

Abstract
Out of BCR‐ABL negative myeloproliferative neoplasm (MPNPh−) patients, 3%–14% display a concomitant monoclonal gammopathy of unknown significance (MGUS). In most cases, the diagnosis of plasma cell dyscrasia is either synchronous with that of MPNPh− or occurs later on. We present a 50‐year‐old patient with type 2 CALR Lys385Asnfs*47 mutation positive essential thrombocythemia (ET) who developed symptomatic multiple myeloma (MM) 13 years after the diagnosis of ET during PEG‐INF2α treatment. The NGS study performed at the time of the MM diagnosis revealed the HRAS Val14Gly/c.41T〉G mutation and the wild type CALR, JAK2 and MPL gene sequence. In the presented case, the complete molecular remission of ET was achieved after 16 months of PEG‐INF2α treatment. The origin of MM cells in MPNPh− patients remains unknown. Published data suggests that type 2 CALRins5 up‐regulate the ATF6 chaperone targets in hematopoietic cells and activate the inositol‐requiring enzyme 1α‐X‐box‐binding protein 1 pathway of the unfolded protein response (UPR) system to drive malignancy. It cannot be excluded that endoplasmic reticulum stress induced by the increased ATF6 resulted in an abnormal redox homeostasis and proteostasis, which are factors linked to MM. The presented case history and the proposed mechanism of mutant CALR interaction with UPR and/or ATF6 should initiate the discussion about the possible impact of the mutant CALR protein on the function and genomic stability of different types of myeloid cells, including progenitor cells.
Keywords: CALR, essential thrombocythemia, HRAS, multiple myeloma
Out of BCR‐ABL negative myeloproliferative neoplasm (MPNPh−) patients, 3%–14% display a concomitant monoclonal gammopathy (MGUS). 1 , 2 The diagnosis of MPN proceeds lymphoproliferative disease (LPD) occurrence in about 50% of patients. The median time between the diagnosis of MPNPh− and LPD was established at 72 months. 3 Herein, we present an unusual outcome of low risk essential thrombocythemia (ET), diagnosed in 2008 in a 50‐year‐old woman. Initially, she was treated with hydroxyurea (HC) 1.5 g/daily orally. Her medical history revealed mitral and aortal valve insufficiency, which was diagnosed in 2018, and the episode of transient ischaemic attack (2018), resulting in foci cerebral ischaemia (MRI). In 2019, due to toxicity, the HC treatment was stopped. The therapy with the pegylated interferonα 2a (Pegasys, PEG‐INFα2a) was started in August 2020. The molecular work‐up showed no JAK2 V617F and MPL exon 10 mutation and the presence of the type 2 CALR mutation‐ LRG_828t1:c.1154_1155insTTGTC, LRG_828p1:p.(Lys385Asnfs*47) (VAF 35%). The bone marrow (BM) biopsy performed in 2020 showed the normocellular BM, locally hypercellular, with an erythroid/granulocytic cell ratio of 1:3, and normal erythroid and granulocytic proliferation index and maturation pattern. The blast cell (CD34+, CD117+) content was determined at 1%–2% of nuclear cells. An increased number of medium‐sized and large megakaryocytes (factor VIII+) was found, without the tendency to form clusters (locally forming loose clusters consisting of 4–8 cells). The BM fibrosis (MF) grade according to the European consensus criteria was 0/1.
In September 2021, she experienced a fracture of the L1 vertebrae of the spine which was treated with Th12‐L1 stabilization with laminectomy. The PET‐CT scan revealed multiple osteolytic lesions in the bones. Immunofixation studies showed the IgG kappa monoclonal protein in the blood at the concentration of 49.6 g/L. The WBS performed in January 2022 documented the fracture of Th9, the presence of an intramedullary tumour in the bottom part of the stern. The MFC of the BM cells showed 65% of abnormal plasma cells. The repeated biopsy documented BM cellularity 60% with about 30% of dispersed plasma cells, locally forming infiltrates. Erythroid cell line content e‐cadherin+ was determined at 30%. The granulocytic cell [CD15+, MPO+] line and megakaryocyte (factor VIII+, CD61+) evaluation showed a normal morphology and maturation pattern. MF +1. Retrospective analysis of a biopsy sample collected in 2020 (before PEG‐INFα2a treatment initiation) confirmed the presence of a small, abnormal plasma cell population with the same immunophenotypic characteristic (Figure 1).
FIGURE 1.
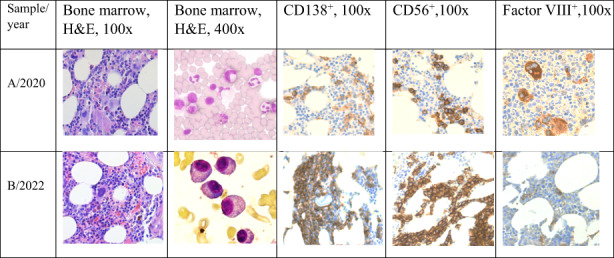
The bone marrow (BM) trephine biopsy sample histopathological evaluation results in type 2 CALR mutation positive essential thrombocythemia patient developing multiple myeloma during disease outcome. Multiple myeloma cells immunophenotype: CD138++, MUM1+, CD56, CD20−, cyclin D1−, Ig‐CISH‐kappa+++/Ig‐lambda‐.
An NGS analysis of the BM cells was performed using a sample collected in January 2022, before MM treatment initiation. The targeted enrichment approach with a custom‐designed gene panel revealed the presence of the Harvey rat sarcoma (HRAS) gene mutation (chr11:000534282‐A > C VAF 14% NM_001130442.1:p.Val14Gly/c.41 T > G) and the wild type of CALR, JAK2 and MPL gene sequence (Figure S1). The analysis of the blood sample collected at the time of PEG‐INF2α treatment initiation using the SS did not confirm the presence of the HRAS variant. The HRAS mutations were also undetectable in the DNA obtained from the blood MNC and buccal swab cells collected immediately before MM treatment initiation (Figure S1).
After the diagnosis of MM, the treatment with bortezomib, thalidomide and dexamethasone (VTD) was introduced. After three VTD therapy cycles, a significant reduction of the protein M concentration in the blood from 49.6 g/L to 3.6 g/L was confirmed (Figure 2).
FIGURE 2.
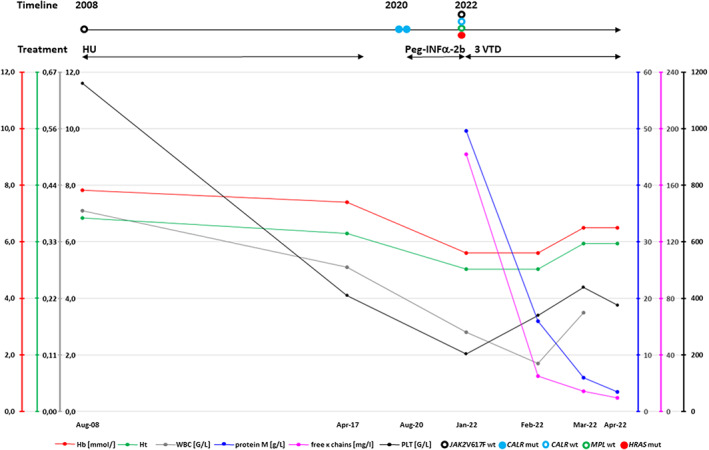
The changes in the basic laboratory parameters in a patient with type 2 CALR mutation positive essential thrombocythemia developing multiple myeloma during disease outcome. The timeline and treatment type are shown.
In the presented case, the mechanism of the CALR mutation positive clone disappearance is not clear. It cannot be ruled out that the PEG‐INFα2a treatment resulted in HSC exhaustion and CALR mutation positive clone eradication. The mentioned hypothesis is in agreement with the published data confirming that INFα treatment resulted in complete molecular remission (CMR) in 10% of ET patients. 4 It is probable that PEG‐INFα targets the type 2 CALR mutation positive haematopoietic stem and progenitor cells preferentially, as was documented by Mosca et al. 5
The published data suggest that the CALRins5 (type 2) in comparison to the CALRdel52 (type 1) differentially activate the inositol‐requiring enzyme 1α (IRE1α)‐X‐box‐binding protein 1 (XBP1) pathway of the unfolded protein response system to drive malignancy. The up‐regulation of the ATF6 chaperone targets specifically in CALRins5 cells suggests that the loss of the chaperone function by CALRins5 may underlie the differential activation of ATF6. 6 In 2019, Salati et al. demonstrated that CALR mutations induce increased sensitivity to oxidative stress, leading to the increase of oxidative DNA damage and the down‐modulation of the oxidation resistance gene 1 in CALR‐mutated cells. 7 The experimental study of bone marrow cells confirmed the presence of the CALR mutation even in multipotent precursors (MPP) of lymphohematopoiesis. 8 Therefore, it is possible that the RE stress due to the CALRins5 in MPP promotes oncogenesis via the abnormal function of IRE1a‐XBP1 axis, even in MPP. Another probable mechanism responsible for secondary malignancies development in MPNPh‐ includes immunosuppressive effects mediated by exon‐9‐mutated CALR released from the malignant cell, resulting in the inhibition of phagocytosis of dying cancer cells by dendritic cells. 9 The above‐mentioned data suggest a possible association between the CALR mutation driven malignancies and an increased risk of secondary neoplasms, including MM. In the systematic review done by Marchetti et al., the coexistence of the type 1 CALRdel52 mutation positive ET and MGUS was reported in 3 patients. 3 Loscocco et al. described a CALRdel52 mutation positive (VAF 57%) ET case who was diagnosed with smouldering MM 1 year after the diagnosis of ET. Four years later, the progression to post‐ET myelofibrosis was documented. After another 2 years, the MGUS evolution to the IgG lambda MM was observed. 10
The occurrence of MM is rare in JAK2 mutation positive PV patients and no case of an MM diagnosis preceding an MPN one has been reported. It was shown that 9% of patients with MPN (36.0% of ET) harboured an M‐protein. ET‐MGUS (IgGκ and IgMκ) was diagnosed in 20% of ET patients, including cases with CALR type 1 and type 2 mutations. 11 The frequency of the coexistence of MM with ET was established at 15%. 3 The mentioned data are in agreement with a previous report documenting the direct role of calnexin, calreticulin, and tapasin abnormalities in the MGUS progression to MM. 12
The presented case is the first reported HRAS Val14Gly/c.41 T > G variant positive haematological malignancy. The HRAS variant detected in our patient had been reported only once in Clinvar (accession number VCV000654373.1) in the case of Costello Syndrome. It is likely pathogenic according to Clinvar and of uncertain significance according to Varsome. The Functional Analysis Through Hidden Markov Models (FATHMM‐MKL) categorizes this change as damaging with a 0.99 coding score. Algorithms developed to predict the effect of missense changes on protein structure and function (SIFT, PolyPhen‐2, Align‐GVGD) all suggest that this variant is likely to be disruptive, but these predictions have not been confirmed by published functional studies (Clinvar).
The presence of NRAS and KRAS mutations was confirmed in 8% and 4% of myeloid malignancies. It was documented that missense mutations at codons 12 and 13 are thought to limit GTPase‐activating protein (GAP) interaction with the GTPase site of RAS proteins, preventing their hydrolysis to an inactive state. It was previously documented that mutations of RAS family genes contribute to myelomagenesis, including the transformation from MGUS to MM. 13 In MM patients, the presence of the NRAS and KRAS gene mutations was confirmed in 19.5% and 24.2% of patients, respectively. 14 The data concerning HRAS mutations in myeloid disorders are limited. The role of HRAS gene mutations in the process of the malignant transformation of plasma cells also remains unknown. In the report of O'Donnell et al., a HRAS mutation [c.181 C > A (p.Q61K)] was identified in 1/67 of patients with MM. 15 Frontzek et al. 16 confirmed the presence of HRAS mutations in 2% of patients with plasmablastic lymphoma.
One still unsolved mystery is that of the origin of MM cells in patients with MPNPh‐. The available data suggest that hydroxyurea or interferonα treatments do not increase the risk of secondary cancers. In the summary of the published case reports and studies prepared by Malhotra et al., it has not yet been definitively shown that these 2 entities arise from a common‐ancestor HSPC. 1 In 2021, Hui W. et al. identified eight proteins that were found to be dysregulated differently in ET patients with mutated CALRdel52 and those with JAK2V617F mutation. The proteomic analysis showed that 20 proteins were altered in ET with the CALRdel52 mutation: those involved in cell signalling (Erk1/2, PTEN, Raf‐B, Rap1, Axin, ERβ, TGF‐β), cell cycle (Cdc42, Cdc2, CyclinD1, p27), apoptosis (Bcl‐Xl, c‐IAP2, NFκB p50, cPKCα, Survivin), transcription factor (eIF4B, SRC‐1), adhesion (E‐cadherin) and DNA repair (TDP1). 17 Similar data, concerning CALR type 2 mutation detected in our patient is not available yet. Therefore, we performed an additional review of The Cancer Genome Atlas (TCGA) database, including datasets of >10,000 samples of 33 cancer types, searching for a possible co‐occurrence of the CALR driver mutations and mutations in the RAS family genes (HRAS, NRAS, and KRAS). We did not find any sample with co‐occurring CALR type 1 or type 2 mutations and mutations in the RAS family genes. The only few CALR mutations co‐occurring with mutations in the RAS genes seem to be random, they were mostly missense mutations randomly distributed in different positions of the CALR gene, detected mostly in cancers (UCEC and COAD) known to be frequently hypermutated due to different mutagenic processes and aberrations in DNA repair machinery (Table S1). We realize that our hypotheses have considerable limitations, as more studies (including functional studies) are necessary to explore the link between the CALR‐driven malignancies and secondary cancer development and origin. For this reason, the precise mechanism responsible for CALRmut‐driven oncogenesis remains unknown and is probably multifactorial.
The question is that of the medical significance of the presented data. In our opinion, more attention should be paid to the monitoring of MPN patients for the presence of MGUS, to allow for an early diagnosis of MM and the initiation of an appropriate treatment. 10 Still, the study of the genetic aberration profile in MPNPh‐patients with coexisting MM is needed to precisely define and attack common molecular target(s) responsible for oncogenesis. 18
AUTHOR CONTRIBUTIONS
Agata Lehmann‐Kopydłowska: Data curation (equal); resources (equal); writing – review and editing (supporting). Zuzanna Kanduła: Data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); visualization (equal); writing – review and editing (equal). Bartłomiej Sankowski: Investigation (equal); writing – review and editing (equal). Marcin Machnicki: Investigation (equal); writing – review and editing (equal). Marta Barańska: Data curation (equal); writing – review and editing (equal). Kinga Gwóźdź‐Bąk: Data curation (equal); investigation (equal); writing – review and editing (equal). Tadeusz Kubicki: Data curation (equal); methodology (equal); writing – review and editing (equal). Anna Płotka: Data curation (equal); investigation (supporting); writing – review and editing (equal). Łucja Przysiecka: Investigation (equal); resources (equal); writing – review and editing (equal). Grzegorz Dworacki: Investigation (equal); writing – original draft (supporting); writing – review and editing (equal). T. Stoklosa: Investigation (equal); methodology (equal); resources (equal); writing – original draft (supporting); writing – review and editing (equal). Piotr Kozlowski: Formal analysis (supporting); validation (supporting); writing – review and editing (supporting).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
INFORMED CONSENT
The study was approved by the Bioethics Committee of the Poznan University of Medical Sciences, Poland (nos. 1056/16, 181/18, 846/21) and was conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from the patient enrolled in this study. The samples were anonymised and then analysed.
Supporting information
FigureS1
TableS1
ACKNOWLEDGEMENT
This work was supported by HARMONIA grant from the National Science Center UMO‐2014/14/M/NZ5/00441 (T. Stoklosa).
Krzysztof L, Agata K, Zuzanna K, et al. HRAS mutation positive multiple myeloma in the type 2 CALR mutation positive essential thrombocythemia: A case report. J Cell Mol Med. 2023;27:299‐303. doi: 10.1111/jcmm.17647
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Malhotra J, Kremyanskaya M, Schorr E, Hoffman R, Mascarenhas J. Coexistence of myeloproliferative neoplasm and plasma‐cell dyscrasia. Clin Lymphoma Myeloma Leuk. 2014. [cited 2022 Apr 16];14(1):31‐36. https://pubmed.ncbi.nlm.nih.gov/24220620/ [DOI] [PubMed] [Google Scholar]
- 2. Le Clech L, Sakka M, Meskar A, et al. The presence of monoclonal gammopathy in Ph‐negative myeloproliferative neoplasms is associated with a detrimental effect on outcomes. Leuk Lymphoma. 2017. [cited 2022 Mar 13];58(11):2582‐2587. https://pubmed.ncbi.nlm.nih.gov/28482711/ [DOI] [PubMed] [Google Scholar]
- 3. Marchetti M, Carobbio A, Capitoni E, Barbui T. Lymphoproliferative disorders in patients with chronic myeloproliferative neoplasms: a systematic review. Am J Hematol. 2018. [cited 2022 Mar 13];93(5):698‐703. https://pubmed.ncbi.nlm.nih.gov/29377227/ [DOI] [PubMed] [Google Scholar]
- 4. Mondello P, Di Mirto C, Cuzzocrea S, Arrigo C, Mian M, Pitini V. Interferon alpha has a strong anti‐tumor effect in Philadelphia‐negative myeloproliferative neoplasms. Clin Lymphoma, Myeloma Leuk. 2019. [cited 2022 Apr 16];19(8):e489‐e495. http://www.clinical‐lymphoma‐myeloma‐leukemia.com/article/S2152265019300874/fulltext [DOI] [PubMed] [Google Scholar]
- 5. Mosca M, Hermange G, Tisserand A, et al. Inferring the dynamics of mutated hematopoietic stem and progenitor cells induced by IFNα in myeloproliferative neoplasms. Blood. 2021. [cited 2022 Mar 13];138(22):2231‐2243. https://pubmed.ncbi.nlm.nih.gov/34407546/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ibarra J, Elbanna Y, Kurylowicz K, et al. Type 1 calreticulin mutations differentially activate the IRE1α‐XBP1 pathway of the unfolded protein response to drive myeloproliferative neoplasms. Blood. 2021;138(Supplement 1):628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salati S, Genovese E, Carretta C, et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci Reports. 2019. [cited 2022 Mar 13];9(1):1‐14. https://www.nature.com/articles/s41598‐019‐46843‐z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mead AJ, Mullally A. Myeloproliferative neoplasm stem cells. Blood. 2017. [cited 2022 Oct 2];129(12):1607‐1616. https://pubmed.ncbi.nlm.nih.gov/28159736/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu P, Zhao L, Loos F, et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol Cell. 2020. [cited 2022 Oct 2];77(4):748‐760.e9. https://pubmed.ncbi.nlm.nih.gov/31785928/ [DOI] [PubMed] [Google Scholar]
- 10. Loscocco GG, Antonioli E, Romano I, et al. Lenalidomide: a double‐edged sword for concomitant multiple myeloma and post‐essential thrombocythemia myelofibrosis. Am J Hematol. 2021. [cited 2022 Mar 13];96(6):749‐754. https://pubmed.ncbi.nlm.nih.gov/33719069/ [DOI] [PubMed] [Google Scholar]
- 11. Javorniczky NR, Wehrle J, Ihorst G, et al. Prevalence and characteristics of myeloproliferative neoplasms with concomitant monoclonal gammopathy. Leuk Res. 2020. [cited 2022 Mar 13];98. https://pubmed.ncbi.nlm.nih.gov/32971364/ [DOI] [PubMed] [Google Scholar]
- 12. Racanelli V, Leone P, Frassanito MA, et al. Alterations in the antigen processing‐presenting machinery of transformed plasma cells are associated with reduced recognition by CD8+ T cells and characterize the progression of MGUS to multiple myeloma. Blood. 2010;115(6):1185‐1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chng WJ, Gonzalez‐Paz N, Price‐Troska T, et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008. [cited 2022 Mar 13];22(12):2280‐2284. https://pubmed.ncbi.nlm.nih.gov/18528420/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mulligan G, Lichter DI, Di BA, et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single‐agent bortezomib therapy. Blood. 2014. [cited 2022 Mar 13];123(5):632‐639. https://pubmed.ncbi.nlm.nih.gov/24335104/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Donnell E, Mahindra A, Yee AJ, et al. Clinical grade “SNaPshot” genetic mutation profiling in multiple myeloma. EBioMedicine. 2014. [cited 2022 Mar 13];2(1):71‐73. https://pubmed.ncbi.nlm.nih.gov/26137536/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frontzek F, Staiger AM, Zapukhlyak M, et al. Molecular and functional profiling identifies therapeutically targetable vulnerabilities in plasmablastic lymphoma. Nat Commun. 2021. [cited 2022 Mar 13];12(1):1‐14. https://www.nature.com/articles/s41467‐021‐25405‐w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hui W, Zhang W, Liu C, Wan S, Sun W, Su L. Alterations of signaling pathways in essential thrombocythemia with calreticulin mutation. Cancer Manag Res. 2021. [cited 2022 Oct 29];13:6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jutzi JS, Marneth AE, Jimenez‐Santos MJ, et al. S191: calreticulin‐mutated hematopoietic cells are vulnerable to the combined inhibition of the proteasome and the IRE1A‐XBP1 axis of the unfolded protein response. HemaSphere. 2022. [cited 2022 Oct 3];6:92‐93. https://journals.lww.com/hemasphere/Fulltext/2022/06003/S191__CALRETICULIN_MUTATED_HEMATOPOIETIC_CELLS_ARE.92.aspx [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigureS1
TableS1
Data Availability Statement
Not applicable.