

Abstract
Background
Emergence of the new SARS-CoV-2 variant B.1.1.529 worried health policy makers worldwide due to a large number of mutations in its genomic sequence, especially in the spike protein region. The World Health Organization (WHO) designated this variant as a global variant of concern (VOC), which was named “Omicron.” Following Omicron’s emergence, a surge of new COVID-19 cases was reported globally, primarily in South Africa.
Objective
The aim of this study was to understand whether Omicron had an epidemiological advantage over existing variants.
Methods
We performed an in silico analysis of the complete genomic sequences of Omicron available on the Global Initiative on Sharing Avian Influenza Data (GISAID) database to analyze the functional impact of the mutations present in this variant on virus-host interactions in terms of viral transmissibility, virulence/lethality, and immune escape. In addition, we performed a correlation analysis of the relative proportion of the genomic sequences of specific SARS-CoV-2 variants (in the period from October 1 to November 29, 2021) with matched epidemiological data (new COVID-19 cases and deaths) from South Africa.
Results
Compared with the current list of global VOCs/variants of interest (VOIs), as per the WHO, Omicron bears more sequence variation, specifically in the spike protein and host receptor-binding motif (RBM). Omicron showed the closest nucleotide and protein sequence homology with the Alpha variant for the complete sequence and the RBM. The mutations were found to be primarily condensed in the spike region (n=28-48) of the virus. Further mutational analysis showed enrichment for the mutations decreasing binding affinity to angiotensin-converting enzyme 2 receptor and receptor-binding domain protein expression, and for increasing the propensity of immune escape. An inverse correlation of Omicron with the Delta variant was noted (r=–0.99, P<.001; 95% CI –0.99 to –0.97) in the sequences reported from South Africa postemergence of the new variant, subsequently showing a decrease. There was a steep rise in new COVID-19 cases in parallel with the increase in the proportion of Omicron isolates since the report of the first case (74%-100%). By contrast, the incidence of new deaths did not increase (r=–0.04, P>.05; 95% CI –0.52 to 0.58).
Conclusions
In silico analysis of viral genomic sequences suggests that the Omicron variant has more remarkable immune-escape ability than existing VOCs/VOIs, including Delta, but reduced virulence/lethality than other reported variants. The higher power for immune escape for Omicron was a likely reason for the resurgence in COVID-19 cases and its rapid rise as the globally dominant strain. Being more infectious but less lethal than the existing variants, Omicron could have plausibly led to widespread unnoticed new, repeated, and vaccine breakthrough infections, raising the population-level immunity barrier against the emergence of new lethal variants. The Omicron variant could have thus paved the way for the end of the pandemic.
Keywords: COVID-19, pandemic, variants, immune escape, transmissibility, virulence, policy, mutations, epidemiology, data, Omicron, virus, transmission, genomic
Introduction
Background
A new variant of SARS-CoV-2 (lineage B.1.1.529) was reported from Botswana, South Africa, and multiple other countries [1], which the World Health Organization (WHO) designated as a global variant of concern (VOC) named “Omicron” [2]. The new variant was classified in the PANGO (Phylogenetic Assignment of Named Global Outbreak) lineage as BA.1. The presence of a large number of mutations in its genomic sequence—especially in the spike protein region, including in the host receptor-binding domain (RBD)—raised speculations that Omicron can prove to be a serious epidemiological threat and contributor to subsequent COVID-19 waves globally [3]. Multiple sublineages of Omicron were then identified with a slightly varying set of mutations [4]. These Omicron subvariants differentially affected the global population, leading to burst waves in various parts of the world [5]. Omicron is currently the predominant strain causing most of the new COVID-19 cases globally [5].
Significance of the Study
Owing to the heterogeneity of previous infections and vaccination coverage across the global population, there has been significant ambiguity in reports on the epidemiological properties of Omicron [6-9]. Specifically, it remains unclear whether the Omicron variant has an epidemiological advantage over existing variants [8]. Many researchers have proposed that Omicron’s emergence has changed the pandemic’s evolutionary course and speculated its end [10-12]. However, contradictory views are also being presented, suggesting against any sooner end of the pandemic and the possibility of the emergence of more lethal variants as the immunity that the global population gained from previous infections and vaccines fades [13]. Therefore, we aimed to resolve the existing ambiguity over the epidemiological properties of the Omicron variant using an integrated approach combining viral genomic sequence analysis and epidemiological data. Integrating viral genomic analysis with epidemiological data is a relatively novel approach; however, its success in predicting the epidemiological properties of SARS-CoV-2 variants and the future course of the COVID-19 pandemic has been validated in recent bioinformatic studies [14,15]. The findings of this study will thus provide concrete insights into the origin and epidemiological attributes of this variant to pave the way for the end of the pandemic.
Objectives
We performed an in silico analysis of the complete genomic sequences of the Omicron BA.1 variant available on the Global Initiative on Sharing Avian Influenza Data (GISAID) platform [16] with the primary objective of predicting the functional impact of the mutations present in this variant on virus-host interactions in terms of viral transmissibility, virulence, and immune-escape capabilities. Moreover, we assessed the relative proportion of the genomic sequences of existing SARS-CoV-2 variants, which was correlated with the rise in new COVID-19 cases in the global geographical location most affected by Omicron to understand whether the new variant had an epidemiological advantage in terms of transmissibility and virulence/lethality over existing variants.
Methods
Data Collection
The SARS-CoV-2 genomic sequence for the Omicron variant and other global VOCs/variants of interest (VOIs) were downloaded from the EpiCoV database of GISAID [16] using the automatic search function feeding information for geographical location, SARS-CoV-2 lineage, sample collection, and sequence reporting dates (up to December 10, 2021). The optimum length and coverage of the downloaded sequences (used for variant comparisons) were obtained by selecting the “complete sequence” and “high coverage” options in the search function.
Data Analysis
Mutational analysis on the genomic sequences was performed, and the 3D structure of the spike protein with amino acid changes in Omicron was generated using the CoVsurver app provided by GISAID [16], employing hCoV-19/Wuhan/WIV04/2019 as the reference strain. Further, a comparative mutational analysis of Omicron with existing global VOCs/VOIs (as per the WHO) [17] was generated using the “compare lineages” function at outbreak.info [18] with GISAID as the source of genomic sequence data. The Expasy Swiss Bioinformatics portal [19] was used for protein sequence translation from the viral genomic sequences. A comparative assessment of the Omicron nucleotide and protein sequences with existing global VOCs/VOIs was performed using the National Center for Biotechnology Information (NCBI) Blast tool [20].
Furthermore, the functional impact of the mutations present at the RBD of the variants was assessed using an open analysis pipeline developed by Starr et al [21], which integrates a yeast-display platform with deep mutational scanning to determine how all possible RBD amino acid mutations affect angiotensin-converting enzyme 2 (ACE2)-binding affinity and protein expression (a correlate of protein folding stability) as compared to the wild-type SARS-CoV-2 strain [22].
The epidemiological correlates of the Omicron variant were assessed based on the comparative analysis of the genomic sequences from GISAID [16] and current epidemiological data (daily new cases and deaths) made available at Worldometer for South Africa [23], as one of the regions most strongly affected by the variant (last date of collection: December 10, 2021). The number of sequences for each SARS-CoV-2 variant was retrieved using an automatic search function feeding information for the lineage and collection dates in the EpiCoV database of GISAID for the period of October 1, 2021, to December 10, 2021. A 3-day sum of the total number of sequences was noted for each variant and their relative proportions were calculated (in percentages). Data were tabulated and the distribution of each variant was charted against the COVID-19 epidemiological data (3-day sum of new cases and deaths). Statistical analysis was performed to appreciate the changes in the relationship between the variables before and after the emergence of Omicron.
Statistical Analysis
An expected (E) value ≤0 was considered significant for the sequence homology match through NCBI Blast. An E value close to 0 or below and a higher Max score indicate a higher sequence homology ranking (see [24] for further details of the statistical methods in predicting significance in similarity scores). For the mutational analysis, only the mutations present in at least 75% of sequenced samples were considered for functional characterization.
For the analysis of epidemiological data, statistical tests were performed to evaluate intergroup differences among SARS-CoV-2 variants in Microsoft Excel 2019 and the R statistical package version 4.2.2. The normality of the data was examined using the Shapiro-Wilk test. Pearson (r) and Spearman (ρ) correlation tests were performed for the normally distributed and skewed data, respectively. A correlation matrix was generated and linear regression analysis was performed between the comparing variables (presented as r values, ranging from 0 to 1, and 95% CIs). Results were considered statistically significant at P≤.05. Graphs were plotted to visualize the data trends.
Ethical Considerations
Approval from the institutional ethics committee was precluded as publicly available/open access databases were used for this study.
Results
Data Summary
A total of 3604 genomic sequences of Omicron from 54 countries were uploaded on GISAID up to December 10, 2021 (see Figure S1 in Multimedia Appendix 1), which were analyzed for mutational characteristics. The mutations found were primarily condensed in the spike protein region (n=28-48) of the virus; however, frequent nonspike mutations were also noted (n=20-26). In this study, we focused on analyzing the genomic sequences of Omicron’s initially most prevalent sublineage (BA.1).
Sequence Homology of Omicron (BA.1) With Wild-Type Strains
Compared to the current list of global VOCs/VOIs (as per the WHO), Omicron showed more sequence variation, specifically in the spike protein (nucleotides 21,563-25,384; amino acids 1-1273), including the receptor-binding motif (RBM; nucleotides 22,869-23,089 and amino acids 438-508), where the riffs were most prominent (Table S1 in Multimedia Appendix 1). The homology of Omicron to the reference strain (hCoV-19/Wuhan/WIV04/2019) for the spike protein sequence varied from 96.23% to 97.8% (28-48 mutations) in the analyzed sequences (Figure 1).
Figure 1.
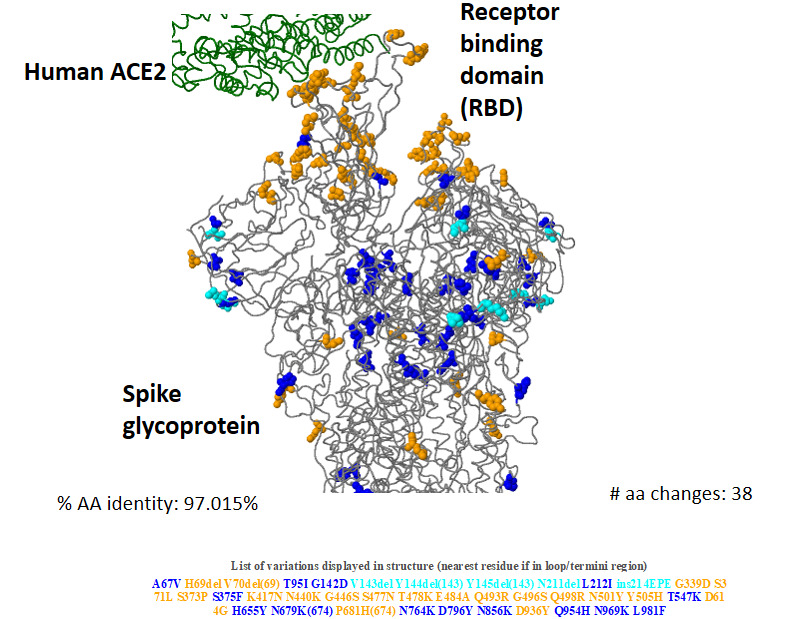
Three-dimensional structure of Omicron (BA.1) spike glycoprotein in the interaction of human angiotensin-converting enzyme 2 (ACE2), showing key amino acid substitutions. (Data source: CoVsurver app from GISAID [16]). AA/aa: amino acid.
Sequence Homology of Omicron (BA.1) With Existing SARS-CoV-2 VOCs/VOIs
The analysis of Omicron’s genomic and protein sequence homology with the reference strain and current global VOCs/VOIs (as per the WHO) showed the highest similarity of Omicron with the Alpha variant for the complete sequence as well as for the RBM. However, the highest similarity for the complete nucleotide and protein sequences for the spike protein were noted with the Beta and Delta variant, respectively (see Table S1 in Multimedia Appendix 1).
Mutational Analysis
Multiple clusters of closely spaced mutations were noted across the sequence, which were most densely located in the spike protein region, particularly in its S1 subunit, including the host RBM (Figure 2, Table 1). Many of the mutations in Omicron are shared with the current global VOCs/VOIs (Figure 3).
Figure 2.
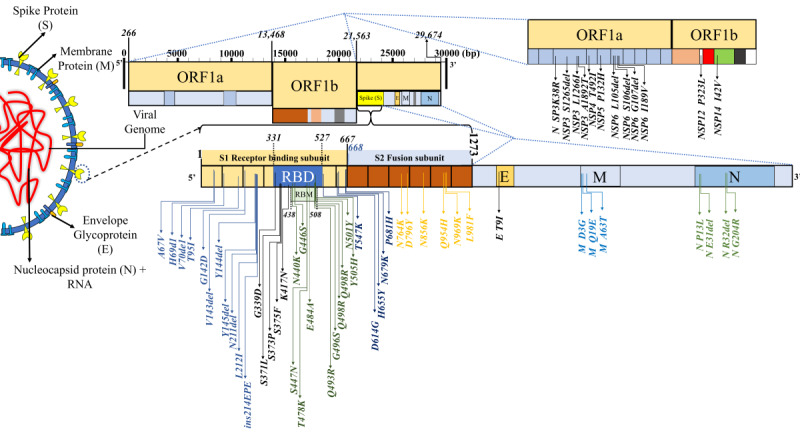
The mutational landscape in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage: BA.1). The analysis of the mutations present at the RBD using a deep mutational scanning pipeline by Starr et al [21] reflected prominent ACE2-binding affinity and protein expression changes (see Table 1). Notably, mutations decreasing the ACE2-binding affinity and protein expression were significantly greater in number. ACE2: angiotensin-converting enzyme 2; ORF: open reading frame; RBD: receptor-binding domain; RBM: receptor-binding motif.
Table 1.
Predicted impact of receptor-binding motif variations in the SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage: BA.1) on interactions with the host.a
| ACE2b binding site mutations | ACE2 binding (Δlog10 KD appc,d) | Protein expression (Δlog mean MFIe,f) | ACE2 contact with SARS-CoV-2 | RSAg bound | SARS-CoV-1 amino acid | RaTG13 amino acid | GD Pangolin-CoV amino acid |
| G339D | 0.06 | 0.30 | false | 0.47 | G | G | G |
| S371L | –0.14 | –0.61 | false | 0.46 | S | S | S |
| S373P | –0.08 | –0.22 | false | 0.48 | F | S | S |
| S375F | –0.55 | –1.81 | false | 0.48 | S | S | S |
| K417N | –0.45 | 0.10 | true | 0.19 | V | K | R |
| N440K | 0.07 | –0.12 | false | 0.68 | N | H | N |
| G446S | –0.20 | –0.40 | true | 0.55 | T | G | G |
| S477N | 0.06 | 0.06 | false | 0.76 | G | S | S |
| T478K | 0.02 | 0.02 | false | 0.48 | K | K | T |
| E484A | –0.07 | –0.23 | false | 0.50 | P | T | E |
| Q493R | –0.09 | –0.06 | true | 0.10 | N | Y | Q |
| G496S | –0.63 | 0.12 | true | 0.04 | G | G | G |
| Q498R | –0.06 | 0.10 | true | 0.00 | Y | Y | H |
| N501Y | 0.24 | –0.14 | true | 0.03 | T | D | N |
| Y505H | –0.71 | 0.16 | true | 0.12 | Y | H | Y |
aBased on the study of Starr et al [21].
bACE2: angiotensin-converting enzyme 2.
cKD app: apparent dissociation constant.
dA positive Δlog10 KD app value relative to the unmutated SARS-CoV-2 receptor-binding domain (3.9 × 10−11 M) indicates stronger binding.
eMFI: mean fluorescence intensity.
fPositive Δlog MFI values relative to the unmutated SARS-CoV-2 receptor-binding domain indicate increased expression.
gRSA: relative solvent accessibility.
Figure 3.

Lineage comparison between Omicron and other global variants of concerns/interest. Only mutations with >75% prevalence in at least one lineage are shown. (Data source: outbreak.info, based on the SARS-CoV-2 genomic sequences uploaded in GISAID until December 6, 2021).
Tables 2-6 summarize the reported mutations in Omicron (BA.1) (present in at least 75% of sequences) and their functional characteristics based on the existing literature [16,21,25-41]. According to the available evidence, these mutations in PANGO lineage BA.1 can be broadly categorized into four major groups: immune escape (n=20) (Table 2), host receptor binding (n=10) (Table 3), virus replication (n=18) (Table 4), and host adaptability (n=3) (Table 5). Mutations outside of the spike protein are summarized in Table 6.
Table 2.
Mutations in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage BA.1) spike protein influencing immune escape via antibody recognition sites and/or antigenic drift.a
| Mutation | Frequency (%)b | Remarks | Reference |
| H69del | 20.35 | H69del+V70del have 2-fold higher infectivity compared to the wild type. H69del+V70del-containing viruses showed reduced neutralization sensitivity to mAbc COVA1-21, targeting an as-yet-undefined epitope outside the RBDd | [16] |
| V70del | 20.37 | H69del+V70del have 2-fold higher infectivity compared to the wild type. H69del+V70del-containing viruses showed reduced neutralization sensitivity to mAb COVA1-21, targeting an as-yet-undefined epitope outside the RBD | [16] |
| V143del | 0.12 | N/Ae | [16] |
| Y144del | 20.94 | Decreased sensitivity to convalescent sera | [25,26] |
| Y145del | 2.33 | Decreased sensitivity to convalescent sera | [16,25,26] |
| G339D | 0.01 | N/A | [16] |
| S371L | 0.00 | N/A | [16,21] |
| S373P | 0.01 | N/A | [16,21] |
| S375F | 0.00 | N/A | [16,27] |
| K417N | 0.83 | N/A | [16,21,28] |
| N440K | 0.17 | N/A | [16,21] |
| G446S | 0.01 | N/A | [16,28] |
| S477N | 1.31 | S477N was also resistant to neutralization by the human convalescent sera tested in this study, but not to vaccine-elicited sera | [16,21,29] |
| E484A | 0.02 | N/A | [27] |
| Q493R | 0.01 | N/A | [30,31] |
| G496S | 0.01 | N/A | [21] |
| Q498R | 0.00 | N/A | [16,27] |
| N501Y | 24.11 | Associated with increased transmissibility and increased affinity for human ACE2f receptor | [16,21,28,32] |
| H655Y | 2.25 | N/A | [34-36] |
Table 6.
Mutations in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage BA.1) outside of the spike protein.a
| Mutations | Frequency (%)b | Effect on virus-host interactions | Remarks | References | |
| Envelope (E) T9I | 0.09 | Viral oligomerization interfaces | N/Ac | [16] | |
| Membrane (M) | |||||
|
|
M D3G | 0.08 | Unknown | N/A | [16] |
|
|
M Q19E | 0.00 | Unknown | N/A | [16] |
|
|
M A63T | 0.01 | Unknown | N/A | [16] |
| Nucleocapsid (N) | |||||
|
|
N P13L | 0.63 | Antigenic drift | P13L variant in B*27:05-restricted CD8+ nucleocapsid epitope, showing complete loss of responsiveness to the T-cell lines evaluated | [16,41] |
|
|
N E31del | 0.00 | Unknown | N/A | [16] |
|
|
N R32del | 0.00 | Unknown | N/A | [16] |
|
|
N G204R | 26.20 | Unknown | N/A | [16] |
| Nonstructural protein (NSP) | |||||
|
|
NSP3 K38R | 0.01 | Unknown | N/A | [16] |
|
|
NSP3 S1265del | 0.02 | Unknown | N/A | [16] |
|
|
NSP3 L1266I | 0.02 | Unknown | N/A | [16] |
|
|
NSP3 A1892T | 0.00 | Unknown | N/A | [16] |
|
|
NSP4 T492I | 47.76 | Viral oligomerization interfaces | N/A | [16] |
|
|
NSP5 P132H | 0.01 | Unknown | N/A | [16] |
|
|
NSP6 L105del | 0.02 | Unknown | N/A | [16] |
|
|
NSP6 S106del | 24.74 | Unknown | N/A | [16] |
|
|
NSP6 G107del | 24.74 | Unknown | N/A | [16] |
|
|
NSP6 I189V | 0.03 | Unknown | N/A | [16] |
|
|
NSP12 P323L | 96.69 | Viral oligomerization interfaces | N/A | [16] |
|
|
NSP14 I42V | 0.00 | Viral oligomerization interfaces | N/A | [16] |
Table 3.
Mutations in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage BA.1) spike protein influencing receptor binding.a
| Mutation | Frequency (%)b | Effect on virus-host interactions | Remarks | Reference |
| G339D | 0.01 | Increased RBDc expression | N/Ad | [16,21] |
| S371L | 0.00 | Increased ACE2e binding | N/A | [16,21] |
| S373P | 0.01 | Increased RBD expression | N/A | [16,21] |
| K417N | 0.83 | Increased RBD expression | N/A | [16,21,28] |
| N440K | 0.17 | Increased ACE2 binding | N/A | [16,21] |
| S477N | 1.31 | Increased ACE2 binding/ increased RBD expression | S477N was also resistant to neutralization by the human convalescent sera tested in this study, but not to vaccine-elicited sera | [16,21,29] |
| T478K | 52.56 | Increased ACE2 binding/increased RBD expression | Decreased sensitivity to convalescent sera | [21,25] |
| Q493R | 0.01 | Host change | N/A | [30,31] |
| G496S | 0.01 | Increased RBD expression | N/A | [21] |
| N501Y | 24.11 | Increased ACE2 binding/host change | Associated with increased transmissibility and increased affinity for human ACE2 receptor | [16,21,28,32] |
| Y505H | 0.00 | Increased RBD expression | N/A | [16,21] |
| D614G | 98.51 | Increased infectivity | Lower cycle threshold values were observed in G614 infections, indicating a higher viral load | [16,25,33] |
Table 4.
Mutations in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage BA.1) spike protein influencing viral oligomerization interfaces.a
| Mutations | Frequency (%)b | Remarks | Reference |
| S371L | 0.00 | N/Ac | [16,21] |
| S373P | 0.01 | N/A | [16,21] |
| S375F | 0.00 | N/A | [16,27] |
| K417N | 0.83 | N/A | [16,21,28] |
| S477N | 1.31 | S477N was also resistant to neutralization by the human convalescent sera tested in this study, but not to vaccine-elicited sera | [16,21,29] |
| Q493R | 0.01 | N/A | [30,31] |
| N501Y | 24.11 | Associated with increased transmissibility and increased affinity for human ACE2d receptor | [16,21,28,32] |
| Y505H | 0.00 | N/A | [16,21] |
| N764K | 0.01 | N/A | [16] |
| D796Y | 0.08 | N/A | [16] |
| N856K | 0.00 | N/A | [16] |
| Q954H | 0.00 | N/A | [16,40] |
| N969K | 0.00 | N/A | [16] |
| L981F | 0.00 | N/A | [16] |
Table 5.
Mutations in SARS-CoV-2 variant B.1.1.529 (Omicron, sublineage BA.1) spike protein influencing host adaptation and other mechanisms.a
| Mutations | Frequency (%)b | Effect on virus-host interactions | Remarks | Reference |
| A67V | 0.36 | Unknown | N/Ac | [16] |
| T95I | 21.32 | Unknown | N/A | [16] |
| G142D | 33.40 | Unknown | N/A | [16] |
| Q954H | 0.00 | Host adaptation (cell culture) | N/A | [16,40] |
| N211del | 0.02 | Unknown | N/A | [16] |
| L212I | 0.01 | Unknown | N/A | [16] |
| ins214EPE | 0.00 | Unknown | N/A | [16] |
| H655Y | 2.25 | Host adaptation (cats); spike glycoprotein fusion efficiency | N/A | [32–34] |
| N679K | 0.09 | Unknown | N/A | [16,37] |
| P681H | 22.73 | Unknown | P681H mutation at the S1/S2 site of the SARS-CoV-2 spike protein may increase its cleavability by furin-like proteases, but this does not translate into increased virus entry or membrane fusion | [16,38,39] |
| T547K | 0.00 | Unknown | N/A | [16] |
| N856K | 0.00 | Ligand binding | N/A | [16] |
Epidemiological Correlates
A total of 4224 SARS-CoV-2 genomic sequences (Delta, n=999; Omicron, n= 2937; and others, n= 288) were uploaded on GISAID from South Africa in the period of study. For the complete duration of the study, Delta correlated negatively with the number of new COVID-19 cases (r=–0.567, P=.004; 95% CI –0.79 to –0.21) but correlated positively with the number of new deaths (r=0.38, P=.07; 95% CI –0.025 to 0.68). The differential analysis of the SARS-CoV-2 genomic sequences from South Africa before and after the emergence of the first case of Omicron (dated November 5, 2021, EPI_ISL_7456440) reflected a sharp change in the dominance of the variant from Delta to Omicron (Figure 4). An inverse correlation of Omicron with Delta variants was noted (r=–0.99, P<.001; 95% CI –0.99 to –0.97) in the period of study. There has been a steep rise in the number of new COVID-19 cases in parallel with the increase in the proportion of Omicron since the first case of Omicron (74%-100% of total genomic sequences after November 15-17, 2021). However, no parallel increase was observed in the death cases, which otherwise showed a reverse trend (r=–0.04, P=.02; 95% CI –0.52 to 0.58) (Figure 4).
Figure 4.
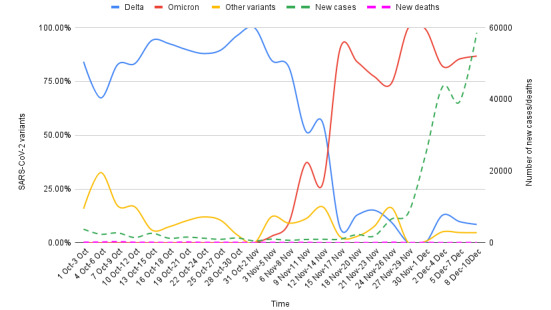
Epidemiological correlates of Omicron and Delta variants genomic sequences reported on GISAID from South Africa for the period of October 1 to December 10, 2021. The proportion of Delta and Omicron variants among the total SARS-CoV-2 genomic sequences were correlated with the new COVID-19 cases and deaths in the study period (3-day sum of each variable). A sharp change in the dominance from Delta to Omicron was observable since the report of the first Omicron case (November 5, 2021). The rise of Omicron cases paralleled the increase in the new COVID-19 cases. In comparison, the Delta variant showed a fall in the same period. Notably, there has been no increase in the number of deaths postemergence of Omicron. (Data sources: GISAID and Worldometer).
Discussion
Principal Findings
Our analysis of the SARS-CoV-2 genomic sequences and epidemiological data from South Africa unravels multiple observations regarding host-virus interactions, which may help to predict the further epidemiological potential of the Omicron variant. We found that compared to the current list of global VOCs/VOIs (as per the WHO), Omicron bears more sequence variation, specifically in the spike protein and RBM. Omicron showed the closest nucleotide and protein sequence homology with the Alpha variant. Further, the mutational analysis showed enrichment for the mutations decreasing ACE2-binding affinity and RBD protein expression, but increased propensity of immune escape. The analysis of the viral genomic sequences and epidemiological data from South Africa reflected an inverse correlation of Omicron with Delta variant infections, with a subsequent decrease. There was a steep rise in the number of new COVID-19 cases in parallel with the increase in the proportion of Omicron since the report of the first case; however, the incidence of deaths did not increase.
Sequence Homology With Wild-Type Strains and Existing SARS-CoV-2 VOCs/VOIs
Our analyses showed that among the existing VOCs and VOIs, Omicron bears the highest homology of the complete sequence and RBM (nucleotide and protein sequences) with the Alpha variant (Table S1 in Multimedia Appendix 1). Interestingly, similar to Alpha variant spike gene target failure, polymerase chain reaction (PCR)-based detection is a sensitive method for detecting Omicron in clinical samples [42].
As Omicron bears key mutations from multiple existing VOCs/VOIs, with approximate sequence homology variation rather than a direct descendance, the numerous recombination events between the variants inside hosts can be a more plausible explanation for its origin.
It will be pertinent to explore the evolutionary mechanisms involved in accumulating such a large number of mutations in Omicron. Speculations were raised that the long-term persistence of SARS-CoV-2 infection in an immunocompromised host could be a probable mechanism behind the origin of Omicron [43-46]. Avanzato et al [43] and Choi et al [45] reported case studies of the persistence of infection and accumulation of novel mutations in the SARS-CoV-2 spike gene and RBD in chronically ill and immunocompromised COVID-19 patients. Another such case was reported by Karim et al [44]. The authors documented the long persistence of SARS-CoV-2 infection (for more than 6 months) in a patient with advanced HIV and antiretroviral treatment failure. Through whole-genome sequencing for SARS-CoV-2 performed at multiple time points from patient samples, the authors demonstrated the early emergence of the E484K substitution, followed by N501Y, K417T, and many other mutations (including some novel mutations) in the spike gene and RBD. An increase in the genomic diversity reflecting the intrahost evolution of SARS-CoV-2 during prolonged infection was also noted in a recent cohort study by Voloch et al [46].
Effect on Virus-Host Interactions
Our analysis shows that Omicron accumulated multiple closely spaced mutations at the RBM with ACE2 (Figure 2). Notably, this variant has many of the mutations common with the earlier VOCs (Figure 3), many of which have been shown to enhance RBD-ACE2 binding in comparison to the wild-type strain [47] (Tables 1 and 3). The selective mutations present at or near the vicinity of the RBM (N440K, S477N, T478K, and N501Y) in most of the Omicron sequences are believed to stabilize binding with ACE2 (Tables 2-3). D614G, a critical mutation in all B.1 descendants [47], is known to stabilize the trimeric structure and create a more open conformation of the RBD, allowing stronger binding with ACE2 [47]. Paradoxically, our analysis suggests that the majority of the novel or rare spike mutations (<0.2% prevalence in the total sequenced samples, Tables 2-6) in Omicron may have a deleterious effect on host interactions owing to their presence at the constrained RBD regions in terms of ACE2 binding (10/15) and/or RBD expression (8/15) (Table 3). Notably, most of the spike mutations that predicted a favorable effect on ACE2 binding, RBD expression, or both are present in current VOCs, primarily the Delta (T478K), Alpha (N501Y), and Beta (K417N) variants. Further, a set of mutations in Omicron that are present inside (P681H) or in the vicinity (D614G, H655Y) of the furin cleavage site of SARS-CoV-2 spike protein—a small stretch of peptide (PRRAR) inserted at the intersection of spike segments S1 and S2 (amino acid residues 681-685)—can enhance proteolytic cleavage of spike protein by a host protease (furin), which is considered to improve its fusion to the host cell membrane [48]. P681H is characteristically present in multiple VOCs/VOIs such as B.1.1.7, P.1, Q.1, and B.1.621 lineage variants [49]. A mutation at the exact location, P681R, has been present in the Delta variant and its emerging sublineages [50]. Characterizing the individual mutations on RBM specifies that Omicron may not have more efficient interactions with the host than existing VOCs/VOIs, specifically Delta. Further assessment of the allosteric influence and dynamic interactions of the mutations present at the RBD and other regions of spike protein and in situ/in vivo studies will be necessary to understand their exact impact on host-receptor binding and its clinical correlates. The clinical data on the severity of the disease indicated a milder illness in Omicron infection than in the existing VOCs [51,52].
Viral Replication
Many of the mutations, especially in the nonspike regions, are linked with viral oligomerization, synthesis, and packaging of the ribonucleic acid core (Tables 4 and 6). These mutations likely have a role in virus replication inside the host cells [53]. The NSP12 P323L mutation located in the RNA-dependent RNA polymerase coding region is of particular interest (Figure 1, Table 6), as this has been a frequently observed mutation in the earlier variants (96.69%) (Table S1 in Multimedia Appendix 1). However, whether these mutations will have a positive or negative impact on viral replication remains unclear. Interestingly, the results of a comparative study [54] that employed ex vivo cultures of SARS-CoV-2 strains isolated from the respiratory tract of infected patients indicated higher replication rates for the Omicron variant. The authors observed that after 24 hours of incubation, Omicron replicated 70 times faster than wild-type and Delta variant strains in the human bronchus. In contrast, it replicated less efficiently (>10 times lower) in the human lung tissue than the wild-type strain and the replication rate was also lower than that of the Delta variant.
Immune Escape
Most spike mutations (18/32) in Omicron have occurred at the known antibody recognition sites (Table 2). Existing studies have established the role of these mutations in immune escape against convalescent sera, vaccine-acquired antibodies, and therapeutically used monoclonal antibodies (Table 2). The evidence from in situ studies indicates potential immune escape by Omicron against convalescent sera, vaccine-acquired antibodies, and therapeutically used monoclonal antibodies [42,55,56]. Interestingly, Omicron contains the K417N and E484A mutations, which are present in multiple existing variants and are believed to contribute to immune escape [47]. Of note, the K417 locus is a known epitope for CB26, a therapeutically used monoclonal antibody in COVID-19 [47]. A more significant number of mutations in Omicron spike protein, specifically in the RBD, may be an evolutionary gain in this variant, providing it with higher immune-escape ability. Support for this notion comes from a study by Nabel et al [57], who demonstrated that SARS-CoV-2 pseudotypes containing up to seven mutations, as opposed to the one to three found in earlier VOCs, were more resistant to neutralization by therapeutic antibodies and serum from vaccine recipients [57].
A nonspike mutation in the nucleocapsid (N) protein (P13L) present in Omicron (Table 6) was shown to cause complete loss of recognition by epitope-specific (B∗27:05-restricted CD8+ nucleocapsid epitope QRNAPRITF9-17) T cells in a cell line–based in situ study [41]. However, no such evidence in human samples is currently available. In another study, Redd et al [58] examined peripheral blood mononuclear cell samples from PCR-confirmed, recovered/convalescent COVID-19 cases (N=30) for their anti-SARS-CoV-2 CD8+ T-cell responses with Omicron. The authors noted that only one low-prevalence (found in 7%) epitope (GVYFASTEK, restricted to HLA*A03:01 and HLA*A11:01) from the spike protein (T95I) region was mutated in Omicron [58]. The presence of these mutations raises concerns about escaping T cell immunity by Omicron [59] and hence should be explored in further detail.
The overall evidence supports Omicron’s very high immune-escape ability [42,55,56,60]. Cele et al [42] tested the ability of plasma from 14 BNT162b2-vaccinated study participants to neutralize Omicron versus the wild-type D614G virus in a live virus neutralization assay. The authors observed that Omicron showed a 41-fold decline in the 50% focus reduction neutralization test geometric mean titer compared to the wild-type D614G virus in subjects without previous infection (6/14). Interestingly, earlier, those with the infection showed relatively higher neutralization titers with Omicron (6/14), which indicated that the last infection, followed by vaccination or booster, might increase the neutralization levels and confer protection from severe disease in cases of Omicron infection.
Epidemiological Correlates: Omicron Versus Delta Variants
The analysis of the SARS-CoV-2 genomic sequences from South Africa indicates that Omicron gained an advantage in terms of transmissibility over the Delta variant (Figure 4). A third COVID-19 wave driven by the Delta variant occurred in South Africa [61]; hence, the epidemiological characteristics of the Delta and Omicron variants in the local population should be analyzed in this backdrop. We observed that before the arrival of Omicron, the Delta variant was dominant locally; by contrast, at present, the majority of new sequences are from Omicron (Omicron vs Delta r=–0.99, P<.001; 95% CI –0.99 to –0.97) (Figure 4). The steep rise in the new COVID-19 cases in South Africa seems to be driven by Omicron, whereas Delta variant–linked cases are seeing a decline (Figure 4). The rapid rise in new COVID-19 cases connected with the emergence of a new SARS-CoV-2 variant strongly indicated the commencement of a new COVID-19 wave in South Africa [14].
Further, death, which is considered a strong indicator of virulence/lethality, showed a negative correlation (r=–0.04, P=.02; 95% CI –0.52 to 0.58) (Figure 4) with the rise in Omicron. However, death correlated positively with the Delta variant in the period postemergence (r=0.38, P=.07; 95% CI –0.025 to 0.68) over the complete study period. This pattern indicates that the reported incidences of death were primarily linked with Delta rather than with Omicron. The significantly reduced lethality of Omicron compared to Delta has been confirmed through recent epidemiological studies [62-64].
An approximately 2.4 (2.0-2.7) times higher transmissibility was suggested with Omicron compared to the Delta variant in the South African population [65]. An estimate from the United Kingdom indicated that Omicron’s risk of spreading the infection to members of a household is 3 times higher than that of the Delta variant [66]. A significantly shorter incubation period and early reaching of the peak have been reported for the Omicron variant [67]. Based on the epidemiological patterns observed in South Africa in our analysis, an epidemiological advantage to Omicron in comparison to Delta can be inferred in terms of transmissibility [66]. However, we found no indications of increased lethality with Omicron compared to Delta and other variants circulating in the South African population.
Notably, the presence of an immunological barrier in the population imparted by the recent COVID-19 wave mediated by the Delta variant could be a likely reason for this variant’s fall in new cases [7,68]. A continuous fall in Delta cases was also noticeable in the period before the emergence of Omicron (Figure 4), further substantiating this notion. The data records showed that a significant proportion of the local population in South Africa was fully vaccinated at the time of Omicron’s emergence (25.2%) [69]. Notably, the high number of immune escape–related mutations in Delta could have contributed to lowered efficacy of the vaccines, immunity from natural infections, and therapeutically used antibodies [47]. As Omicron contains a much higher number of immune escape–related mutations, including many shared with Delta (Figure 3), Omicron might have added potential for vaccine breakthrough infections and reinfections. Similar speculations were presented by other authors and global health regulatory bodies [2,70,71]. A higher risk of reinfections with Omicron was indicated by Pulliam et al [72] based on a retrospective analysis of routine epidemiological surveillance data to examine whether SARS-CoV-2 reinfection risk has changed over time in South Africa in the context of the emergence of the consecutive variants: Beta, Delta, and Omicron. The authors noted that as compared to the first wave driven by wild-type strains, subsequent waves by Beta and Delta variants had a lower estimated hazard ratio for reinfection versus primary infection (relative hazard ratio for wave 2 versus wave 1: 0.75, 95% CI 0.59-0.97; for wave 3 versus wave 1: 0.71, 95% CI 0.56-0.92) in comparison to Omicron (Omicron surge for the period of November 1-27, 2021, versus wave 1: 2.39, 95% CI 1.88-3.11).
Study Limitations
We analyzed a limited number of genomic sequences and epidemiological data from specific geographical regions affected by Omicron. Further, the relative frequency of specific lineage-characterizing mutations in the Omicron variant may have varied since the study’s inception. Both of these limitations may have an impact on the quality of the results.
Conclusion
In silico analysis of viral genomic sequences suggests that the Omicron variant has more remarkable immune-escape ability than the existing VOCs/VOIs, including Delta, but reduced virulence/lethality than other reported variants. The higher power for immune escape for Omicron was a likely reason for the resurgence in COVID-19 cases and its soon becoming a globally dominant strain. Being more infectious but less lethal than the existing variants, Omicron could have plausibly led to widespread unnoticed new, repeated, and vaccine breakthrough infections, raising the population-level immunity barrier against the emergence of new lethal variants. The Omicron variant could have thus paved the way for the end of the pandemic.
Abbreviations
- ACE2
angiotensin-converting enzyme 2
- GISAID
Global Initiative on Sharing Avian Influenza Data
- NCBI
National Center for Biotechnology Information
- PANGO
Phylogenetic Assignment of Named Global Outbreak
- PCR
polymerase chain reaction
- RBD
receptor-binding domain
- RBM
receptor-binding motif
- VOC
variant of concern
- VOI
variant of interest
- WHO
World Health Organization
Global spread of the SARS-CoV-2 Omicron variant (Figure S1). Nucleotide and protein sequence homology of Omicron (BA.1) with wild-type SARS-CoV-2 and other global variants of concern/interest (Table S1).
Data Sharing
Primary data used for this study are publicly available on the GISAID database [16] for SARS-CoV-2 genomic sequences and Worldometer [23] for epidemiological data. The categorized data for the study period can be availed from the corresponding author upon reasonable request.
Footnotes
Authors' Contributions: AK, GK, and PD collected samples and analyzed data. AK wrote the first draft. AA and HNS performed the statistical analysis. MAF, SK, RKN, RKJ, CS, MK, PP, KS, KK, and SNP reviewed and edited the paper. All authors consented to submit the final draft.
Conflicts of Interest: None declared.
References
- 1.Tracking of hCoV-19 variants. GISAID. [2021-07-20]. https://www.gisaid.org/hcov19-variants/
- 2.Update on Omicron. World Health Organization. 2021. Nov 28, [2021-12-04]. https://www.who.int/news/item/28-11-2021-update-on-omicron .
- 3.Callaway E. Heavily mutated Omicron variant puts scientists on alert. Nature. 2021 Dec 25;600(7887):21. doi: 10.1038/d41586-021-03552-w.10.1038/d41586-021-03552-w [DOI] [PubMed] [Google Scholar]
- 4.Omicron variant report. outbreak.info. [2022-11-26]. https://outbreak.info/situation-reports/omicron .
- 5.One year since the emergence of COVID-19 virus variant Omicron. World Health Organization. 2022. Nov 25, [2022-11-26]. https://www.who.int/news-room/feature-stories/detail/one-year-since-the-emergence-of-omicron .
- 6.Nealon J, Cowling BJ. Omicron severity: milder but not mild. Lancet. 2022 Jan 29;399(10323):412–413. doi: 10.1016/S0140-6736(22)00056-3. https://europepmc.org/abstract/MED/35065007 .S0140-6736(22)00056-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madhi SA, Kwatra G, Myers JE, Jassat W, Dhar N, Mukendi CK, Nana AJ, Blumberg L, Welch R, Ngorima-Mabhena N, Mutevedzi PC. Population immunity and Covid-19 severity with Omicron variant in South Africa. N Engl J Med. 2022 Apr 07;386(14):1314–1326. doi: 10.1056/NEJMoa2119658. https://europepmc.org/abstract/MED/35196424 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharyya RP, Hanage WP. Challenges in inferring intrinsic severity of the SARS-CoV-2 Omicron variant. N Engl J Med. 2022 Feb 17;386(7):e14. doi: 10.1056/NEJMp2119682. [DOI] [PubMed] [Google Scholar]
- 9.Strasser Z, Hadavand A, Murphy S, Strasser Z, Hadavand PA, Murphy S. SARS-CoV-2 Omicron variant is as deadly as previous waves after adjusting for vaccinations, demographics, and comorbidities. Research Square Preprints. 2022. May 02, [2022-12-19]. https://assets.researchsquare.com/files/rs-1601788/v1_covered.pdf?c=1651763984 .
- 10.Daria S, Islam MR. The SARS-CoV-2 omicron wave is indicating the end of the pandemic phase but the COVID-19 will continue. J Med Virol. 2022 Jun 04;94(6):2343–2345. doi: 10.1002/jmv.27635. https://europepmc.org/abstract/MED/35098543 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray CJL. COVID-19 will continue but the end of the pandemic is near. Lancet. 2022 Jan 29;399(10323):417–419. doi: 10.1016/S0140-6736(22)00100-3. https://europepmc.org/abstract/MED/35065006 .S0140-6736(22)00100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson D, Doshi P. The end of the pandemic will not be televised. BMJ. 2021 Dec 14;375:e068094. doi: 10.1136/bmj-2021-068094. [DOI] [PubMed] [Google Scholar]
- 13.Das S, Samanta S, Banerjee J, Pal A, Giri B, Kar SS, Dash SK. Is Omicron the end of pandemic or start of a new innings? Travel Med Infect Dis. 2022 Jul;48:102332. doi: 10.1016/j.tmaid.2022.102332. https://europepmc.org/abstract/MED/35472451 .S1477-8939(22)00078-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar A, Asghar A, Dwivedi P, Kumar G, Narayan RK, Jha RK, Parashar R, Sahni C, Pandey SN. A bioinformatics tool for predicting future COVID-19 waves based on a retrospective analysis of the second wave in India: model development study. JMIR Bioinform Biotech. 2022 Sep 22;3(1):e36860. doi: 10.2196/36860. https://europepmc.org/abstract/MED/36193192 .v3i1e36860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Hoffer A, Vatani S, Cot C, Cacciapaglia G, Chiusano ML, Cimarelli A, Conventi F, Giannini A, Hohenegger S, Sannino F. Variant-driven early warning via unsupervised machine learning analysis of spike protein mutations for COVID-19. Sci Rep. 2022 Jun 03;12(1):9275. doi: 10.1038/s41598-022-12442-8.10.1038/s41598-022-12442-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.GISAID. [2022-11-26]. https://gisaid.org/
- 17.Tracking SARS-CoV-2 variants. World Health Organization. [2021-06-17]. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
- 18.SARS-CoV-2 data explorer. outbreak.info. [2022-11-26]. https://outbreak.info/
- 19.Expasy. Swiss Institute of Bioinformatics. [2022-12-19]. https://www.expasy.org/
- 20.Basic Local Alignment Search Tool. National Library of Medicine. [2022-11-26]. https://blast.ncbi.nlm.nih.gov/Blast.cgi .
- 21.Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, King NP, Veesler D, Bloom JD. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020 Sep 03;182(5):1295–1310. doi: 10.1016/j.cell.2020.08.012. https://linkinghub.elsevier.com/retrieve/pii/S0092-8674(20)31003-5 .S0092-8674(20)31003-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.SARS-CoV-2 RBD DMS. JBloom Lab. 2022. Mar 03, [2022-12-19]. https://jbloomlab.github.io/SARS-CoV-2-RBD_DMS/
- 23.South Africa COVID - Coronavirus Statistics. Worldometer. [2022-11-26]. https://www.worldometers.info/coronavirus/country/south-africa .
- 24.The statistics of similarity scores. National Center for Biotechnology Information (NCBI) [2022-11-26]. https://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html .
- 25.Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, Zhao C, Zhang Q, Liu H, Nie L, Qin H, Wang M, Lu Q, Li X, Sun Q, Liu J, Zhang L, Li X, Huang W, Wang Y. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell. 2020 Sep 03;182(5):1284–1294. doi: 10.1016/j.cell.2020.07.012. https://linkinghub.elsevier.com/retrieve/pii/S0092-8674(20)30877-1 .S0092-8674(20)30877-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen L, Bard JD, Triche TJ, Judkins AR, Biegel JA, Gai X. Rapidly emerging SARS-CoV-2 B.1.1.7 sub-lineage in the United States of America with spike protein D178H and membrane protein V70L mutations. Emerg Microbes Infect. 2021 Dec 26;10(1):1293–1299. doi: 10.1080/22221751.2021.1943540. https://www.tandfonline.com/doi/full/10.1080/22221751.2021.1943540 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, Hilton SK, Huddleston J, Eguia R, Crawford KH, Dingens AS, Nargi RS, Sutton RE, Suryadevara N, Rothlauf PW, Liu Z, Whelan SP, Carnahan RH, Crowe JE, Bloom JD. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021 Jan 13;29(1):44–57. doi: 10.1016/j.chom.2020.11.007. https://linkinghub.elsevier.com/retrieve/pii/S1931-3128(20)30624-7 .S1931-3128(20)30624-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, Wang M, Yu J, Zhang B, Kwong PD, Graham BS, Mascola JR, Chang JY, Yin MT, Sobieszczyk M, Kyratsous CA, Shapiro L, Sheng Z, Huang Y, Ho DD. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021 May 08;593(7857):130–135. doi: 10.1038/s41586-021-03398-2.10.1038/s41586-021-03398-2 [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Zhang L, Zhang Y, Wang H, Ding R, Nie J, Li Q, Liu S, Yu Y, Yang X, Duan K, Qu X, Wang Y, Huang W. The antigenicity of epidemic SARS-CoV-2 variants in the United Kingdom. Front Immunol. 2021 Jun 17;12:687869. doi: 10.3389/fimmu.2021.687869. https://europepmc.org/abstract/MED/34220844 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greaney AJ, Loes AN, Crawford KH, Starr TN, Malone KD, Chu HY, Bloom JD. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021 Mar 10;29(3):463–476. doi: 10.1016/j.chom.2021.02.003. https://linkinghub.elsevier.com/retrieve/pii/S1931-3128(21)00082-2 .S1931-3128(21)00082-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu K, Peng G, Wilken M, Geraghty RJ, Li F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J Biol Chem. 2012 Mar 16;287(12):8904–8911. doi: 10.1074/jbc.M111.325803. https://linkinghub.elsevier.com/retrieve/pii/S0021-9258(20)60852-3 .S0021-9258(20)60852-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, Lambson BE, de Oliveira T, Vermeulen M, van der Berg K, Rossouw T, Boswell M, Ueckermann V, Meiring S, von Gottberg A, Cohen C, Morris L, Bhiman JN, Moore PL. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat Med. 2021 Apr 02;27(4):622–625. doi: 10.1038/s41591-021-01285-x.10.1038/s41591-021-01285-x [DOI] [PubMed] [Google Scholar]
- 33.Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, Sheffield COVID-19 Genomics Group. McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020 Aug 20;182(4):812–827. doi: 10.1016/j.cell.2020.06.043. https://linkinghub.elsevier.com/retrieve/pii/S0092-8674(20)30820-5 .S0092-8674(20)30820-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braun KM, Moreno GK, Halfmann PJ, Hodcroft EB, Baker DA, Boehm EC, Weiler AM, Haj AK, Hatta M, Chiba S, Maemura T, Kawaoka Y, Koelle K, O'Connor DH, Friedrich TC. Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. PLoS Pathog. 2021 Feb 26;17(2):e1009373. doi: 10.1371/journal.ppat.1009373. https://boris.unibe.ch/id/eprint/152793 .PPATHOGENS-D-20-02782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dieterle ME, Haslwanter D, Bortz RH, Wirchnianski AS, Lasso G, Vergnolle O, Abbasi SA, Fels JM, Laudermilch E, Florez C, Mengotto A, Kimmel D, Malonis RJ, Georgiev G, Quiroz J, Barnhill J, Pirofski L, Daily JP, Dye JM, Lai JR, Herbert AS, Chandran K, Jangra RK. A replication-competent vesicular stomatitis virus for studies of SARS-CoV-2 spike-mediated cell entry and its inhibition. Cell Host Microbe. 2020 Sep 09;28(3):486–496. doi: 10.1016/j.chom.2020.06.020. https://linkinghub.elsevier.com/retrieve/pii/S1931-3128(20)30361-9 .S1931-3128(20)30361-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, Giordano S, Lanza K, Negron N, Ni M, Wei Y, Atwal GS, Murphy AJ, Stahl N, Yancopoulos GD, Kyratsous CA. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020 Aug 21;369(6506):1014–1018. doi: 10.1126/science.abd0831. https://www.science.org/doi/abs/10.1126/science.abd0831?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub0pubmed .science.abd0831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabir DK. Analysis of SARS-COV2 spike protein variants among Iraqi isolates. Gene Rep. 2022 Mar;26:101420. doi: 10.1016/j.genrep.2021.101420. https://europepmc.org/abstract/MED/34754982 .S2452-0144(21)00404-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voss C, Esmail S, Liu X, Knauer M, Ackloo S, Kaneko T, Lowes L, Stogios P, Seitova A, Hutchinson A, Yusifov F, Skarina T, Evdokimova E, Loppnau P, Ghiabi P, Haijan T, Zhong S, Abdoh H, Hedley BD, Bhayana V, Martin CM, Slessarev M, Chin-Yee B, Fraser DD, Chin-Yee I, Li SS. Epitope-specific antibody responses differentiate COVID-19 outcomes and variants of concern. JCI Insight. 2021 Jul 08;6(13):e148855. doi: 10.1172/jci.insight.148855.e148855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lubinski B, Fernandes MH, Frazier L, Tang T, Daniel S, Diel DG, Jaimes JA, Whittaker GR. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. iScience. 2022 Jan 21;25(1):103589. doi: 10.1016/j.isci.2021.103589. https://linkinghub.elsevier.com/retrieve/pii/S2589-0042(21)01559-5 .S2589-0042(21)01559-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramirez S, Fernandez-Antunez C, Galli A, Underwood A, Pham LV, Ryberg LA, Feng S, Pedersen MS, Mikkelsen LS, Belouzard S, Dubuisson J, Sølund C, Weis N, Gottwein JM, Fahnøe U, Bukh J. Overcoming culture restriction for SARS-CoV-2 in human cells facilitates the screening of compounds inhibiting viral replication. Antimicrob Agents Chemother. 2021 Jun 17;65(7):e0009721. doi: 10.1128/AAC.00097-21. https://journals.asm.org/doi/abs/10.1128/AAC.00097-21?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub0pubmed .AAC.00097-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Silva TI, Liu G, Lindsey B, Dong D, Moore S, Hsu N, Shah D, Wellington D, Mentzer AJ, Angyal A, Brown R, Parker MD, Ying Z, Yao X, Turtle L, Dunachie S, COVID-19 Genomics UK (COG-UK) Consortium. Maini MK, Ogg G, Knight JC, ISARIC4C Investigators. Peng Y, Rowland-Jones SL, Dong T. The impact of viral mutations on recognition by SARS-CoV-2 specific T cells. iScience. 2021 Nov 19;24(11):103353. doi: 10.1016/j.isci.2021.103353. https://linkinghub.elsevier.com/retrieve/pii/S2589-0042(21)01322-5 .S2589-0042(21)01322-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cele S, Jackson L, Khoury DS, Khan K, Moyo-Gwete T, Tegally H, San JE, Cromer D, Scheepers C, Amoako D, Karim F, Bernstein M, Lustig G, Archary D, Smith M, Ganga Y, Jule Z, Reedoy K, Hwa S, Giandhari J, Blackburn JM, Gosnell BI, Abdool Karim SS, Hanekom W, NGS-SA. COMMIT-KZN Team. von Gottberg A, Bhiman JN, Lessells RJ, Moosa MYS, Davenport MP, de Oliveira T, Moore PL, Sigal A. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022 Feb;602(7898):654–656. doi: 10.1038/s41586-021-04387-1. https://europepmc.org/abstract/MED/35016196 .10.1038/s41586-021-04387-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avanzato VA, Matson MJ, Seifert SN, Pryce R, Williamson BN, Anzick SL, Barbian K, Judson SD, Fischer ER, Martens C, Bowden TA, de Wit E, Riedo FX, Munster VJ. Case study: Prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell. 2020 Dec 23;183(7):1901–1912. doi: 10.1016/j.cell.2020.10.049. https://linkinghub.elsevier.com/retrieve/pii/S0092-8674(20)31456-2 .S0092-8674(20)31456-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karim F, Moosa M, Gosnell B, Cele S, Giandhari J, Pillay S. Persistent SARS-CoV-2 infection and intra-host evolution in association with advanced HIV infection. medRxiv. 2021. Jun 04, [2022-12-19]. https://www.medrxiv.org/content/10.1101/2021.06.03.21258228v1#:~:text=While%20most%20people%20effectively%20clear,HIV%20and%20antiretroviral%20treatment%20failure .
- 45.Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, Solomon IH, Kuo H, Boucau J, Bowman K, Adhikari UD, Winkler ML, Mueller AA, Hsu TY, Desjardins M, Baden LR, Chan BT, Walker BD, Lichterfeld M, Brigl M, Kwon DS, Kanjilal S, Richardson ET, Jonsson AH, Alter G, Barczak AK, Hanage WP, Yu XG, Gaiha GD, Seaman MS, Cernadas M, Li JZ. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N Engl J Med. 2020 Dec 03;383(23):2291–2293. doi: 10.1056/NEJMc2031364. https://europepmc.org/abstract/MED/33176080 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voloch C, da Silva Francisco R, de Almeida LGP, Brustolini OJ, Cardoso CC, Gerber AL, de C Guimarães AP, de Carvalho Leitão I, Mariani D, Ota VA, Lima CX, Teixeira MM, Dias ACF, Galliez RM, Faffe DS, Pôrto LC, Aguiar RS, Castiñeira TMPP, Ferreira OC, Tanuri A, de Vasconcelos ATR. Intra-host evolution during SARS-CoV-2 prolonged infection. Virus Evol. 2021 Sep 29;7(2):veab078. doi: 10.1093/ve/veab078. https://europepmc.org/abstract/MED/34642605 .veab078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar A, Parashar R, Kumar S, Faiq MA, Kumari C, Kulandhasamy M, Narayan RK, Jha RK, Singh HN, Prasoon P, Pandey SN, Kant K. Emerging SARS-CoV-2 variants can potentially break set epidemiological barriers in COVID-19. J Med Virol. 2022 Apr;94(4):1300–1314. doi: 10.1002/jmv.27467. https://europepmc.org/abstract/MED/34811761 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar A, Prasoon P, Kumari C, Pareek V, Faiq MA, Narayan RK, Kulandhasamy M, Kant K. SARS-CoV-2-specific virulence factors in COVID-19. J Med Virol. 2021 Mar;93(3):1343–1350. doi: 10.1002/jmv.26615. [DOI] [PubMed] [Google Scholar]
- 49.S:P681H mutation report. outbreak.info. [2021-12-08]. https://outbreak.info/situation-reports?muts=S%3AP681H .
- 50.S:P681R mutation report. outbreak.info. [2021-12-08]. https://outbreak.info/situation-reports?muts=S%3AP681R .
- 51.Esper F, Adhikari T, Tu Z, Cheng Y, El-Haddad K, Farkas D, Bosler D, Rhoads D, Procop GW, Ko JS, Jehi L, Li J, Rubin BP. Alpha to Omicron: disease severity and clinical outcomes of major SARS-CoV-2 variants. J Infect Dis. 2022 Oct 10;:jiac411. doi: 10.1093/infdis/jiac411. https://europepmc.org/abstract/MED/36214810 .6754902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Consolazio D, Murtas R, Tunesi S, Lamberti A, Senatore S, Faccini M, Russo AG. A comparison between Omicron and earlier COVID-19 variants' disease severity in the Milan area, Italy. Front Epidemiol. 2022 Jun 28;2:891162. doi: 10.3389/fepid.2022.891162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naqvi A, Fatima K, Mohammad T, Fatima U, Singh I, Singh A, Atif SM, Hariprasad G, Hasan GM, Hassan MI. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 2020 Oct 01;1866(10):165878. doi: 10.1016/j.bbadis.2020.165878. https://linkinghub.elsevier.com/retrieve/pii/S0925-4439(20)30226-X .S0925-4439(20)30226-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hui KPY, Ho JCW, Cheung M, Ng K, Ching RHH, Lai K, Kam TT, Gu H, Sit K, Hsin MKY, Au TWK, Poon LLM, Peiris M, Nicholls JM, Chan MCW. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature. 2022 Mar 01;603(7902):715–720. doi: 10.1038/s41586-022-04479-6.10.1038/s41586-022-04479-6 [DOI] [PubMed] [Google Scholar]
- 55.Wilhelm A, Widera M, Grikscheit K, Toptan T, Schenk B, Pallas C, Metzler M, Kohmer N, Hoehl S, Marschalek R, Herrmann E, Helfritz FA, Wolf T, Goetsch U, Ciesek S. Limited neutralisation of the SARS-CoV-2 Omicron subvariants BA.1 and BA.2 by convalescent and vaccine serum and monoclonal antibodies. EBioMedicine. 2022 Aug;82:104158. doi: 10.1016/j.ebiom.2022.104158. https://linkinghub.elsevier.com/retrieve/pii/S2352-3964(22)00339-5 .S2352-3964(22)00339-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu J, Peng P, Cao X, Wu K, Chen J, Wang K, Tang N, Huang A. Increased immune escape of the new SARS-CoV-2 variant of concern Omicron. Cell Mol Immunol. 2022 Feb 11;19(2):293–295. doi: 10.1038/s41423-021-00836-z. https://europepmc.org/abstract/MED/35017716 .10.1038/s41423-021-00836-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nabel KG, Clark SA, Shankar S, Pan J, Clark LE, Yang P, Coscia A, McKay LGA, Varnum HH, Brusic V, Tolan NV, Zhou G, Desjardins M, Turbett SE, Kanjilal S, Sherman AC, Dighe A, LaRocque RC, Ryan ET, Tylek C, Cohen-Solal JF, Darcy AT, Tavella D, Clabbers A, Fan Y, Griffiths A, Correia IR, Seagal J, Baden LR, Charles RC, Abraham J. Structural basis for continued antibody evasion by the SARS-CoV-2 receptor binding domain. Science. 2022 Jan 21;375(6578):eabl6251. doi: 10.1126/science.abl6251. https://www.science.org/doi/abs/10.1126/science.abl6251?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub0pubmed .10.1126/science.abl6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Redd A, Nardin A, Kared H, Bloch E, Abel B, Pekosz A, Laeyendecker O, Fehlings M, Quinn TC, Tobian AA. Minimal cross-over between mutations associated with Omicron variant of SARS-CoV-2 and CD8+ T cell epitopes identified in COVID-19 convalescent individuals. bioRxiv. 2021. Dec 09, [2022-12-19]. https://www.biorxiv.org/content/10.1101/2021.12.06.471446v1 . [DOI] [PMC free article] [PubMed]
- 59.Grifoni A, Sidney J, Vita R, Peters B, Crotty S, Weiskopf D, Sette A. SARS-CoV-2 human T cell epitopes: adaptive immune response against COVID-19. Cell Host Microbe. 2021 Jul 14;29(7):1076–1092. doi: 10.1016/j.chom.2021.05.010. https://linkinghub.elsevier.com/retrieve/pii/S1931-3128(21)00238-9 .S1931-3128(21)00238-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.SARS-CoV-2 variants of concern and variants under investigation in England. Variant of concern: Omicron, VOC21NOV-01 (B.1.1.529). Technical briefing 30. UK Health Security Agency. 2021. Dec 03, [2022-12-19]. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1038404/Technical_Briefing_30.pdf .
- 61.Tegally H, Wilkinson E, Althaus C, Giovanetti M, San J, Giandhari J. Rapid replacement of the Beta variant by the Delta variant in South Africa. medRxiv. 2021. Sep 27, [2022-12-19]. https://www.medrxiv.org/content/10.1101/2021.09.23.21264018v1 .
- 62.Sigal A, Milo R, Jassat W. Estimating disease severity of Omicron and Delta SARS-CoV-2 infections. Nat Rev Immunol. 2022 May 12;22(5):267–269. doi: 10.1038/s41577-022-00720-5. https://europepmc.org/abstract/MED/35414124 .10.1038/s41577-022-00720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ward IL, Bermingham C, Ayoubkhani D, Gethings OJ, Pouwels KB, Yates T, Khunti K, Hippisley-Cox J, Banerjee A, Walker AS, Nafilyan V. Risk of covid-19 related deaths for SARS-CoV-2 omicron (B.1.1.529) compared with delta (B.1.617.2): retrospective cohort study. BMJ. 2022 Aug 02;378:e070695. doi: 10.1136/bmj-2022-070695. http://www.bmj.com/lookup/pmidlookup?view=long&pmid=35918098 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nyberg T, Ferguson NM, Nash SG, Webster HH, Flaxman S, Andrews N, Hinsley W, Bernal JL, Kall M, Bhatt S, Blomquist P, Zaidi A, Volz E, Aziz NA, Harman K, Funk S, Abbott S, COVID-19 Genomics UK (COG-UK) consortium. Hope R, Charlett A, Chand M, Ghani AC, Seaman SR, Dabrera G, De Angelis D, Presanis AM, Thelwall S. Comparative analysis of the risks of hospitalisation and death associated with SARS-CoV-2 omicron (B.1.1.529) and delta (B.1.617.2) variants in England: a cohort study. Lancet. 2022 Apr 02;399(10332):1303–1312. doi: 10.1016/S0140-6736(22)00462-7. https://linkinghub.elsevier.com/retrieve/pii/S0140-6736(22)00462-7 .S0140-6736(22)00462-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearson C, Silal S, Dushoff J, Abbott S, van Schalkwyk C, Bingham J, Meyer-Rath G, Jamieson L, Glass A, Wolter N, Moultrie H, Pulliam J, on behalf of the South African COVID-19 Modelling Consortium Google Drive. [2021-12-04]. https://drive.google.com/file/d/1hA6Mec2Gq3LGqTEOj35RqSeAb_SmXpbI/view .
- 66.SARS-CoV-2 variants of concern and variants under investigation in England. Technical briefing 31. UK Health Security Agency. 2021. Dec 10, [2022-12-19]. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1040076/Technical_Briefing_31.pdf .
- 67.Lundberg AL, Lorenzo-Redondo R, Ozer EA, Hawkins CA, Hultquist JF, Welch SB, Prasad PV, Oehmke JF, Achenbach CJ, Murphy RL, White JI, Havey RJ, Post LA. Has Omicron changed the evolution of the pandemic? JMIR Public Health Surveill. 2022 Jan 31;8(1):e35763. doi: 10.2196/35763. https://publichealth.jmir.org/2022/1/e35763/ v8i1e35763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lundberg AL, Lorenzo-Redondo R, Hultquist JF, Hawkins CA, Ozer EA, Welch SB, Prasad PVV, Achenbach CJ, White JI, Oehmke JF, Murphy RL, Havey RJ, Post LA. Overlapping Delta and Omicron outbreaks during the COVID-19 pandemic: dynamic panel data estimates. JMIR Public Health Surveill. 2022 Jun 03;8(6):e37377. doi: 10.2196/37377. https://publichealth.jmir.org/2022/6/e37377/ v8i6e37377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coronavirus (COVID-19) vaccinations. Our World in Data. [2021-12-06]. https://ourworldindata.org/covid-vaccinations?country=~ZAF)
- 70.Threat Assessment Brief: Implications of the further emergence and spread of the SARS-CoV-2 B.1.1.529 variant of concern (Omicron) for the EU/EEA - first update. European Centre for Disease Prevention and Control. 2021. Dec 02, [2021-12-05]. https://www.ecdc.europa.eu/en/publications-data/covid-19-threat-assessment-spread-omicron-first-update .
- 71.Science Brief: Omicron (B.1.1.529) Variant. Centers for Disease Control and Prevention. 2021. Dec 02, [2021-12-05]. https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html#6 . [PubMed]
- 72.Pulliam J, van Schalkwyk C, Govender N, von Gottberg A, Cohen C, Groome M, Dushoff J, Mlisana K, Moultrie H. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science. 2022 May 06;376(6593):eabn4947. doi: 10.1126/science.abn4947. https://www.science.org/doi/abs/10.1126/science.abn4947?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub0pubmed . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Global spread of the SARS-CoV-2 Omicron variant (Figure S1). Nucleotide and protein sequence homology of Omicron (BA.1) with wild-type SARS-CoV-2 and other global variants of concern/interest (Table S1).