

Abstract
BACKGROUND:
Endothelial dysfunction in sepsis is a pathophysiological feature of septic organ failure. Endothelial cells (ECs) exhibit specific metabolic traits and release metabolites to adapt to the septic state in the blood to maintain vascular homeostasis.
METHODS:
Web of Science and PubMed were searched from inception to October 1, 2022. The search was limited to the English language only. Two reviewers independently identified studies related to EC metabolism in sepsis. The exclusion criteria were duplicate articles according to multiple search criteria.
RESULTS:
Sixty articles were included, and most of them were cell and animal studies. These studies reported the role of glycolysis, oxidative phosphorylation, fatty acid metabolism, and amino acid metabolism in EC homeostasis. including glycolysis, oxidative phosphorylation, fatty acid metabolism and amino acid metabolism. However, dysregulation of EC metabolism can contribute to sepsis progression.
CONCLUSION:
There are few clinical studies on EC metabolism in sepsis. Related research mainly focuses on basic research, but some scientific problems have also been clarified. Therefore, this review may provide an overall comprehension and novel aspects of EC metabolism in sepsis.
Keywords: Sepsis, Endothelium, Metabolism, Glycolysis, Oxidative phosphorylation, Fatty acid metabolism, Amino acid metabolism
INTRODUCTION
Sepsis, defined as life-threatening organ dysfunction caused by dysregulation of the body’s response to infection, is one of the most fatal and epidemic syndromes in intensive care units and emergency clinics.[1,2] Over 48.9 million people suffered from sepsis, of which 11 million died from 1990 to 2017. The enormous number of patients and high morbidity and mortality make sepsis a malignant syndrome that seriously threatens public health.[3] Sepsis is characterized as an urgent disease that has rapid progression and a high disability rate. The global burden of sepsis has been grossly underestimated over the past few decades. Elderly individuals with hypoimmunity are vulnerable to sepsis.
Endothelial dysfunction is a key step in sepsis-induced organ dysfunction. Vascular leakage caused by endothelial dysfunction can be regarded as the pathophysiological basis of sepsis progression, and these pathological changes further trigger systemic organ damage.[4] Therefore, the reconstruction of endothelial function is a major direction for the treatment of sepsis and septic organ dysfunction.
Endothelial cells (ECs) are a unique multifunctional cell type with critical basal and inducible metabolic and synthetic functions. Many pathological states, such as sepsis, are hallmarks of endothelial activation and dysfunction. The pathogenesis of sepsis is a result of a complex network of events, including the interaction of inflammatory, immune suppression, endocrine, metabolic dysregulation, and coagulation systems.
Recently, more studies have been devoted to exploring the metabolic changes of ECs in sepsis and related pathological dysfunctions, hoping to provide new therapeutic strategies for sepsis based on endothelial metabolism. This review aims to provide insights into different metabolic aspects of ECs in sepsis and to discuss current and unexplored EC metabolism-centric therapeutic avenues.
METHODS
We systematically performed a search of Web of Science and PubMed from inception to October 1, 2022. The search was limited to the English language only. We combined title/abstract keywords and MeSH, such as “endothelial cell”, “endothelium”, “sepsis”, “sepsis shock”, “metabolism”, “glycolysis”, “fatty acid metabolism”, “amino acid”, and key enzyme in metabolism pathways to identify all studies related to endothelial dysfunction in sepsis.
Two authors independently identified articles for inclusion based on titles and abstracts, browsed the full texts, and reviewed the bibliographies of each article to identify additional studies.Duplicate articles according to multiple search criteria were excluded.
RESULTS
Endothelial cell metabolism in sepsis
ECs display a remarkable capability to switch rapidly from a quiescent state to a highly inflammatory and activated state during sepsis. Exogenous pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs) in sepsis are critical triggers to activate ECs. The activation of pathogen recognition receptors would drive ECs to reprogram themselves into a proinflammatory phenotype, manifested by increased secretion of cytokines, chemokines, and procoagulant factors as well as the increased expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin. These processes in ECs aim to recruit leukocytes, eliminate bacteria, and limit pathogen spread.[5,6] However, hyperactivated cellular reactions may lead to damage to blood flow and tissue hypoperfusion, ultimately resulting in organ failure in patients with sepsis.[7]
ECs in sepsis have the following pathophysiological features: glycocalyx damage, impairment of adherent junctions, capillary leakage, excessive intercellular chemokine production, and recruitment of leukocytes, all of which could alter the metabolic profile of ECs.[4] Metabolomics is widely utilized to detect changes in metabolic reprogramming in septic patients. Liu et al[8] collected serum samples from surviving and non-surviving septic shock adult patients before clinical intervention at admission. The serum samples were analyzed by liquid chromatography-mass spectrometry to determine metabolic profiles in sepsis. Citrulline, carnitine, batanine, valine, leucine, and isoleucine were defined as primary discriminators differentiating the survivors from the non-survivors at the early stage of septic shock. Thus, the authors emphasized that the levels of amino acids or fatty acids (FAs) can differentiate septic survivors from non-survivors. However, this study focused on serum metabolites but not on ECs. Toral et al[9], using a proteomics-based metabolic model, found that as the vascular network fully formed, fatty acid oxidation (FAO) increased and glycolysis decreased in ECs, and inhibition of carnitine palmitoyl transferase 1A, which transports FA into mitochondria for oxidation, increased EC permeability. In another study, McGarrity et al[10] established an EC-specific metabolic model and found that the increase in permeability and glycocalyx shedding in ECs after inflammatory stimulation was associated with glycan production and FA metabolism.
Above all, these studies indicated that ECs may change their metabolism to adapt to the septic state. Although EC metabolism has been studied for decades, its role in sepsis has been gradually strengthened during the last few years. Here, we will focus on EC metabolism in sepsis development. General overview of metabolic pathways in physiological ECs and septic ECs have been showed in Figure 1.
Figure 1.
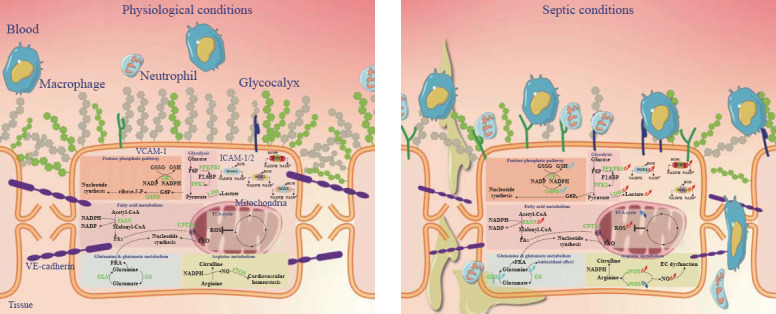
General overview of metabolic pathways in physiological ECs (left) and septic ECs (right).
Glycolysis is enhanced in ECs during sepsis
Although ECs readily reach oxygen in the blood, they use glycolytic flux as the predominant way to generate cellular adenosine triphosphate (ATP).[11,12] Accumulating evidence indicates that almost 98% of glucose is metabolized into lactate, while little is oxidized by oxidative phosphorylation in angiogenic ECs.[13] In the pathogenesis of inflammation, enhanced glycolysis or reduced oxidative phosphorylation occurs in ECs;[14] and high levels of serum lactate, the final product of glycolysis, have been recognized as a critical biomarker for sepsis prognosis.[15] Furthermore, it results in a vicious cycle, whereby lactate-induced extracellular signal-regulated kinase 2 (ERK2) phosphorylation could promote ERK2 disassociation from vascular endothelial-cadherin (VE-cadherin), thus destroying the vascular endothelial cadherin complex on the surface of ECs and leading to increased vascular permeability.[16] In addition, lactate reduces the expression of heat shock protein, which is required for VE-cadherin-mediated tight junctions in ECs. Inhibition of G protein-coupled receptor 81, a specific receptor for lactate, attenuates vascular permeability and reverses the inhibited expression of heat shock protein in septic mice.[17]
Recent evidence suggests that 6-phosphofructo-2-kinase (PFKFB3), a key glycolytic activator, is significantly upregulated in septic lungs. Moreover, endothelial-specific PFKFB3 deficiency or inhibition can diminish mortality in lipopolysaccharide (LPS)-treated mice owing to the reduction of neutrophil infiltration and endothelial permeability.[18] PFKFB3 can upregulate fructose-2,6-bisphosphate (F-2,6-P2) in ECs under septic conditions. Furthermore, F-2,6-P2 allosterically activates 6-phosphofructo-1-kinase (PFK1), the pyruvate dehydrogenase (PDC) second rate-limiting enzyme in glycolysis.[12] Similarly, septic mice treated with an inhibitor of hexokinase, another rate-limiting enzyme in glycolysis, show improved survival outcomes.[19] Briefly, restriction of aberrant glycolytic flux in ECs can maintain endothelial function during sepsis. PDC catalyzes pyruvate into acetyl-CoA to promote the tricarboxylic acid cycle (TCA). Mao et al[20] found that the activated PDC complex reversed lipopolysaccharide (LPS)-stimulated glycolysis flux, leading to accelerated aerobic oxidation and decreased lactate production. Interestingly, the activated PDC complex reduces high permeability and improves endothelial function of vascular ECs. In the last step of glycolysis, pyruvate is metabolized into lactic acid by lactate dehydrogenase (LDH), a biomarker to evaluate the severity of sepsis. The accumulation of lactate can lead to EC hyper-permeability and dysfunction.[21] Glycolysis is enhanced in septic ECs, and its inhibition can maintain EC integrity and reduce EC polarization to a proinflammatory phenotype.
Pentose phosphate pathway (PPP) and antioxidation in sepsis
Glucose-6-phosphate dehydrogenase (G6PD or G6PDH) and transketolase (TK) are two rate-limiting enzymes in the PPP. ECs show low migratory ability and viability if either or both enzymes are inhibited.[22,23] The products in the PPP are involved in lipid or nucleotide synthesis and in the maintenance of intracellular oxidation-reduction equilibrium.[11] Spolarics and his team[24,25] found that, unlike hepatic parenchymal cells, Kupffer cells and hepatic ECs significantly upregulate G6PD activity when stimulated with LPS. Therefore, the PPP might exert an important regulatory effect on ECs to produce adaptive responses during infections.
The PPP-derived nicotinamide adenine dinucleotide phosphate (NADPH) is not only a vital cofactor for endothelial nitric oxide synthase (eNOS)[26] but is also essential for converting oxidized glutathione disulfide (GSSG) to reduce glutathione (GSH). When NADPH and GSH are in the normal physiological range, ECs can retain a benign oxidative defense response.[27] In general, the PPP provides antioxidative precursors for ECs and plays a vital role in subsequent metabolic reactions. In addition, human vasculature expresses four isoforms of NADPH oxidases (NADPH oxidase 1 [NOX1], NOX2, NOX4, and NOS5) in ECs, and these enzymes catalyze NADPH into NADP+ and produce reactive oxygen species (ROS). Under physiological conditions, NOX4 has beneficial effects and maintains endothelial function in ECs.[28] However, NOX4 is expressed rapidly at first, then NOX1 and NOX2 are upregulated, but the increase of them just lasts for 2 h in septic mice. Calcium/calmodulin-dependent protein kinase II (CaMKII)/ERK/myosin light-chain kinase (MLCK) pathway activation in ECs leads to the consequent loss of tight junction proteins (zonula occludens-1 [ZO-1] and occludin) and results in barrier dysfunction. NOX4 knockdown attenuates the activation of the CaMK/ERK/MLCK pathway in ECs, which can protect ECs from the oxidative stress response and maintain the integrity of the EC barrier.[29] In an independent study, NOX1 knockdown had similar effects.[30] The migratory process is mediated by the generation of ROS through the activation of NOX,[31] and polymorphonuclear leukocyte recruitment and transendothelial migration also rely on endothelial NOX activation.[32] Moreover, NOX-induced oxidative stress can regulate calpain-dependent caspase activity and cause apoptosis in ECs during sepsis.[33] An intensive study showed that NOX2, which decreases nuclear factor-kappa B (NF-κB) transcription of toll-like receptor 4 (TLR4) in ECs, can control the inflammatory response and hypotension.[34] Thus, some therapies targeting NOX in ECs may be regarded as new strategies for sepsis.[35,36]
Another side branch of glycolysis, the hexosamine biosynthesis pathway (HBP), uses glycolytic intermediate fructose-6-phosphate (F6P) to produce UDP-N-acetylglucosamine (UDP-GlcNAc) for protein N- and O-glycosylation. Therefore, the HBP was suggested to function as a nutrient sensor that modulates EC function, since the glycosylation status co-determines the functionality of endothelial nitric oxide synthase (eNOS). However, the study of HBP in septic ECs remains a relatively unworked area and is underestimated at present. Therefore, HBP in septic ECs is worthy of further investigation.
Fatty acid metabolism affects endothelial function in sepsis
In addition to glucose, FAs represent another fuel source for ECs. Due to their hydrophobicity, intracellular FAs are either complexed with FA-binding proteins or are free for transportation to the destination. FAs are transported into ECs by passive diffusion or facilitated diffusion, whereby FAs are loaded on fatty acid translocase (FAD)/cluster of differentiation 36 (CD36). However, very long-chain FAs are preferentially transported by fatty acid transport protein (FATP).[37]
Interestingly, exogenous short-chain fatty acids (SCFAs) appear to have a regulatory function in ECs. SCFAs can inhibit histone deacetylases or activate G-protein coupled receptor 109A. Free FA receptor type 2 plays a role in modulating immune and inflammatory responses.[38] Voltolini et al[39] found that sodium propionate (an SCFA) could reduce the secretion of IL-8 and IL-6 upon LPS stimulation in human myometrium vascular and fetal membrane ECs. Additionally, some SCFAs can reduce the expression of VCAM-1 and attenuate the recruitment of monocytes and lymphocytes in ECs.[40] Polyunsaturated fatty acids (PUFAs) induce ROS synthesis; treatment with PUFAs can reduce the number of ECs positive for ICAM-1.[41] Metabolites of FAO, such as arachidonic acid and ceramide, affect ECs in several ways to maintain the integrity of ECs.[42-44] SCFAs and other FAs have unique functions in ECs.
FAO is a definitive metabolic pathway that provides ATP to ECs in addition to glycolysis.[45] FAO is also an essential fuel source for deoxy-ribonucleoside triphosphate (dNTP) synthesis in ECs. Quiescent ECs sustain redox homeostasis through NADPH regeneration. Therefore, upregulating FAO in ECs is presumably modest for energy production.[46] Carnitine palmitoyltransferase-1 is required for the transportation of FAs through the mitochondrial membrane, and its deficiency in ECs can induce endothelial dysfunction. When carnitine palmitoyltransferase-1 is acutely inhibited, calcium-dependent signaling in ECs is inhibited. As a result, the permeability of ECs increases.[9] Fatty acid synthase mediates lipid synthesis and is mainly involved in the synthesis of saturated long-chain FAs. The expression of fatty acid synthase increases in septic mice, and its downregulation reduces EC apoptosis in septic lung tissues.[47] However, the authors did not elucidate the specific metabolic changes and the mechanisms by which fatty acid synthase attenuates apoptosis in ECs. Research on endothelial sepsis in the light of FAO is scant from the perspective of metabolic pathways, and this warrants attention.
Oxidative phosphorylation (OXPHOS) reduces EC oxidation in sepsis
Although modestly contributing to ATP generation, OXPHOS plays an antioxidant role in ECs. Mitochondrial antioxidants can alleviate nitrosative and oxidative stresses in ECs under LPS stimulation.[48] Quiescent ECs generate low levels of ROS from OXPHOS to maintain normal cellular physiological function through autophagy. Therefore, the OXPHOS-autophagy system in ECs provides ROS resistance, thus ameliorating LPS-induced damage.[49] Sun et al[50] found that phosphocreatine, an exogenous energy substance, enhanced OXPHOS levels in ECs, suppressed the permeability transition of mitochondria, reduced the release of cytochrome C and ROS, and protected against EC apoptosis under LPS stimulation. Thus, OXPHOS shows an antioxidative function in ECs during infection.
Glutamine metabolism in ECs during sepsis
The non-essential amino acid glutamine is known to contribute both its carbons and nitrogens to EC metabolism. Intracellular glutamine can be catalyzed into glutamate by glutaminases (GLS); conversely, glutamine synthetase can catalyze glutamate back to glutamine in ECs. Glutaminolysis is important for energy production pathways, and inhibition of GLS can result in premature senescence in ECs.[51] Moreover, the relative importance of glutaminolysis in vessel sprouting and regulation of blood pressure has been demonstrated.[11,52] In addition, glutamine has been shown to reduce cardiometabolic risk in the circulatory system by inducing the expression of heat shock protein, heme oxygenase-1, and glutathione.[53] Along with enhanced GLS activity, accelerated intracellular glutamine biosynthesis is observed in rat pulmonary microvascular ECs during sepsis, which might cause an increase in glutamine in the lungs during sepsis.[54,55]
Arginine metabolism is a key to nitric oxide synthesis
Arginine (Arg) is another widely studied amino acid in ECs and can be either hydrolyzed by arginases to yield urea and ornithine or converted by endothelial nitric oxide synthase (eNOS) to citrulline and nitric oxide (NO), an endogenous gaseous signaling molecule that has a wide variety of biological properties that maintain vascular homeostasis.[56] The decrease in the activity of arginase may significantly enhance the production of eNOS-dependent NO.[57] There are three subtypes of NOS: eNOS, inducible NOS (iNOS), and neuronal NOS (nNOS). Particularly, quiescent ECs cannot express iNOS unless ECs are exposed to stimuli of inflammation and pathogens.[58] iNOS-derived NO is implicated in immune symptoms and is a cause of fatality during septic shock, whereas nNOS-derived NO is involved in neurotransmitter and blood pressure control.[26] Since NO plays an important role in ECs, eNOS- and iNOS-mediated production of NO make arginine metabolism a research hotspot.
Arg is a non-essential amino acid in the human body but promotes a semi-essential function during sepsis. Intravenous injection of Arg after cecal ligation and puncture (CLP) can increase the release of C-X-C motif chemokine Ligand 12 (CXCL-12), vascular endothelial growth factor (VEGF), and NO, which mobilize proangiogenic cells in lung tissues. Arg can maintain homeostasis of the angiopoietin/Tie-2 signaling pathway to sustain endothelial barrier functions.[59] Arginase plays an important role in the regulation of NO production in immune responses or the cardiovascular system, and arginase-1 is characterized as a protective factor in ECs. In a previous study, Wijnands et al[60] demonstrated that endothelial- and macrophage-specific deletion of arginase, on the one hand, shows increased plasma Arg and NO production in septic mice and, on the other hand, leads to upregulated iNOS activity and proinflammatory response in macrophages. Thus, arginase-deficient mice exhibit more aggravated microcirculation damage after LPS stimulation. Although this study does not allow differentiation between the effects of arginase-1 depletion in macrophages and vascular ECs in vivo, it still implies that a balance between the activities of arginase-1 and iNOS can be the key to developing a therapeutic target against endothelial dysfunction during sepsis. eNOS activity in ECs is also an important factor in sepsis. One mechanism for eNOS uncoupling related to asymmetric dimethylarginine (ADMA) is that, as an arginine analog, it binds to the catalytic site of eNOS and thereby competes with Arg to antagonize NO production. Inhibition of dimethylarginine dimethylaminohydrolase 1, a hydrolase for ADMA, can reduce NO production in ECs. The endogenous accumulation of ADMA provides an alternative therapeutic mechanism to regulate NO overproduction and vascular hemodynamics in septic shock.
Future perspectives
The circulating EC count in non-survivors of sepsis or septic shock is greater than that in survivors of sepsis, and thus, it can serve as a biomarker to predict the prognosis of sepsis, especially for elderly individuals.[61] In conclusion, herein, we reviewed the different metabolic changes during sepsis or infection. Pulmonary microvascular ECs reveal high glycolysis and rapid growth, while pulmonary arterial ECs show high oxidation and slower growth.[62] ECs in different organs or different parts of vessels may exhibit differential metabolism. The heterogeneity of ECs poses a challenge in metabolic research. This has not been confirmed by research, and this review can provide directions for future investigations.
A deeper knowledge of EC metabolism and changes between different phenotypes of ECs in sepsis is necessary in this emerging field. Explicitly addressing this scientific problem may provide the basis for developing novel therapeutic strategies.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China (82272236) and Key Emergency Medical Disciplines and Specialities Program of Guangzhou (2021-2023).
Ethical approval: Not needed.
Conflicts of interest: The authors have no conflicts of interest to disclose.
Contributors: JXW conceived the study concept and design and was involved in the drafting and critical revision of the manuscript. All authors contributed to the design and interpretation of the study and to further drafts.
REFERENCES
- 1.Duan LW, Qu JL, Wan J, Xu YH, Shan Y, Wu LX, et al. Effects of viral infection and microbial diversity on patients with sepsis:A retrospective study based on metagenomic next-generation sequencing. World J Emerg Med. 2021;12(1):29–35. doi: 10.5847/wjem.j.1920-8642.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin J, Chen Y, Huang JL, Yan L, Kuang ZS, Xue MM, Sun S, Xiang H, Hu YY, Dong ZM, Tong CY, Bai CX, Song ZJ. Prognosis-related classification and dynamic monitoring of immune status in patients with sepsis:A prospective observational study. World J Emerg Med. 2021;12(3):185–191. doi: 10.5847/wjem.j.1920-8642.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, |y1990-2017:analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–11. doi: 10.1016/S0140-6736(19)32989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361–70. doi: 10.1164/rccm.201910-1911TR. [DOI] [PubMed] [Google Scholar]
- 5.Colbert JF, Schmidt EP. Endothelial and microcirculatory function and dysfunction in sepsis. Clin Chest Med. 2016;37(2):263–75. doi: 10.1016/j.ccm.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fry DE. Sepsis, systemic inflammatory response, and multiple organ dysfunction:the mystery continues. Am Surg. 2012;78(1):1–8. [PubMed] [Google Scholar]
- 7.Aird WC. Phenotypic heterogeneity of the endothelium. Circ Res. 2007;100(2):174–90. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 8.Liu ZC, Yin PY, Amathieu R, Savarin P, Xu GW. Application of LC-MS-based metabolomics method in differentiating septic survivors from non-survivors. Anal Bioanal Chem. 2016;408(27):7641–9. doi: 10.1007/s00216-016-9845-9. [DOI] [PubMed] [Google Scholar]
- 9.Toral M, Romero M, Jiménez R, Mahmoud AM, Barroso E, Gómez-Guzmán M, et al. Carnitine palmitoyltransferase-1 up-regulation by PPAR-β/δprevents lipid-induced endothelial dysfunction. Clin Sci. 2015;129(9):823–37. doi: 10.1042/CS20150111. [DOI] [PubMed] [Google Scholar]
- 10.McGarrity S, Anuforo Ó, Halldórsson H, Bergmann A, Halldórsson S, Palsson S, et al. Metabolic systems analysis of LPS induced endothelial dysfunction applied to sepsis patient stratification. Sci Rep. 2018;8(1):6811. doi: 10.1038/s41598-018-25015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eelen G, Zeeuw de P, Treps L, Harjes U, Wong BW, Carmeliet P. Endothelial cell metabolism. Physiol Rev. 2018;98(1):3–58. doi: 10.1152/physrev.00001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teuwen LA, Draoui N, Dubois C, Carmeliet P. Endothelial cell metabolism:an update anno 2017. Curr Opin Hematol. 2017;24(3):240–7. doi: 10.1097/MOH.0000000000000335. [DOI] [PubMed] [Google Scholar]
- 13.Bock KD, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–63. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 14.Tang CY, Mauro C. Similarities in the metabolic reprogramming of immune system and endothelium. Front Immunol. 2017;8:837. doi: 10.3389/fimmu.2017.00837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis:therapeutic targets at last? EMBO Mol Med. 2018;10(8):e∈. doi: 10.15252/emmm.201708712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang K, Fan M, Wang XH, Xu JJ, Wang YN, Gill PS, et al. Lactate induces vascular permeability via disruption of VE-cadherin in endothelial cells during sepsis. Sci Adv. 2022;8(17):eabm8965. doi: 10.1126/sciadv.abm8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan M, Yang K, Wang XH, Zhang X, Xu JJ, Tu F, et al. Lactate impairs vascular permeability by inhibiting hspa12b expression via gpr81-dependent signaling in sepsis. Shock. 2022;58(4):304–12. doi: 10.1097/SHK.0000000000001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang LN, Cao YP, Gorshkov B, Zhou YQ, Yang QH, Xu JA, et al. Ablation of endothelial PFKFB3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol Res. 2019;146:104292. doi: 10.1016/j.phrs.2019.104292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng ZB, Ma H, Zhang X, Tu F, Wang XH, Ha TZ, et al. Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis. 2017;215(9):1396–406. doi: 10.1093/infdis/jix138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao LF, Sun MM, Chen ZF, Zeng ZH, Wu J, Chen ZQ, et al. The pyruvate dehydrogenase complex mitigates LPS-induced endothelial barrier dysfunction by metabolic regulation. Shock. 2022;57(6):308–17. doi: 10.1097/SHK.0000000000001931. [DOI] [PubMed] [Google Scholar]
- 21.Ekaney ML, Otto GP, Sossdorf M, Sponholz C, Boehringer M, Loesche W, et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit Care. 2014;18(5):543. doi: 10.1186/s13054-014-0543-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vizán P, Sánchez-Tena S, Alcarraz-Vizán G, Soler M, Messeguer R, Pujol MD, et al. Characterization of the metabolic changes underlying growth factor angiogenic activation:identification of new potential therapeutic targets. Carcinogenesis. 2009;30(6):946–52. doi: 10.1093/carcin/bgp083. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem. 2000;275(51):40042–7. doi: 10.1074/jbc.M007505200. [DOI] [PubMed] [Google Scholar]
- 24.Spolarics Z, Stein DS, Garcia ZC. Endotoxin stimulates hydrogen peroxide detoxifying activity in rat hepatic endothelial cells. Hepatology. 1996;24(3):691–6. doi: 10.1002/hep.510240336. [DOI] [PubMed] [Google Scholar]
- 25.Spolarics Z, Navarro L. Endotoxin stimulates the expression of glucose-6-phosphate dehydrogenase in Kupffer and hepatic endothelial cells. J Leukoc Biol. 1994;56(4):453–7. doi: 10.1002/jlb.56.4.453. [DOI] [PubMed] [Google Scholar]
- 26.Förstermann U, Sessa WC. Nitric oxide synthases:regulation and function. Eur Heart J. 2011;33(7):829–37. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chon J, Stover PJ. Field MS Targeting nuclear thymidylate biosynthesis. Mol Aspects Med. 2017;53:48–56. doi: 10.1016/j.mam.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J. 2016;37(22):1753–61. doi: 10.1093/eurheartj/ehv564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang JY, Huang K, Xu SQ, Garcia JGN, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020;36:101638. doi: 10.1016/j.redox.2020.101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyoshi T, Yamashita K, Arai T, Yamamoto K, Mizugishi K, Uchiyama T. The role of endothelial interleukin-8/NADPH oxidase 1 axis in sepsis. Immunology. 2010;131(3):331–9. doi: 10.1111/j.1365-2567.2010.03303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarmiento D, Montorfano I, Cáceres M, Echeverría C, Fernández R, Cabello-Verrugio C, et al. Endotoxin-induced vascular endothelial cell migration is dependent on TLR4/NF-κB pathway, NAD(P)H oxidase activation, and transient receptor potential melastatin 7 calcium channel activity. Int J Biochem Cell Biol. 2014;55:11–23. doi: 10.1016/j.biocel.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 32.Wang ZF, Rui T, Yang M, Valiyeva F, Kvietys PR. Alveolar macrophages from septic mice promote polymorphonuclear leukocyte transendothelial migration via an endothelial cell Src kinase/NADPH oxidase pathway. J Immunol. 2008;181(12):8735–44. doi: 10.4049/jimmunol.181.12.8735. [DOI] [PubMed] [Google Scholar]
- 33.Gill SE, Rohan M, Mehta S. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res. 2015;16(1):109. doi: 10.1186/s12931-015-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trevelin SC, Sag CM, Zhang M, Alves-Filho JC, Cunha TM, Santos CXD, et al. Endothelial Nox2 limits systemic inflammation and hypotension in endotoxemia by controlling expression of toll-like receptor 4. Shock. 2021;56(2):268–77. doi: 10.1097/SHK.0000000000001706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu F, Schuster DP, Tyml K, Wilson JX. Ascorbate inhibits NADPH oxidase subunit p47phox expression in microvascular endothelial cells. Free Radic Biol Med. 2007;42(1):124–31. doi: 10.1016/j.freeradbiomed.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 36.Kröller-Schön S, Knorr M, Hausding M, Oelze M, Schuff A, Schell R, et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc Res. 2012;96(1):140–9. doi: 10.1093/cvr/cvs246. [DOI] [PubMed] [Google Scholar]
- 37.Yu CW, Liang XL, Lipsky S, Karaaslan C, Kozakewich H, Hotamisligil GS, et al. Dual role of fatty acid-binding protein 5 on endothelial cell fate:a potential link between lipid metabolism and angiogenic responses. Angiogenesis. 2016;19(1):95–106. doi: 10.1007/s10456-015-9491-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M, van Esch BCAM, Wagenaar GTM, Garssen J, Folkerts G, Henricks PAJ. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur J Pharmacol. 2018;831:52–59. doi: 10.1016/j.ejphar.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Voltolini C, Battersby S, Etherington SL, Petraglia F, Norman JE, Jabbour HN. A novel antiinflammatory role for the short-chain fatty acids in human labor. Endocrinology. 2012;153(1):395–403. doi: 10.1210/en.2011-1457. [DOI] [PubMed] [Google Scholar]
- 40.Li M, van Esch BCAM, Henricks PAJ, Garssen J, Folkerts G. Time and concentration dependent effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-induced endothelial activation. Front Pharmacol. 2018;9:233. doi: 10.3389/fphar.2018.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trommer S, Leimert A, Bucher M, Schumann J. Impact of unsaturated fatty acids on cytokine-driven endothelial cell dysfunction. Int J Mol Sci. 2017;18(12):E2739. doi: 10.3390/ijms18122739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lawton SK, Xu FY, Tran A, Wong E, Prakash A, Schumacher M, et al. N-arachidonoyl dopamine modulates acute systemic inflammation via nonhematopoietic TRPV1. J Immunol. 2017;199(4):1465–75. doi: 10.4049/jimmunol.1602151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rittchen S, Rohrer K, Platzer W, Knuplez E, Bärnthaler T, Marsh LM, et al. Prostaglandin D2 strengthens human endothelial barrier by activation of E-type receptor 4. Biochem Pharmacol. 2020;182:114277. doi: 10.1016/j.bcp.2020.114277. [DOI] [PubMed] [Google Scholar]
- 44.Wang YF, Hsu YJ, Wu HF, Lee GL, Yang YS, Wu JY, et al. Endothelium-derived 5-methoxytryptophan is a circulating anti-inflammatory molecule that blocks systemic inflammation. Circ Res. 2016;119(2):222–36. doi: 10.1161/CIRCRESAHA.116.308559. [DOI] [PubMed] [Google Scholar]
- 45.Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520(7546):192–7. doi: 10.1038/nature14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Brüning U, et al. Quiescent endothelial cells upregulate fatty acid β-oxidation for vasculoprotection via redox homeostasis. Cell Metab. 2018;28(6):881–94e13. doi: 10.1016/j.cmet.2018.07.016. [DOI] [PubMed] [Google Scholar]
- 47.Gao XL, Li JQ, Dong YT, Cheng EJ, Gong JN, Qin YL, et al. Upregulation of microRNA-335-5p reduces inflammatory responses by inhibiting FASN through the activation of AMPK/ULK1 signaling pathway in a septic mouse model. Cytokine. 2018;110:466–78. doi: 10.1016/j.cyto.2018.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Apostolova N, Garcia-Bou R, Hernandez-Mijares A, Herance R, Rocha M, Victor VM. Mitochondrial antioxidants alleviate oxidative and nitrosative stress in a cellular model of sepsis. Pharm Res. 2011;28(11):2910–9. doi: 10.1007/s11095-011-0528-0. [DOI] [PubMed] [Google Scholar]
- 49.Magalhaes-Novais S, Blecha J, Naraine R, Mikesova J, Abaffy P, Pecinova A, et al. Mitochondrial respiration supports autophagy to provide stress resistance during quiescence. Autophagy. 2022;18(10):2409–26. doi: 10.1080/15548627.2022.2038898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Z, Lan X, Ahsan A, Xi Y, Liu S, Zhang Z, et al. Phosphocreatine protects against LPS-induced human umbilical vein endothelial cell apoptosis by regulating mitochondrial oxidative phosphorylation. Apoptosis. 2016;21(3):283–97. doi: 10.1007/s10495-015-1210-5. [DOI] [PubMed] [Google Scholar]
- 51.Unterluggauer H, Mazurek S, Lener B, Hütter E, Eigenbrodt E, Zwerschke W, et al. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology. 2008;9(4):247–59. doi: 10.1007/s10522-008-9134-x. [DOI] [PubMed] [Google Scholar]
- 52.Sen S, Roy S, Bandyopadhyay G, Scott B, Xiao DL, Ramadoss S, et al. γ-aminobutyric acid is synthesized and released by the endothelium:potential implications. Circ Res. 2016;119(5):621–34. doi: 10.1161/CIRCRESAHA.116.308645. [DOI] [PubMed] [Google Scholar]
- 53.Durante W. The emerging role of l-glutamine in cardiovascular health and disease. Nutrients. 2019;11(9):E2092. doi: 10.3390/nu11092092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pan M, Wasa M, Ryan U, Souba W. Inhibition of pulmonary microvascular endothelial glutamine transport by glucocorticoids and endotoxin. JPEN J Parenter Enteral Nutr. 1995;19(6):477–81. doi: 10.1177/0148607195019006477. [DOI] [PubMed] [Google Scholar]
- 55.Abcouwer SF, Lukascewicz GC, Ryan US, Souba WW. Molecular regulation of lung endothelial glutamine synthetase expression. Surgery. 1995;118(2):325–34. doi: 10.1016/s0039-6060(05)80341-1. ;discussion335. [DOI] [PubMed] [Google Scholar]
- 56.Leo F, Suvorava T, Heuser SK, Li JJ, LoBue A, Barbarino F, et al. Red blood cell and endothelial eNOS independently regulate circulating nitric oxide metabolites and blood pressure. Circulation. 2021;144(11):870–89. doi: 10.1161/CIRCULATIONAHA.120.049606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol (|y1985) 2009;107(4):1249–57. doi: 10.1152/japplphysiol.91393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez E, Fukuda S, Modis K, Fujiwara O, Enkhtaivan B, Trujillo-Abarca R, et al. Arginine vasopressin receptor 2 activation promotes microvascular permeability in sepsis. Pharmacol Res. 2021;163:105272. doi: 10.1016/j.phrs.2020.105272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yeh CL, Pai MH, Shih YM, Shih JM, Yeh SL. Intravenous arginine administration promotes proangiogenic cells mobilization and attenuates lung injury in mice with polymicrobial sepsis. Nutrients. 2017;9(5):E507. doi: 10.3390/nu9050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wijnands KA, Hoeksema MA, Meesters DM, van den Akker NM, Molin DG, Briedé JJ, et al. Arginase-1 deficiency regulates arginine concentrations and NOS2-mediated NO production during endotoxemia. PLoS One. 2014;9(1):e8613–5. doi: 10.1371/journal.pone.0086135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoo JW, Moon JY, Hong SB, Lim CM, Koh Y, Huh JW. Clinical significance of circulating endothelial cells in patients with severe sepsis or septic shock. Infect Dis (Lond) 2015;47(6):393–8. doi: 10.3109/00365548.2014.1001999. [DOI] [PubMed] [Google Scholar]
- 62.Parra-Bonilla G, Alvarez DF, Al-Mehdi AB, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L513–22. doi: 10.1152/ajplung.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]