

Abstract
Trypanosoma cruzi invasion of host cells involves several discrete steps: attachment, parasite internalization mediated by recruitment and fusion of host cell lysosomes, and escape from the parasitophorous vacuole to liberate amastigotes to multiply freely in the cytosol. This report describes the initial characterization of the LYT1 gene and the demonstration that the gene product is involved in cell lysis and infectivity. Mutational analysis demonstrated that deletion of LYT1 resulted in attenuation of infection, which was associated with diminished hemolytic activity. Reintroduction of LYT1 restored infectivity in null mutants, confirming the critical role of LYT1 in infection. Additionally, in vitro stage transition experiments with LYT1-deficient lines showed that these parasites converted to extracellular amastigote-like cells and metacyclic trypomastigotes more rapidly than wild-type parasites, suggesting that the diminished infectivity was not a result of the LYT1 deficiency that affected the parasite's ability to complete the life cycle.
Parasitic protozoa of the family Trypanosomatidae cause disease on a worldwide scale in a variety of vertebrate, invertebrate, and plant species. Included in this family is Trypanosoma cruzi, the etiologic agent of Chagas' disease, which is endemic throughout much of South and Central America, affecting over 20 million people. T. cruzi undergoes a biphasic life cycle, comprised of several distinct developmental stages in both the reduviid beetle vector and the mammalian host. In the beetle, the flagellated epimastigote proliferates in the midgut before differentiating into the nondividing but infectious metacyclic trypomastigote found in the vector's hindgut. Following its introduction into the mammalian blood, the parasite infects host cells, differentiates into an amastigote, and initiates replication in the cytosol of the infected cell. Ultimately, the amastigotes develop into nondividing bloodstream trypomastigotes, which can either initiate another round of infection or be taken up by the reduviid vector during a blood meal. The life cycle is completed upon development of the epimastigote from the bloodstream trypomastigote.
T. cruzi invasion of host cells is a complex event, which has only recently begun to be unraveled (7). This process appears to involve several discrete steps, beginning with the attachment of the parasite to the host cell. Immediately after attachment but probably prior to parasite internalization, host cell lysosomes are recruited to the site of attachment, where they transiently fuse with the plasma membrane (18). Then, in a rapid series of events, the parasite is internalized concomitant with stable fusion of the recruited lysosomes to the plasma membrane, resulting in the formation of the parasitophorous vacuole (3, 4). Ultimately, the amastigotes escape from the parasitophorous vacuole and, thus liberated, they multiply freely in the cytosol. Although the host cell machinery involved in internalization is reasonably well understood, little is known of the parasite molecules involved in the process.
Though many parasite proteins are undoubtedly important for T. cruzi infection and successful completion of the life cycle, surprisingly few have been identified experimentally. One parasite factor likely to be involved is TC-TOX, a secreted acid-stable hemolytic protein (2). This protein has membrane pore-forming activity at low pH levels and cross-reacts with monoclonal antibodies directed against C9, and it has been postulated that it mediates the escape of T. cruzi from the parasitophorous vacuole into the cytosol (1, 5). Another protein shown to be involved in infection was a trypomastigote-secreted peptidyl-prolyl cis or trans isomerase (17), but its specific target on the host cell remains to be elucidated. A third T. cruzi protein, which has been shown to play an important role in host cell invasion, is oligopeptidase B. Using a targeted gene replacement approach it was demonstrated that this enzyme mediated production of a signaling agonist for mammalian cells that is required for efficient invasion and infectivity (8).
The present study describes the cloning of LYT1, which was isolated from a T. cruzi cDNA library based on the cross-reactivity of its gene product to antibodies against the C9 component of the membrane attack complex of complement. Searches of all available DNA and protein databases failed to identify significant homology of proteins or potential translation products to the LYT1 protein. In order to gain insight in the possible role of the gene product, LYT1 deletion mutants were generated by targeted gene replacements. Using these genetic methodologies we have shown that LYT1 is not required for viability of epimastigotes; however, LYT1-deficient parasites exhibit three distinct phenotypes. These parasites exhibit accelerated in vitro development, demonstrate reduced infectivity, and have diminished hemolytic activity. Reintroduction of LYT1 reconstituted infectivity for the null parasites, demonstrating that the LYT1 gene plays an important role in infection.
MATERIALS AND METHODS
Cells and parasites.
NIH 3T3 and NRK fibroblasts were maintained in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% glutamine, and 5 μg of penicillin-streptomycin (pen-strep) per ml at 37°C in a humidified atmosphere containing 5% CO2. Epimastigotes from the T. cruzi Cl-Brener and Y strains were maintained in liver infusion tryptose medium containing 10% FBS (LIT) at 28°C (13). Mid-log-phase cultures containing from 5 × 106 to 2 × 107 parasites ml−1 were used in all experiments. Transformed clones were isolated from the G418- and hygromicin B-resistant T. cruzi population by limiting dilution in the absence of selection, as described by Hariharan et al. (13). Trypomastigotes were obtained from supernatant of infected monolayers of NIH 3T3 fibroblasts. Amastigotes were obtained from the supernatant of infected monolayers of NIH 3T3 fibroblasts or from in vitro stage transition experiments. The amastigotes were separated from trypomastigotes or epimastigotes using the amastigote-specific antibody 2C2B6, specific for the Ssp-4 surface antigen of amastigotes (6).
Isolation and sequence of LYT1 cDNA and genomic clones.
A T. cruzi Y strain amastigote cDNA expression library constructed in λgt11 (12) was screened with antibodies against human complement component C9. Using the 0.8-kb insert from a positive clone (λgt11-LYT1-0.8) as a probe, a λ EMBL-3 genomic library (11) was screened. A positive clone (λEMBL-3LYT1-20) with a 20-kb insert was digested with different restriction enzymes and probed by Southern blotting to obtain a 5.8-kb AatII fragment containing the LYT1 gene and flanking sequences. Following gel purification, the 5.8-kb fragment was ligated into the AatII site of pGEM5Zf(+). The pGEM5Zf(+)LYT1-5.8 clone insert was sequenced by the dideoxy chain termination method (19). After gel purification, a 2,888-bp BsiWI fragment from pGEM5zfLYT1a-5.8 was treated with T4 DNA polymerase to generate flush ends and then ligated into the HincII site of pBS. The resultant plasmid, pBSLYT1a-BsiWI, contains the complete LYT1a coding sequence and 644 and 591 bp of the 5′ and 3′ flanking sequences, respectively. Using a 587-bp PCR fragment (1,496 to 2,083 bp from pGEM5Zf(+)LYT1-5.8) as the probe, a 4.3-kb BsiWI genomic clone containing the LYT1a allele from the Cl-Brener strain was isolated and partially sequenced (19). The LYT1b allele was cloned by PCR using genomic DNA from the C1-Brener strain and the primers TcL1 (5′-CGAGCCCGAACGATGAACAT) and TcL4 (5′-TTTGCGAGCCTCTGCATTTT) and sequenced.
Expression of LYT1 in yeast.
The full coding sequence of LYT1 plus 597 bp of downstream sequence was amplified by PCR using the primers ARTX-8 (5′-CGGGATCCATGCGGAAGA) and ARTX-1 (5′-TGAAAGAGAAGAAAGTC) and pGEM5Zf(+)LYT1-5.8 as the template. After BamHI digestion (BamHI sites are located 9 bp upstream of the initiator codon and 199 bp upstream of ARTX-1), the fragment was ligated to dephosphorylated BamHI-digested pREP3X. After transformation of Escherichia coli XL1-blue, the plasmid was purified and used to transform Schizosaccharomyces pombe by electroporation. Transformants were grown in Edimburgh minimal medium with thiamine (5 μg/ml) to repress expression of LYT1. Expression was achieved by growth of the transformed yeast in the absence of thiamine, and the presence of LYT1 was tested in whole extracts by Western blotting. Extracts for measurements of hemolytic activity were obtained by grinding 2 × 109 yeast cells in a mortar with a quantity of sea sand that was double the weight. The ground material was resuspended in 4 ml of acid buffer plus protease inhibitors (ABPI) (50 mM NaCl, 100 mM sodium acetate [pH 5.5], 0.1% d-glucose, 50 mM EDTA, 1 mM phenylmethylsulfonyl fluoride 1 μg of leupeptin per ml, 1 μg of pepstatin per ml) and clarified by centrifugation.
Western blots.
Samples were electrophoresed under reducing conditions in 7.5% polyacrylamide gels (15) and transferred for 2 h at 80 V to polyvinylidine difluoride membrane (Immobilon-P; Millipore). Membranes were blocked overnight with 5% nonfat milk in a solution containing 10 mM Tris-HCl [pH 8], 150 mM NaCl, and 0.05% Tween 20 (TBST) plus 0.02% NaN3 and incubated for 1 h at room temperature with 1:200 dilutions of rabbit anti-C9 serum or rabbit serum against truncated LYT1 (approximately 250 amino-terminal amino acids) expressed in E. coli and purified by affinity chromatography (E. González-Rey and A. González, unpublished results). The membranes were exhaustively washed with TBST and then incubated with 1:1,000 alkaline phosphatase-conjugated goat anti-rabbit immunoglobulin for 1 h at room temperature. The membranes were washed and developed with 5-bromo-4-chloro-3-indolyl phosphate–nitroblue tetrazolium (Sigma).
Construction of pGEM5Zf(+)hyg::LYT1 and pGEM5Zf(+)neo::LYT1.
A 7,253-bp fragment containing the 5′ and 3′ LYT1 flanking sequences and the complete pGEM5Zf(+) sequence was generated by PCR amplification using pGEM5Zf(+)LYT1-5.8 as the template and the primers ARTX-9 (5′-TCTAGAGCGAGCACG [reverse primer, nucleotides −1 to −13 upstream from the first LYT1 ATG and terminal XbaI site]) and ARTX-10 (5′-CCCGGGCAGCTAGA [nucleotides 85 to 95 downstream of the LYT1 stop codon, followed by 2 additional nucleotides as part of a terminal SmaI site]). The PCR conditions used were as follows: 1.5 min of denaturation at 94°C, 3 min of annealing at 50°C, and 7 min of polymerization at 72°C (20 cycles). The PCR product was treated with T4 DNA polymerase to generate flush ends and then religated. The resultant plasmid was digested with XbaI and SmaI, gel purified after electrophoresis, and used to ligate the hygr and neor coding sequences, which have been generated by digestion of pBSSK-hyg1f8 and pBSSK-neo1f8 (21) with XbaI/StuI and XbaI/SmaI, respectively. The resulting plasmids were pGEM5Zf(+)hyg::LYT1 and pGEM5Zf(+)neo::LYT1 containing, respectively, hygr and neor coding sequence, as well as 714 and 2,854 bp of the 5′ and 3′ LYT1 flanking sequences, respectively.
Generation of LYT1 gene knockouts.
For LYT1 sequence replacement, a 4,515-bp LYT1(Neo) fragment was generated, as was a 4,773-bp LYT1(Hyg) fragment, by digestion of pGEM5Zf(+)neo::LYT1 and pGEM5Zf(+)hyg::LYT1 with BglI. Approximately 25 μg of each fragment was purified and used to transform the Cl-Brener epimastigotes following established procedures (9, 10, 13, 14). Forty-eight hours after electroporation, the cultures were exposed to antibiotic selection using G418 (250 μg/ml) and hygromycin B (250 μg/ml). Once antibiotic-resistant growth cultures were established, clonal derivatives were isolated from each population by limiting dilution and analyzed as described below.
Southern and Northern hybridization.
The blotting, hybridization, and washing conditions used in Southern and Northern analysis were precisely as described previously (13). For Northern analysis total cellular RNA was isolated by the guanidium-cesium chloride method and size fractionated on 1.1% agarose gels containing 2.2 M formaldehyde (16). Probes were generated by PCR. The oligonucleotides used to amplify the LYT1 coding sequence were LYTEXP1 ( 5 ′ - CGCGATATC TAAGAAGGAGATATACATATGCGGAAGAAAGCCGCAGCATT [an EcoRV restriction site sequence followed by nucleotides 1 to 23 of LYT1 coding sequence]) and LYT3′ (5′-ACGTGGATCCCAGTGGCGGAGCAGCACTATTCGC [complementary to nucleotides 648 to 672 of the LYT1 coding sequence downstream of the BamHI site]). The oligonucleotides used for the 5′ probe were LYTEXP1 and LYT2 (5′-CGTGCGACTGAGATGTCACC [complementary to nucleotides 239 to 259 of the LYT1 coding sequence]). The oligonucleotides used for the 3′ probe were either LYT4 (5′-GCAGGATTTGCCAGCGATGC [nucleotides 392 to 411 of the LYT1 coding sequence]) and LYT3′ or LYT7 (5′-GCGACAACATACCCGACCCCGCGGA [nucleotides 1058 to 1082 of the LYT1 coding sequence]) and LYT8 (5′-GTCATCCCTAATGCCAAAGACTTC [complementary to nucleotides 3 to 26 downstream of the LYT1 translation stop codon]).
Infectivity assay.
Monolayers of NIH 3T3 cells grown to 50% confluency in DMEM supplemented with 2% fetal calf serum were infected with 2 × 106 mid-log-phase epimastigotes per ml derived from wild-type Cl-Brener as well as each of the LYT1 mutant lines cultured in LIT media plus 10% FBS at 28°C. Forty-eight hours later the cells were washed, and they were subsequently washed every 2 days with DMEM to remove nonadherent parasites, after which fresh DMEM plus 2% fetal calf serum was added. For the secondary infection experiments, wild-type and LYT1 mutant trypomastigotes (2 × 104 trypomastigotes/ml) resulting from the first infection were purified as described above and used to infect NIH 3T3 cells (grown to 50% confluency) over 2 h. NIH 3T3 cells were washed every 2 days, and fresh DMEM plus 2% fetal calf serum was added. A host cell was considered infected if replicating amastigotes were observed inside the cell. Infections were monitored daily, and the number of amastigotes and trypomastigotes in the supernatant was determined. The percentage of infected cells determined by microscopic observation was calculated by comparing the number of cells containing parasites to the total number of cells.
Reconstitution of LYT1 null mutants.
For LYT1 reconstitution of null mutant parasites, a 1,656-bp fragment was generated from the pBSLYT1a-BsiWI genomic clone. After electrophoresis and gel purification, 100 or 200 μg of fragment was used to transform wild-type and null mutant parasites. Forty-eight hours after electroporation, the same numbers of wild-type and mutant parasites as described in the infectivity assay were used to infect NIH 3T3 cells.
In vitro stage transition.
Epimastigotes were grown in LIT medium plus 10% FBS at 28°C until they reach mid-log phase (about 1 × 107 epimastigotes/ml). Parasites were then either transferred to DMEM plus 2% FBS–1% glutamine–5-μg/ml penicillin-streptomycin at 37°C or were allowed to progress to stationary phase in the original LIT medium at 28°C. Cultures were monitored daily, and the percentage of amastigotes in DMEM or of metacyclic trypomastigotes in LIT medium was determined by microscopic observation.
Hemolytic assays.
For yeast extract, 0.9 ml of yeast extract was incubated for 2 h at 37°C with 0.1 ml of ABPI containing 5 × 107 rabbit erythrocytes. After centrifugation, the results were quantified by monitoring optical adsorbance at 545 nm. To obtain values for 0 and 100% lysis, the same numbers of erythrocytes were incubated in ABPI and water, respectively, over the same time course as the experimental samples.
For measuring hemolytic activity with whole parasites, parasites and horse erythrocytes (PML Microbiologicals, Tualatin, Oreg.) were pelleted by centrifugation, washed, and resuspended in acid buffer (10 mM NaOAc [pH 5.4], 200 mM NaCl, 0.2% d-glucose) or neutral buffer (10 mM NaOAc [pH 7], 150 mM NaCl, 0.2% d-glucose). Washed red blood cells (2 × 107 cells) were incubated with parasites (2 × 107 parasites) in 0.5 ml of acid or neutral buffer at 37°C and for various periods of time as indicated below.
Nucleotide sequence accession numbers.
The nucleotide sequence accession numbers for pGEM5Zf(+)LYT1-5.8, LYT1a, and LYT1b are AF253317, AF263616, and AF320626, respectively (all are GenBank accession numbers).
RESULTS
Cloning, characterization, and yeast expression of LYT1.
A LYT1 cDNA clone was isolated from a Y strain T. cruzi amastigote expression library by virtue of its cross-reactivity to antibodies against human complement component C9 (Fig. 1A). The 0.8-kb insert from the LYT1 cDNA clone (γgt11-LYT1-0.8) was subsequently used as a probe to isolate γEMBL-3LYT1-20 from a T. cruzi Y strain genomic library. From the genomic clone, a 5.8-kb AatII restriction fragment was subcloned (pGEM5Zf(+)LYT1-5.8) and sequenced. Using the pGEM5Zf(+)LYT1-5.8 sequence, a set of primers was designed to isolate and sequence both the LYT1a and LYT1b alleles from the Cl-Brener strain. A 1,653-bp open reading frame encoding a 552-amino-acid protein was identified, as were 5′ and 3′ flanking sequences. Figure 1B shows the deduced amino acid sequence derived from the LYT1a coding sequence of the Y strain and from the LYT1a and LYT1b coding sequences of the Cl-Brener strain. The derived amino acid sequence of LYT1 predicts a 61,400-dalton polypeptide with an isoelectric point of 11.04.
FIG. 1.

Isolation and sequencing of LYT1 cDNA clone. (A) Western blot of isopropyl-β-d-thiogalactopyraroside-induced λ-infected E. coli lysates showing reaction with rabbit anti-C9 antibodies. Lane 1, λgt11-LYT1-0.8; lane 2, λgt11. (B) Deduced amino acid sequences of LYT1a of the Y strain and LYT1a and LYT1b of the Cl-Brener strain. Differences in the Cl-Brener strain are shown above (for LYT1a) or below (for LYT1b) the Y strain sequence.
Computer analysis of LYT1 failed to show any significant homology to C9 but did predict the amino-terminal sequence as a signal peptide for secretion. Searches of all available DNA and protein databases also failed to identify significant homology to known proteins or potential translation products. Since the size of the protein, the presence of a leader peptide, and the cross-reactivity to C9 suggest a possible relationship to TC-TOX, we aimed at determining the potential lytic activity of LYT1 by expression in yeast. Figure 2A shows that transformed S. pombe was able to express LYT1. As shown in Fig. 2B, extracts from LYT1-expressing yeast lyse rabbit erythrocytes at pH 5.5 more than twice as efficiently as nonexpressing or nontransformed controls.
FIG. 2.

Expression of LYT1 in yeast. (A) Western blot of extracts of S. pombe transformed with pREP3X (lane 1) or pREP3X-LYT1 (lane 2), grown in the absence of thiamin to induce expression. The filter was reacted with rabbit antibodies against recombinant LYT1 produced in E. coli. (B) Hemolytic activity of extracts of S. pombe transformed with pREP3X and grown in the presence of thiamin (bar 1), transformed with pREP3X-LYT1 and grown in the presence of thiamin (bar 2 [control]), or transformed with pREP3X-LYT1 and grown in the absence of thiamin (bar 3) was determined using rabbit erythrocytes. The results presented have been corrected for spontaneous lysis (13%).
Genomic Southern analysis using several restriction enzymes indicated that LYT1 was a single-copy gene (data not shown). The genomic organization of the genes was confirmed by the gene knockout experiments described below (Fig. 3). Chromosomal blot hybridization also showed that the allelic copies of LYT1 gene are located in chromosomes VI and XII, of 1.03 and 1.60 Mb, respectively (9) (data not shown).
FIG. 3.

Genomic Southern blot analysis of wild-type Cl-Brener strain (lane 1), LYT1 double knockout L16 (lane 2), and LYT1 single knockout L14 (lane 3). The PstI (P) restriction maps of a and b LYT1 loci are shown beneath the gel image. Restriction fragments hybridizing with the LYT1 probe (hatched rectangles) were the expected sizes, indicating that correct integration had occurred. The LYT1 probe failed to hybridize with the LYT1 double knockout, demonstrating that both alleles of the LYT1 gene were replaced.
Generation of LYT1 mutants.
As mentioned above, LYT1 lacks homology to known proteins or hypothetical translation products. Consequently, a genetic strategy aimed at generating single and double knockouts of both LYT1 alleles in the Cl-Brener strain of T. cruzi was used to gain insight into the possible role of the gene product. Two plasmids were designed consisting of either the neomycin phosphotransferase II gene (neor) or the hygromycin B phosphotransferase gene (hygr), conferring resistance to G418 and hygromycin B, respectively, bounded by LYT1 5′ and 3′ flanking sequences. The gene replacement constructs were targeted to the wild-type LYT1 loci by homologous recombination. These two LYT1 loci can be distinguished in the Cl-Brener strain by virtue of the absence (allele a) or presence (allele b) of a PstI restriction site at position 1588 in the nucleotide coding sequence.
Single (clone L14)- and double (clone L16)-knockout parasites were obtained. To characterize the gene replacements, comparative genomic Southern analysis of PstI-digested DNA isolated from wild-type and mutant parasites was carried out (Fig. 3). Hybridization with a probe from nucleotides 1 to 672 of the LYT1 coding region revealed that the restriction patterns were consistent with deletions of the LYT1 loci. The wild-type restriction fragments hybridizing to this probe included fragments of 3.7, 2.5, and 1.2 kb. The 2.5-kb band is common to both alleles, the 1.2-kb band is specific for the LYT1b allele, and the 3.7-kb band is specific for the LYT1a allele. As expected, the double gene knockout L16 lacks all hybridizing bands, demonstrating that both LYT1 sequences were deleted. The single gene knockout L14 carries a deletion of the LYT1a allele as evidenced by the lack of the 3.7-kb restriction fragment. The successful generation of LYT1 null mutant line indicates that LYT1 is not essential for the viability of epimastigotes.
LYT1 transcripts in wild-type and mutant lines were analyzed by Northern blotting of epimastigote nRNA. As shown in Fig. 4, hybridization with a LYT1 probe revealed a single band of approximately 1.8 kb in RNA isolated from wild-type parasites. With L14 RNA, a band of identical size but weaker intensity appeared. This result indicates that L14 expresses the retained single LYT1b allele. As expected, no hybridization was observed with L16 RNA.
FIG. 4.
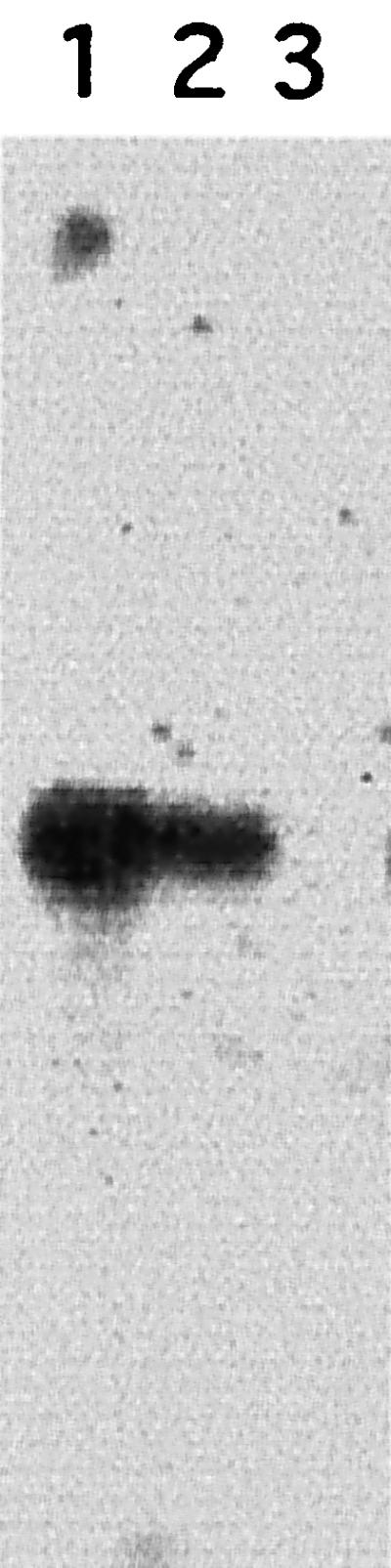
Northern blot analysis of LYT1 expression in wild-type and mutant parasites. Total RNA (10 μg) from the wild-type Cl-Brener (lane 1), the single knockout L14 (lane 2), and the double knockout L16 (lane 3) was hybridized to a LYT1 probe. A single 1.8-kb band is observable in lanes 1 and 2.
LYT1 deficiency reduces the efficiency of in vitro infection.
Since LYT1 appears to have hemolytic activity and LYT1 was originally cloned based on its cross-reactivity to anti-C9 antibodies, we aimed to determine whether LYT1 plays a role in host cell infection. Monolayers of NIH 3T3 cells were infected with mid-log-phase epimastigotes derived from strain Cl-Brener and each of the LYT1 mutant lines. When using these lines, we typically used mid-log-phase epimastigotes rather than metacyclic trypomastigotes to initiate infections because of the inefficiency of in vitro metacyclogenesis in the wild-type parasite line.
Subsequently, secondary-infection experiments were carried out using trypomastigotes isolated following the first infections, as described in Material and Methods. The results of these experiments are shown in Fig. 5 and indicate that each LYT1 mutant line exhibited significantly reduced infectivity. In the first infection experiments, wild-type parasites infected 100% of the cells at day 16 while each of the mutant parasite lines exhibited barely detectable infectivity (Fig. 5A). By the time wild-type parasites reached 100% infectivity, the infection rates for L14 and L16 were approximately 9 and 14%, respectively. Consistent with these results, the secondary-infection experiments, using a multiplicity of infection of one, also showed that the LYT1 mutant lines exhibited significantly reduced infectivity. In this case, by the time wild-type parasites reached the peak of infection (60%) at day 14, L14 and L16 both exhibited reduced infectivity, with rates approximately half (27%) and 13 times less (4%), respectively (Fig. 5B). A similar gene dosage effect was observed when amastigotes and trypomastigotes were quantified. Figure 5C shows that the wild-type strain, Cl-Brener, reached 17 × 106 parasites by day 14, while the mutant lines reached approximately 11 × 106 parasites and 4 × 106 parasites (L14 and L16, respectively). Trypomastigotes counted at different points after infection showed a similar pattern.
FIG. 5.

Single and double LYT1 allele replacements decrease the in vitro infectivity of T. cruzi. NIH 3T3 cells were infected with wild-type Cl-Brener (■), L14 (●), or L16 (▵), parasites and the infections were monitored as described in Material and Methods. The results shown are the averages of three independent experiments. (A and B) Percent infected cells 48 h after infection with epimastigotes and 2 h after infection with trypomastigotes, respectively; (C) Number of parasites 2 h following infection with trypomastigotes.
Reconstitution of infectivity in LYT1 null parasites.
The additive effect of the LYT1 deletions in L14 (single deletion) and L16 (double deletion) is consistent with the notion that the reduced-infectivity phenotype observed in these lines is a result of the LYT1 deletions. To provide further proof of the association between LYT1 and infection rates, we reconstituted the infectivity of the null mutant parasite by reintroducing the native allele LYT1a. Reconstitution was carried out using a BsiWI restriction fragment that includes the native LYT1a allele and approximately 600 bp of flanking regions. Purified DNA fragment (100 μg) was used to transform wild-type Cl-Brener epimastigotes, and 100 or 200 μg of purified DNA fragment was used to transform L16 epimastigotes. As a negative control electroporation was also carried out in the absence of added DNA. Infection of NIH 3T3 monolayers was monitored for 21 days. The results are shown in Fig. 6. Because of the killing incurred during electroporation (70 to 80% death), electroporated wild-type parasites exhibited reduced infectivity compared to nonelectroporated parasites (compare approximately 15% infected cells in this experiment at day 19 with 100% on day 16 [Fig. 5A]). L16 electroporated with buffer alone exhibited no increase in infectivity over previous experiments (2% at day 19). In contrast, L16 electroporated with 100 or 200 μg of LYT1a restriction fragment showed significantly enhanced infectivity (about 20% at day 19) comparable with the wild-type strain, Cl-Brener, plus DNA (20% at day 19).
FIG. 6.

Reconstitution of infectivity in LYT1 null mutant by transient LYT1a allele transformation. Wild-type and LYT1 double-knockout epimastigotes were electroporated with LYT1a coding sequence. Transformed parasites were used to infect NIH 3T3 cells, and the infection was monitored at different times as described in Material and Methods. The percentage of infected cells was determined by microscopic observation by comparing the number of cells containing parasites to total cells. Symbols: ■, wild type plus TE; ●, wild type plus 100 μg of LYT1a DNA; ▵, L16 plus TE; ◊, L16 plus 100 μg of LYT1a DNA; ▿, L16 plus 200 μg of LYT1a DNA. (A) Representative experiment of two different experiments; (B) Average results from two different experiments, determined at point when the maximum levels of infection were reached (19 days).
LYT1 deficiencies accelerate in vitro stage transition.
To successfully complete an infection cycle, parasites must efficiently transition through the different developmental stages. Consequently, it might be possible that the reduced infectivity of LYT1-deficient parasites is an indirect effect of the mutant parasite's inability to complete the life cycle. To determine how the LYT1 mutation effects the parasite's ability to complete the developmental cycle, an in vitro stage transition experiment was carried out. The results presented in Fig. 7A show that, following transfer to DMEM medium, the deficient lines rapidly converted to extracellular amastigote-like parasites (20) characterized by a nearly spherical shape and lack of visible flagella and by the fact that they were quantitatively precipitated by the monoclonal antibody 2C2B6, specific for the Ssp-4 surface antigen of amastigotes. On day 9, when wild-type parasites reach 5% conversion, the mutant lines exhibited approximately 68% (L14) and 65% (L16) infectivity. Similar results were obtained when epimastigotes were allowed to progress to stationary phase in the original LIT medium (Fig. 7B). The deficient lines rapidly converted to metacyclic trypomastigotes: 32% for L14 and 18% for L16 at day 13. In contrast, less than 3% of the wild-type epimastigotes had converted to this form. Over the time course of the experiment the overall titer of each parasite culture continued to increase, eliminating the possibility that LYT1 mutant epimastigotes were less viable in DMEM than wild-type epimastigotes. Why an increase in metacyclic trypomastigote titer was not seen in DMEM is unclear. It is possible that under our experimental conditions amastigotes developed directly from epimastigotes or the parasite proceeded through the metacyclic trypomastigote stage very rapidly.
FIG. 7.

LYT1 gene deletion accelerates in vitro stage transition. Transition efficiency of wild-type (■), LYT1 single-knockout (●), and LYT1 double-knockout (▵) epimastigotes was determined by their ability to convert to extracellular amastigote-like cells in DMEM (A) or metacyclic trypomastigotes in LIT medium (B). The results plotted are the means ± the standard deviations of three different experiments.
LYT1 deficiency reduces hemolytic activity.
The marked reduction in host cell infection ability by LYT1 null mutants could be predicted as a consequence of decreased parasite-mediated lytic activity. To test this possibility, the effect of LYT1 mutations on the hemolytic activity of the parasite was assessed. As a first step the hemolytic activity of the wild-type parasite during the three developmental stages of the parasite was tested under acid and neutral conditions. The results showed that after 6 h of incubation no hemolytic activity was obtained at a neutral pH in any of the three developmental stages, while a strong lysis activity was observed when the red cells were incubated in the presence of amastigotes at an acid pH (Fig. 8). Subsequently, the hemolytic activities of wild-type and single- and double-knockout parasites were evaluated at the time point at which wild-type parasites lysed 50% of the red blood cells at pH 5.4 (defined as 100% lytic activity). The results presented in Fig. 9 show that each mutant line expressed less lytic activity than wild-type parasites. At the time that wild-type amastigotes had lysed 100% of the erythrocytes, L14 and L16 amastigotes had lysed only 41 and 36% of the red blood cells, respectively (Fig. 9A). Although lytic activity has been primarily associated with amastigotes, epimastigotes have also been shown to express low levels of lytic activity (1). Consequently, the lytic activity of wild-type and LYT1 mutant parasites was assessed, with the result that in each case mutant epimastigotes expressed less lytic activity than wild-type epimastigotes did (Fig. 9B). The mutant phenotype was, however, less striking in epimastigotes than in amastigotes (Fig. 8). At the time that wild-type epimastigotes reached 100% hemolysis, L14 (single knockout) exhibited 84% lytic activity and L16 (double knockout) exhibited 58% lytic activity. Similar to the results of the in vitro infection experiments, these results suggest a possible gene dosage effect since the single knockout retains more activity that the null line.
FIG. 8.

Hemolytic activity of the various stages of T. cruzi in neutral and acid pH. The lysis of erythrocytes by metacyclic trypomastigotes (T), epimastigotes (E), and amastigotes (A) was determined in acid (filled bars) or neutral (empty bars) conditions at 37°C for 6 h. The spontaneous lysis (SL) and 100% lysis (C) of erythrocytes in the two different pH buffers and water, respectively, under the same sample conditions, were determined. The results shown are averages from three independent experiments.
FIG. 9.

LYT1 single- and double-knockout mutants exhibit reduced hemolytic activity at pH 5.4. The lysis of erythrocytes by amastigotes (A) and epimastigotes (B) was determined at 37°C after various periods of time, and the results were normalized by defining 100% hemolysis as the time point at which wild-type parasites lysed 50% of the red blood cells. The average percentage of lysis of three different experiments was calculated from the released hemoglobin where the spontaneous lysis has been substracted, using wild-type (WT), LYT1 single-knockout (L14), and LYT1 double-knockout (L16) parasites.
DISCUSSION
T. cruzi infection of mammalian cells is a complex event involving multiple host and parasite proteins. To date, only three parasite proteins, TC-TOX (1, 2, 5), oligopeptidase B (8), and TcMIP (17), have been convincingly shown to be involved in host cell invasion. The present study focused on the LYT1 gene that encodes a protein that, based on genetic evidence, is required for efficient infection since deletions of the gene result in significantly decreased infectivity in vitro. The LYT1 gene was originally cloned from a cDNA expression library on the basis of cross-reactivity to antibodies specific for the C9 component of the complement cascade. Subsequent computer analysis failed, however, to detect homology to C9 and in fact failed to detect homology to any known protein or possible translation product. The only clues pointing to a possible function consisted of circumstantial evidence suggesting the protein could be, at least structurally, related to TC-TOX, a secreted protein exhibiting hemolytic activity expressed in T. cruzi (1). This evidence consisted of the fact that the two proteins are of similar size and both cross-reacted with antibodies directed towards C9. The notion of a functional relationship between the two proteins was strengthened by the finding of hemolytic activity in extracts from yeast transformed to express LYT1 and by the analysis of mutant parasites which demonstrated that parasites carrying LYT1 deficiencies exhibited significantly less TC-TOX-associated hemolytic activity than wild-type parasites. Moreover, the kinetics of the lytic activity we attribute to LYT1 were indistinguishable from those reported for TC-TOX (1), which is consistent with the idea that the molecules are at the very least involved in the same pathway.
The central role of LYT1 in the parasite life cycle was illustrated by the three phenotypes associated with LYT1 deficiencies. Each of these phenotypes, decreased infectivity, decreased hemolysis, and enhanced development, also displayed a gene dosage effect, further supporting the involvement of LYT1. The involvement of LYT1 in infection was formally demonstrated by the LYT1-dependent reconstitution of infectivity in null mutants. Whether the decreased infectivity of LYT1-deficient parasites was directly related to their decreased hemolytic activity remains uncertain.
The results presented here also demonstrate that the reduced infectivity of the LYT1-deficient parasites was not a consequence of an inability to complete the life cycle, since the mutant epimastigotes converted to metacyclic trypomastigotes and amastigotes more efficiently than wild-type parasites. The accelerated stage conversion associated with LYT1 deficiencies is consistent with a common model for both amastigote and metacyclic trypomastigote development in which the LYT1 gene product acts as a suppressor of stage transition in epimastigotes. This would effectively maintain the epimastigote gene expression profile while suppressing expression of proteins specific for either of the other two stages. Inhibition of LYT1 expression, either through normal processes or mutation, would relieve suppression, permitting expression of either amastigote- or metacyclic trypomastigote-specific proteins, depending on the culture media.
The diverse phenotypes associated with LYT1 deficiencies raise the question of how a single protein could be involved in processes that are both extracellular (e.g., hemolysis) and intracellular (e.g., regulation of stage transition). The simplest explanation would be one that places LYT1 in a cell-signaling pathway common to both processes. An alternative possibility is that different forms of the protein are expressed and are responsible for the different phenotypes. Support for this later possibility comes from preliminary experiments demonstrating that, as a result of alternative trans splicing, derivatives of LYT1 may be expressed (R. Manning-Cela and J. Swindle, unpublished results). One derivative would consist of the complete 552-amino-acid protein encoded by the LYT1 open reading frame containing a computer-predicted 15-amino-acid leader peptide. The second would carry an amino-terminal truncation of 29 amino acids eliminating the putative secretion signal. Therefore, it is possible that two forms of the protein are produced, with the secreted form involved in pore-forming activity and the cytosolic derivative responsible for suppression of stage transition.
ACKNOWLEDGMENTS
This work was supported by USPHS grant A126578 awarded to J.S. and grants PM95-0100 and PB98-0479 awarded to A.G. by the Spanish Ministry of Education and Culture. R.M.-C. is the recipient of a Fogarty Postdoctoral Fellowship (1F05TW05274-01). A.C. and E.G.-R. receive doctoral fellowships from FPI/MEC (Spain).
Rebeca Manning-Cela and Arantxa Cortés each contributed significantly to the published work.
We thank Norma Andrews (Yale University) for advice and for the generous gift of 2C2B6 antibodies and Juan Jiménez (Universidad de Málaga) for guidance during the yeast expression experiments.
REFERENCES
- 1.Andrews N A, Whitlow M B. Secretion by Trypanosoma cruzi of a hemolysin at low pH. Mol Biochem Parasitol. 1989;33:249–256. doi: 10.1016/0166-6851(89)90086-8. [DOI] [PubMed] [Google Scholar]
- 2.Andrews N W. The acid-active hemolysin of Trypanosoma cruzi. Exp Parasitol. 1990;71:241–244. doi: 10.1016/0014-4894(90)90027-a. [DOI] [PubMed] [Google Scholar]
- 3.Andrews N W. From lysosomes into the cytosol: the intracellular pathway of Trypanosoma cruzi. Braz J Med Biol Res. 1994;27:471–475. [PubMed] [Google Scholar]
- 4.Andrews N W. Living dangerously: how Trypanosoma cruzi uses lysosomes to get inside host cells, and then escapes into the cytoplasm. Biol Res. 1993;26:65–67. [PubMed] [Google Scholar]
- 5.Andrews N W, Abrams C K, Slatin S L, Griffiths G. A T. cruzi-secreted protein immunologically related to the complement component C9: evidence for membrane pore-forming activity at low pH. Cell. 1990;61:1277–1287. doi: 10.1016/0092-8674(90)90692-8. [DOI] [PubMed] [Google Scholar]
- 6.Andrews N W, Hong K S, Robbins E S, Nussenzweig V. Stage-specific surface antigens expressed during the morphogenesis of vertebrate forms of Trypanosoma cruzi. Exp Parasitol. 1987;64:474–484. doi: 10.1016/0014-4894(87)90062-2. [DOI] [PubMed] [Google Scholar]
- 7.Burleigh B A, Andrews N W. The mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu Rev Microbiol. 1995;49:175–200. doi: 10.1146/annurev.mi.49.100195.001135. [DOI] [PubMed] [Google Scholar]
- 8.Caler E V, Vaena de Avalos S, Haynes P A, Andrews N W, Burleigh B A. Oligopeptidase B-dependent signaling mediates host cell invasion by Trypanosoma cruzi. EMBO J. 1998;17:4975–4986. doi: 10.1093/emboj/17.17.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung S H, Gillespie R D, Swindle J. Analyzing expression of the calmodulin and ubiquitin-fusion genes of Trypanosoma cruzi using simultaneous, independent dual gene replacements. Mol Biochem Parasitol. 1994;63:95–107. doi: 10.1016/0166-6851(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 10.Gillespie R D, Ajioka J, Swindle J. Using simultaneous, tandem gene replacements to study expression of the multicopy ubiquitin-fusion (FUS) gene family of Trypanosoma cruzi. Mol Biochem Parasitol. 1993;60:281–292. doi: 10.1016/0166-6851(93)90139-o. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez A, Lerner T J, Huecas M, Sosa-Pineda B, Noqueira N, Lizardi P M. Apparent generation of a segmented mRNA from two separate tandem gene families in Trypanosoma cruzi. Nucleic Acids Res. 1985;13:5789–5804. doi: 10.1093/nar/13.16.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez A, Rosales J L, Ley V, Diaz C. Cloning and characterization of a gene coding for a protein (KAP) associated with the kinetoplast of epimastigotes and amastigotes of Trypanosoma cruzi. Mol Biochem Parasitol. 1990;40:233–243. doi: 10.1016/0166-6851(90)90045-n. [DOI] [PubMed] [Google Scholar]
- 13.Hariharan S, Ajioka J, Swindle J. Stable transformation of Trypanosoma cruzi: inactivation of the PUB12.5 polyubiquitin gene by targeted gene disruption. Mol Biochem Parasitol. 1993;57:15–30. doi: 10.1016/0166-6851(93)90240-x. [DOI] [PubMed] [Google Scholar]
- 14.La F A C, Buckner F, Swindle J, Ajioka J, Barrett L, Van V W C. Engineering cytokine secretion from Trypanosoma cruzi. Mem Inst Oswaldo Cruz. 1994;89:650–651. doi: 10.1590/s0074-02761994000400025. [DOI] [PubMed] [Google Scholar]
- 15.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 16.Maniatis T, Fritsch E F, Sambrook J. Molecular cloning: a laboratory manual. 1st ed. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1982. [Google Scholar]
- 17.Moro A, Ruiz-Cabello F, Fernandez-Cano A, Stock R P, Gonzalez A. Secretion by Trypanosoma cruzi of a peptidyl-prolyl cis-trans isomerase involved in cell infection. EMBO J. 1995;14:2483–2490. doi: 10.1002/j.1460-2075.1995.tb07245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez A, Samoff E, Rioult M G, Chung A, Andrews N W. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol. 1996;134:349–362. doi: 10.1083/jcb.134.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teixeira M R S, Otsu K, Hill K L, Kirchoff L V, Donelson J E. Expression of a marker for intracellular Trypanosoma cruzi amastigotes in extracellular spheromastigotes. Mol Biochem Parasitol. 1999;98:265–270. doi: 10.1016/s0166-6851(98)00158-3. [DOI] [PubMed] [Google Scholar]
- 21.Thomas M C, Gonzalez A. A transformation vector for stage-specific expression of heterologous genes in Trypanosoma cruzi epimastigotes. Parasitol Res. 1997;83:151–156. doi: 10.1007/s004360050225. [DOI] [PubMed] [Google Scholar]