was used to evaluate the genetic ties between index cases and their epidemiologically linked partners identified through contact tracing | In 673 total participants, pol sequences were on average 5.1% genetically different. 25% of participant sequences clustered when combining the SDC and FCP cohorts, with the largest cluster consisting of 12 participants. 24 of the 36 (67%) epidemiologically linked partners were supported by phylogenetic analysis. Epi-linked partner sequences clustered with 60% of the clustering FCP and SDC sequences and were grouped into 15 clusters |
| Schneider et al. | Abstract: Do Partner Services Initiated from Molecular Clusters Yield New or Viremic HIV Cases? | 2020 | Cohort | Chicago, IL | 2012–2016 | Chicago Department of Public Health (CDPH) | 1015 newly diagnosed HIV+ index cases reported to the CDPH from 2012 to 2016 with available HIV sequences. Characteristics for the 336 index cases with phylogenetically clustered sequences: Average age 28, 47% Black, 29% Latinx, 89% MSM | Newly diagnosed HIV+ index cases were offered partner services by CDPH at time of diagnosis. Partner notification data was extracted from CDPH records | Molecular clusters were constructed using HIV-TRACE at a pairwise genetic distance threshold of 0.5% | Integration of partner notification data and phylogenetic analysis was used to assess the difference in yields between clustered and nonclustered index cases and identifying sex/drug-use partners with new HIV diagnoses or viremia at time of diagnosis | Out of 1015 index cases, 336 (33%) had HIV sequences that clustered. 96 index cases named a total of 539 sex/drug-use partners. Out of all listed partners, 162 (37%) were linked to clustered index cases. 20% of the listed partners were either new HIV diagnoses or viremic at the time of diagnosis. There was no significant different in the yield of new or viremic HIV partners linked to clustered vs. nonclustered index cases |
| Ssemwanga et al. | HIV Type 1 Subtype Distribution, Multiple Infections, Sexual Networks, and Partnership Histories in Female Sex Workers in Kampala, Uganda | 2012 | Cohort | Kampala, Uganda | 2008–2009 | Good Health for Women Project (GHWP) | 324 HIV+ women who were a part of a larger cohort of 1027 high-risk sex and local entertainment facility workers in Kampala enrolled between March 2008 and April 2009 who also provided blood samples | At 3 months intervals, participants completed sociodemographic and behavioral questionnaires. Information on women’s’ life stories and sexual partnerships was also elicited during a study featuring a subset of the cohort (n = 200) who were found to have phylogenetically similar viruses and be at high risk of contracting STIs | HIV-1 gp-41 and pol sequences were obtained from 210 participants. Reference sequences from the Los Alamos Sequence database were used, and Clustal X was used to align the sequences. Phylogenetic trees were constructed using neighbor joining method. Clusters with bootstrap values > 90% were considered phylogenetically similar | Life stories were used to determine when/where sex work was performed. The location of sex work was compared for women with phylogenetically similar viruses | In the 210 participants, HIV subtype A (~60%) was the most prevalent subtype. Sexual networks of 6 pairs and one triplet of participants with similar viral sequences were identified. Four dyads of women with phylogenetically similar viruses shared similar sex work venues. Five cases of multiple infections (9%) were also discovered |
| Tordoff et al. | Combining Traditional and Molecular Epidemiology Methods to Quantify Local HIV Transmission among Foreign- Born Residents | 2021 | Cohort | King County, WA | 2010–2018 | HIV/STD Program, Public Health – Seattle & King County, National HIV Surveillance Program | 2409 HIV+ residents of King County, Washington diagnosed between January 1st, 2010, and December 31st, 2018. 33% foreign-born | Partner services was offered to all newly diagnosed HIV+ residents of King County as defined by the National HIV Surveillance System. All interviews were conducted by Public Health—Seattle & King County | HIV-1 pol sequences were obtained for 1448 study participants. Distance-based clustering analyses were used to identify clusters of 2+ individuals using Tamura-Nei pairwise genetic distance with a threshold of 0.02 substitutions per site. Sensitivity analyses were also conducted using thresholds of 0.025, 0.015, and 0.01 substitutions per site. A phylogenetic tree was constructed using FastTree approximate maximum likelihood method | Molecular cluster and partner services data were combined to infer the location of HIV acquisition for foreign-born individuals | Among individuals with available sequences (n = 1448), 1104 (76%) clustered into 295 genetically similar clusters. Combined partner services and molecular data were used to infer the HIV acquisition location for 611 (77%) of all 798 HIV+ foreign born residents, with 254 (32%) presumably acquiring HIV outside King County, 394 (49%) outside of the US, 205 (26%) in King County, and 13 (2%) elsewhere in the US. Partner services data alone inferred HIV acquisition locations for 100 (13%) participants, and molecular cluster data inferred acquisition locations for 258 (32%) participants |
| Wertheim et al. | Social and Genetic Networks of HIV-1 Transmission in New York City | 2017 | Cohort | New York, NY | 2006–2012 | New York City Department of Health and Mental Hygiene | 756 HIV+ index cases who were reported to the NYC Department of Public Health. 52% Black, 41% Hispanic, 45% MSM, 7% IDUs; 586 HIV+ sex and IDU partners of the index cases | HIV+ index cases were interviewed by the NY Field Services Unit for the names of their sex and IDU partners over the previous 12 months. Named partners were contacted and referred to testing and care. Index cases and named partners were classified by their primary transmission risk factor (i.e. IDU) | HIV-1 sequences were obtained for index cases and HIV+ named partners (n = 1342) through the NYC Department of Health. HIV-TRACE was used to construct the genetic transmission network. Sequences were aligned to the HXB2 reference sequence and pairwise Tamura-Nei 93 genetic distances between sequences were calculated. Pairs of sequences falling below a range of distance thresholds were then linked, with connected components forming transmission clusters | Molecular and sexual/IDU network data were used to create two separate and one merged HIV transmission network. These networks were used to determine a range of genetic distance thresholds suitable for identifying potential transmission partners as well as to determine factors associated with genetic linkage in possible transmission partners | Based on 756 index cases and 586 HIV+ identified partners, plausible thresholds for genetic distance were calculated to be between 0.01 and 0.02 substitutions/site. 43–65% of genetic linkage clusters were supported by contact tracing data while 37–49% of contact tracing networks were supported by genomic analysis. Genomic data supported HIV transmission along 310 edges between 651 partner names in the contact tracing network, while contact tracing supported 388 out of 736 edges in the genomic network. Contact tracing did not identify possible transmission in over half of HIV cases across distance thresholds. 449 out of 756 (59%) index cases were genetically linked to at least one named partner. Bidirectionally named partners had increased odds for being genetically linked than unidirectionally named partners |
Year of publication
City and/or state abbreviation
MSM Men who have sex with men, HIV human immunodeficiency virus, IDU injection drug user, CDC centers for disease control and prevention, BLAST basic local alignment search tool, BRAI bio-rad avidity incidence, IVDU intravenous drug user, STD sexually transmitted disease, TB tuberculosis, ECVC Encuesta Centroamericano de Vigilancia de VIH y Comportamiento en Poblaciones Vulnerables, FSW female sex worker, STI sexually transmitted infection, Hx history; NC-DHHS North Carolina Department of Health and Human Services, NS-STAT North Carolina Screening and Tracing for Active HIV-1 Transmission, AHI acute HIV infection, RHI recent HIV infection, DIS disease intervention specialists; iMAN integrated molecular & affiliation network analysis of HIV transmission, AIC Akaike’s Information Criterion, RDS respondent-driven sampling, DPH department of public health, TRACE secure HIV transmission cluster engine, PLWHA people living with HIV/AIDS, PCR polymerase chain reaction; PWID people who inject drugs; COMET context-based modeling for expeditious typing, GRT genotypic resistance testing, CDPH Chicago Department of Public Health, MUSCLE multiple sequence comparison by log-expectation, HCV hepatitis C virus, SD-PIRC San Diego Primary Infection Resource Consortium, CNICS center for AIDS research network of integrated clinical systems, RTPCR reverse transcription polymerase chain reaction, AIEDRP acute infection and early disease research program, FCP first choice program, ART antiretroviral therapy, GHWP good health for women project, UCSD University of California San Diego, SDC San Diego County, NYCDOH New York City Department of Health
The most common use of molecular data in the context of social network data was to assess purported transmission links, either by using the data to support or refute specific partnerships [28, 32, 34, 36, 44, 56, 57] or to calculate the proportion of social ties that were genetically supported [29, 33, 37]. For instance, a study by Dennis et al. created a social network based on relationships identified through RDS and then utilized sequences from HIV-positive members of the RDS chains to assess how many social links between HIV-positive members also represented genetically supported transmission pairs [33]. A review of all studies characterizing social/sexual and molecular network overlap demonstrated that 14–73% of socially linked contacts were genetically supported [28–32, 37, 43].
A subset of studies also reported on whether molecular cluster analysis revealed new partnerships not identified through social network data and whether those additional genetic ties spanned previously unconnected social or sexual clusters [30, 32, 35]. A study by Pasquale et al. generated a large social network from state-wide DIS interview data and integrated this network with the molecular cluster analysis of 15,246 HIV pol sequences from the largest reference laboratory in the state [30]. Using this analysis, the authors reported that the molecular cluster data identified new, previously undocumented links in 59% of the transmission networks they had identified based on DIS interviews. Another study by Campbell et al. identified an additional 102 putative transmission links among a social network by utilizing phylogenetic data [32].
Instead of utilizing molecular analysis to validate or add ties, some studies generated transmission clusters from HIV sequence data and then used social network data to characterize these molecular clusters by determining whether they contained socially, sexually, or venue-connected individuals [20, 38, 43, 45, 47, 49]. For instance, a study by Kostaki et al. generated molecular clusters based on sequences from HIV-positive participants in an HIV prevention cohort study. Utilizing a combination of contact tracing interviews and RDS data, they assessed the proportion of individuals with social network ties within their respective molecular cluster [47]. They then compared these proportions to what would be expected under simulated random social mixing, demonstrating that individuals in molecular clusters are more likely to be socially connected to other cluster members. Overall, studies that examined social ties within genetically-based transmission clusters found that 43–65% of genetically linked ties within clusters were also socially connected [31, 47].
Outbreak Investigations
Six studies were identified that utilized molecular cluster analysis and social network data to surveille a specific HIV outbreak (Table 3) [27, 39, 40, 42, 51, 54]. Of these studies, Dennis et al. focused on understanding the dynamics behind a large country-wide HIV outbreak [42], while Nett et al. and Hayman et al. focused more narrowly on supporting or refuting purported transmission links within one or two small outbreak clusters [27, 54]. Nett et al. in particular drew conclusions about the initial source of the outbreak [27]. Samoff et al. compared social network to molecular cluster data within the context of an HIV outbreak [51], and Monterosso et al. used both molecular and social data sources to construct the outbreak cluster itself [39]. All studies utilized contact tracing data from partner services and/or DIS interviews. Reported outcomes involving the integration of social network and HIV sequence data included analyzing the number of links between network members found by contact tracing data compared with molecular analysis as well as differences in composition of social clusters compared with molecular clusters (i.e., size, density, demographics).
Table 3.
Studies combining phylogenetic and social network analysis in the context of an HIV outbreak investigation (N = 6)
| Authors | Title | Pub. yeara | Study design | Locationb | Time period | Data source/project | Population characteristics | Social/sexual network data | Molecular cluster analysis | Data integration | Resultsc |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Dennis et al. | Integration of Contact Tracing and Phylogenetics in an Investigation of Acute HIV Infection | 2018 | Outbreak investigation | NC | 2013–2014 | NC Department of Health and Human Services (NC-DHHS), North Carolina Screening and Tracing for Active HIV-1 Transmission (NC-STAT) program | 68 AHI/RHI index cases: 85% male, median age of 25, 66% Black. 210 reported contacts: 189 first degree, 21 degree. All sexual (93%) or social (7%) contacts, no IDU contacts | In response to new foci of AHI cases in early 2014 detected by NC-STAT, all AHI or RHI cases in NC in 2014 were investigated. DIS interviewed all index cases and followed-up with all high-risk sexual contacts. Contact networks were constructed using contact tracing data for all index cases, their reported contacts, and second-degree contacts if linked first-degree contact had a new HIV diagnosis or an HIV diagnosis in 2013 or 2014 | NC-DHHS requested HIV-1 pol sequences for all index cases and HIV-infected contacts. Partial pol sequences (n = 1672) from UNC Center for AIDS Research HIV Clinical Cohort were included as background references. Maximum-likelihood phylogenies were constructed in RAxML with general time reversible model of nucleotide substitution. Transmission clusters were defined as clades with bootstrap support >98% that had one or more index case sequences with<0.015 substitutions/site pairwise genetic distance from another case | Independent phylogenetic and contacttracing networks were constructed. These networks were compared by the study | Statewide contact tracing network had 58 independent components, with mean degree of 1.7 and overall network density of 1.2%. Mean index node degrees by contacts’ HIV statuses were 0.8 for HIV-infected contacts, 1.0 for HIV-negative contacts, and 1.3 for contacts with unknown HIV status. HIV-1 pol sequences were available for 56% of index cases and 48% of HIV-infected contacts. 13 identified transmission clusters involved at least two index cases or an index and a contact. 31% included a previously diagnosed contact. 4 clusters revealed links not apparent from contact tracing. The largest component of the contact network included members from distinct phylogenetic clusters |
| Hayman et al. | Phylogenetic Analysis of Multiple Heterosexual Transmission Events Involving Subtype B of HIV Type 1 | 2001 | Outbreak investigation | Doncaster, United Kingdom | 1996–1999 | Not identified | 13 individuals who tested HIV+ in the town of Doncaster between 1996 and 1999. 31% male, 69% female | Initial epidemiological follow-up of an HIV index case identified in 1996 found a cluster of previously undiagnosed cases. Further contact tracing demonstrated the cluster of 13 cases were linked by both sexual and venue-based histories | Frozen blood samples from the 13 cases were amplified by PCR and the same C1 to C4 region of the HXB2 peptide (930 bp) within the HIV genome was sequenced for phylogenetic analysis. Phylogeny was assessed using two methods, both maximum likelihood and neighbor joining, and both indicated the same phylogeny. Sequences were also compared to those available in GenBank | Phylogenetic relationships and known social contact links (sexual or IDU) were compared. Phylogenetic analysis was primarily used to assess the ability to discern relationships that were unclear from contact tracing due to multiple interlinked contacts among the 13 cases | Sequencing indicated three separate HIV strains among the 13 sequences. Ten of the sequences could be linked to the index case but could also be delineated into two separate clusters. One group was more closely genetically related to itself and the index case sequence (this group included the index case and five other cases), while the other consisted of six cases that were less related to the index and to one another. Contact tracing postulated sexual, social, or an indirect IDU contact between the index case and 10 other cases. The set of 11 cases connected by contact tracing was the same set connected by phylogenetic analysis apart from one possibly distantly related sequence |
| Monterosso et al. | Identifying and investigating a Rapidly Growing HIV Transmission Cluster in Texas | 2017 | Outbreak investigation | TX | 2015–2016 | Texas Department of State Health Services | 27 newly diagnosed HIV+ individuals constituting the initial outbreak group and 112 HIV+ sex/IDU partners and social contacts. Of the 76 partners with available records, 100% men, 89% MSM, 87% Hispanic, 78% aged 13–29 | Partner services were used to elicit index cases’ sex and IDU partners, the partners of identified partners, and social network contacts, along with information on demographics, risk behaviors, and partner meeting sites | Phylogenetic analysis conducted on HIV sequences collected through the National HIV Surveillance System identified a rapidly growing molecular cluster in Texas | Partner services data was integrated with molecular data to identify sex/IDU partners and social contacts without molecular sequences who could have composed part of the rapidly growing cluster | An initial cluster of 27 cases was identified via phylogenetic analysis. Twelve (44%) of the 27 cases were linked via social or sexual partnerships. From the original 27, an additional 112 cases were identified via partner services information |
| Nett et al. | Two Clusters of HIV-1 Infection, Rural Idaho, USA, 2008 | 2010 | Outbreak investigation | ID | 2008 | Idaho Department of Health and Welfare | 15 newly diagnosed HIV+ individuals comprising two outbreak clusters in southeastern Idaho. Median age of cluster members 24 and 26, respectively. Overall, 73% MSM, 27% suspected IDU | Participant demographics, drug use, and sexual activity was elicited through epidemiological investigation. This information was used to construct sexual and drug-use networks | HIV pol sequences for 10 participants were obtained from commercial laboratories. An additional sequence from the same region but not linked via contact tracing was also included. Control sequences were provided by 2 Idaho HIV clinics. Sequences were grouped into phylogenetic clusters using multiple sequence alignment, neighbor joining, and maximum likelihood tree analysis | Contact tracing and molecular data were combined to determine possible sources of HIV transmission for both outbreak clusters | The 11 sequences clustered into two phylogenetic groupings. The first grouping contained 4 sequences from outbreak cluster A and the sequence that was not linked to either outbreak cluster. The second grouping contained 5 sequences from outbreak cluster B. The two groupings had an average pol genetic distance of 4.8%, which was similar to that of the control group (5.1%), suggesting no linkage between the two clusters |
| Samoff et al. | HIV Outbreak Control with Effective Access to Care and Harm Reduction in North Carolina, 2017–2018 | 2020 | Outbreak investigation | Western NC | 2017–2018 | North Carolina Department of Public Health | 7 HIV+ individuals diagnosed with HIV from 2017 to early 2018 in western NC and their primary, secondary, and tertiary IDU contacts. 100% IDU, 62% male, 96% White, median age 36 | Disease intervention specialists conducted contact tracing interviews with the 7 individuals part of the initial HIV outbreak as well as their listed sexual and IDU contacts (primary contacts). DIS then performed contact tracing on their partners (secondary contacts) as well as the partners of secondary contacts (tertiary contacts). Network diagrams were constructed using partnership information | New HIV nucleotide sequences were reported to reference laboratories monthly. Molecular clusters were constructed using a TN-93 pairwise genetic distance of 1.5% or less. BEAST version 2.4.8 was used to build phylogenetic trees for outbreak-associated clusters using the GTR+ gamma model, Bayesian skyline coalescent prior, and a relaxed log-normal molecular clock | The number of sexual and IDU contacts newly diagnosed with HIV was compared with the number of new diagnoses in genetic clusters. Markers of linkage to care (ex: pertinent laboratory and appointment dates) were used to determine success in decreasing HIV exposure within the transmission networks | DIS successfully contacted 6 out of 7 individuals part of the initial outbreak as well as 96 IDU contacts linked to the initial outbreak members. Of those 96, 14(15%) were HIV+. Five outbreak group members and 1 newly diagnosed HIV+ individual had sequences that grouped with a cluster of 14 individuals diagnosed between 2011 and 2018, 6 of whom were IDUs. As of March 2019 (12 months follow-up), 5 of the initial outbreak members remained virally suppressed, as did both individuals newly diagnosed during the investigation |
| Sizemore et al. | Using an Established Outbreak Response Plan and Molecular Epidemiology Methods in an HIV Transmission Cluster Investigation, Tennessee, January–June 2017 | 2020 | Outbreak investigation | TN | 2017 | Tennessee Department of Health | 31 individuals in eastern TN diagnosed with HIV after January 1, 2017, and their 107 named sex/drug-use partners. For index cases: 45% MSM, 42% recently incarcerated, 31% IDU | Index cases completed interviews and questionnaires detailing their sex/drug-use partnerships as well as high risk sexual and drug-use history. Social network analysis was performed to visualize the linkages between index cases and contacts | HIV pol sequences were amplified via PCR. COMET was used to determine Tamura-Nei pairwise distances and subtyping. A genetic distance threshold of under 1.5% was used to establish molecular clusters | Social and molecular data were both utilized to describe HIV transmission among study participants | Partner services interviews with 31 index cases resulted in 107 unique sex and drug-use partners. HIV testing of partners resulted in 2 new HIV+ diagnoses. 14 HIV-1 pol sequences were obtained from 7 of the 8 IDUs as well as 7 other participants. Molecular analysis revealed 3 clusters, 1 of which included 3 of the IDUs |
Year of publication
City and/or state abbreviation
MSM Men who have sex with men, HIV human immunodeficiency virus, IDU injection drug user, CDC centers for disease control and prevention, BLAST basic local alignment search tool, BRAI bio-rad avidity incidence, IVDU intravenous drug user, STD sexually transmitted disease, TB tuberculosis, ECVC Encuesta Centroamericano de Vigilancia de VIH y Comportamiento en Poblaciones Vulnerables, FSW female sex worker, STI sexually transmitted infection, Hx history; NC-DHHS North Carolina Department of Health and Human Services, NS-STAT North Carolina Screening and Tracing for Active HIV-1 Transmission, AHI acute HIV infection, RHI recent HIV infection, DIS disease intervention specialists; iMAN integrated molecular & affiliation network analysis of HIV transmission, AIC Akaike’s Information Criterion, RDS respondent-driven sampling, DPH department of public health, TRACE secure HIV transmission cluster engine, PLWHA people living with HIV/AIDS, PCR polymerase chain reaction; PWID people who inject drugs; COMET context-based modeling for expeditious typing, GRT genotypic resistance testing, CDPH Chicago Department of Public Health, MUSCLE multiple sequence comparison by log-expectation, HCV hepatitis C virus, SD-PIRC San Diego Primary Infection Resource Consortium, CNICS center for AIDS research network of integrated clinical systems, RTPCR reverse transcription polymerase chain reaction, AIEDRP acute infection and early disease research program, FCP first choice program, ART antiretroviral therapy, GHWP good health for women project, UCSD University of California San Diego, SDC San Diego County, NYCDOH New York City Department of Health
Response Interventions
Six studies were identified that described interventions informed by the use of molecular and social network analyses (Table 4) [19, 41, 46, 50, 52, 53]. All six studies described interventions executed within the context of HIV outbreaks and associated investigations, five of which occurred largely among people who inject drugs [41, 46, 50, 52, 53]. While all the studies utilized DIS to collect social network data, Poon et al. additionally featured a “near real-time” HIV surveillance method that utilized monthly reports from a government-run drug resistance database to identify and monitor transmission clusters as sequences were added, prompting initial recognition of the HIV outbreak [19]. Multiple studies further compared linkage networks to identify links supported by molecular data, social network data, or both sources [46, 50, 52].
Table 4.
Interventions Informed by social network and molecular cluster analysis during outbreak investigations (N = 6)
| Authors | Title | Pub. yeara | Study design | Location | Time period | Data source/project | Population characteristics | Social network and molecular cluster analysis | Intervention informed by molecular and social data | Resultsb |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Alpren et al. | Opioid Use Fueling HIV Transmission in an Urban Setting: An Outbreak of HIV Infection Among People Who Inject Drugs— Massachusetts, 2015–2018 | 2020 | Outbreak Investigation | Lowell and Lawrence, MA | 2015–2018 | Massachusetts Department of Public Health | 129 total IDUs or homeless individuals newly diagnosed with HIV+ from January 2015 to June 2018 in northeastern MA, their HIV+ sex/drug-use partners, and molecularly linked individuals. 57% male, 73% aged 20–39, 67% non-Hispanic White, 86% IDUs | Partner services was offered to all newly diagnosed HIV+ individuals to elicit all sex and drug-use partners. Social network data was utilized to suggest possible transmission routes. HIV-1 pol sequences were derived from drug resistance testing upon initial diagnosis. HIV-TRACE was used to group individuals into clusters linked at a genetic distance of 1.5% or less as well as 0.5% or less |
Field epidemiologist staffing was doubled and enhanced DIS follow-up was extended to all new HIV+ diagnoses. Hours of existing syringe exchange programs were increased, and an additional syringe exchange program was established in Lowell. HIV testing services were expanded to emergency departments, homeless shelters, and jails. Total new HIV-related expenditures by the Massachusetts Department of Public Health exceeded $1.7 million | Out of 113 individuals with available pol sequences, 102 clustered with at least one other case at a genetic distance of 1.5%, with the largest cluster consisting of 56 people. At a distance of 0.5%, 93 sequences clustered with at least one other case. 27 (21%) cases were only linked through social network data, 29 (22%) were linked only though molecular data, and 73 (57%) were linked through both data sources. 37 additional IDU-related HIV diagnoses were made between June 2018 and June 2019, with only 7 outbreak-linked diagnoses reporting from January to March 2019, suggesting a decline in new IDU-related HIV diagnoses |
| Golden et al. | Outbreak of Human Immunodeficiency Virus Infection Among Heterosexual Persons Who Are Living Homeless and Inject Drugs — Seattle, Washington, 2018 | 2019 | Outbreak Investigation | Seattle, WA | 2018 | Public Health – Seattle and King County | 14 HIV+ IDUs and their sex/drug-use partners diagnosed between February and mid-November, 2018. 100% homeless, 86% IDUs, 78% women, 64% sex workers | Partner services was used to elicit the sex/drug-use partnerships of individuals involved in the outbreak. Social network data was used to suggest possible HIV transmission routes. Molecular analysis was performed using HIV-TRACE. A genetic distance threshold of 1.5% and under was used to molecularly link cases to one another as well as previous clusters. All sequences with HIV strains unrelated to the cluster were ultimately excluded |
Local emergency departments and hospitals increased HIV testing among IDUs and homeless individuals, some through systematic, risk-based opt-out-HIV screening programs. King county jail instituted optout testing during health assessments at 10–14 days after jail admission and occasionally upon jail admission pending resource availability. PHSKC further expanded outreach testing, condom distribution, and syringe services among North-Seattle homeless populations | In 2018, 14 homeless individuals living within a 3-mile radius were diagnosed with HIV. Four individuals were socially linked, while three were molecularly linked. Molecular analysis showed 7 of those individuals clustered with 8 cases from 2008 to 2017. As of November 2018, the cluster contained a total of 23 cases. Emergency room screening identified 1 of the 14 HIV+ diagnoses composing the HIV outbreak. Jail HIV screening identified 1 HIV+ diagnosis not linked to the outbreak. PHSKC HIV tested 534 people, identifying 4 of the 14 HIV+ individuals composing the outbreak |
| Metcalfe et al. | From Hospital to the Community: Redesigning the Human Immunodeficiency Virus (HIV) Clinical Service Model to Respond to an Outbreak of HIV Among People Who Inject Drugs | 2020 | Outbreak Investigation | Glasgow, Scotland | 2012–2019 | West of Scotland Specialist Virology Centre | 156 newly diagnosed HIV+ people associated with the 2014–2019 Glasgow HIV outbreak identified largely through contact tracing. 66% male, 51% aged 35–44, 64% homeless, 35% recently incarcerated, 81% IDU | Partner services was offered to all newly diagnosed HIV+ individuals to identify sex and IDU partners. Identified partners were similarly tested for HIV and offered partner services and linkage-to-care if HIV+. HIV pol sequences were obtained from all individuals diagnosed between mid-2012 through July 2019 with subtype C sequences (n = 151). Additional sequences were provided by the UK HIV Drug Resistance Database (n = 28). HIV-TRACE was used to genetically link the sequences associated with the outbreak through social network data at a linkage threshold range of 0.5–2.0% |
Educational outreach initiatives targeting affected populations such as IDU, homeless, and recently incarcerated populations were developed with community input. Content included public promotional materials describing available resources and specialized trainings for healthcare/ auxiliary staff who frequently work with these populations. HIV services were enhanced in a central homeless health building and provided alongside substance-use treatment. Nurses assisted with patient outreach and connection to auxiliary resources. ART provision was dispensed daily in all community pharmacies | 175 of the 179 sequences associated with the outbreak via social network data genetically linked at a threshold of 2.0% with the formation of three subclusters. New diagnoses were observed in all subclusters, though a spike occurred in one particular subcluster in 2019. Of the 156 HIV+ outbreak-associated individuals who remained alive and in Glasgow (n = 149), all participants initially commenced ART. Viral suppression in this cohort increased steadily from 18% in 2015 to 86% in 2019. Median time from HIV diagnosis to ART initiation reduced from 264 days in 2015 to 23 days in 2019. Self-reported HIV testing among IDUs increased from 30 to 50% by 2017–2018 |
| Peters et al. | HIV Infection Linked to Injection Use of Oxymor-phone in Indiana, 2014–2015 | 2016 | Outbreak Investigation | Scott County, IN | 2014–2015 | Indiana State Department of Health, Centers for Disease Control and Prevention | 181 HIV+ index cases residing in Scott County diagnosed after October 2014 and their 536 sex and drug-use contacts. Among index cases, 87.8% IDUs | All HIV+ index cases were interviewed and asked to name sexual contacts, injection drug-use partners, and anyone they believed might benefit from HIV testing. Contact tracing networks were mapped using this data. Viral sequences from the CDC were analyzed for persons who met the case definition. BLAST was used to compare pol sequences against GenBank, commercial databases, and CDC’s Molecular HIV Surveillance database. Sequences were aligned via MEGA6 software, and phylogenetic trees were constructed using FastTree. Clusters were defined as >97% identical sequence |
A public health emergency was declared in March 2015. 5 new HIV and HCV testing sites were opened. HIV testing was newly offered in jails in 8 surrounding counties. 42 additional DIS were deployed to enhance partner services efforts. An emergency community outreach center was established to provide syringe exchange, vaccinations, insurance, enrollment, and HIV testing. A substance-use organization increased their presence in Scott County. Education was provided to local primary care physicians on substance use and HIV treatment. HIV educational infographics were further distributed in the community | 159 index cases with available blood specimens clustered into 2 groups, one with 157 sequences (98.7%), of which 48 sequences shared 100% nucleotide identity. 113 sequences (90.4%) had infections classified as recent. DIS identified 536 partners. HIV+ index cases were more likely to be named as IDU partners than those who were HIV−. Through the response efforts, 465 individuals were enrolled in immediate health insurance. Hepatitis B vaccinations were provided to 454 people. 114 individuals received outpatient mental health and substance use services. HIV testing increased from 23 in November 2014 to 1834 in March 2015. Reactive tests declined from 7.7% in March 2015 to 0.8% in May 2015. 582 inmates in county jails were tested for HIV, 2 of which were newly diagnosed (0.3%), both which were phylogenetically and socially linked to the outbreak. Out of 176 trackable HIV+ patients, 152 (86.4%) attended an HIV care appointment, 107 (60.8%) initiated ART. 277 IDUs enrolled in a needle exchange program, and over 97,000 sterile syringes were distributed and returned |
| Poon et al. | Near Real-Time Monitoring of HIV Transmission Hotspots from Routine HIV Genotyping: an Implementation Case Study | 2016 | Outbreak Investigation | British Colombia, Canada | 2014–2015 | British Columbia Drug Treatment Program | 8839 HIV+ individuals in British Columbia (BC) whose genetic sequences were deposited in the Center for Excellence (CFE) Laboratory Program | New HIV diagnoses were in part identified through contact tracing procedures extensively embedded in Canada’s healthcare infrastructure. 32,505 HIV genotype records were deposited in the CFE Laboratory Program. The monitoring system queried the program database hourly and the detection of new genotype records prompted database reanalysis. New Records were integrated into old phylogenetic clusters or prompted the creation of new clusters. Phylogenetic clusters were defined as 5+ paired individuals that fulfilled selective criteria |
Beginning in February 2014, monthly and quarterly reports on the growth and characteristics of active clusters were distributed to the BC Centre for Disease Control and medical health officers at the five BC regional health authorities. Rapidly growing clusters prompted enhanced public health follow-up with an emphasis on linkage-to-care, ART initiation, and partner notification and testing | 218 phylogenetic clusters were detected by the monitoring system between October 2014 and October 2015. Nine out of the top 10 most rapidly growing clusters were MSM predominant, with variation among clusters by age and prevalence of transmitted drug resistance. In June 2014, the monitoring system detected the expansion of an MSM-majority cluster by 11 new cases over 3 months, 8 of whom had drug-resistant strains. Enhanced public health follow-up involved 9 HIV+ individuals, three of which subsequently started ART. Over 1 year of subsequent follow up, the cluster grew by 12 cases, but with decreased transmission of drug-resistant strains |
| Tookes et al. | Rapid Identification and Investigation of an HIV Risk Network Among People Who Inject Drugs –Miami, FL, 2018 | 2019 | Outbreak Investigation | Miami, FL | 2018 | Syringe Services Program, Florida Department of Health | 7 IDUs diagnosed with HIV from mid-February to December 2018 and their sexual/drug-use partners and social contacts. 59% male, 65% White, 65% homeless | DIS contacted newly diagnosed HIV+ individuals to elicit their sexual, drug-use, and other social partnerships. This social data was used to epidemiologically link participants and help infer transmission routes. HIV pol sequences were amplified via RTPCR and assessed at the 1.5% and 0.5% genetic distance thresholds using TRACE. Study sequences were compared with 38,395 Florida-based reference sequences uploaded to TRACE |
Patients testing HIV+ were linked to care at Jackson Memorial hospital via the Department of Health “Test and Treat” program. Patients received a 30-day supply of ART and were given 60 days to enroll in the AIDS Drug Assistance Program. Syringe service program staff accompanied patients during their initial appointments with a case manager, clinician, phlebotomist, and a pharmacist | 7 individuals met the case definition for the initial HIV outbreak, and an additional 10 individuals were listed as social partners. All 7 index cases were socially linked to at least 1 other index case. Two index cases clustered at a molecular distance of 1.5%, while another 2 clustered at 0.5%. One of the socially linked partners clustered with 2 index sequences at a genetic distance of 0.5%. All 7 individuals were linked to care (mean of 20 days), started on ART, and achieved viral suppression (mean of 70 days). Among 32 previously diagnosed HIV+ individuals affiliated with the syringe service program, only 17 (53%) achieved viral suppression by the end of the investigation period |
Year of publication
MSM Men who have sex with men, HIV human immunodeficiency virus, IDU injection drug user, CDC centers for disease control and prevention, BLAST basic local alignment search tool, BRAI bio-rad avidity incidence, IVDU intravenous drug user, STD sexually transmitted disease, TB tuberculosis, ECVC Encuesta Centroamericano de Vigilancia de VIH y Comportamiento en Poblaciones Vulnerables, FSW female sex worker, STI sexually transmitted infection, Hx history; NC-DHHS North Carolina Department of Health and Human Services, NS-STAT North Carolina Screening and Tracing for Active HIV-1 Transmission, AHI acute HIV infection, RHI recent HIV infection, DIS disease intervention specialists; iMAN integrated molecular & affiliation network analysis of HIV transmission, AIC Akaike’s Information Criterion, RDS respondent-driven sampling, DPH department of public health, TRACE secure HIV transmission cluster engine, PLWHA people living with HIV/AIDS, PCR polymerase chain reaction; PWID people who inject drugs; COMET context-based modeling for expeditious typing, GRT genotypic resistance testing, CDPH Chicago Department of Public Health, MUSCLE multiple sequence comparison by log-expectation, HCV hepatitis C virus, SD-PIRC San Diego Primary Infection Resource Consortium, CNICS center for AIDS research network of integrated clinical systems, RTPCR reverse transcription polymerase chain reaction, AIEDRP acute infection and early disease research program, FCP first choice program, ART antiretroviral therapy, GHWP good health for women project, UCSD University of California San Diego, SDC San Diego County, NYCDOH New York City Department of Health, PHSKC Public Health Department of Seattle and King County
Included interventions varied greatly in focus and scope. Interventions broadly fit into the following categories: (1) enhanced partner services, (2) education for providers, patients, or community members, (3) increased auxiliary services, and (4) expanded HIV testing or treatment. More granularly, studies discussed the following intervention: (1) increased DIS staffers or field epidemiologists [41, 50], (2) enhanced DIS follow-up with an emphasis on antiretroviral initiation, partner notification, and HIV testing [19, 50, 52], (3) educational outreach initiatives geared towards patients, community members, healthcare providers, or auxiliary service providers [41, 53], (4) increased syringe services via expanded hours of operation or operating sites [41, 46, 50], (5) expanded HIV testing in emergency rooms and hospitals [46, 50], homeless shelters [50], jails [41, 46, 50], or other sites [41, 46], (6) improved antiretroviral care provision or access [52, 53], and (7) enhanced linkage to auxiliary services such as insurance enrollment, vaccinations, drug assistance payment services, syringe services, condoms, etc. [41, 46, 52, 53]. Most studies featured multimodal interventions. For example, Metcalfe et al. detailed the impact of targeted education outreach initiatives, enhanced HIV service provision, and increased nursing outreach and linkage-to-care on HIV treatment and testing outcomes among a cohort of HIVpositive persons who inject drugs [53].
Reported outcomes for interventions varied greatly, but largely included the following: (1) the number of HIV tests completed [41, 46, 53], (2) the number of individuals newly diagnosed with HIV and over what time period [19, 41, 46, 50], (3) the number of individuals linked to HIV care [52, 53], initiated antiretroviral therapy [19, 41, 52, 53], and obtained viral suppression [52, 53], (4) the amount of elapsed time from HIV diagnosis to antiretroviral therapy initiation [53] or viral suppression [52], and (5) the number of individuals provided auxiliary services such as insurance enrollment, Hepatitis B vaccinations, mental health resources, substance use treatment, or syringe services [41]. No studies, however, incorporated a control arm making it impossible to assess the relative efficacy of any listed intervention. In Metcalfe et al., a multifaceted intervention was utilized to successfully initiate all trackable participants on antiretroviral therapy. Viral suppression in this cohort increased steadily from 18% in 2015 to 86% in 2019, and median time from HIV diagnosis to antiretroviral therapy initiation reduced from 264 days in 2015 to 23 days in 2019. Yet due to the lack of control arm, the study’s intervention could not be explicitly compared to standard partner services and linkage-to-care programming.
Discussion
Our systematic review of published literature identified 32 descriptive studies that incorporated both HIV molecular cluster analysis and network data capturing social, sexual, or venue-based relationships between individuals. Most studies were exploratory and focused on examining the overlap between social and molecular networks by using molecular data to support or refute purported transmission ties, assessing the proportion of a social network with genetic links, comparing the number of purported transmission ties identified by each type of network data, or examining the social or venue-based connections within a genetically linked network. Six studies analyzed molecular and social network data in the context of a specific HIV outbreak [27, 39, 40, 42, 51, 54]. Another six studies discussed socially and molecularly informed interventions implemented along the HIV care continuum during particular outbreaks, but their efficacies were not rigorously assessed [19, 41, 46, 50, 52, 53].
While health departments and researchers have integrated HIV molecular cluster analyses with social and sexual network data for over 20 years [58], the majority of studies employing both a molecular and social network approach were published within the last 6 years. This recent emphasis on cluster detection and response is likely influenced by decreasing cost and increasing feasibility of rapid sequencing, as well as recent growth in funding for phylogenetic analysis from the CDC and National Institutes of Health [59, 60]. Our review showed little standardization in how researchers and health departments integrated molecular data with existing surveillance methods. Though molecular cluster analysis was most often used to validate existing links, suggest new linkages, or characterize transmission clusters, research aims and outcomes varied widely given the exploratory nature of the studies. Researchers further employed varying thresholds for defining molecular linkage and utilized different statistical methods of conducting molecular cluster analysis, most commonly maximum-likelihood or neighbor-joining trees.
Despite the variability among studies characterizing social and molecular network overlap, our review demonstrated that 14–73% of individuals within social networks were genetically linked [28–32, 37, 43] and 43–65% of individuals within molecular cluster networks were socially linked [31, 47]. The significant overlap between molecular and social network linkages suggests that combining both data sources may prove useful in positing transmission pathways and identifying individuals who serve to benefit from partner services or linkage-to-care. As methods for combining molecular cluster analysis with social network data continue to improve, health departments could utilize integrated molecular and social networks to better characterize transmission routes and ultimately intervene in actively growing molecular or social/sexual network clusters [15, 19], though no studies we reviewed provided practical recommendations for how to do so.
Prior reviews have suggested that molecular cluster analysis, when combined with social network surveillance methods, may be used to inform cluster-specific interventions [15, 19], evaluate the efficacy of ongoing interventions, or determine how future interventions should be structured [9, 61]. In addition, given the stabilizing rates of HIV transmission in many regions, including the United States [62], combined with steady improvements in cluster detection and response techniques, potential interventions based upon molecular cluster analysis should be increasingly manageable at the local jurisdictional level. However, our review demonstrated that there is little published evidence discussing the comparative effectiveness of combined molecular and social network analyses in identifying new HIV cases or improving HIV outcomes compared to traditional surveillance methods. Six studies discussed targeted interventions employed during HIV outbreaks that featured enhanced partner services, educational initiatives, auxiliary services, and improved HIV testing/treatment programs [19, 41, 46, 50, 52, 53]. These studies further presented quantitative HIV intervention outcomes on the number of HIV tests completed, new individuals diagnosed and linked to care, auxiliary services provided, and the timeline for achieving viral load suppression. Yet none of the studies included control groups enabling comparisons with standard HIV prevention methods. Though the nature of partner services presents logistical challenges to rigorous study, comparing newer network-based approaches with standard partner notification and surveillance practices is essential in determining whether the addition of cluster detection and response to current network-based interventions produces superior HIV case finding and transmission reduction.
Our systematic review identified six studies that described the integration of molecular and social network data in the context of investigating HIV outbreaks [27, 39, 40, 42, 51, 54]. Recent DHHS guidance suggests that all individuals with new HIV diagnoses should receive resistance testing [18], which has led to increased access to the viral sequences required to conduct molecular cluster analysis. Additionally, the CDC has recommended that molecular cluster detection and response be used as a tool to monitor active clusters and help prioritize resources [1]. Despite the novelty of rapid sequencing technology [63, 64], it appears health departments are currently conducting surveillance or even interventions utilizing both molecular and social network data, but are not yet systematically documenting, evaluating, and disseminating how they integrate these data sources into existing HIV outbreak investigation and prevention strategies. Additionally, the personnel and resources required to conduct this integrated surveillance approach have not been adequately addressed given flat budgets and ethical concerns. Successful community engagement further requires deliberate consideration and work tailored to each context, in addition to proper training and access to the novel sequencing technologies. Rigorous evaluation and increased dissemination of this work must be undertaken in order to develop standardized protocols that can be tested for efficacy against accepted surveillance and prevention methods.
Given the lack of standardized methods of using HIV molecular cluster analysis, a focus on implementation science frameworks [65, 66] may be necessary to best incorporate molecular analysis into existing network approaches or design socially and molecularly-informed interventions. Implementation science frameworks focus on metrics critical to programmatic success including acceptability, feasibility, sustainability, and cost, which could aid researchers in transitioning from descriptive to experimental HIV surveillance studies [66]. Implementation science may further prove essential in identifying suitable uses for cluster detection and response given ethical concerns such as erroneously implicating individuals in transmission events, risking privacy breaches via publicly accessible phylogenetic databases and limited privacy laws, and ultimately leaving vulnerable populations increasingly open to persecution [23]. A first step in implementing integrated molecular cluster/social network interventions may be, for example, to work with stakeholders to determine the level of integration of data across health departments and front-line intervention agencies. Health departments have a more complete view and ownership of the molecular cluster data but must work intentionally to secure community buy-in and engagement. Determining how data could be shared safely and efficiently would seem to be an important first step.
There are several limitations to our systematic review. First, we were unable to rate the quality of the evidence presented given that all studies were exploratory with no testable hypothesis or sampling frame. In addition, we excluded non-HIV infectious agents which could have yielded some insights into the possible benefits of molecular investigation. However, we wished to focus on HIV and its unique surveillance systems in order to identify lessons specific to designing and implementing future HIV interventions.
There is no current evidence demonstrating that combined molecular and social network approaches are superior to traditional HIV contact tracing in identifying new HIV cases, facilitating linkage-to-care, or reducing onward HIV transmission. Future research should focus not only on documenting current utilization of integrated network analysis, but on implementing reproducible interventions that compare this innovative approach to current standards for partner services or related interventions. Further evidence is necessary to build recommendations for or against the integration of molecular cluster detection and response into current surveillance and intervention methods. Molecular cluster analysis may prove indispensable in ending HIV transmission, but only rigorous testing and standardization will demonstrate potential benefits and allow for widespread dissemination.
Supplementary Material
Funding
This work was supported by grants from the National Institute of Allergy and Infectious Disease, National Institute of Health (Project # 1R01AI136056-01) and the Third Coast Center for AIDS Research (CFAR), an NIH funded center (P30 AI117943).
Footnotes
Conflict of interest The authors have no conflicts of interest to declare that are relevant to the content of this article.
Code Availability Not applicable.
Ethical Approval This study was a review of published articles and did not involve human subjects, thus ethics approval was not required.
Consent to Participate Not applicable.
Consent for Publication Not applicable.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s10461-021-03525-0.
Data Availability
The EndNote library used for the systematic review can be made available upon request.
References
- 1.Centers for Disease Control & Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Division of HIV/AIDS Prevention. Detecting and responding to HIV transmission clusters—a guide for health departments. 2018. https://www.cdc.gov/hiv/pdf/funding/announcements/ps18-1802/CDC-HIV-PS18-1802-AttachmentE-Detecting-Investigating-and-Responding-to-HIV-Transmission-Clusters.pdf. Accessed 06 Jan 2020.
- 2.Centers for Disease Control and Prevention (CDC). Recommendations for partner services programs for HIV infection, syphilis, gonorrhea, and chlamydial infection. MMWR Recomm Rep Morb Mortal Wkly Rep Recomm Rep. 2008;57:1–83. [PubMed] [Google Scholar]
- 3.Hogben M, McNally T, McPheeters M, Hutchinson AB. The effectiveness of HIV partner counseling and referral services in increasing identification of HIV-positive individuals a systematic review. Am J Prev Med. 2007;33:S89–100. [DOI] [PubMed] [Google Scholar]
- 4.Hochberg CH, Berringer K, Schneider JA. Next-generation methods for HIV partner services: a systematic review. Sex Transm Dis. 2015;42:533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seth P, Wang G, Collins NT, Belcher L, Centers for Disease Control and Prevention (CDC). Identifying new positives and linkage to HIV medical care–23 testing site types, United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;64:663–7. [PMC free article] [PubMed] [Google Scholar]
- 6.Rayment M, Curtis H, Carne C, McClean H, Bell G, Estcourt C, et al. An effective strategy to diagnose HIV infection: findings from a national audit of HIV partner notification outcomes in sexual health and infectious disease clinics in the UK. Sex Transm Infect. 2017;93:94–9. [DOI] [PubMed] [Google Scholar]
- 7.Khanna A, Goodreau SM, Dan Wohlfeiler M, Daar E, Little S, Gorbach PM. Individualized diagnosis interventions can add significant effectiveness in reducing HIV incidence among men who have sex with men (MSM): insights from Southern California. Ann Epidemiol. 2015;25:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nichols BE, Götz HM, van Gorp ECM, Verbon A, Rokx C, Boucher CAB, et al. Partner notification for reduction of HIV-1 transmission and related costs among men who have sex with men: a mathematical modeling study. PLoS ONE. 2015. 10.1371/journal.pone.0142576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Vijver DAMC, Boucher CAB. Insights on transmission of HIV from phylogenetic analysis to locally optimize HIV prevention strategies. Curr Opin HIV AIDS. 2018;13:95–101. [DOI] [PubMed] [Google Scholar]
- 10.Ciccozzi M, Lai A, Zehender G, Borsetti A, Cella E, Ciotti M, et al. The phylogenetic approach for viral infectious disease evolution and epidemiology: an updating review. J Med Virol. 2019;91:1707–24. [DOI] [PubMed] [Google Scholar]
- 11.German D, Grabowski MK, Beyrer C. Enhanced use of phylogenetic data to inform public health approaches to HIV among men who have sex with men. Sex Health. 2017;14:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avila D, Keiser O, Egger M, Kouyos R, Böni J, Yerly S, et al. Social meets molecular: combining phylogenetic and latent class analyses to understand HIV-1 transmission in Switzerland. Am J Epidemiol. 2014;179:1514–25. [DOI] [PubMed] [Google Scholar]
- 13.Chan PA, Hogan JW, Huang A, DeLong A, Salemi M, Mayer KH, et al. Phylogenetic investigation of a statewide HIV-1 epidemic reveals ongoing and active transmission networks among men who have sex with men. J Acquir Immune Defic Syndr. 1999;70:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhodes T, Singer M, Bourgois P, Friedman SR, Strathdee SA. The social structural production of HIV risk among injecting drug users. Soc Sci Med. 1982;61:1026–44. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Wu Y, Mao L, Xia W, Zhang W, Dai L, et al. Targeting HIV prevention based on molecular epidemiology among deeply sampled subnetworks of men who have sex with men. Clin Infect Dis Off Publ Infect Dis Soc Am. 2015;61:1462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Little SJ, Kosakovsky Pond SL, Anderson CM, Young JA, Wertheim JO, Mehta SR, et al. Using HIV networks to inform real time prevention interventions. PLoS ONE. 2014;9:e98443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buthelezi UE, Davidson CL, Kharsany AB. Strengthening HIV surveillance: measurements to track the epidemic in real time. Afr J AIDS Res AJAR. 2016;15:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panel on antiretroviral guidelines for adults and adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents with HIV. Department of Health and Human Services. Available athttps://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultandAdolescentGL.pdf. Accessed June 2021. [Google Scholar]
- 19.Poon AFY, Gustafson R, Daly P, Zerr L, Demlow SE, Wong J, et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: an implementation case study. Lancet HIV. 2016;3:e231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SS, Tam DKP, Tan Y, Mak WL, Wong KH, Chen JHK, et al. An exploratory study on the social and genotypic clustering of HIV infection in men having sex with men. AIDS Lond Engl. 2009;23:1755–64. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira A, Young T, Mathews C, Zunza M, Low N. Strategies for partner notification for sexually transmitted infections, including HIV. Cochrane Database Syst Rev. 2013. 10.1002/14651858.CD002843.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanoni BC, Elliott RJ, Neilan AM, Haberer JE. Screening for HIV and linkage to care in adolescents: insights from a systematic review of recent interventions in high- versus low- and middle-income settings. Adolesc Health Med Ther. 2018;9:211–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mutenherwa F, Wassenaar DR, de Oliveira T. Ethical issues associated with HIV phylogenetics in HIV transmission dynamics research: a review of the literature using the Emanuel framework. Dev World Bioeth. 2019;19:25–35. [DOI] [PubMed] [Google Scholar]
- 24.Moher D, Liberati A, Tetzlaff J, Altman DG, Group TP. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters MDJ, Godfrey CM, Khalil H, McInerney P, Parker D, Soares CB. Guidance for conducting systematic scoping reviews. Int J Evid Based Healthc. 2015;13:141–6. [DOI] [PubMed] [Google Scholar]
- 26.The World Bank. World Bank country and lending groups. 2020. https://datahelpdesk.worldbank.org/knowledgebase/articles/906519-world-bank-country-and-lending-groups. Accessed 26 June 2020.
- 27.Nett RJ, Bartschi JL, Ellis GM, Hachey DM, Frenkel LM, Roscoe JC, et al. Two clusters of HIV-1 infection, rural Idaho, USA, 2008. Emerg Infect Dis. 2010;16:1807–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin H, He N, Sujuan Z, Ding Y, Qiu D, Zhang T, et al. Behavioral and molecular tracing of risky sexual contacts in a sample of Chinese HIV-infected men who have sex with men. Am J Epidemiol. 2013. 10.1093/aje/kws256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grande K, Schumann C, Ocfemia M, Vergeront J, Wertheim J, Oster A. Transmission patterns in a low HIV-morbidity state—Wisconsin, 2014–2017. MMWR Morb Mortal Wkly Rep. 2019;68:149–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasquale DK, Doherty IA, Sampson LA, Hue S, Leone PA, Sebastian J, et al. Leveraging phylogenetics to understand HIV transmission and partner notification networks. J Acquir Immune Defic Syndr. 2018. 10.1097/QAI.0000000000001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wertheim J, Pond S, Forgione L, Mehta S, Murrell B, Shah S, et al. Social and genetic networks of HIV-1 transmission in New York City. PLOS Pathog. 2017;13:e1006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell EM, Patala A, Shankar A, Li J-F, Johnson JA, Westheimer E, et al. Phylodynamic analysis complements partner services by identifying acute and unreported HIV transmission. Viruses. 2020;12:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dennis AM, Murillo W, de Maria Hernandez F, Guardado ME, Nieto AI, Lorenzana de Rivera I, et al. Social network-based recruitment successfully reveals HIV-1 transmission networks among high-risk individuals in El Salvador. J Acquir Immune Defic Syndr. 2013;63:135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujimoto K, Coghill L, Weier C, Hwang L-Y, Kim J, Schneider J, et al. Lack of support for socially connected HIV-1 transmission among young adult black MSM. AIDS Res Hum Retrovir. 2017. 10.1089/aid.2016.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan E, Skaathun B, Schneider J. Sexual, social, and genetic network overlap: a socio-molecular approach toward public health intervention of HIV. Am J Public Health. 2018;108:e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pines HA, Wertheim JO, Liu L, Garfein RS, Little SJ, Karris MY. Concurrency and HIV transmission network characteristics among MSM with recent HIV infection. AIDS. 2016;30:2875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith D, May S, Tweeten S, Drumright L, Pacold M, Pond S, et al. A Public health model for the molecular surveillance of HIV transmission in San Diego, Califonia. AIDS Lond Engl. 2009;23:225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepej SZ, Vrakela IB, Poljak M, Bozicevic I, Begovac J. Phylogenetic analysis of HIV sequences obtained in a respondent-driven sampling study of men who have sex with men. AIDS Res Hum Retrovir. 2009;25:1335–8. [DOI] [PubMed] [Google Scholar]
- 39.Monterosso A, Minnerrly S, Goings S, Morriss A, France AM, Dasgupta S, et al. Identifying and investigating a rapidly growing HIV transmission cluster in Texas. Conf Retrovir Oppor Infect. https://www.croiconference.org/abstract/identifying-and-investigating-rapidly-growing-hiv-transmission-cluster-texas/.Accessed 11 July 2021
- 40.Sizemore L, Fill M-M, Mathieson SA, Black J, Brantley M, Cooper K, et al. Using an established outbreak response plan and molecular epidemiology methods in an HIV transmission cluster investigation, Tennessee, January-June 2017. Public Health Rep Wash DC. 1974;2020(135):329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters PJ, Pontones P, Hoover KW, Patel MR, Galang RR, Shields J, et al. HIV infection linked to injection use of oxymorphone in Indiana, 2014–2015. N Engl J Med. 2016;375:229–39. [DOI] [PubMed] [Google Scholar]
- 42.Dennis A, Pasquale D, Billock R, Beagle S, Mobley V, Cope A, et al. Integration of contact tracing and phylogenetics in an investigation of acute HIV infection. Sex Transm Dis. 2018;45:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider JA, Hayford CS, Tabidze I, Wertheim JO, Hallmark C, Khanna A, et al. Do partner services initiated from molecular clusters yield new or Viremic Hiv cases?. Boston, Massachusetts. 2020. https://www.croiconference.org/abstract/do-partner-services-initiated-from-molecular-clusters-yield-new-or-viremic-hiv-cases/. Accessed 25 June 2020 [Google Scholar]
- 44.Deng W, Fu P, Bao L, Vidal N, He Q, Qin C, et al. Molecular epidemiological tracing of HIV-1 outbreaks in Hainan island of southern China. AIDS Lond Engl. 2009;23:977–85. [DOI] [PubMed] [Google Scholar]
- 45.Ssemwanga D, Ndembi N, Lyagoba F, Bukenya J, Seeley J, Vandepitte J, et al. HIV type 1 subtype distribution, multiple infections, sexual networks, and partnership histories in female sex workers in Kampala, Uganda. AIDS Res Hum Retrovir. 2011;28:357–65. [DOI] [PubMed] [Google Scholar]
- 46.Golden MR, Lechtenberg R, Glick SN, Dombrowski J, Duchin J, Reuer JR, et al. Outbreak of human immunodeficiency virus infection among heterosexual persons who are living homeless and inject drugs—Seattle, Washington, 2018. MMWR Morb Mortal Wkly Rep. 2019;68:344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kostaki E, Nikolopoulos G, Pavlitina E, Williams L, Magiorkinis G, Schneider J, et al. Molecular analysis of HIV-1 infected individuals in a network-based intervention (TRIP): phylogenetics identify HIV-1 infected individuals with social links. J Infect Dis. 2018. 10.1093/infdis/jiy239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monroe-Wise A, Mbogo L, Guthrie B, Bukusi D, Sambai B, Chohan B, et al. Peer-mediated HIV assisted partner services to identify and link to care HIV-positive and HCV-positive people who inject drugs: a cohort study protocol. BMJ Open. 2021;11:e041083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pilon R, Leonard L, Kim J, Vallee D, De Rubeis E, Jolly AM, et al. Transmission patterns of HIV and hepatitis C virus among networks of people who inject drugs. PLoS ONE. 2011;6:e22245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alpren C, Dawson EL, John B, Cranston K, Panneer N, Fukuda HD, et al. Opioid use fueling HIV transmission in an urban setting: an outbreak of HIV infection among people who inject drugs—Massachusetts, 2015–2018. Am J Public Health. 2020;110:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samoff E, Mobley V, Hudgins M, Cope AB, Adams ND, Caputo CR, et al. HIV outbreak control with effective access to care and harm reduction in North Carolina, 2017–2018. Am J Public Health. 2020;110:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tookes H, Bartholomew TS, Geary S, Matthias J, Poschman K, Blackmore C, et al. Rapid identification and investigation of an HIV risk network among people who inject Drugs -Miami, FL, 2018. AIDS Behav. 2020;24:246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Metcalfe R, Ragonnet-Cronin M, Bradley-Stewart A, McAuley A, Stubbs H, Ritchie T, et al. From hospital to the community: redesigning the human immunodeficiency virus (HIV) clinical service model to respond to an outbreak of HIV among people who inject drugs. J Infect Dis. 2020;222:S410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayman A, Moss T, Simmons G, Arnold C, Holmes E, Naylor-Adamson L, et al. Phylogenetic analysis of multiple heterosexual transmission events involving subtype B of HIV type 1. AIDS Res Hum Retrovir. 2001;17:689–95. [DOI] [PubMed] [Google Scholar]
- 55.Mehta SR, Wertheim JO, Delport W, Ene L, Tardei G, Duiculescu D, et al. Using phylogeography to characterize the origins of the HIV-1 subtype F epidemic in Romania. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2011;11:975–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Resik S, Lemey P, Ping L-H, Kouri V, Joanes J, Pérez J, et al. Limitations to contact tracing and phylogenetic analysis in establishing HIV type 1 transmission networks in Cuba. AIDS Res Hum Retrovir. 2007;23:347–56. [DOI] [PubMed] [Google Scholar]
- 57.Tordoff DM, Buskin S, Lechtenberg R, Golden MR, Kerani RP, Herbeck JT. Combining traditional and molecular epidemiology methods to quantify local HIV transmission among foreign-born residents. AIDS Lond Engl. 2021;35:655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Centers for Disease Control and Prevention (CDC). Cluster of HIV-positive young women–New York, 1997–1998. MMWR Morb Mortal Wkly Rep. 1999;48:413–6. [PubMed] [Google Scholar]
- 59.National Institutes of Health. PAR-17–048: Phylodynamic tracking of HIV transmission (R01). 2016. https://grants.nih.gov/grants/guide/pa-files/par-17-048.html. Accessed 9 June 2021
- 60.Centers for Disease Control and Prevention. Funding Opportunity Announcement (FOA) PS18–1802: Integrated Human Immunodeficiency Virus (HIV) Surveillance and Prevention Programs for Health Departments. 2019. https://www.cdc.gov/hiv/funding/announcements/ps18-1802/index.html. Accesseed 9 June 2021
- 61.Ratmann O, van Sighem A, Bezemer D, Gavryushkina A, Jurriaans S, Wensing A, et al. Sources of HIV infection among men having sex with men and implications for prevention. Sci Transl Med. 2016. 10.1126/scitranslmed.aad1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Centers for Disease Control & Prevention. Estimated HIV incidence and prevalence in the United States, 2010–2016. HIV Surveill Suppl Rep. 2019;24:89. [Google Scholar]
- 63.Parikh UM, McCormick K, van Zyl G, Mellors JW. Future technologies for monitoring HIV drug resistance and cure. Curr Opin HIV AIDS. 2017;12:182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramamurthy M, Sankar S, Kannangai R, Nandagopal B, Sridharan G. Application of viromics: a new approach to the understanding of viral infections in humans. VirusDisease. 2017;28:349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glasgow RE, Eckstein ET, ElZarrad MK. Implementation science perspectives and opportunities for HIV/AIDS research: integrating science, practice, and policy. JAIDS J Acquir Immune Defic Syndr. 2013;63:S26. [DOI] [PubMed] [Google Scholar]
- 66.Cox J, Gutner C, Kronfli N, Lawson A, Robbins M, Nientker L, et al. A need for implementation science to optimise the use of evidence-based interventions in HIV care: a systematic literature review. PLoS ONE. 2019. 10.1371/journal.pone.0220060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The EndNote library used for the systematic review can be made available upon request.