

Abstract
PURPOSE:
The purpose of this study was to identify gene mutation and phenotype correlations in a cohort of Taiwanese patients with Stickler syndrome.
MATERIALS AND METHODS:
Patients clinically diagnosed with Stickler syndrome or suspected Stickler syndrome were enrolled. DNA was extracted from venous blood samples. For the targeted next-generation sequencing (NGS) approach, specific primers were designed for all COL2A1, COL11A1, COL11A2, COL9A1, and COL9A2 exons and flanking intron sequences.
RESULTS:
Twenty-three patients from 12 families were enrolled in this study. The myopia power in these 23 cases (35 eyes) ranged from −4.625 to −25.625 D, with a median of −10.00 D. Four patients had retinal detachment. Fourteen patients had a cleft palate. These 23 patients and 13 healthy controls were enrolled in the NGS study. Three families had significant single nucleotide variants (SNVs) in COL2A1. The mutation rates in this survey were 25% (3/12 families) and 35% (8/23 cases). The SNV of family #1, located at exon 27, c.1753G >T, p. Gly585Val, was novel and has not yet been reported in the ClinVar database. Families #10 and #11 had the same SNV, located in exon 33, c.2101C >T, p. Arg701X. Both variants were classified as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines.
CONCLUSION:
Genetic mutations in COL2A1 were found in 25% of Taiwanese families with Stickler syndrome. One novel variant was identified using NGS, which expanded the COL2A1 mutation spectrum. Molecular genetic analysis is helpful to confirm the clinical diagnosis of patients with suspected Stickler syndrome.
Keywords: Cleft palate, COL2A1 gene, myopia, next-generation sequencing, single nucleotide variant, stickler syndrome
Introduction
Stickler syndrome (hereditary arthro-ophthalmopathy) is a genetic disorder affecting multiple organ systems that was first described by Stickler et al. in 1965.[1] Stickler syndrome is a clinically and genetically heterogeneous disorder of heritable connective tissue disorders, characterized by joint hypermobility, premature joint degeneration, high myopia, retinal detachment, conductive and/or sensorineural hearing loss, midfacial hypoplasia, micrognathia, and palatal defects.[1,2,3] Variable phenotypic expression of Stickler syndrome occurs both within and among families; interfamilial variability is partially explained by locus and allelic heterogeneity.[3] The diagnosis of Stickler syndrome is clinically based; however, no consensus exists.[2]
Stickler syndrome is the principal cause of pediatric rhegmatogenous retinal detachment.[4,5] The ocular features associated with Stickler syndrome include congenital axial myopia, vitreous degeneration, retinal degeneration, and a high incidence of retinal detachments.[5,6] Pediatric retinal detachment is always the most difficult case for repair with a poor prognosis.[5,6,7,8] Early detection of Stickler syndrome and prophylactic treatment of ocular complications associated with high myopia are important.[9,10]
Pathogenic variants in six genes (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3) have been reported to be associated with Stickler syndrome type 1-6. Type 1 Stickler syndrome, characterized by a “membranous” vitreous and mild-to-moderate high-frequency sensorineural hearing loss, is the most common type (85%) and is caused by mutations in the COL2A1 gene.[11] The features of type 2 Stickler syndrome, present in approximately 10% of Stickler patients, in which a COL11A1 mutation can be found, include a “beaded” vitreous and pronounced sensorineural hearing loss affecting all frequencies.[12,13,14,15,16,17,18,19] The rare type 3 Stickler syndrome, caused by a COL11A2 mutation, is similar to type 2 in terms of hearing loss but can be distinguished by the absence of ocular anomalies.[20,21] Types I, II, and III have autosomal dominant (AD) inheritance, whereas the very rare types IV, V, and VI have autosomal recessive inheritance.[22]
Traditional sanger sequencing, which has routinely been used to identify disease-causing mutations, is laborious, expensive, and time-consuming, especially for large genes similar to COL2A1. The development of high-throughput next-generation sequencing (NGS) has allowed for a more efficient diagnosis of diseases by combining clinical characteristics and gene mutation screening.[23] The purpose of this study was to determine the phenotype-genotype correlation in Taiwanese patients with Stickler syndrome. We conducted targeted NGS in 12 families.
Materials and Methods
The enrolled patients were individuals listed in our craniofacial patients database who were clinically diagnosed with Stickler syndrome or were highly suspected of having the syndrome. Most were enrolled by craniofacial surgeon F. Huang and a few by ophthalmologists due to congenital or early onset high myopia. High myopia was defined as myopia <-5.0 D.[24] In total, 23 patients from 12 families signed informed consent forms, and 13 healthy parents of the included cases without the features of Stickler syndrome were invited as controls for the NGS study. Asymptomatic parents were good candidates as controls for result comparison. The diagnostic criteria of Stickler syndrome were modified on a scale system for type 1 Stickler syndrome proposed by Rose et al., which considers clinical features, family history, and molecular genetic data.[2] The major manifestations (2-points) included cleft palate, characteristic vitreous or retinal findings, and high-frequency sensorineural hearing loss. Minor (1-point) manifestations included characteristic facial features (hypoplastic malar region, broad or flat nasal bridge, and micrognathia) and skeletal anomalies (femoral head failure, early osteoarthritis, and scoliosis). Patients with ≥5 points and at least one major manifestation were diagnosed with Stickler syndrome. In the study of Hoornaert et al., sensorineural hearing loss was observed more frequently in the mutation-negative group than mutation-positive group. The presence of vitreous anomalies, retinal tears or detachments, cleft palate and a positive family history were shown to be good indicators for a COL2A1 defect.[11] Since most of our patients were children at their first visit, hearing tests and skeletal anomalies were not available for everyone; therefore, we modified the grading system, including orofacial and ocular abnormalities, and family history. The orofacial criteria included cleft palate (major, 2 points) and characteristic facial features (minor, 1 point). The ocular abnormalities included characteristic vitreous/retinal findings or retinal detachment (major, 2 points) and congenital high myopia with long axial length (minor, 1 point). The family history of first-degree relatives meeting the criteria was 1 point. Stickler syndrome included confirmed cases with 4 points (2 + 2 or 2 + 1 + 1) and highly suspected cases with 3 points (2 + 1 or 1 + 1 + 1). This study was approved by the Institutional Review Board of Chang-Gung Medical Foundation (No. 106-4526C), according to the principles of the Declaration of Helsinki.
Clinical investigations
All index patients enrolled in this study underwent orofacial evaluation or surgery. They were referred to the ophthalmology department for eye examinations. A complete ophthalmologic examination included refraction, best-corrected visual acuity testing with Landolt's C chart, dilated slit-lamp examination with stereo biomicroscopy, fundus examination with indirect ophthalmoscopy, and axial length measurement with IOLMaster (Carl Zeiss Meditec, Germany). The hearing test was optional, depending on patient age and cooperation. A chart review was performed to collect the clinical features.
Mutation detection
All patients and controls were recruited for this study. Genomic DNA was extracted from venous blood samples using a ZYMO Quick-DNA Miniprep Plus Kit (Zymo Research, Irvine, CA, USA). For the targeted NGS approach, specific primers were designed for all COL2A1 (NM_001844), COL11A1 (NM_001854), COL11A2 (NM_080680), COL9A1 (NM_001851), and COL9A2 (NM_001852) exons, and flanking intronic sequences based on the UCSC genome browser (https://genome.ucsc.edu/). COL9A3 was not included due to the limitation of grant budget and it was very rare. The subsequent workflows of library preparation and exome enrichment were performed according to the TruSeq Exome Library Prep Reference Guide (Illumina, Cat#FC-150-1001). The exome libraries were sequenced with a 2 × 75 bp read length using the Illumina NextSeq platform (Illumina, San Diego, USA). The generated sequence reads were compared with the reference sequences. Single nucleotide variants (SNVs) and small insertion/deletion were called using the Genome Analysis Toolkit, which was reported by McKenna A in 2010. The variants in the target regions were annotated against several databases, including dbSNP, 1000 Genomes, gnom AD, ExAC, Taiwan Biobank, and ClinVar. Two main algorithms, PolyPhen and SIFT, were used to predict the effects of the functional consequences of the changed sequences. All services were quality controlled by Insight Genomics Co., Tainan, Taiwan. The results were interpreted by the inheritance-pediatrician TJ Wang. The description of pathogenicity of variants follows the guidelines of the American College of Medical Genetics and Genomics (ACMG).[25]
Results
Clinical profiles and phenotypes
A total of 23 patients from 12 families were enrolled in this study. The study included 12 men and 11 women. The age at enrollment ranged from 1 to 47 years, with a median age of 11 years (Q1, 4.75, Q3 31.25). The myopia power in these 23 cases (35 eyes) ranged from −4.625 to −25.625 D, with a median of-10.00 D (Q1-6.6875, Q3-16.1875). The axial length of eyeballs (29 eyes) was from 24.34 to 36.22 mm, with a median of 27.34 mm (Q1 26.10, Q3 29.99). Four patients had retinal detachment. Five patients had significant retinal degeneration and underwent cryotherapy or laser photocoagulation. Fourteen patients had a cleft palate. Six patients had hearing tests, and five of them had mild to severe high-frequency hearing loss [Table 1].
Table 1.
Clinical profiles
| Family number | Age | Refraction (diopter) | Axial length (mm) | Retina Detachment or degeneration | Cleft palate and midface anomaly | 1. Stickler 2. Suspected stickler |
||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| OS | OD | OS | OD | |||||
| F1-mother | 40 | S/P LASIK | Phthisis | NA | NA | Retinal detachment | - | 1 |
| F1-son 1 | 12 | Pseudophakia | Pseudophakia | NA | NA | Retinal detachment | Cleft palate | 1 |
| F1-son 2 | 7 | −16.5 | −16.875 | 28.77 | 29.25 | Retinal degeneration | Small recessive chin | 1 |
| F2-father | 40 | Pseudophakia | −16.875 | 28.42 | 29.98 | - | Small recessive chin | 1 |
| F2-son 1 | 5 | −9.125 | −8.875 | 26.07 | 26.22 | - | Small recessive chin | 1 |
| F2-son 2 | 5 | −13.375 | −10.875 | 26.88 | 26.1 | - | Incomplete cleft palate | 1 |
| F3-mother | 47 | −4.875 | −5.5 | NA | NA | - | Small recessive chin | 2 |
| F3-daughter | 18 | −17 | −16.75 | 28.58 | 28.57 | Retinal detachment | Cleft palate | 1 |
| F4-daughter | 7 | −7.375 | −7.125 | 24.99 | 25.12 | - | Cleft palate | 1 |
| F5-daughter | 10 | −7.125 | −7.875 | 26.92 | 27.35 | Retinal degeneration | Cleft palate | 1 |
| F6-daughter | 18 | −5.5 | −5.75 | NA | NA | - | Cleft palate | 1 |
| F7-mother | 31 | −11.25 | −10.25 | NA | NA | - | - | 2 |
| F7-son | 3 | −23.875 | −25.625 | 30.37 | 30.29 | - | Cleft palate | 1 |
| F8-father | 47 | Phthisis | Surgical aphakia | NA | 36.22 | Retinal detachment | Cleft palate | 1 |
| F8-son | 17 | −16.125 | −16.25 | 32.69 | 32.48 | Retinal degeneration | Cleft palate | 1 |
| F8-daughter | 15 | −6.25 | −4.625 | 26.95 | 26.09 | - | - | 2 |
| F9-son | 9 | −7 | −10.5 | 26.12 | 27.23 | - | Cleft palate | 1 |
| F10-son 1 | 1 | NA | NA | NA | NA | - | Incomplete cleft palate | 2 |
| F10-son 2 | 1 | NA | NA | NA | NA | - | Cleft palate | 2 |
| F11-mother | 32 | −10 | −10 | NA | NA | Retinal degeneration | - | 2 |
| F11-son | 6 | −9.875 | −10.125 | 27.45 | 27.34 | Retinal degeneration | - | 2 |
| F11-daughter | 4 | −6.375 | −6.375 | 24.47 | 24.34 | - | Cleft palate | 1 |
| F12-daughter | 4 | −24.125 | −19.875 | 29.99 | 30.16 | - | Cleft palate | 1 |
N/A: Not available
Genotype
A total of 23 patients and 13 healthy controls were enrolled in the NGS study. The variant on the exon was treated as significant when the variant caused an amino acid change, nonsynonymous consequence, and allele frequency <0.01%. The variant on the intron was treated as significant only when this was located at the splicing site.
Three families had significant SNVs in the COL2A1 gene, including families #1, #10, and #11. The positive rate of this survey was 25% (3/12 families) and 35% (8/23 cases) [Table 2]. In family #1, the coordinates of the SNV were 48378857, located at exon 27, c. 1753G > T, p. Gly585Val. The SIFT prediction marked this as deleterious change while PolyPhen2 indicated this was probably damaging. This SNV was novel and has not been reported in the ClinVar database. According to the ACMG guidelines, this variant is classified as likely pathogenic (PM2, PP3, PP1, PM6). In families #10 and #11, the coordinates of SNV were 48376723, located at exon 33, c. 2101C >T and p. Arg701X. This variant has been previously reported.[17] This variant was reported to be pathogenic for type 1 Stickler syndrome in the ClinVar database in May 2019. It was classified as likely pathogenic (PM2, PS1, and PP1).
Table 2.
Genotypes
| Family number | Gene | c.HGVS | Amino_Acids | Variant | Exon | Consequence | ACMG classification | Allele_frequency | ClinVar |
|---|---|---|---|---|---|---|---|---|---|
| F1-mother | COL2A1 | c.1753G>T | p.Gly585Val | C>C/A | 27 | Nonsynonymous SNV | Likely pathogenic | 0.00 | - |
| F1-son1 | COL2A1 | c.1753G>T | p.Gly585Val | C>C/A | 27 | Nonsynonymous SNV | Likely pathogenic | 0.00 | - |
| F1-son2 | COL2A1 | c.1753G>T | p.Gly585Val | C>C/A | 27 | Nonsynonymous SNV | Likely pathogenic | 0.00 | - |
| F10-son1 | COL2A1 | c.2101C>T | p.Arg701X | G>G/A | 33 | Stopgain SNV | Likely pathogenic | 0.00 | Pathogenic |
| F10-son2 | COL2A1 | c.2101C>T | p.Arg701X | G>G/A | 33 | Stopgain SNV | Likely pathogenic | 0.00 | Pathogenic |
| F11-mother | COL2A1 | c.2101C>T | p.Arg701X | G>G/A | 33 | Stopgain SNV | Likely pathogenic | 0.00 | Pathogenic |
| F11-son | COL2A1 | c.2101C>T | p.Arg701X | G>G/A | 33 | Stopgain SNV | Likely pathogenic | 0.00 | Pathogenic |
| F11-daughter | COL2A1 | c.2101C>T | p.Arg701X | G>G/A | 33 | Stopgain SNV | Likely pathogenic | 0.00 | Pathogenic |
HGVS=Human Genome Variation Society, ACMG=American College of Medical Genetics and Genomics, SNV=Single nucleotide variant
Case presentations
Family #1 is a family of spondyloepiphyseal dysplasia congenita (SEDC) combined with Stickler syndrome. The father was normal. The phenotypes of the mother and their two sons (ages 12 and 7) included short stature [Figure 1], micrognathia, and high myopia. X-ray examination revealed features of SEDC with delayed ossification of the bilateral femoral head [Figure 2]. The mother's parents were normal. The mother underwent bilateral total hip replacement and right total knee replacement. The right eye was blind because of retinal detachment. The elder brother was a case of Pierre Robin sequence with an incomplete cleft of the secondary palate and underwent palate repair at 12 months of age and surgical correction for velopharyngeal insufficiency at 4 years of age. This patient also had retinal detachment and underwent vitrectomy with silicone oil tamponade (OU); the axial length of the eyeball was 28.69 mm/27.69 mm, compatible with excessive myopia. The younger brother underwent cryotherapy for peripheral retinal degeneration (OU). The axial length of the eyeball was 28.65 mm/29.12 mm. A novel SNV was identified in this family and was located in exon 27, c.1753G >T, p. Gly585Val. The SIFT prediction was deleterious, and PolyPhen prediction was probably damaging.
Figure 1.

Family #1 (spondyloepiphyseal dysplasia congenital) and the author HK Kuo. (with permission from the family)
Figure 2.
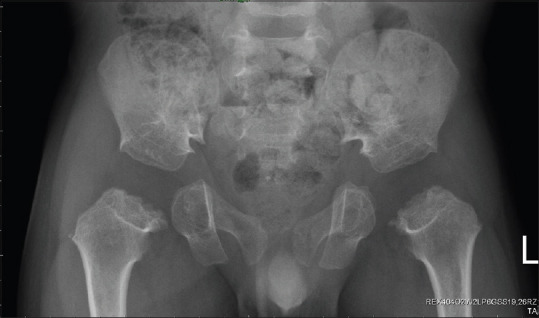
X-ray examination of son 1 at 3 years of age showed features of spondyloepiphyseal dysplasia congenita with delayed ossification of the bilateral femoral head
The parents were normal in family #10. The index cases included twins. Both had cleft palate. Further ocular examinations and hearing tests were not available due to their young age. The father was normal in family #11. The mother, son, and daughter had high myopia. The daughter had a cleft palate. Both nonrelated families had the same variant, c.2101C >T, p. Arg701X, a stop-gain SNV. The pedigrees of these three families are illustrated in Figure 3.
Figure 3.
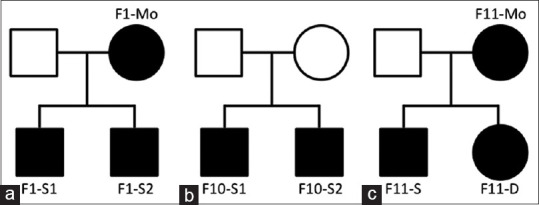
Pedigrees of three families with pathogenic single nucleotide variants in the COL2A1 gene. Square symbols denote males, and circle symbols denote females. The solid symbols indicate individuals with Stickler syndrome, while the open symbols indicate unaffected family members. (a) Family 1 (b) family 10 (c) family 11
Discussion
Twelve Taiwanese families with Stickler syndrome or highly suspected Stickler syndrome were enrolled in this gene survey, which was conducted using the targeted NGS method. The positive rate of this survey was 25% (3/12 families). All variants were found in COL2A1 but not in COL11A1. Type 1 Stickler syndrome caused by mutations in the COL2A1 gene is the most common type (85%), based on the studies from the Europeans and North Americans.[2,11,17] Few studies on Stickler genetics have been reported in Asia. In a recent review by Wang et al., only 107 type 1 Stickler patients with one heterozygous variant in the COL2A1 gene have been reported in East Asia.[26] In a COL2A1 and COL11A1 mutation study from Southern China, five mutations in COL2A1 were identified in six of 16 unrelated families (37.5%), and putative pathogenic mutations of the COL11A1 gene were absent in a cohort of patients with Stickler syndrome.[27] The low positive detection rate was similar to that observed in the present study.
Two SNVs in COL2A1 were identified in the present study. In the review by Wang et al., the variants c. 1833 + 1G >A, c. 1693C >T, c. 3106C >T, c. 2353C >T, and c. 1957C >T were common in the East Asian population.[26] Our variants were different, but families #10 and #11 had the same mutation, c. 2101C >T. COL2A1 encodes the alpha-1 chain of type II collagen. Type II collagen, which adds structure and strength to connective tissues, is found primarily in the cartilage, vitreous of the eye, inner ear, and center portion of the discs between the vertebrae in the spine (nucleus pulposus).[2,3] Stickler syndrome was the most frequent form of type II collagenopathies. Type II collagenopathies include a wide variety of skeletal dysplasias, ranging from lethal disorders, such as achondrogenesis, mild disorders, and adult early onset osteoarthritis.[28] In family #1, the mother and two sons had Stickler syndrome and SEDC. The SNV was located in exon 27, causing a glycine (Gly) to valine (Val) substitution. This variant is however classified as a likely pathogenic variant according to the ACMG guidelines. The other variants might exist if whole-genome sequencing is used to investigate this family. In the report of Richards et al., they also included three cases of SEDC combined with Stickler syndrome.[17] The other families in this survey had Stickler syndrome without severe skeletal dysplasia.
The limitation and possible reasons for the relatively low mutation-detection rate in this targeted NGS study are the inclusion criteria and investigation methods. First, Stickler syndrome includes a wide variety of clinically and genetically heterogeneous heritable connective tissue disorders. Since most enrolled cases in our study were children, we modified the grading system and did not include auditory and skeletal manifestations. The relatively loose inclusion criteria may have lowered the detection rate. Furthermore, NGS as a tool for the identification of mutations has proven to be efficient and cost-effective.[23] Whole-exome sequencing, on the other hand, has enabled the investigation of genes other than the known candidate genes.[29] Here, we used a targeted NGS approach to investigate five candidate genes. This might miss other possible causal genes and explains the relatively low mutation-detection rate. Besides all that, we only reported pathogenic or likely pathogenic variants by the ACMG criteria.
Patients with Stickler syndrome have congenital degenerative vitreous and high myopia. Myopia is a common and severe health issue in East Asia.[30] The prevalence of high myopia can be estimated at approximately 20% of the myopia prevalence in adults.[31] Patients with idiopathic high myopia gradually have elongated eyeballs and degenerative vitreous cavities. Both of these conditions are similar, and Stickler syndrome may be under-diagnosed in East Asian countries. In a study by Wang et al., 115 patients with high myopia were analyzed by NGS, and five Stickler patients from four unrelated Chinese families were identified.[32] Clinicians should be aware of the possibility of Stickler syndrome in patients with congenital high myopia, and therefore, genotype surveys are important for precise diagnosis.
Conclusion
In conclusion, we found variants in the COL2A1 gene in 25% of Taiwanese families with Stickler syndrome or highly suspected Stickler syndrome. One novel variant was identified using NGS analysis, which expanded the spectrum of the COL2A1 gene mutation spectrum. Molecular genetic analysis is important for confirming the clinical diagnosis and further genetic consultation for patients with suspected Stickler syndrome.
Ethics approval and consent to participate
The study adhered to the Declaration of Helsinki and was approved by the Institutional Review Board of Chang Gung Memorial Hospital (study reference number: 106-4526C).
Consent for publication
This manuscript has not been published elsewhere and is not under consideration by any other journal. The contents of this manuscript will not be copyrighted, submitted, or published elsewhere, while acceptance by your journal is under consideration.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Financial support and sponsorship
This work was supported by a grant from CMRPG8G1111 from Chang-Gung Memorial Hospital, Taiwan.
Conflicts of interest
The authors declare that there are no conflicts of interests of this paper.
Acknowledgments
This work was supported by a grant from CMRPG8G1111 from Chang-Gung Memorial Hospital, Taiwan
The lab work was supported by Insight Genomics Co., Tainan, Taiwan.
References
- 1.Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc. 1965;40:433–55. [PubMed] [Google Scholar]
- 2.Rose PS, Levy HP, Liberfarb RM, Davis J, Szymko-Bennett Y, Rubin BI, et al. Stickler syndrome: Clinical characteristics and diagnostic criteria. Am J Med Genet A. 2005;138A:199–207. doi: 10.1002/ajmg.a.30955. [DOI] [PubMed] [Google Scholar]
- 3.Snead MP, Yates JR. Clinical and Molecular genetics of Stickler syndrome. J Med Genet. 1999;36:353–9. [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards AO. Clinical features of the congenital vitreoretinopathies. Eye (Lond) 2008;22:1233–42. doi: 10.1038/eye.2008.38. [DOI] [PubMed] [Google Scholar]
- 5.Reddy DN, Yonekawa Y, Thomas BJ, Nudleman ED, Williams GA. Long-term surgical outcomes of retinal detachment in patients with Stickler syndrome. Clin Ophthalmol. 2016;10:1531–4. doi: 10.2147/OPTH.S111526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang F, Kuo HK, Hsieh CH, Lai JP, Chen PK. Visual complications of Stickler syndrome in paediatric patients with Robin sequence. J Craniomaxillofac Surg. 2007;35:76–80. doi: 10.1016/j.jcms.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Alshahrani ST, Ghazi NG, Al-Rashaed S. Rhegmatogenous retinal detachments associated to Stickler syndrome in a tertiary eye care center in Saudi Arabia. Clin Ophthalmol. 2016;10:1–6. doi: 10.2147/OPTH.S91444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith JM, Ward LT, Townsend JH, Yan J, Hendrick AM, Cribbs BE, et al. Rhegmatogenous retinal detachment in children: Clinical factors predictive of successful surgical repair. Ophthalmology. 2019;126:1263–70. doi: 10.1016/j.ophtha.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fincham GS, Pasea L, Carroll C, McNinch AM, Poulson AV, Richards AJ, et al. Prevention of retinal detachment in Stickler syndrome: The Cambridge prophylactic cryotherapy protocol. Ophthalmology. 2014;121:1588–97. doi: 10.1016/j.ophtha.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Coussa RG, Sears J, Traboulsi EI. Stickler syndrome: Exploring prophylaxis for retinal detachment. Curr Opin Ophthalmol. 2019;30:306–13. doi: 10.1097/ICU.0000000000000599. [DOI] [PubMed] [Google Scholar]
- 11.Hoornaert KP, Vereecke I, Dewinter C, Rosenberg T, Beemer FA, Leroy JG, et al. Stickler syndrome caused by COL2A1 mutations: Genotype-phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010;18:872–80. doi: 10.1038/ejhg.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards AJ, Yates JR, Williams R, Payne SJ, Pope FM, Scott JD, et al. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet. 1996;5:1339–43. doi: 10.1093/hmg/5.9.1339. [DOI] [PubMed] [Google Scholar]
- 13.Annunen S, Körkkö J, Czarny M, Warman ML, Brunner HG, Kääriäinen H, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65:974–83. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin S, Richards AJ, Yates JR, Scott JD, Pope M, Snead MP. Stickler syndrome: Further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur J Hum Genet. 1999;7:807–14. doi: 10.1038/sj.ejhg.5200377. [DOI] [PubMed] [Google Scholar]
- 15.Majava M, Hoornaert KP, Bartholdi D, Bouma MC, Bouman K, Carrera M, et al. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am J Med Genet A. 2007;143A:258–64. doi: 10.1002/ajmg.a.31586. [DOI] [PubMed] [Google Scholar]
- 16.Melkoniemi M, Koillinen H, Männikkö M, Warman ML, Pihlajamaa T, Kääriäinen H, et al. Collagen XI sequence variations in nonsyndromic cleft palate, Robin sequence and micrognathia. Eur J Hum Genet. 2003;11:265–70. doi: 10.1038/sj.ejhg.5200950. [DOI] [PubMed] [Google Scholar]
- 17.Richards AJ, McNinch A, Martin H, Oakhill K, Rai H, Waller S, et al. Stickler syndrome and the vitreous phenotype: Mutations in COL2A1 and COL11A1. Hum Mutat. 2010;31:E1461–71. doi: 10.1002/humu.21257. [DOI] [PubMed] [Google Scholar]
- 18.Richards AJ, McNinch A, Whittaker J, Treacy B, Oakhill K, Poulson A, et al. Splicing analysis of unclassified variants in COL2A1 and COL11A1 identifies deep intronic pathogenic mutations. Eur J Hum Genet. 2012;20:552–8. doi: 10.1038/ejhg.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vijzelaar R, Waller S, Errami A, Donaldson A, Lourenco T, Rodrigues M, et al. Deletions within COL11A1 in Type 2 stickler syndrome detected by Multiplex Ligation-Dependent Probe Amplification (MLPA) BMC Med Genet. 2013;14:48. doi: 10.1186/1471-2350-14-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sirko-Osadsa DA, Murray MA, Scott JA, Lavery MA, Warman ML, Robin NH. Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the alpha2(XI) chain of type XI collagen. J Pediatr. 1998;132:368–71. doi: 10.1016/s0022-3476(98)70466-4. [DOI] [PubMed] [Google Scholar]
- 21.Vuoristo MM, Pappas JG, Jansen V, Ala-Kokko L. A stop codon mutation in COL11A2 induces exon skipping and leads to non-ocular Stickler syndrome. Am J Med Genet A. 2004;130A:160–4. doi: 10.1002/ajmg.a.30111. [DOI] [PubMed] [Google Scholar]
- 22.Nixon TR, Alexander P, Richards A, McNinch A, Bearcroft PW, Cobben J, et al. Homozygous Type IX collagen variants (COL9A1, COL9A2, and COL9A3) causing recessive Stickler syndrome-Expanding the phenotype. Am J Med Genet A. 2019;179:1498–506. doi: 10.1002/ajmg.a.61191. [DOI] [PubMed] [Google Scholar]
- 23.Desai AN, Jere A. Next-generation sequencing: Ready for the clinics? Clin Genet. 2012;81:503–10. doi: 10.1111/j.1399-0004.2012.01865.x. [DOI] [PubMed] [Google Scholar]
- 24.Holden BA, Fricke TR, Wilson DA, Jong M, Naidoo KS, Sankaridurg P, et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050. Ophthalmology. 2016;123:1036–42. doi: 10.1016/j.ophtha.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang DD, Gao FJ, Hu FY, Zhang SH, Xu P, Wu JH. Mutation spectrum of stickler syndrome type i and genotype-phenotype analysis in east Asian population: A systematic review. BMC Med Genet. 2020;21:27. doi: 10.1186/s12881-020-0963-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Jia X, Xiao X, Li S, Li J, Li Y, et al. Mutation survey and genotype-phenotype analysis of COL2A1 and COL11A1 genes in 16 Chinese patients with Stickler syndrome. Mol Vis. 2016;22:697–704. [PMC free article] [PubMed] [Google Scholar]
- 28.Barat-Houari M, Dumont B, Fabre A, Them FT, Alembik Y, Alessandri JL, et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet. 2016;24:992–1000. doi: 10.1038/ejhg.2015.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acke FR, Malfait F, Vanakker OM, Steyaert W, De Leeneer K, Mortier G, et al. Novel pathogenic COL11A1/COL11A2 variants in Stickler syndrome detected by targeted NGS and exome sequencing. Mol Genet Metab. 2014;113:230–5. doi: 10.1016/j.ymgme.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 30.Wu PC, Tsai CL, Wu HL, Yang YH, Kuo HK. Outdoor activity during class recess reduces myopia onset and progression in school children. Ophthalmology. 2013;120:1080–5. doi: 10.1016/j.ophtha.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Wu PC, Huang HM, Yu HJ, Fang PC, Chen CT. Epidemiology of myopia. Asia Pac J Ophthalmol. 2016;5:386–93. doi: 10.1097/APO.0000000000000236. [DOI] [PubMed] [Google Scholar]
- 32.Wang DD, Gao FJ, Hu FY, Li JK, Zhang SH, Xu P, et al. Next-generation sequencing-aided precise diagnosis of Stickler syndrome type I. Acta Ophthalmol. 2020;98:e440–6. doi: 10.1111/aos.14302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.