

Abstract
The design and synthesis of a series of peptide derivatives based on a short ACE2 α-helix 1 epitope and subsequent [i - i+4] stapling of the secondary structure resulted in the identification of a 9-mer peptide capable to compete with recombinant ACE2 towards Spike RBD in the micromolar range. Specifically, SARS-CoV-2 Spike inhibitor screening based on colorimetric ELISA assay and structural studies by circular dichroism showed the ring-closing metathesis cyclization being capable to stabilize the helical structure of the 9-mer 34HEAEDLFYQ42 epitope better than the triazole stapling via click chemistry. MD simulations showed the stapled peptide being able not only to bind the Spike RBD, sterically interfering with ACE2, but also showing higher affinity to the target as compared to parent epitope.
Keywords: Peptidomimetics, Protein-protein interaction, COVID-19, Infectious diseases, Molecular modeling, Ring-closing metathesis
Graphical abstract

1. Introduction
The Respiratory Syndrome caused by Coronavirus SARS-CoV-2 that appeared at the end of 2019 [1,2] has put in serious difficulty the health systems globally and raised health concerns as well as unprecedented economic challenges. The restriction measures that were necessary to counteract the spread of the contagion have caused serious losses to the economies and the budget for health has significantly increased to allow the recovery of a high number of people mainly through hospitalization in intensive care units. As of October 2022, the COVID-19 outbreak has been responsible for over 627 million infections and 6,5 million confirmed deaths worldwide [3]. While the introduction of vaccines (12,7 billion vaccine doses administered) [3] has made it possible to keep most acute infections under control (0.1% serious and critical cases among 45 million actually infected people, as of October 2022) [4], it is also true that immunosuppressed or unvaccinated subjects still represent an easily attackable target, in particular with the onset of new and potentially more contagious and infectious genetic variants of SARS-CoV-2 that continue to emerge and spread [5]. If mass vaccination represents an excellent emergency response, a normal situation would only have positive aspects to be drawn from the presence of targeted treatments to counter the effects of lung infection in infected subjects without the need of expensive treatments such as intubation and hospitalization in intensive care.
To date, a series of therapies are being implemented for the treatment of COVID-19 [6], and drug classes currently used include antiviral agents, inflammation inhibitors, antirheumatic drugs, plasma and therapeutic antibodies. Given the nature of the coronavirus, the greatest efforts are focused on key events linked to the infection, the blocking of the Spike-ACE2 (angiotensin-converting enzyme 2) interaction and the inhibition of the viral proteins necessary for replication, such as the 3CLPro protease [7,8] and the RNA-dependent RNA polymerase (RdRp) [9]. The first process represents the initial contact between the virus and the host cells to promote fusion with the host cell membrane for subsequent release of the genetic material necessary for viral replication.
The viral entry for SARS-CoV-2 is a key event mediated by the Spike transmembrane protein that forms homotrimers, each structure consisting of a S1 subunit that binds to ACE2 of the host cell via a receptor-binding domain (RBD) to initiate infection, a S2 subunit that mediates virus fusion with host cells, and a transmembrane domain. The RBD-Spike S1-ACE2 protein-protein interaction at the surface of epithelial cells has been identified as the molecular event causing the infection of human respiratory cells [[10], [11], [12], [13]]. Given the pivotal role of such proteins for promoting viral entry and replication, SARS-CoV-2 monoclonal antibodies (mAbs) have been developed successfully for their capability to bind to the Spike receptor-binding domain of the SARS-CoV-2, preventing viral entry into host cells, taking advantage of high specificity that limits their “off-target” toxicity, although showing reduced antiviral potency with the occurrence of viral variants [14]. Complimentary to this therapeutic approach, small molecule entry inhibitors impairing virus particles from infecting human cells could be used to prevent SARS-CoV-2 infection and to shorten the outcome of COVID-19 infections.
The structure and characterization of the ACE2-fragment S1 complex of the SARS-CoV-2 Spike protein obtained with the cryo-EM technique was soon reported (Fig. 1 , left) [15,16], allowing to identify the structural determinants responsible for protein-protein interaction, particularly those involving the spike RBD directly interacting with the exposed domain of ACE2 consisting of the N- and C-terminal regions of the helix α1 and small areas of the helix α2 (Fig. 1, right).
Fig. 1.

Left: trimeric Spike protein (cyan) complexed to two ACE2 fragments (purple, PDB: 7A97); right: close-up of RBD (cyan)-ACE2 (purple) interaction and highlight of the selected ACE2 α-helix 1 epitope (green, PDB: 6M0J).
Specifically, the central segment of ACE2 α-helix 1 is involved in strong interactions mainly engaging polar residues. At the N-terminal end of α-helix 1, Y41, Q42 and D38 experience a hydrogen bond network with Q498, T500 and N501 spike residues, whereas in the middle of the α-helix 1 bridge, H34 interacts with Y453 of Spike RBD. In this view, A36, L39 and F40 in this helical epitope are not directly involved in the interaction with the spike binding region (Fig. 2 ).
Fig. 2.

Magnification of RBD (cyan)-ACE2 (purple) interaction, highlighting the selected ACE2 α-helix 1 epitope (green, PDB: 6M0J), the key interacting amino acids and the selected amino acids for the [i-i+4] stapling region.
Taking advantage of such structural biology data published with unprecedented speed, several research groups considered the structure-based design of peptides and stapled constructs as mimetics of ACE2 α-helix 1 motif, capable of disrupting the key Spike RBD-ACE2 protein-protein interaction. The absence of binding pockets as hot spots in this PPI that develops on a rather extensive surface made the identification of small molecule PPI inhibitors rather difficult [17,18].
Based on the structure of the RBD-ACE2 complex, a first-in-class 23-mer peptide capable of binding RBD with nanomolar affinity according to interferometry bio-layer experiments was reported [19]. Successively, Karoyan et al. developed a ACE2-derived 27-mer mimetic of the N-terminal of the α-helix 1, capable of binding the Spike RBD with IC50 in the nM range as measured by ELISA test and suggesting a potential application as a nasal or oral spray to overcome the limited biodistribution and bioavailability of peptides [20]. Starting from the evidence of peptidomimetics as promising and more druggable inhibitors of PPIs [21,22], stapled peptidomimetics of the ACE2 α-helix 1 were proposed to block the bioactive helical conformation using chemical linkers. Peptide stapling increases the helicity in solution of small unstructured peptides, improves resistance against proteolysis, increases potency and often improves cell penetration [[23], [24], [25], [26], [27], [28], [29]]. Accordingly, Mecinović et al. reported the synthesis of 35-mer stapled peptides based on ACE2 α-helix 1 displaying μM activity in ELISA based screening assays [30]. Debnath et al., based on the α-helix 1 and the identified key interactions, designed 30-mer peptides with double stapling that showed a high percentage of helicity and the capability to inhibit the activity of SARS-CoV-2 through in vitro pseudovirus and viral infectivity assays. Interestingly, the less active compounds showed a sequence with the key Q42 being replaced with an alkenyl amino acid useful for the stapling [31]. Similarly, Jamieson et al. reported a stapling array of 10- to 23-mer α-helix 1 ACE2 peptidomimetics [32]. Although possessing the helix structure in solution, as confirmed by circular dichroism, none of them showed activity in fluorescence polarization and neutralization assays. In view of developing short peptidomimetics capable of acting as entry inhibitors [33], we got interested in the work by Larue et al. [34] that rationally designed and tested a series of small peptide inhibitors of the Spike-ACE2 interaction, based on the minimal conserved 6-mer 37EDLFYQ42 epitope of ACE2 α-helix 1 interacting domain, resulting in the identification of two peptides possessing mM inhibition potency towards SARS-CoV-2 infection. Starting from this epitope, we devised selecting the longer 9-mer 34HEAEDLFYQ42 epitope to introduce a minimal [i-i+4] stapling and maintaining Q42 as the C-terminal amino acid, following the observation that the presence of the two following serine residues (43SS44) in the peptide may affect the peptide flexibility and activity, as reported [34]. From the structural data on the Spike RBD-ACE2 interaction (Fig. 2) the two amino acids A36 and F40 were identified for the stapling region, due to their orientation at the opposite site of the helix interacting with the RBD and at the right distance for the [i-i+4] helix pitch (Fig. 2).
2. Results and discussion
2.1. Synthesis, structural and biological assays
All the peptides were synthesized using a microwave-assisted solid-phase synthesis protocol [31,35]. The peptide epitope 34HEAEDLFYQ42 1 and its N-acetyl derivative 2 were synthesized as reference compounds (Table 1 ). Briefly, the peptides were synthesized using a ChemMatrix Rink Amide resin, with a 0.5 mmol/g loading capacity. DIC/Oxyma [36] was used as the activating mixture for coupling Fmoc-amino acids, and morpholine was used as a Fmoc deblocking agent. 95% TFA containing water and TIPS as scavengers, each in 2.5% amount was used as the cleaving mixture. Peptides were precipitated using cold diethyl ether and purified by reverse-phase HPLC and characterized by analytical HPLC and ESI-MS.
Table 1.
Sequence and inhibitory activity of selected ACE2 epitopes and peptidomimetics.a
| Entry | Sequence/Structure | Yield, % | % inhib. at 30 μM |
|---|---|---|---|
| 1 | H-HEAEDLFYQ-NH2 | 28 | 20 |
| 2 | Ac-HEAEDLFYQ-NH2 | 39 | 12 |
| 3 | 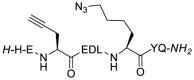 |
46 | 6 |
| 4 | 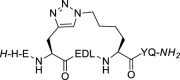 |
12 | 30 |
| 5 | 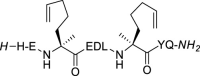 |
35 | 8 |
| 6 | 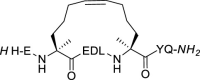 |
15 | 55 (21 ± 7 μM)b |
Mean from three different assays, errors were in the range of 5–10% of the reported values.
IC50 value was retrieved from dose-response assays as the concentration of compound required for 50% inhibition and estimated by non-linear correlation using GraphPad Prism software.
The stapling between A36 to F40 was achieved using both click chemistry [37] and ring-closing metathesis (RCM) [31,38] on solid-phase. The linear peptide precursors 3 and 5 were synthesized as controls (Table 1). The formation of a triazole through the Cu-catalyzed azide-alkyne cycloaddition (CuAAC) reaction as a click chemistry approach was achieved by replacing A36 with Fmoc-Pra-OH (Fmoc-propargyl-Gly-OH) and F40 with Fmoc-Lys(N3)–OH (Fmoc-azidolysine) in the peptide sequence. The fully protected resin-bound peptide was treated with CuBr and sodium ascorbate in a DMF/DMSO mixture containing DIPEA and 2,6-lutidine, stirring the resin for 5 min at room temperature, then under microwave irradiation at 55 °C for 10 min. After final Fmoc deprotection and peptide cleavage, subsequent purification via semi-preparative HPLC allowed to obtain peptide 4 in 12% overall yield, with a macrocyclization yield of 26%, as assumed with respect to that of linear peptide 3. The achievement of the triazole ring was confirmed by ESI-MS and HPLC analysis (see Supplementary Material), and also by comparison of the 1H NMR spectra of the stapled peptide 4 and linear precursor 3, which showed the disappearance of the triplet NMR signal at 2.29 ppm (attributable at the terminal alkynyl hydrogen of the propargyl-Gly moiety) and the appearance of the diagnostic singlet signal at 7.61 ppm of the triazole CH atom (Fig. 3 , left). 1H NMR proton signals were assigned by 2D experiments performed on peptide 4 (and parent peptide 1) in 90% HDO under water suppression technique with excitation sculpting [39] and were found to be in agreement with those of other triazole containing peptides as reported in the literature [40]. For the synthesis of the stapled peptide via RCM, the two amino acids A36 and F40 were replaced with the quaternary amino acid Fmoc-(S)-2-(4-pentenyl)Ala-OH. The metathesis was carried out on the fully protected resin-bound peptide using 1st generation Grubbs catalyst (15 mg for 0.05 mmol resin-bound peptide) in 1,2-dichloroethane. After final Fmoc deprotection and peptide cleavage, subsequent purification by semi-preparative HPLC allowed to obtain peptide 6 in 15% overall yield, with a macrocyclization yield of 43%, determined with respect to that of linear peptide 5. The achievement of the ring closing metathesis was confirmed by ESI-MS and HPLC analysis (see Supplementary Material) and by the comparison of the 1H NMR spectra of the stapled peptide 6 and the linear precursor 5. As showed in Fig. 3, right, the two multiplet signals at 5.66–5.57 ppm and 5.52–5.41 ppm of the terminal alkenyl hydrogen atoms were replaced by the diagnostic broad singlet signal at 5.13 ppm attributable to the newly formed internal double bond, which showed a preference for the Z isomeric form, as reported for other RCM of (S)-(4-pentenyl)Ala at positions i and i+4 present in the literature [41,42].
Fig. 3.

Left: plots of 1H NMR spectra of (a) acyclic peptide 3 and (b) stapled peptide 4 with box highlighting the key signals of alkyne and triazole protons. Right: plots of 1H NMR spectra of (c) acyclic peptide 5 and (b) stapled peptide 6 with box highlighting the key signals of terminal alkenes and internal double bond protons. Spectra were recorded at 400 MHz in D2O.
The synthesized compounds were tested to preliminary assess their biological activity using a competitive ELISA assay, which measures the binding of the RBD of the Spike protein from SARS-CoV-2 to its human receptor ACE2 (Table 1). The SARS-CoV-2 inhibitor in the samples competes with ACE2 to combine with immobilized SARS-CoV-2 Spike RBD. The signal colour becomes lighter as the content of SARS-CoV-2 inhibitor increases. Concerning the selected peptide epitope 1 and the N-acetyl derivative 2, the competitive ELISA assay at 30 μM concentration did not show a significant inhibition of the spike RBD-ACE2 interaction with respect to biotinylated ACE2. Concerning stapled peptides with respect to the linear precursors, a significant inhibitory activity was observed for the RCM stapled peptide 6, whereas the triazole-stapled peptide 4 showed minor activity, though better than the corresponding linear precursor 3 and the reference peptide 1. The RCM stapling as in 6 allowed to constrain the peptide epitope in the bioactive conformation, taking advantage of the two quaternary amino acids as the stapling elements, capable of promoting a higher degree of helicity and providing a flexible tether for the modulation of the conformation to the bioactive shape. A dose-response assay was carried out for 6 to achieve an inhibition curve of SARS-CoV-2 Spike Protein (RBD) in the range of 10 nM–300 μM, resulting in a IC50 value of 21 μM, suggesting the importance of RCM stapling to improve the inhibition potency of a minimal ACE2 epitope (Fig. 4 ).
Fig. 4.

Inhibition curve of 6 on Spike RBD-ACE2 interaction.
Circular dichroism analyses were performed in the range from 190 nm to 250 nm on peptides 4, 6 and the corresponding linear precursors 3 and 5 as 500 and 100 μM PBS solutions to determine the secondary structure of the peptides in PBS buffer (Fig. 5 ). Both stapled peptides 4 and 6 exhibited an appreciable α-helix degree. On the other hand, the linear precursor 3 of the stapled peptide 4 showed a spectrum attributable to random coil. The unexpected result obtained for the linear peptide 5 was attributed to the presence of disubstituted α,α-amino acids that recall the structure of α-aminoisobutyric acid, already widely characterized as a helicity inducer in linear peptides [43]. Nevertheless, such helical character of 5 was not sufficient to achieve a significant binding to RBD.
Fig. 5.

Circular dichroism spectra of triazole-stapled peptide 4 and RCM-stapled peptide 6 along with linear precursors 3 and 5, respectively, at 500 μM in PBS. Spectra were recorded between 190 and 250 nm.
2.2. Plasma stability assay
Resistance to proteolysis was determined for compounds 1 and 6 by treating the peptides with human plasma at 37 °C and recording peptide degradation over time using LC-MS/MS (Fig. 6 ). Interestingly, the data showed both compounds remaining highly stable over time. At 48 h peptide 6 was observed to be comparatively more stable than the natural sequence, which is likely due to the stabilizing modification imposed by the introduction of the staple.
Fig. 6.

The plasma stability of 1 and 6 at 37 °C recording peptide degradation over time using LC-MS/MS.
2.3. PepGaMD simulations
The SpikeRBD:1 and SpikeRBD:6 models were generated by protein-protein docking within MOE software. Poses were selected to have the peptide bound to the Spike RBD in the same region where the native sequence in ACE2 interacts (green in Fig. 7 A and Table S5). The two complexes (Fig. S2) were then minimized, and 1 μs long Peptide Gaussian Accelerated Molecular Dynamics (PepGaMD) simulations were performed.
Fig. 7.

A) 3D structure of SpikeRBD:ACE2 complex (PDB entry: 6M0J). Spike RBD is represented in light grey ribbon, ACE2 is rendered in dark grey ribbon. Sequence of the protein consisting of WT peptide is highlighted in cyan; B–C) PMFs obtained from distances of CA atoms of residues Y449 and K487 to CA atoms of B) 1 and C) 6. PMF is shown in color bar (0.0–20.0 kcal/mol range) from dark blue to red; D) Lowest energy conformation obtained from the simulation of SpikeRBD:1 complex. SpikeRBD is represented in light grey, WT is rendered in green; E) Lowest energy conformation obtained from the simulation of SpikeRBD:6 complex. SpikeRBD is represented in light grey, 6 is rendered in purple; F) Superposition of 1 and 6 to SpikeRBD:ACE2 complex. Spike RBD is represented in light grey ribbon ACE2 is rendered in dark grey ribbon. Sequence of the protein consisting of 1 is highlighted in cyan, 1 is rendered in green, 6 is rendered in purple; G) Site view of the interactions of SpikeRBD:1 PMF = 0 geometry. Spike RBD amino acids within 4.5 Å of WT are represented in grey sticks. 1 residues are rendered in green. Spike RBD is represented in light grey ribbon; H) Site view of the interactions of SpikeRBD:6 PMF = 0 geometry. ACE2 is rendered in dark grey ribbon Spike RBD amino acids within 4.5 Å of 6 are represented in purple sticks. Peptide 6 residues are rendered in purple. Spike RBD is represented in light grey ribbon.
From the two simulations, the lowest energy conformations were predicted by evaluating the potential of the mean force (PMF, Fig. 7B and C) using the distances between the center of mass of all the Cα of each peptide and the Cα of RBD Y449 and K417, involved in SpikeRBD:ACE2 binding, as reaction coordinates [44]. As shown by Fig. 7D–F, the lowest energy structure (PMF = 0) of 1 interacts with the Spike RBD in a region not involved in the SpikeRBD:ACE2 interaction (Fig. 7G and Fig. S3A). Conversely, peptide 6 resulted as stably bound to a region, different from the starting one, where it can hamper the SpikeRBD:ACE2 interaction (Fig. 7H and Fig. S3B). In fact, 6 forms H-bonds with side chains of K458 and E484, H-bonds with backbone of G485 and C488, and arene-H interaction with Y498 (Fig. S3B) and, as shown by Fig. 7F and H, it can sterically interfere with ACE2. A classical MD simulation (4 ns, NPT) was performed starting from the PMF = 0 geometry of each complex, as well as from the SpikeRBD:ACE2 complex, to compute binding energies by Nwat-MMGBSA (Table 2 ) [45,46]. The energies reported in Table 2 show that peptide 6 possess higher affinity to Spike as compared to 1. Therefore, 6 is not only able to bind the Spike RBD, sterically interfering with ACE2, but it also shows higher affinity to the target compared to 1.
Table 2.
SpikeRBD::1 and SpikeRBD::6 binding energies (kcal/mol) calculated by Nwat-GBSA with 0, 5, 30 or 60 explicit water molecules.
| PMF0 1 | ΔEbind | Std. Dev. | Std. Err. Of Mean |
|---|---|---|---|
| NWAT = 0 | −12.4 | 3.3 | 0.3 |
| NWAT = 5 | −14.0 | 5.5 | 0.5 |
| NWAT = 30 | −62.0 | 11.0 | 1.1 |
| NWAT = 60 |
−100.8 |
13.8 |
1.4 |
|
PMF0 6 |
ΔEbind |
Std. Dev. |
Std. Err. Of Mean |
| NWAT = 0 | −32.7 | 2.8 | 0.3 |
| NWAT = 5 | −33.1 | 5.5 | 0.5 |
| NWAT = 30 | −70.7 | 9.5 | 0.9 |
| NWAT = 60 | −112.4 | 9.9 | 1.0 |
2.4. T-REMD simulations
Additionally, we evaluated the folding behaviour of both 1 and 6 in silico, by running 200 ns of T-REMD simulations. Results show that 1 prefers a disordered secondary structure, while a α-helix was preferred by 6, as shown by both cluster and DSSP analyses (Fig. S4 and Table S6), in line with the experimental results obtained by CD.
3. Conclusion
In conclusion, the synthesis of short stapled peptides based on a α-helix 1 ACE2 epitope was achieved, resulting in the identification of a RCM-stapled 9-mer peptide capable to compete with recombinant ACE2 towards Spike RBD in the micromolar range, according to competitive ELISA assay on immobilized SARS-CoV-2 Spike RBD. Structural studies by circular dichroism showed RCM cyclization being capable to stabilize the helical structure better than triazole stapling via click chemistry, also taking advantage of quaternary amino acids promoting peptide helicity. Plasma stability assays by LC-MS/MS demonstrated the stapled peptide possessing higher stability at 48 h than the natural sequence, likely due to the stabilizing modification imposed by the introduction of the staple. Molecular dynamics and energy calculations suggested peptide 6 possessing structural rigidity and higher affinity to Spike as compared to 1, as well as the ability to bind the Spike RBD in a region more appropriate to hamper ACE2 interaction. An in-depth biological characterization of stapled peptide 6 will be performed in due course to determine its binding affinity for the Spike protein, by measuring the dissociation constant or by performing precipitation assays. The results are preliminary for subsequent development of a small molecule helix peptidomimetic capable of interfering with the key ACE2-Spike S1 protein-protein interaction.
4. Experimental section
All commercially available reagents and solvents were used as received. Solid-phase peptide synthesis was carried out using an automated single-mode microwave synthesizer (Initiator Sixty, Biotage AB) using sealed reaction vessels and built-in internal pressure and temperature sensors. HPLC analyses were performed on synthesized peptides using Dionex Ultimate 3000 system. ESI-MS were carried out on methanol/formic acid solutions by direct inlet on a LCQ Fleet™ Ion Trap LC/MS system (Thermo Fisher Scientific) using electrospray (ES + or ES-) ionization techniques. 1H-water-ES, TOCSY-ES and ROESY-ES spectra of peptides 1, 4 and 6, obtained in 4 mM 90% HDO under water suppression technique with excitation sculpting are reported in the Supplementary Material.
4.1. General procedure for peptide synthesis
The peptides were synthesized on the ChemMatrix Rink Amide resin, with a 0.5 mmol/g loading capacity and bead size of 100–200 mesh. Peptide cleavage from the resin was achieved under acidic conditions. The resin was swelled in dichloromethane (DCM) for 20 min, under magnetic stirring prior to peptide synthesis. Then, the solution was filtered and washed twice with N,N-dimethylformamide (DMF). Peptide coupling were carried out using 5 eq. the Fmoc-amino acids as 0.2 M DMF solution and 10 eq. DIC/Oxyma, both 1 M in DMF as the activating mixture by heating at 90 °C for 3 min, followed by DMF washings (x3). 20% Morpholine was used as Fmoc deblocking agent by heating at 90 °C for 2 min (x2), each cycle followed by DMF washings (x3).
Final resin washing before acidic cleavage were carried out with DMF (x3) and CH2Cl2 (x2). A mixture of 2.5% H2O and 2.5% TIPS in TFA was added into the peptidyl resin, and the suspension was shaken for 2 h followed by filtration. Cold diethyl ether (10 mL) was added to the filtrate, and peptide was precipitated. The mixture was centrifuged for 3 min at 2500 rpm, and the ether layer was separated. The procedure was repeated three times, then a second treatment with TFA containing water and TIPS was repeated for 15 min, followed by peptide precipitation with cold ether. Then, the peptide was dried under vacuum overnight to give the crude peptide.
4.2. HPLC purification and analysis
Peptides were analyzed and purified using Dionex Ultimate 3000 system equipped with a reverse-phase analytical column Synergi 4 μm Fusion-RP 80 Å (150 × 4.6 mm) or semi-preparative column Synergi 10 μm Fusion-RP 80 Å (250 × 10.0 mm) and using acetonitrile (0.1% TFA) in H2O (0.1% TFA) at room temperature, 5–95% linear gradient over 20 min for analytical and semi-preparative runs or alternatively 5–30%/3′, 30–50%/27′ and 50–95%/2’ as gradient for semi-preparative runs. A flow rate of 1 and 5 mL/min were used for analytical and semi-preparative runs, respectively, and peak detection was achieved at 223 nm. All crude peptides were obtained in >95% purity. The molecular weight of all peptides was confirmed by electrospray mass spectrometry. Analytical samples were prepared as 1 mg/mL conc. by dissolving the dry peptides in 0.1% H2O/HCOOH (v/v).
Compound 1. The linear peptide 1 was achieved in 28% yield using 100 mg of ChemMatrix Rink Amide resin and following the general procedures for peptide synthesis and HPLC purification. HPLC: 90% purity, rt = 10.36 min; ESI-MS: m/z 1151.20 [M+H]+.
Compound 2. The linear peptide 2 was achieved in 39% yield using 100 mg of ChemMatrix Rink Amide resin and following the general procedures for peptide synthesis and HPLC purification. Acetylation of the N-terminal amino acid was carried out on solid phase using a DMF solution of Ac2O (20 eq) and DIPEA (20 eq) for 30 min at room temperature. HPLC: 95% purity, rt = 10.65 min; ESI-MS: m/z 1192.31 [M+H]+.
Compound 3. The linear peptide 3 was achieved in 46% yield using 100 mg of ChemMatrix Rink Amide resin and following the general procedures for peptide synthesis and HPLC purification. Fmoc-Pra-OH (Fmoc-propargyl-Gly-OH) and Fmoc-Lys(N3)–OH were used in i and i+4 relative positions in place of Ala and Phe of the reference peptide 1, respectively. HPLC: 92% purity, rt = 10.22 min; ESI-MS: m/z 1179.43 [M − H]-.
Compound 4. The triazole-stapled peptide 4 was achieved in 12% yield using 100 mg of ChemMatrix Rink Amide resin and following the general procedures for peptide synthesis and HPLC purification. Fmoc-Pra-OH (Fmoc-propargyl-Gly-OH) and Fmoc-Lys(N3)–OH were used in i and i+4 relative positions in place of Ala and Phe of the reference peptide 1, respectively. The cyclization was carried out on resin and before the deprotection of the last Fmoc-amino acid. In a test tube, Cu(I)Br and sodium ascorbate were dissolved in a DMF/DMSO, then DIPEA and 2,6-Lutidine were added. This solution was then transferred to the reactor containing the peptide resin and nitrogen was bubbled into the mixture for 5 min at room temperature and then heated under microwave irradiation at 55 °C for 10 min, followed by resin washing by DMF (x2), CH2Cl2 (x2) and DMF (x2). Final Fmoc deprotection and peptide cleavage from the resin was carried out as described in the general procedure for peptide synthesis. HPLC: 99% purity, rt = 8.64 min; ESI-MS: m/z 1181.47 [M+H]+.
Compound 5. The linear peptide 5 was achieved in 35% yield using 100 mg of ChemMatrix Rink Amide resin and following the general procedures for peptide synthesis and HPLC purification. Fmoc-(S)-2-(4-pentenyl)alanine was used in i and i+4 relative positions in place of Ala and Phe of the reference peptide 1. HPLC: 89% purity, rt = 12.27 min; ESI-MS: m/z 1210.41 [M+H]+.
Compound 6. The RCM-stapled peptide 6 was achieved in 15% yield using 100 mg of ChemMatrix Rink Amide resin and following the general procedures for peptide synthesis and HPLC purification. Fmoc-(S)-2-(4-pentenyl)alanine was used in i and i+4 relative positions in place of Ala and Phe of the reference peptide 1. The cyclization was carried out on resin and before the deprotection of the last Fmoc-amino acid. Initially, the resin was washed 3 times with CH2Cl2. Then 5 mL (maximum capacity of the reactor used) of 1,2-dichloroethane (DCE) in the reactor and the solution was bubbled with nitrogen. After 15 min, 15 mg (amount for 0.05 mmol resin-bound peptide) of 1st-generation Grubbs catalyst was added and the solution was bubbled with nitrogen for 4 h. Then, the solution was filtered, and the resin was successively washed with MeOH (x2), CH2Cl2 (x2) and MeOH (x2). Final Fmoc deprotection and peptide cleavage from the resin were carried out as described in the general procedure for peptide synthesis. HPLC: 94% purity, rt = 11.67 min; ESI-MS: m/z 1181.50 [M − H]-.
4.3. Circular dichroism spectroscopy
Selected peptide molecules were analyzed with respect to the secondary structure by means of circular dichroism spectroscopy (CD). The spectra were acquired at the Jasco j-810 spectropolarimeter, in the range from 190 nm to 250 nm. Samples were prepared at a concentration of 500 and 100 μM in phosphate-buffered saline (PBS) solution.
4.4. SARS-CoV-2 Spike/hACE2 inhibitor screening assays
The synthesized peptides were screened for inhibition of the SARS-CoV-2 Spike/hACE2 complex formation using a commercially available SARS-CoV-2 Spike Inhibitor Screening Assay Kit (AdipoGen Life Sciences, Inc., San Diego, USA) based on the colorimetric ELISA assay, which measures the binding of the RBD of the Spike S protein from SARS-CoV-2 to its human receptor ACE2. All the measurements were performed in triplicates in 96-well plates following the manufacturer's instructions. Briefly, the wells were coated by adding 100 μL/well of diluted SPIKE (1 μg/mL) to the 96-well ELISA microplate. After leaving the covered plate overnight at 4 °C, the liquid was removed by the wells by inverting the plate and blotting it against clean absorbent paper. Then, the plate was blocked by adding 200 μL of blocking buffer for 2 h at room temperature. Following liquid removal and washing of coated wells, 100 μL/well of diluted compounds to be tested were added to the wells and the plate covered and incubated for 1 h at 37 °C. 100 μL/well of inhibitory control ACE2 (human) mAb was used as a positive control. Then, 100 μL/well of diluted HRP labeled streptavidin was added and the plate covered and incubated again for 1 h at room temperature. After liquid removal and subsequent washings, substrate development was conducted by addition of 100 μL of ready-to-use TMB to each well for 5 min at room temperature, then the reaction was stopped by adding 50 μL of Stop Solution, the OD values measured at 450 nm using a BMG Labtech Fluostar Optima microplate reader and the collected data were analyzed using Graphpad 5.0 Software Package (Graphpad Prism, San Diego, CA). All the compounds were screened for inhibition at a single concentration (30 μM) in PBS, and the IC50 value of the active compound was obtained by dose-response measurements using inhibitor range of concentration 0.01–300 μM.
4.5. Plasma stability assay
Analyses were performed on a HPLC LC-20AD XR Prominence (Shimadzu Corporation, Kyoto, Japan). The system was interfaced with a 4000 Qtrap mass spectrometer (ABSciex, Framingham, MA, U.S.A.), equipped by an electrospray ion source and ESI configuration consisting of ion source temperature 260 °C, declustering potential 155 V, curtain gas 20 psi, ion source gas 1 55 psi, ion source gas 2 60 psi and ion spray voltage 5500 V. Chromatographic separation was achieved on a XDB-C18 (50 mm × 4.6 mm, 1.8 μm) (Agilent, Santa Clara, CA, USA) column thermostated at 25 °C with an operating flow rate of 0.5 mL/min. The elution gradient program was obtained using a mobile phase A (water with 0.1% formic acid) and a mobile phase B (acetonitrile with 0.1% of formic acid) provides the following program: 0–1 min (90% A), 1–5 min (90-10% A), 5–6 min (10% A), 6–6.5 (10–90% A) and 6.5–9.5 (90% A). The analysis by LC-MS/MS was performed in Multiple Reaction Monitoring (MRM) mode, in positive polarity with a dwell time of 50 ms, using m/z 1151 → 1007 and 1151 → 843 and 1183 → 1037 and 1183 → 875 as qualifier ions and 1151 → 1135 and 1183 → 1167 as quantifier ions respectively for 1 and 6. Data were acquired using Analyst 1.5.1 software and were processed using Multiquant 2.1.1 software. Human plasma pools were used for calibrators and controls preparation and for stability assay of peptides. Two samples were prepared by spiking freshly collected human plasma with an amount of the working solutions respectively of 1 and 6 to obtain a 200 μg/mL final concentration. The obtained samples were incubated at 37 °C for up to 48 h. At each time point (0, 10, 20, 30, 60, 120, 240, 480, 1440, 2880 min) 25 μL of the samples were picked up in triplicate, added with 125 μL of a mixture of the Internal Standard and a zinc sulfate solution in H2O:MeOH (1:4), vortexed for 1 min, followed by centrifugation at 14000 rpm and 25 °C for 10 min. The supernatant was injected in LC-MS/MS system (40 μL). The calibration standards and the quality controls samples were prepared in the same way. The two peptides were used one as internal standard of the other. The percent remaining of the peptides after incubation in plasma was calculated using the following equation: % remaining = 100 x (Am (t)/Am (t0)), where Am (t) is the average of the peak areas of each peptide at appointed time and Am (t0) is the average of the peak area of each peptide at t0.
4.6. In silico calculations
Crystal structure of Spike RBD bound to ACE2 was prepared starting from the PDB entry 6M0J [40] and used for docking and molecular dynamics (MD) simulations. All the water molecules and ions were deleted. The complex was prepared by using the Structure Prepare module of the MOE software v.2020 [47] and all the glycosylation sites were removed. Subsequently, the system was protonated at pH = 7 (temperature of 300 K and salt concentration of 0.1 M) and energy-minimized within MOE, using the Amber10:EHT force field and the Born implicit solvation model [48,49], up to a gradient of 0.1 kcal/mol Å−2. Then ACE2 was deleted and the spike RBD alone was saved for docking simulations.
The wild type peptide (WT, 1) was generated by deleting ACE2 from the complex, except for the sequence H34-Q42 forming the peptide (H-HEAEDLFYQ-NH 2). The C-terminus was capped by amidation while the N-terminus was left uncapped. Compound 6 was generated starting from 1 by modifying residues A36 and F40 into the non-natural α-methyl amino acids (ABU, Fig. S1) stapled by a Z-4-octene chain linked to the α-carbon to give a S stereochemical configuration.
Parametrization of ABU. A conformational search for 6 was done by using the MOE LowModeMD method, with the MMFF94x force field [50] and the Born solvation model, to generate a pool of low-energy conformations to be used for charge parameterization. Backbone atoms were constrained during the search, while side chains were unconstrained. Rejection limit, iteration limit, MM iteration limit and conformation limit were set to 1000, 100000, 5000 and 100000, respectively. Charges were calculated using R.E.D. program [51] by averaging the RESP point charges derived from two spatial orientation of the lowest energy conformation and another low energy conformation differing in the side chain orientation. The file generated was subsequently used in LEaP and antechamber [45] to generate the parameters based on ff15ipq-m force field [46] (force field parameters for ABU are given in the SI).
Protein-protein docking. The system containing the Spike RBD alone was used to run docking simulation by using the protein-protein docking simulation tool of MOE, rigid body refinement and default setting. Pose seven for 1 and pose six for 6, resembling the binding mode of the original SpikeRBD:ACE2 complex, were then saved in complex with the Spike RBD, minimized and used as the starting point for MD simulations.
PepGaMD simulations. To evaluate the peptides binding mode, PepGaMD simulations [52] of the two systems containing SpikeRBD:1 and SpikeRBD:6 complexes were run by using AMBER20 [48,53] package and ff15ipq-m force field. Systems were solvated in a TIP3P water octahedral box extending up to 10 Å from the solute. Systems were relaxed by geometry minimization of hydrogen atoms first (1000 steps of steepest descent and 4000 steps of conjugate gradient) and solvent and hydrogen atoms later (2000 steps of steepest descent 3000 steps of conjugate gradient) applying a restrain of 50 kcal/mol on the other atoms. Two equilibration steps, 100 ps constant volume (NVT) followed by 100 ps constant pressure (NPT, Berendsen barostat), were run to equilibrate the solvent, restraining all non-solvent atoms (restrains applied = 50 kcal/mol). Subsequently, two more minimizations, 5000 steps each (2500 steepest descent and 2500 conjugated gradient), were performed by restraining the backbone atoms only. In the first simulation restrains were set to 25 kcal/mol, in the second they were reduced to 10 kcal/mol. These minimizations were followed by six steps of 5 ps long NVT simulations (Langevin thermostat) each, where temperature was increased from 0 K to 300 K (50 K of increase at each cycle). During these steps, backbone restrains were reduced from 10 to 5 kcal/mol. A NVT equilibration 100 ps long (Langevin thermostat, T = 300, SHAKE), maintaining the same restrains, was then performed to stabilize temperature, followed by 400 ps NPT simulations (Langevin thermostat, Berendsen barostat, T = 300, SHAKE) where the restrains were decreased to 2 kcal/mol. The last two steps of equilibration consisted of 500 ns long NPT simulations with the same protocol as above but reducing the restrains to 1 kcal/mol and then to 0 kcal/mol. At the end of the equilibration 20 ns of MD simulation were performed in NPT environment (300 K, 1 atm, Langevin thermostat, Berendsen barostat, SHAKE). The average potential energy of the peptide from the second half of this simulation was then used to calculate the boost to be applied to the 1 μs long PepGaMD simulation (igamd = 14) that followed. During the first 40 ns the boost was constant, then it updated automatically every 200 ps until the end of the simulation. Sigma0P and sigma0D were set to 4.0 and 6.0, respectively, as suggested in literature [51]. Simulations were performed by using pmemd.cuda on workstations equipped with GPU cards [54]. Geometrical analyses were performed by using cpptraj implemented in AmberTools20. The PepGaMD trajectories were reweighted using a 10th order Maclaurin series expansion, and a discretization of 1 for both X and Y dimension, to compute accurate potentials of mean force (PMF). Bidimensional and 3D plots were generated by using the corresponding PyReweighting scripts by Miao et al. [55], available at https://miaolab.ku.edu/PyReweighting/.
Binding energy calculations. A 4ns long NPT MD simulation was performed, after equilibration as described above, by starting from the lowest energy conformations predicted by PepGaMD (structures corresponding to the PMF = 0 kcal/mol from the reweighting procedure). Binding energies were then calculated by using the Nwat-MMGBSA method, keeping 0, 5, 30 or 60 explicit waters [56,57], and selecting 100 evenly spaced frames from the last nanosecond of trajectory. Cluster analyses were also performed by using cpptraj with the hierarchical agglomerative clustering method. We requested to save 10 clusters, using RMSD calculated on backbone atoms as the metrics (average linker).
REMD simulations. To evaluate the folding behaviour of unbounded peptides, 200 ns of Temperature Replica Exchange MD (REMD) [58] were performed by using ff99SBildn force field, GBNeck2 implicit solvation model for water as suggested in previous works [[59], [60], [61]]. Twelve parallel MD replicas were performed at different temperatures (300.0 K, 330.69 K, 364.36 K, 401.31 K, 441.89 K, 486.42 K, 535.31 K, 588.95 K, 647.82 K, 712.41 K, 783.26 K, 860.93 K) and cluster analysis and DSSP calculation were performed on the 300.0 K trajectory using cpptraj.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
“Fondo di Beneficenza ed opere di carattere sociale e culturale di Intesa Sanpaolo”, project B/2020/0125 “Sviluppo di peptidomimetici inibitori dell'interazione ACE2-proteina Spike S per il trattamento di infezioni da Coronavirus”. Alessandro Contini and Crescenzo Coppa thank Fondazione Invernizzi for their financial support through the LIB_FOND_COVID_19_01 project. We thank Prof. Lucia Banci for support to circular dichroism studies. Prof. Antonio Cassone is gratefully acknowledged for insightful references on SARS-CoV-2 biology and mechanism.
Footnotes
Supplementary data to this article (copies of HPLC chromatograms and ESI-MS spectra for compounds 1–6, 1H NMR spectra of 1, 3–6 in D2O, 1H-water-ES and TOCSY-ES/ROESY-ES spectra and proton assignment tables for 1, 4, 6, LC-MS/MS chromatograms and calibration curves for 1 and 6, and in silico calculation data) can be found online at https://doi.org/10.1016/j.ejmech.2023.115118.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
Data availability
Data will be made available on request.
References
- 1.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO, Coronavirus disease (COVID-19) pandemic. https://www.who.int/emergencies/diseases/novel-coronavirus-2019
- 4.Worldometer, Cases Coronavirus. https://www.worldometers.info/coronavirus/coronavirus-cases/#daily-cases
- 5.Callaway E. Beyond Omicron: what's next for COVID's viral evolution. Nature. 2021;600:204–207. doi: 10.1038/d41586-021-03619-8. [DOI] [PubMed] [Google Scholar]
- 6.Robinson P.C., Liew D.F.L., Tanner H.L., Grainger J.R., Dwek R.A., Reisler R.B., Steinman L., Feldmann M., Ho L.-P., Hussell T., Moss P., Richards D., Zitzmann N. COVID-19 therapeutics: challenges and directions for the future. Proc. Natl. Acad. Sci. U.S.A. 2022;119 doi: 10.1073/pnas.2119893119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu Y., Xu K. Functional studies of the coronavirus nonstructural proteins. STEMedicine. 2020;1:e39. [Google Scholar]
- 8.Vandyck K., Deval J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021;49:36–40. doi: 10.1016/j.coviro.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Painter G.R., Natchus M.G., Cohen O., Holman W., Painter W.P. Developing a direct acting, orally available antiviral agent in a pandemic: the evolution of molnupiravir as a potential treatment for COVID-19. Curr. Opin. Virol. 2021;50:17–22. doi: 10.1016/j.coviro.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J. Med. Chem. 2020;63:12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., Müller M.A., Drosten C., Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson C.B., Farzan M., Chen B., Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021:1–18. doi: 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor P.C., Adams A.C., Hufford M.M., de la Torre I., Winthrop K., Gottlieb R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021;21:382–393. doi: 10.1038/s41577-021-00542-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benton D.J., Wrobel A.G., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588:327–330. doi: 10.1038/s41586-020-2772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheng C., Dong G., Miao Z., Zhang W., Wang W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015;44:8238–8259. doi: 10.1039/c5cs00252d. [DOI] [PubMed] [Google Scholar]
- 18.Geppert T., Hoy B., Wessler S., Schneider G. Context-based identification of protein-protein interfaces and "hot-spot" residues. Chem. Biol. 2011;18:344–353. doi: 10.1016/j.chembiol.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Zhang G., Pomplun S., Loftis A.R., Tan X., Loas A., Pentelute B.L. 2020. Investigation of ACE2 N-Terminal Fragments Binding to SARS-CoV-2 Spike RBD. bioRxiv. 2020.03.19.999318. [DOI] [Google Scholar]
- 20.Karoyan P., Vieillard V., Gómez-Morales L., Odile E., Guihot A., Luyt C.-E., Denis A., Grondin P., Lequin O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 2021;4:197. doi: 10.1038/s42003-021-01736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lenci E., Trabocchi A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020;49:3262–3277. doi: 10.1039/d0cs00102c. [DOI] [PubMed] [Google Scholar]
- 22.Ali A.M., Atmaj J., Van Oosterwijk N., Groves M.R., Dömling A. Stapled peptides inhibitors: a new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019;17:263–281. doi: 10.1016/j.csbj.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charoenpattarapreeda J., Tan Y.S., Iegre J., Walsh S.J., Fowler E., Eapen R.S., Wu Y., Sore H.F., Verma C.S., Itzhaki L., Spring D.R. Targeted covalent inhibitors of MDM2 using electrophile-bearing stapled peptides. Chem. Commun. 2019;55:7914–7917. doi: 10.1039/c9cc04022f. [DOI] [PubMed] [Google Scholar]
- 24.Dougherty P.G., Wen J., Pan X., Koley A., Ren J.G., Sahni A., Basu R., Salim H., Appiah Kubi G., Qian Z., Pei D. Enhancing the cell permeability of stapled peptides with a cyclic cell-penetrating peptide. J. Med. Chem. 2019;62:10098–10107. doi: 10.1021/acs.jmedchem.9b00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowell J.K., Teng Y., Bendzunas N.G., Ara R., Arbab A.S., Kennedy E.J. Suppression of breast cancer metastasis using stapled peptides targeting the WASF regulatory complex. Cancer Growth Metastasis. 2017;10 doi: 10.1177/1179064417713197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dietrich L., Rathmer B., Ewan K., Bange T., Heinrichs S., Dale T.C., Schade D., Grossmann T.N. Cell permeable stapled peptide inhibitor of wnt signaling that targets β-catenin protein-protein interactions. Cell Chem. Biol. 2017;24:958–968.e5. doi: 10.1016/j.chembiol.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Gaillard V., Galloux M., Garcin D., Eléouët J.F., Le Goffic R., Larcher T., Rameix-Welti M.A., Boukadiri A., Héritier J., Segura J.M., Baechler E., Arrell M., Mottet-Osman G., Nyanguile O. A short double-stapled peptide inhibits respiratory syncytial virus entry and spreading. Antimicrob. Agents Chemother. 2017;61 doi: 10.1128/AAC.02241-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cromm P.M., Spiegel J., Grossmann T.N. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015;10:1362–1375. doi: 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- 29.Verdine G.L., Hilinski G.J. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 30.Maas M.N., Hintzen J.C.J., Löffler P.M.G., Mecinović J. Targeting SARS-CoV-2 spike protein by stapled hACE2 peptides. Chem. Commun. 2021;57:3283–3286. doi: 10.1039/d0cc08387a. [DOI] [PubMed] [Google Scholar]
- 31.Curreli F., Victor S.M.B., Ahmed S., Drelich A., Tong X., Tseng C.K., Hillyer C.D., Debnath A.K. Stapled peptides based on human angiotensin-converting enzyme 2 (ACE2) potently inhibit SARS-CoV-2 infection in vitro. mBio. 2020;11 doi: 10.1128/mBio.02451-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan D.C., Morris C., Mahindra A., Blair C.M., Tejeda G., Herbert I., Turnbull M.L., Lieber G., Willett B.J., Logan N., Smith B., Tobin A.B., Bhella D., Baillie G., Jamieson A.G. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization. Pept. Sci. 2021 doi: 10.1002/pep2.24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tedesco F., Calugi L., Lenci E., Trabocchi A. Peptidomimetic small-molecule inhibitors of 3CLPro activity and spike–ACE2 interaction: toward dual-action molecules against coronavirus infections. J. Org. Chem. 2022;87:12041–12051. doi: 10.1021/acs.joc.2c01047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larue R.C., Xing E., Kenney A.D., Zhang Y., Tuazon J.A., Li J., Yount J.S., Li P.-K., Sharma A. Rationally designed ACE2-derived peptides inhibit SARS-CoV-2. Bioconj. Chem. 2021;32:215–223. doi: 10.1021/acs.bioconjchem.0c00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedersen S.L., Tofteng A.P., Malik L., Jensen K.J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 2012;41:1826–1844. doi: 10.1039/c1cs15214a. [DOI] [PubMed] [Google Scholar]
- 36.Subirós-Funosas R., Prohens R., Barbas R., El-Faham A., Al-bericio F. Oxyma: an efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion. Chem. Eur J. 2009;15:9394–9403. doi: 10.1002/chem.200900614. [DOI] [PubMed] [Google Scholar]
- 37.D'Ercole A., Sabatino G., Pacini L., Impresari E., Capecchi I., Papini A.M., Rovero P. On-resin microwave-assisted copper-catalyzed azide-alkyne cycloaddition of H1-relaxin B single chain ‘stapled’ analogues. Pept. Sci. 2020;112 [Google Scholar]
- 38.Barragán F., Moreno V., Marchán V. Solid-phase synthesis and DNA binding studies of dichloroplatinum(ii) conjugates of dicarba analogues of octreotide as new anticancer drugs. Chem. Commun. 2009;31:4705–4707. doi: 10.1039/b909698a. [DOI] [PubMed] [Google Scholar]
- 39.Hwang T.L., Shaka A.J. Water suppression that works. excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson., Ser. A. 1995;112:275–279. [Google Scholar]
- 40.Oueis E., Jaspars M., Westwood N.J., Naismith J.H. Enzymatic macrocyclization of 1,2,3-triazole peptide mimetics. Angew Chem. Int. Ed. Engl. 2016;55:5842–5845. doi: 10.1002/anie.201601564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schafmeister C.E., Po J., Verdine G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000;122:5891–5892. [Google Scholar]
- 42.Ueda A., Makura Y., Kakazu S., Kato T., Umeno T., Hirayama K., Doi M., Oba M., Tanaka M. E-selective ring-closing metathesis in α-helical stapled peptides using carbocyclic α,α-disubstituted α-amino acids. Org. Lett. 2022;24:1049–1054. doi: 10.1021/acs.orglett.1c04256. [DOI] [PubMed] [Google Scholar]
- 43.Biondi B., Casciaro B., Di Grazia A., Cappiello F., Luca V., Crisma M., Mangoni M.L. Effects of Aib residues insertion on the structural-functional properties of the frog skin-derived peptide esculentin-1a(1-21)NH2. Amino Acids. 2017;49:139–150. doi: 10.1007/s00726-016-2341-x. [DOI] [PubMed] [Google Scholar]
- 44.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 45.Wang J., Wang W., Kollman P.A., Case D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Bogetti A.T., Piston H.E., Leung J.M.G., Cabalteja C.C., Yang D.T., Degrave A.J., Debiec K.T., Cerutti D.S., Case D.A., Horne W.S., Chong L.T. A twist in the road less traveled: the AMBER ff15ipq-m force field for protein mimetics. J. Chem. Phys. 2020;153 doi: 10.1063/5.0019054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molecular Operating Environment (Moe) Chemical Computing Group ULC; Montreal: 2020. 09; Chemical Computing Group ULC, Montreal, Canada. Canada 2020. [Google Scholar]
- 48.Case D.A., Ben-Shalom I.Y., Brozell S.R., Cerutti D.S., Cheatham T.E., Cruzeiro V.W.D., III, D T.A., Duke R.E., Ghoreishi D., Gilson M.K., Gohlke H., Goetz A.W., Greene D., Harris R., Homeyer N., H Y., Izadi S., Kovalenko A., Kurtzman T., Lee T.S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., J D., Mermelstein K.M., Merz Y., Miao G., Monard C., Nguyen H., Nguyen I., Omelyan A., Onufriev F., Pan R., Qi D.R., Roe A., Roitberg C., Sagui S., Schott-Verdugo J., Shen C.L., Simmerling J., Smith R., Salomon Ferrer J., Swails R.C., Walker J., Wang H., Wei R.M., Wolf X., Wu L., Xiao D.M.Y., K P.A. University of California; San Francisco: 2020. AMBER 2020. [Google Scholar]
- 49.Onufriev A.V., Case D.A. Generalized Born implicit solvent models for biomolecules. Annu. Rev. Biophys. 2019;48:275–296. doi: 10.1146/annurev-biophys-052118-115325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halgren T.A. Merck molecular force field. J. Comput. Chem. 1996;17:61–67. [Google Scholar]
- 51.Dupradeau F.Y., Pigache A., Zaffran T., Savineau C., Lelong R., Grivel N., Lelong D., Rosanski W., Cieplak P. The R.E.D. Tools: advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010;12:7821–7839. doi: 10.1039/c0cp00111b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J., Miao Y. Peptide Gaussian accelerated molecular dynamics (Pep-GaMD): enhanced sampling and free energy and kinetics calculations of peptide binding. J. Chem. Phys. 2020;153 doi: 10.1063/5.0021399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber P.R., Müller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 1995;9(3):251–268. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- 54.Kaus J.W., Pierce L.T., Walker R.C., McCammon J.A. Improving the efficiency of free energy calculations in the amber molecular dynamics package. J. Chem. Theor. Comput. 2013;9:4131–4139. doi: 10.1021/ct400340s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miao Y., Sinko W., Pierce L., Bucher D., Walker R.C., McCammon J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theor. Comput. 2014;10:2677–2689. doi: 10.1021/ct500090q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maffucci I., Hu X., Fumagalli V., Contini A. An efficient implementation of the nwat-MMGBSA method to rescore docking results in medium-throughput virtual screenings. Front. Chem. 2018;6:43. doi: 10.3389/fchem.2018.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maffucci I., Contini A. Improved computation of protein-protein relative binding energies with the nwat-MMGBSA method. J. Chem. Inf. Model. 2016;56:1692–1704. doi: 10.1021/acs.jcim.6b00196. [DOI] [PubMed] [Google Scholar]
- 58.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 59.Maffucci I., Pellegrino S., Clayden J., Contini A. Mechanism of stabilization of helix secondary structure by constrained cα-tetrasubstituted α-amino acids. J. Phys. Chem. B. 2015;119:1350–1361. doi: 10.1021/jp510775e. [DOI] [PubMed] [Google Scholar]
- 60.Maffucci I., Clayden J., Contini A. Origin of helical screw sense selectivity induced by chiral constrained cα-tetrasubstituted α-amino acids in aib-based peptides. J. Phys. Chem. B. 2015;119:14003–14013. doi: 10.1021/acs.jpcb.5b07050. [DOI] [PubMed] [Google Scholar]
- 61.Maffucci I., Contini A. An updated test of AMBER force fields and implicit solvent models in predicting the secondary structure of helical, β-hairpin, and intrinsically disordered peptides. J. Chem. Theor. Comput. 2016;12:714–727. doi: 10.1021/acs.jctc.5b01211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.