

CONSPECTUS:
SARS-CoV-2 is the etiological pathogen of the COVID-19 pandemic, which led to more than 6.5 million deaths since the beginning of the outbreak in December 2019. The unprecedented disruption of social life and public health caused by COVID-19 calls for fast-track development of diagnostic kits, vaccines, and antiviral drugs. Small molecule antivirals are essential complements of vaccines and can be used for the treatment of SARS-CoV-2 infections. Currently, there are three FDA-approved antiviral drugs, remdesivir, molnupiravir, and Paxlovid. Given the moderate clinical efficacy of remdesivir and molnupiravir, the drug-drug interaction of Paxlovid, and the emergence of SARS-CoV-2 variants with potential drug resistant mutations, there is a pressing need for additional antivirals to combat current and future coronavirus outbreaks.
In this account, we describe our efforts in developing covalent and non-covalent main protease (Mpro) inhibitors, and the identification of nirmatrelvir resistant mutants. We initially discovered GC376, calpain inhibitors II and XII, and boceprevir as dual inhibitors of Mpro and host cathepsin L from a screening of protease inhibitor library. Given the controversy of targeting cathepsin L, we subsequently shifted the focus to designing Mpro specific inhibitors. Specifically, guided by the X-ray crystal structures of these initial hits, we designed non-covalent Mpro inhibitors such as Jun8-76-3R that are highly selective towards Mpro over host cathepsin L. Using the same scaffold, we also designed covalent Mpro inhibitors with novel cysteine reactive warheads containing di- and tri-haloacetamides, which similarly had high target specificity. In parallel to our drug discovery efforts, we developed the cell based FlipGFP Mpro assay to characterize the cellular target engagement of our rationally designed Mpro inhibitors. The FlipGFP assay was also applied to validate the structurally disparate Mpro inhibitors reported in the literature. Lastly, we introduce recent progress in identifying naturally occurring Mpro mutants that are resistant to nirmatrelvir from genome mining of the nsp5 sequences deposited in the GISAID database. Collectively, the covalent and non-covalent Mpro inhibitors and the nirmatrelvir resistant hot spot residues from our studies provide insightful guidance for future work aimed at developing orally bioavailable Mpro inhibitors that do not have overlapping resistance profile with nirmatrelvir.
Graphical Abstract
INTRODUCTION
SARS-CoV-2 is an enveloped, positive-sense, and single-stranded RNA virus that belongs to the β-lineage of the coronavirus.5 The β lineage also contains SARS-CoV and MERS-CoV, both of which have caused outbreaks with significant mortalities.6 SARS-CoV-2 shares ~82% of sequence identity as SARS-CoV. However, unlike SARS-CoV, SARS-CoV-2 has evolved to be more transmissible with a large percentage of infected individuals being asymptomatic, rendering it challenging for contact tracing and disease containment. Nonetheless, the knowledge gained through studying SARS-CoV pathogenicity in the past two decades paved the way for the fast-track development of SARS-CoV-2 vaccines and antiviral drugs.
The cell attachment of SARS-CoV-2 is mediated through the interactions between viral spike protein and the cell surface angiotensin converting enzyme-2 (ACE2) receptor (Figure 1).7–8 Subsequently, the spike protein is cleaved by the host transmembrane protease TMPRSS2, leading to the fusion and release of viral RNA into the cytoplasm. In tissues with low expression levels of TMPRSS2, SARS-CoV-2 enters cell through endocytosis and the cleavage of spike protein is mediated by host proteases including cathepsin L, cathepsin B, and calpains.8 The released viral RNA then hijacks host cell ribosome to synthesize viral polyproteins pp1a and pp1ab through frame shifting translation. The polyproteins are subsequently cleaved by the viral main protease (Mpro, also called 3-chymotrypsin-like protease (3CLpro)) and papain-like protease (PLpro) to produce functional viral proteins for the assembly of viral replication transcription complex. The newly synthesized viral RNAs are then packed into progeny virions, which are released from the infected cells for the next cycle of replication.
Figure 1.
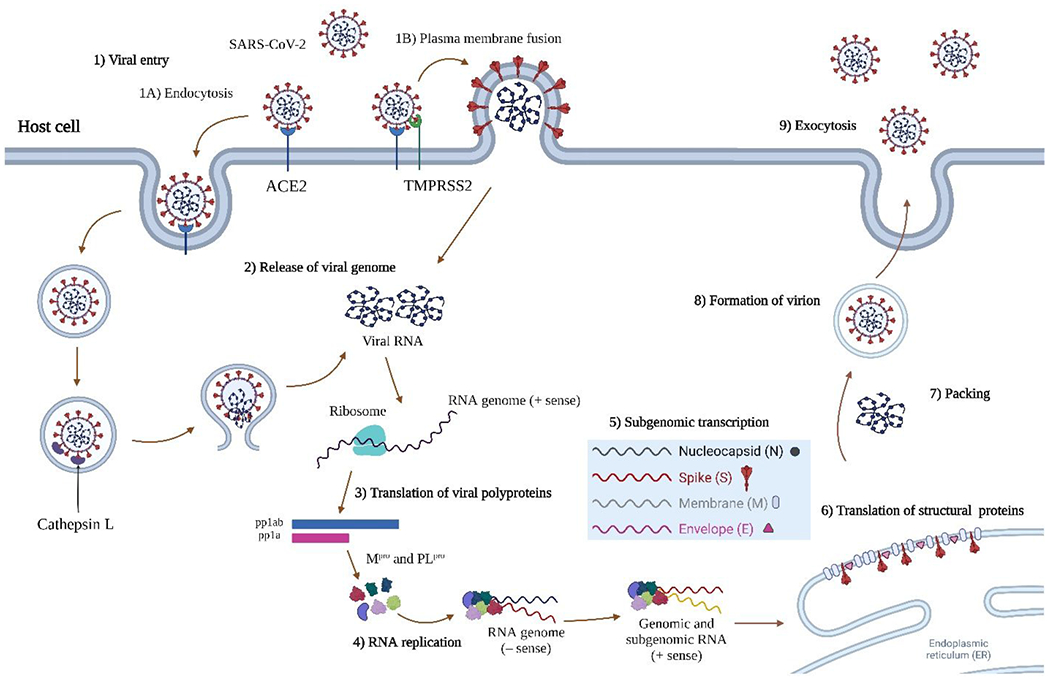
SARS-CoV-2 viral entry and replication cycle. SARS-CoV-2 enters cell through two main pathways, direct plasma membrane fusion and endocytosis, depending on the expression level of TMPRSS2. The single-stranded positive-sense RNA is translated by host ribosome to produce viral polyproteins pp1a and pp1ab, which are cleaved by Mpro and PLpro to produce non-structural proteins (Nsps). The Nsps assemble to form replication transcription complex, which mediates the replication of genomic and subgenomic RNAs. The viral RNAs and proteins assemble to form progeny virions, which are released by exocytosis. Figure was created with Biorender.com.
There are three FDA-approved antiviral drugs, remdesivir, molnupiravir, and Paxlovid.9 Remdesivir and molnupiravir are viral RNA-dependent RNA polymerase (RdRp) inhibitors that were discovered from drug repurposing approach. Paxlovid is a combination of the SARS-CoV-2 Mpro inhibitor nirmatrelvir and its metabolic enhancer ritonavir. SARS-CoV-2 encodes two viral cysteine proteases, the Mpro and PLpro, both of which are essential for the viral replication and are high-profile drug targets. Nirmatrelvir is a rationally designed covalent inhibitor of Mpro containing a nitrile reactive warhead with a high target selectivity, but its suboptimal pharmacokinetic (PK) properties requires the co-administration of metabolic enhancer ritonavir.10
Protease inhibitors have strong clinical precedence for blocking viral replication. The FDA-approved HIV and HCV protease inhibitors are essential components in the combination therapy cocktails.11 SARS-CoV-2 Mpro and PLpro are cysteine proteases that cleave the viral polyproteins pp1a and pp1ab at 11 and 3 sites during viral replication (Figure 2A, B).12 The SARS-CoV-2 Mpro shares 96% overall sequence similarity and 100% sequence identity at the active site with the SARS-CoV Mpro. Mpro is also highly conserved among coronaviruses and picornaviruses, rendering it a high-profile antiviral drug target. The X-ray crystal structure of SARS-CoV-2 Mpro was solved at the beginning of the pandemic, which greatly facilitated drug design and mechanistic studies.13 Mpro adopts a chymotrypsin-like fold consisting of three domains I, II and III (Figure 2C). The C-terminal domain regulates dimerization of the enzyme. The catalytic dyad Cys145-His41 locates in a cleft between the two N-terminal domains I and II, and the active site composes of four subsites S1’, S1, S2, and S4 (Figure 2D).
Figure 2.
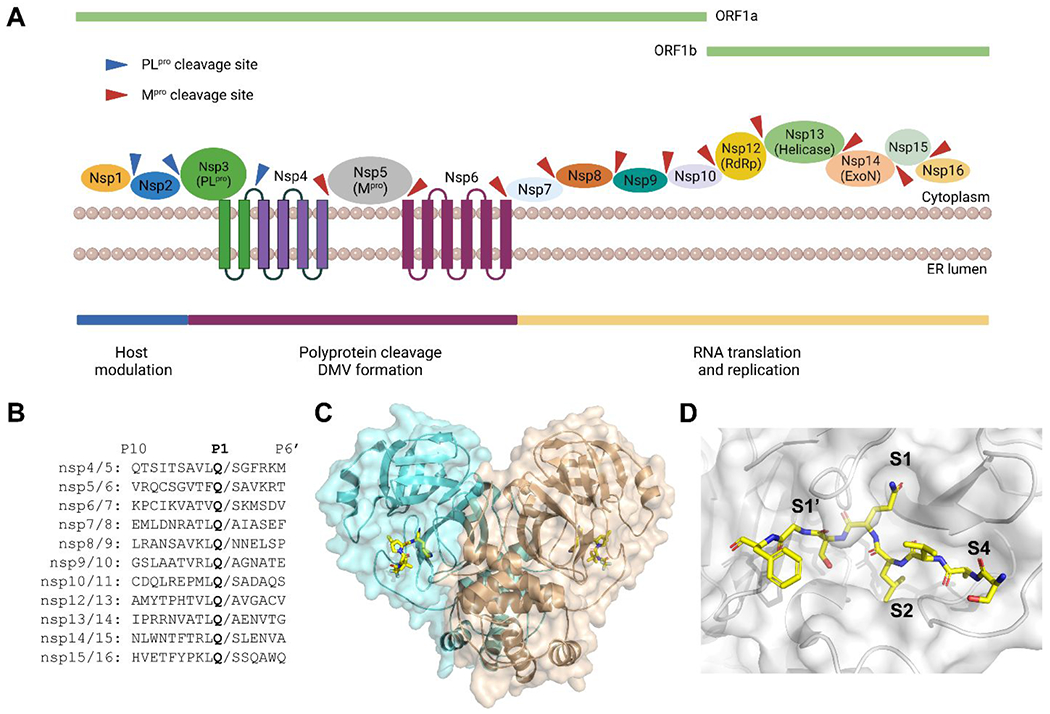
SARS-CoV-2 Mpro function, cleavage sites, and structure. (A) Cartoon representation of the Mpro and PLpro cleavage sites in viral polyproteins. (B) Sequence alignment of the 11 cleavage sites of Mpro. (C) X-ray crystal structure SARS-CoV-2 Mpro with nirmatrelvir (PDB: 7SI9). (D) X-ray crystal structure of SARS-CoV-2 Mpro C145A mutant with the peptide substrate SAVLQSGF (PDB: 7MGS). Figure (A) was created with Biorender.com.
Among the various viral proteins, we chose to target the Mpro because of our relevant experience in viral cysteine proteases.14–15 Mpro functions as a dimer and has a high substrate preference of glutamine at the P1 position (Figure 2B). My group has been working on developing antivirals targeting the enterovirus D68 2A and 3C proteases, both are viral cysteine proteases, since 2016.14 As such, we are experienced with protease protein expression, the FRET enzymatic assay, the thermal shift binding assay, and antiviral assays. We also have assembled an in-house protease inhibitor library that is ready for screening. Given similarities of enterovirus 3C protease and SARS-CoV-2 Mpro,15 we were convinced that targeting SARS-CoV-2 Mpro is likely lead to broad-spectrum antivirals against coronaviruses and enteroviruses.
In this account, we outline the history of our work in Mpro inhibitor screening, drug design, assay development, and drug resistance studies. We initiated this project in January 2020 and conducted a screening of a library of protease inhibitors against SARS-CoV-2 Mpro.1–2 Following the hits discovered and their X-ray crystal structures, we designed both covalent and non-covalent Mpro inhibitors with enhanced target specificity toward Mpro over host proteases including cathepsin L.3–4 To facilitate the prioritization of compounds for the SARS-CoV-2 antiviral assays and to validate many other reported Mpro inhibitors, we developed the cell-based FlipGFP Mpro assay to characterize their cellular target engagement.16–17 Recently, we have discovered naturally occurring Mpro mutants that are resistant to nirmatrelvir.18 Overall, this body of work provides a guidance for the design of next generation of Mpro inhibitors with high target specificity, increased genetic barrier to drug resistance, and favorable pharmacokinetic (PK) properties.
DUAL INHIBITORS OF MPRO AND HOST CATHEPSIN L
GC376, calpain inhibitors II and XII, and boceprevir
Our initial screening of a focused library of protease inhibitors against SARS-CoV-2 Mpro led to the discovery of GC376, boceprevir, and calpain inhibitors II and XII with potent enzymatic inhibition and antiviral activity (Figure 3).1 The discovery of GC376 was expected as it is known to have broad-spectrum antiviral activity against multiple viruses including SARS-CoV, MERS-CoV, enteroviruses, and norovirus.19 GC376 is a veterinary drug candidate for cats infected with feline infectious peritonitis virus (FIPV),20 a coronavirus that belongs to the α-lineage. With the COVID-19 outbreak, GC376 became an apparent drug candidate for SARS-CoV-2.21 Several groups independently predicted and validated the antiviral activity of GC376 against SARS-CoV-2.1, 21–23 In collaboration with Yu Chen at USF, we solved the X-ray crystal structure of Mpro with GC376. Interestingly, the structure revealed that the C145 thiol can attack the aldehyde from either the front or the back side, resulting in the covalent hemithioacetal in either the R or S configuration (PDB: 6WTT).1 The (S)-hemithioacetal conformation is the energetically preferred confirmation as the hydroxyl group engages two hydrogen bonds with G143 and N142 (Figure 3A). In collaboration with Daniel Perez at Georgia, we then tested the in vivo antiviral efficacy of GC376 in a K18-hACE2 mouse model for SARS-CoV-2 infection.24 The results were somewhat disappointing: mice in the GC376 treatment group showing slight improvement in survival (20% compared to 0% in vehicle control) and no differences in clinical symptoms. A follow up study by Shi et al. confirmed this finding.25 Nevertheless, GC376 treatment group that survived viral challenging had milder tissue lesions, reduced inflammation, and reduced viral loads especially in the brain. Collectively, these results suggest GC376 needs further optimization to improve the in vivo antiviral efficacy.
Figure 3.
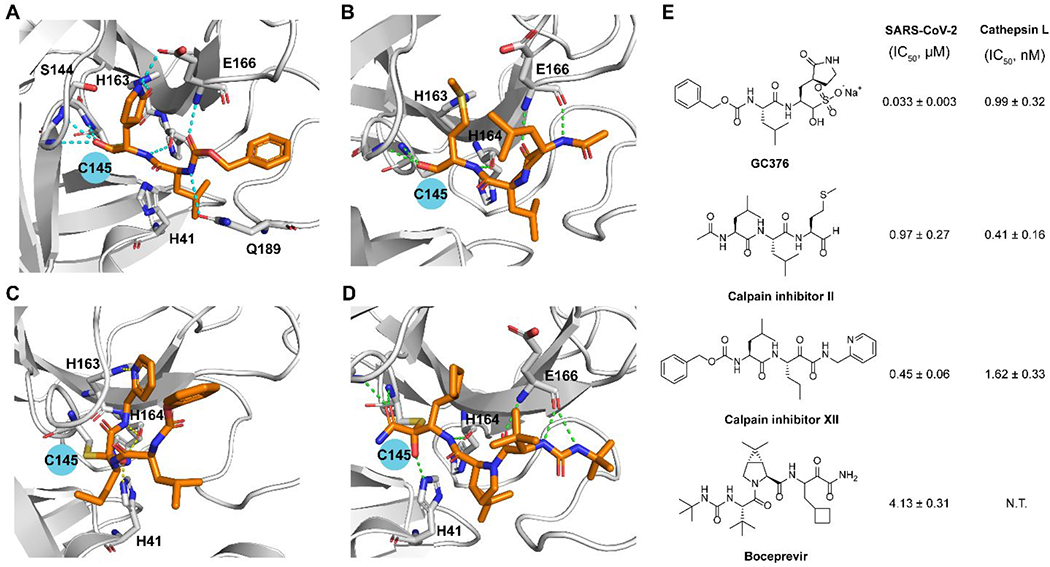
SARS-CoV-2 Mpro inhibitors identified from screening a protease inhibitor library. X-ray crystal structures of SARS-CoV-2 Mpro with GC376 (PDB: 6WTT) (A), calpain inhibitor II (PDB: 6XA4) (B), calpain inhibitor XII (PDB: 6XFN) (C), and boceprevir (PDB: 6XQU) (D). (E) Chemical structures of GC376, calpain inhibitors II and XII, and boceprevir and their enzymatic inhibitory activity against SARS-CoV-2 Mpro and host cathepsin L. N.T. = not tested.
Calpain inhibitors II and XII inhibited Mpro with IC50 values of 0.97 and 0.45 μM and SARS-CoV-2 viral replication in Vero E6 cells with EC50 values of 2.07 and 0.49 μM.1 Our follow up study solved the X-ray crystal structures of Mpro with calpain inhibitors II and XII.2 The binding pose of calpain inhibitor II in the X-ray crystal structure is as expected: the P1 methionine fits in the S1 pocket with the sulfur forms a hydrogen bond with the His163 side chain imidazole; the P2 leucine fits in the S2 hydrophobic pocket; while the P3 leucine is solvent exposed (Figure 3B). Interestingly, calpain inhibitor XII binds in a reverted conformation with the pyridine fits in the S1 pocket (Figure 3C), not the norvaline as one might predict from its chemical structure. The pyridine nitrogen forms a critical hydrogen bond with the His163 side chain imidazole NH. In corroborating with this binding mode, replacing the pyridyl with phenyl led to a significant loss of the enzymatic inhibition (64.5 vs 0.45 μM).2
The HTS hit list also includes HCV protease inhibitors simeprevir (IC50 = 13.74 μM), boceprevir (IC50 = 4.13 μM), narlaprevir (IC50 = 5.73 μM), and telaprevir (31% inhibition at 20 μM).1 Boceprevir inhibited SARS-CoV-2 viral replication in Vero E6 cells in the primary CPE and secondary viral yield reduction assays with EC50 values of 1.31 and 1.95 μM, respectively. In parallel, Fu et al. also reported boceprevir as a SARS-CoV-2 Mpro inhibitor with an enzymatic inhibition IC50 of 8.0 μM and an antiviral EC50 of 15.57 μM.23 They solved the X-ray crystal structure of Mpro with boceprevir, revealing a covalent modification of the C145 thiol by the ketoamide (PDB: 7BRP and 7C6S).
Due to the unsuccessful attempts to gain access to the BSL-3 facility at the University of Arizona, we opted to develop surrogate antiviral assays to test the antiviral activity of our Mpro inhibitors. Mpro is conserved among coronaviruses and Mpro inhibitors are expected to have broad-spectrum antiviral activity. We established the cytopathic effect assay, plaque assay, and immunofluorescence assay for human coronaviruses 229E (HCoV-229E), OC43 (HCoV-OC43), and NL63 (HCoV-NL63), and showed that GC376, calpain inhibitors II and XII, and boceprevir had broad-spectrum antiviral activity against these common human coronaviruses.26–27 In collaboration with Brett Hurst at USU, we further demonstrated that GC376, calpain inhibitors II and XII, and boceprevir inhibited SARS-CoV and MERS-CoV in addition to SARS-CoV-2.26
Mechanistic studies using the enzymatic assay and SARS-CoV-2 pseudovirus assay revealed that the antiviral activity of GC376 and calpain inhibitors II and XII against SARS-CoV-2 in Vero cells involves not only Mpro inhibition but also cathepsin L inhibition.2, 26 As stated earlier, cathepsin L is a critical host factor for SARS-CoV-2 entry in TMPRSS2-negative cell lines (Figure 1). Several groups also reported the antiviral activity of cathepsin L inhibitors in inhibiting SARS-CoV-2 in cell culture and animal model.28–31 For reviews of the roles of cathepsin L in COVID-19, please refer to recent reviews.32–33
However, with the advent of nirmatrelvir and several Mpro clinical candidates, it appears that selective inhibitors with minimal or no effect on cathepsin L might be preferred. For example, nirmatrelvir and the Mpro clinical candidates such as ensitrelvir (S-217622) are all selective towards Mpro over cathepsin L.10, 34 In this context, we shifted our strategy towards designing specific Mpro inhibitors. Nonetheless, the debate of targeting cathepsin L as a strategy for COVID-19 treatment is ongoing. Recent studies showed that Omicron variants prefer endosomal entry to cell-surface fusion,35–36 implying that dual inhibitors of Mpro and cathepsin L might be more effective in inhibiting Omicron variants. Additional studies are needed to justify whether the therapeutic benefit might outweigh the potential side effects of inhibiting cathepsin L.
In addition to our own work, we also contributed to the development of Pfizer’s intravenous clinical candidate PF-07304814.37 Pfizer reached us through the referral of NIH in March 2020 and we were tasked with testing a collection of protease inhibitors in our established FRET assay, thermal shift assay, and cytotoxicity assay.37 This work led to the advance of PF-07304814 to clinical trials.
Covalent boceprevir- and telaprevir-derived hybrid Mpro inhibitors
Although boceprevir (IC50 = 4.13 μM), narlaprevir (IC50 = 5.73 μM), and telaprevir (31% inhibition at 20 μM) are relatively moderate SARS-CoV-2 Mpro inhibitors,1 the significance of their discovery cannot be understated. The X-ray crystal structures of SARS-CoV-2 Mpro with boceprevir, narlaprevir, and telaprevir23, 38 laid the foundation for the following design of more potent and selective Mpro inhibitors including Pfizer’s nirmatrelvir,10 which similarly contains a dimethylcyclopropylproline at the P2 position as boceprevir and narlaprevir. Superimposing the X-ray crystal structures of Mpro with boceprevir, telaprevir, and GC376 revealed that the dimethylcyclopropylproline in boceprevir and cyclopentylproline in telaprevir bind to the same S2 pocket as leucine in GC376 (Figures 4A, D). Subsequently, we designed telaprevir-derived UAWJ9-36-1 (Figure 4B) and boceprevir-derived UAWJ9-36-3 (Figure 4E) with cyclopentylproline and dimethylcyclopropylproline at P2 positions, respectively.16 UAWJ9-36-1 and UAWJ9-36-3 inhibited SARS-CoV-2 Mpro with IC50 values of 51 and 54 nM, comparable to GC376 (IC50=41 nM). Encouragingly, UAWJ9-36-3 had improved antiviral activity than GC376 in both the Vero E6 (EC50=0.37 vs 0.96 μM) and Caco2-hACE2 (EC50=1.06 vs 2.90 μM) cells. Furthermore, UAJW9-36-1 and UAWJ9-36-3 had broad-spectrum antiviral activity against common human coronaviruses HCoV-OC43, HCoV-NL63, and HCoV-229E by targeting their viral Mpro. Target selectivity profiling revealed that UAWJ9-36-3 had improved selectivity than GC376 against host proteases cathepsin L and calpain 1, but not cathepsin K. The X-ray crystal structures of Mpro with UAWJ9-36-1 and UAWJ9-36-3 revealed superimposable poses as telaprevir and boceprevir at the active site (Figures 4C, F), validating our design hypothesis.
Figure 4.
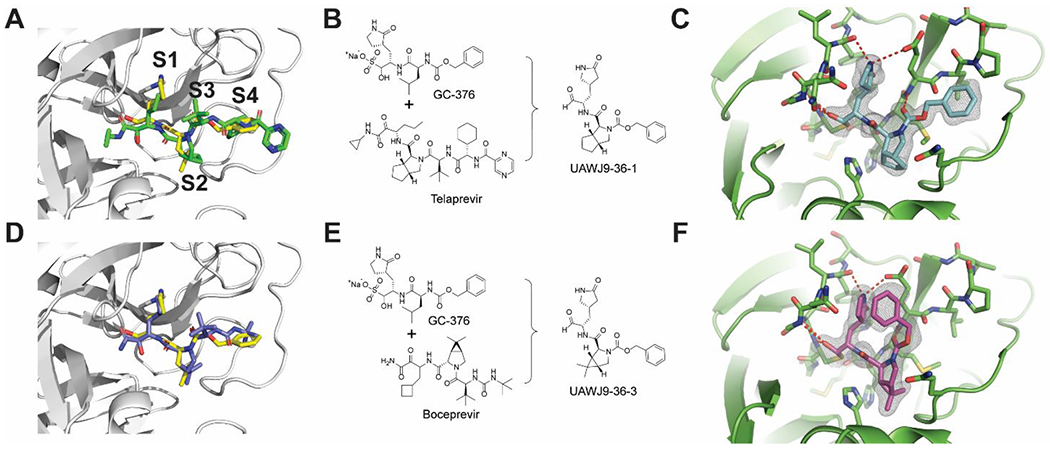
Telaprevir- and boceprevir-derived hybrid SARS-CoV-2 Mpro inhibitors UAWJ9-36-1 and UAWJ9-36-3. (A) Superimposed X-ray crystal structures of SARS-CoV-2 Mpro with GC376 and telaprevir (PDBs: 6WTT and 6XQS). (B) Structure-based design of UAWJ9-36-1 as a hybrid of GC376 and telaprevir. (C) X-ray crystal structure of SARS-CoV-2 Mpro with UAWJ9-36-1 (PDB: 7LYH). (D) Superimposed X-ray crystal structures of SARS-CoV-2 Mpro with GC376 and boceprevir (PDBs: 6WTT and 6XQU). (E) Structure-based design of UAWJ9-36-3 as a hybrid of GC376 and boceprevir. (F) X-ray crystal structure of SARS-CoV-2 Mpro with UAWJ9-36-3 (PDB: 7LYI). Figures were adapted with permission from ref (16). Copyright 2022 American Chemical Society.
In parallel to our study, several other groups also reported the design of hybrid Mpro inhibitors with potent antiviral activity and improved target specificity.39–40 Notably, MI-09 and MI-30 had favorable in vivo pharmacokinetic properties and in vivo antiviral efficacy in a SARS-CoV-2 infection human ACE2 transgenic mouse model.40
NON-COVALENT MPRO INHIBITORS WITH ENHANCED TARGET SPECIFICITY
To solve the cathepsin L off target issue with GC376 and calpain inhibitors II and XII, we sought to design non-covalent Mpro inhibitors. Surveying the literature revealed a promising candidate ML188, which was originally developed for SARS-CoV Mpro.41 Interestingly, structural analysis showed that the pyridine ring from ML188 and calpain inhibitor XII binds to the same position in Mpro.2, 42 We then resynthesized ML188 and confirmed that it is a weak inhibitor of SARS-CoV-2 Mpro with an IC50 of 10.96 μM.3 The superimposed X-ray crystal structures of SARS-CoV-2 Mpro with GC376, ML188, and UAWJ247 suggest that the P2 and P3 substitutions in ML188 can be extended to achieve greater active site occupancy at the S2 and S4 subsites (Figures 5A, B). Next, a library of non-covalent Mpro inhibitors was designed (Figure 5C). The most potent compound Jun8-76-3R inhibited Mpro with an IC50 of 0.2 μM, representing a 54.8-fold increase compared to ML188. Jun8-76-3R also inhibited SARS-CoV-2 replication in both Vero E6 and Calu-3 cells with EC50 values of 1.27 and 3.03 μM, respectively. X-ray crystal structure of Mpro with Jun8-76-3R revealed a ligand induced subpocket located between S2 and S4 where the benzyl group fits (Figures 5D, E), which is unexpected from our design perspective.3 Selectivity profiling showed that Jun8-76-3R had an increased selectivity than the covalent inhibitor GC376 towards Mpro over host proteases including cathepsin L (Figure 5F).
Figure 5.
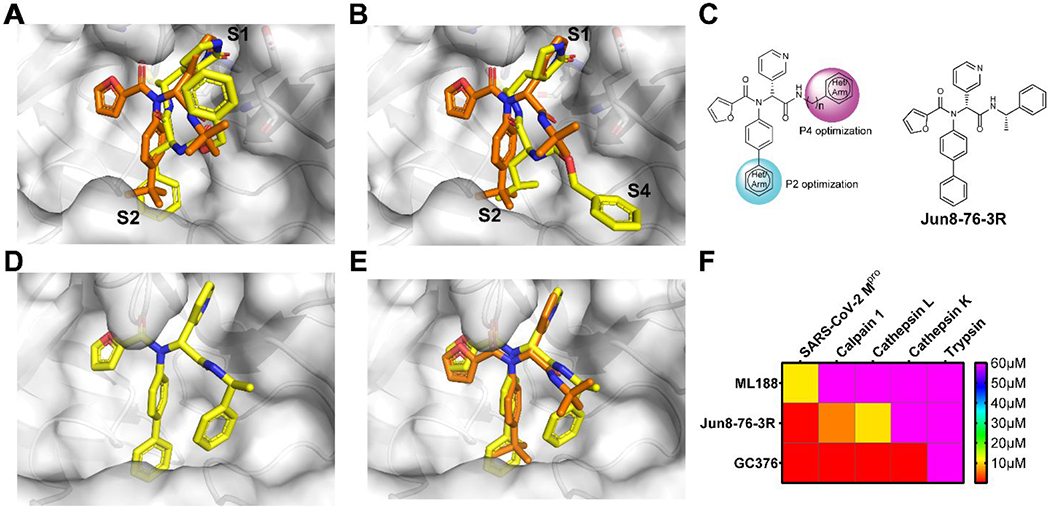
Rational design of non-covalent SARS-CoV-2 Mpro inhibitor Jun8-76-3R with improved target selectivity. (A) Superimposed X-ray crystal structures of SARS-CoV Mpro/ML188 (PDB: 3V3M) and SARS-CoV-2 Mpro/UAWJ247 (PDB: 6XBH). (B) Superimposed X-ray crystal structures of SARS-CoV Mpro/ML188 (PDB: 3V3M) and SARS-CoV-2 Mpro/GC376 (PDB: 6WTT). (C) Structure-based design of ML188 analogs with P2 and P4 optimizations. (D) X-ray crystal structure of SARS-CoV-2 Mpro with Jun8-76-3R (PDB: 7KX5). (E) Superimposed X-ray crystal structures of SARS-CoV-2 Mpro/Jun8-76-3R (PDB: 7KX5) and SARS-CoV Mpro/ML188 (PDB: 3V3M). (F) Selectivity heat map of ML188, Jun8-76-3R and GC376 against host proteases.
The expeditious assembly of Mpro inhibitors using the one pot Ugi four-component reaction also inspired other researchers to design Mpro inhibitors with the same scaffold.43–44
COVALENT MPRO INHIBITORS WITH NOVEL REACTIVE WARHEADS
The X-ray crystal structure of SARS-CoV-2 Mpro with the non-covalent Mpro inhibitor Jun8-76-3R revealed that the P1’ furyl substitution is close to the catalytic C145 (3.4 Å) (Figure 6A), indicating that replacing furyl with a cysteine reactive group will lead to covalent Mpro inhibitors. Guided by this design hypothesis, we first installed several established cysteine warheads at the P1’ position including acrylamide, 2-butynamide, and chloroacetamide. Compound Jun9-54-1 with the chloroacetamide warhead had potent Mpro inhibition with an IC50 of 0.17 μM, comparable to 23R (IC50 = 0.20 μM) (Figure 6B). However, Jun9-54-1 was toxic to both Vero and Calu-3 cells (CC50 < 5 μM). To mitigate the cytotoxicity, we then explored a series of di- and tri-haloacetamides as C145 reactive warheads (Figure 6B). Dibromoacetamide Jun9-89-2R (IC50 = 0.08 μM, CC50 = 8.94 μM), 2-bromo-2,2-dichloroacetamide Jun9-89-3 (IC50 = 1.20 μM, CC50 = 32.43 μM), 2-chloro-2,2-dibromoacetamide Jun9-89-4R (IC50 = 0.05 μM, CC50 = 8.41 μM), and the tribromide Jun9-88-6R (IC50 = 0.08 μM, CC50 = 5.48 μM) all had potent Mpro inhibition but failed to improve the cellular cytotoxicity. Gratifyingly, compound Jun9-62-2R with the dichloroacetamide had both potent Mpro inhibition (IC50 = 0.43 μM) and reduced cytotoxicity (Vero CC50 > 100 μM). The mechanism of action of Jun9-62-2R was characterized by native mass spectrometry and X-ray crystallography, both revealing the displacement of one chloro from the dichloroacetamide warhead by the C145 sulfhydryl (Figure 6C). Jun9-62-2R had potent antiviral activity against SARS-CoV-2 in three different cell lines, Vero E6 (EC50 = 0.90 μM), Caco2-hACE2 (EC50 = 2.05 μM), and Calu-3 (EC50 = 2.00 μM). Target selectivity profiling showed that Jun9-62-2R is highly selective towards Mpro and had no effect on calpain I, cathepsins B, K, L, and caspase-3 (IC50 > 20 μM) (Figure 6D). The relatively weaker reactivity, coupled with its pharmacological compliance, renders dichloroacetamide a promising candidate for the designing cysteine reactive covalent inhibitors.
Figure 6.
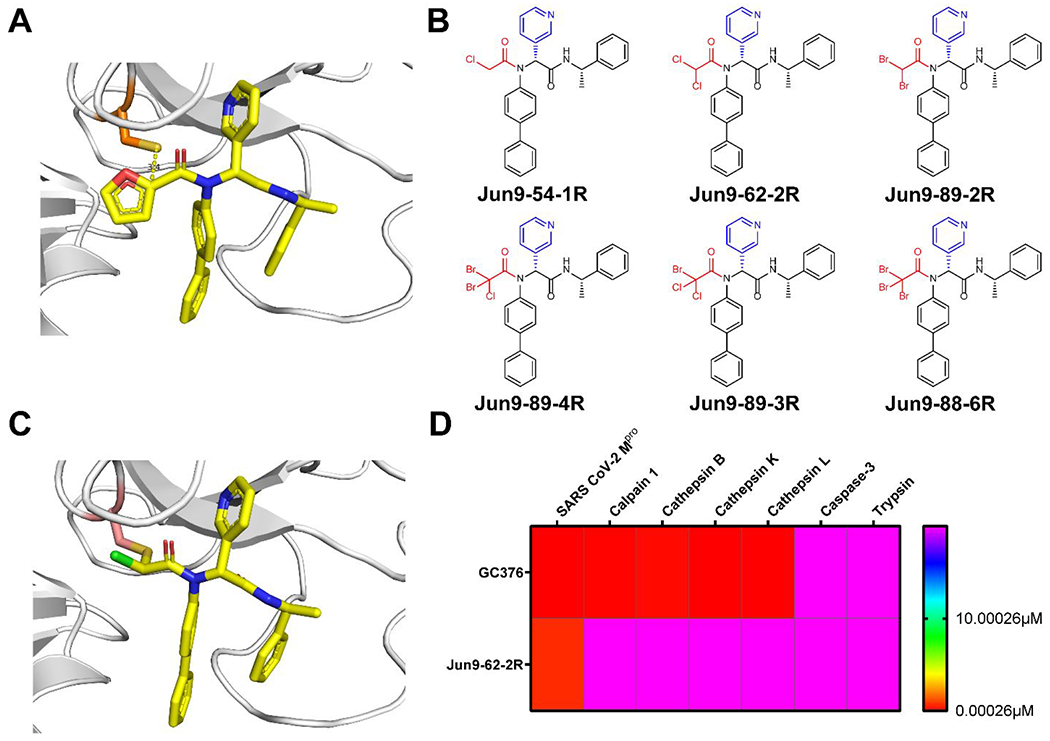
Rational design of covalent SARS-CoV-2 Mpro inhibitor Jun9-62-2R with improved target selectivity. (A) X-ray crystal structures of SARS-CoV-2 Mpro with Jun8-76-3R (PDB: 7KX5). (B) Covalent SARS-CoV-2 Mpro inhibitors with chloroacetamide, dihaloacetamide and trihaloacetamide warheads. (C) X-ray crystal structure of SARS-CoV-2 Mpro with dichloracetamide Jun9-62-2R (PDB: 7RN1). (D) Selectivity heat map of Jun9-62-2R and GC376 against host proteases.
SARS-CoV-2 MPRO ASSAY DEVELOPMENT
In addition to the rationally design Mpro inhibitors with validated mechanisms of action, structurally disparate compounds have also been reported as Mpro inhibitors including repurposed drugs (chloroquine, lopinavir, nelfinavir, etc), natural products (shikonin, baicalein, etc), and even promiscuous compounds (ebselen, disulfiram, carmofur, etc), some of which also showed antiviral activity against SARS-CoV-2 in cell culture, casting confusions on their mechanisms of action.9, 17 We therefore were interested in validating these hits so medicinal chemists can focus their efforts on hits with translational potential. Mpro is a cysteine protease and a reduced C145 is vital for its enzymatic activity. Promiscuous compounds that either non-specifically oxidize or alkylate C145 will show as false positives. As such, it is recommended that reducing reagents dithiothreitol (DTT), β-mercaptoethanol, glutathione (GSH) or tris(2-carboxyethyl)phosphine) (TCCP) are included in the protease assay buffer.45 In addition, it is expected that the enzymatic assay results can be used to predict the cellular antiviral activity. For this, there is a need of Mpro-specific cell-based assay to mimic SARS-CoV-2 replication in cells. The cellular Mpro assay has additional advantages of ruling out compounds that are cytotoxic, membrane impermeable, or metabolically unstable. We therefore developed the FlipGFP Mpro assay and validated the assay with positive control GC376 and negative control GRL0617 (Figures 7A, B).16 Next, employing the FlipGFP assay, together with the enzymatic assay and thermal shift binding assay, we systematically validated the Mpro inhibitors reported in the literature (Figure 7C).17, 46–47 A positive correlation was observed between the results of FlipGFP assay and the enzymatic assay for Mpro inhibitors that were identified in the presence of reducing reagents, but not for compounds that were claimed as Mpro inhibitors based on the enzymatic assay results in the absence of reducing reagents.17 In addition, the establishment of the FlipGFP assay greatly facilitates our lead optimization and we routinely rely on the FlipGFP assay results to prioritize Mpro inhibitors for the SARS-CoV-2 antiviral assay.
Figure 7.
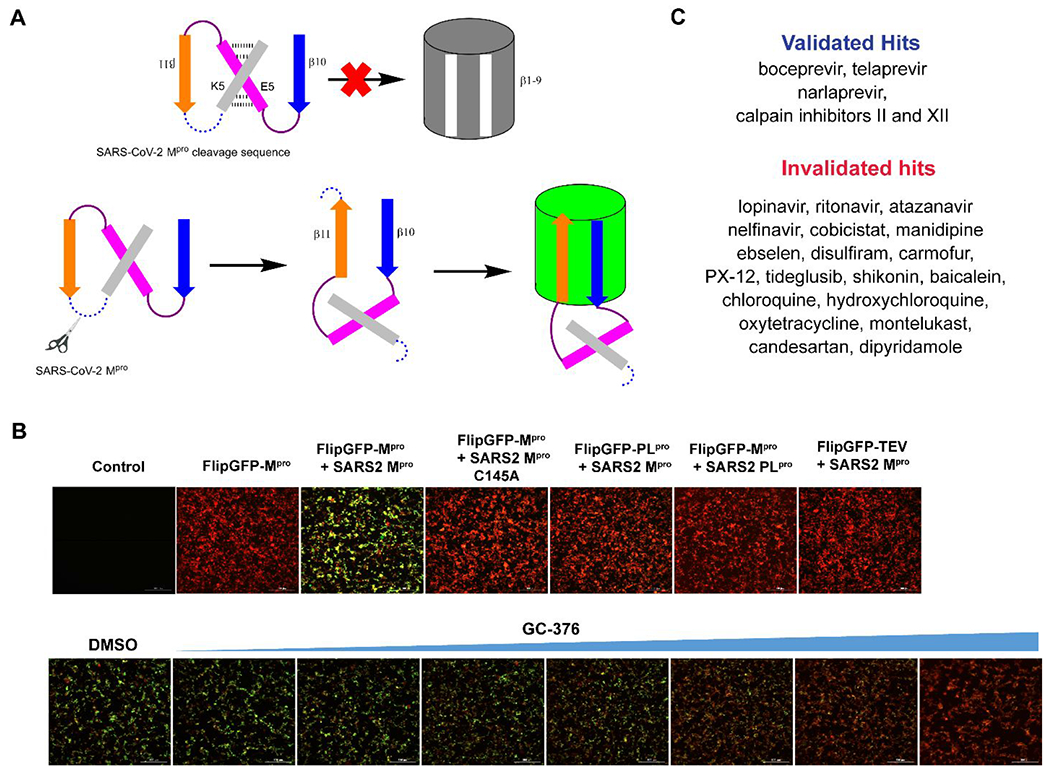
Cellular FlipGFP SARS-CoV-2 Mpro assay. (A) Assay principle of the FlipGFP assay. Cleavage of the linker in the β10-11 fragments leads to restoration of the GFP signal. (B) FlipGFP assay optimization. GFP signal is only observed with a match between the protease and the substrate. (C) List of validated and invalidated SARS-CoV-2 Mpro inhibitors using the FlipGFP assay. Figures were adapted with permission from ref (16). Copyright 2022 American Chemical Society.
SARS-CoV-2 MPRO DRUG RESISTANCE STUDY
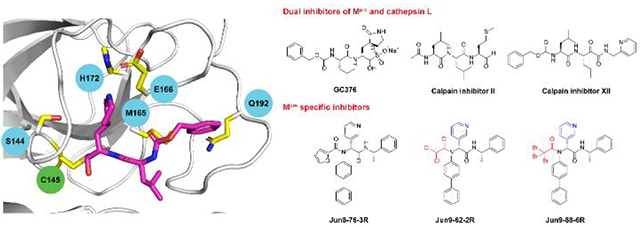
Paxlovid is currently prescribed as a monotherapy for COVID-19 patients, which is unusual for protease-targeting antivirals. Both HIV and HCV protease inhibitors are prescribed as combination therapy together with viral polymerases inhibitors and others.11 With the continuous prescription of Paxlovid and the emerging of delta and Omicron variants, there is a concern that drug resistance might evolve to evade nirmatrelvir inhibition. Studying drug resistance is also a critical component in antiviral drug discovery to validate the mechanism of action. Learning from our experience in studying the drug resistance mechanism of influenza M2-S31N channel blockers, drug resistance can evolve both with and without drug selection pressure.48 The standard approach of evolving drug resistance is to perform serial viral passage experiments in cell culture with escalating drug selection pressure. However, this is not an option for us since we do not have access to the BSL-3 facility. Instead, we turned our attention to identifying naturally occurring Mpro mutants through genome sequence mining.18, 49 Analyzing the nsp5 gene, which encodes Mpro, deposited in the Global Initiative on Sharing Avian Influenza Data (GISAID) database revealed several high frequency mutations including P132H, G15S, T21I, L89F, and K90R, all are located distal to the nirmatrelvir binding site. We first characterized the P132H mutation, which is predominant in current circulating Omicron variants, and found that P132H remains sensitive to nirmatrelvir and has comparable enzymatic parameters as wild type Mpro.49 X-ray crystal structure of P132H with GC376 is nearly identical to the WT Mpro. Results from others also independently confirmed our findings.50–51 Next, we focused on 12 active site residues that are located within 6 Å of the nirmatrelvir binding site. We expressed, purified, and characterized over 100 naturally occurring Mpro mutants based on the GISAID sequence analysis.18 To prioritize Mpro mutants with clinical relevance, we focus on mutants that have comparable enzymatic activity as the WT (catalytic efficacy kcat/Km decrease less than 10-fold) and increased drug resistance (inhibitory constant Ki increase by more than 10-fold). This led to the identification of 20 high profile nirmatrelvir-resistant Mpro mutants located at 5 hotspot residues including S144M/F/A/G/Y, M165T, E166G, H172Q/F, and Q192T/S/L/A/I/P/H/V/W/C/F (Figures 8A, B). In collaboration with Yu Chen at USF, we solved the X-ray crystal structures of six representative mutants, providing a structural explanation for the drug resistance. In collaboration with Xufang Deng at Oklahoma State, we generated recombinant SARS-CoV-2 viruses with nsp5-S144A, H172Y, S144M, and H172Q mutants. Viral growth kinetics showed that SARS-CoV-2 Nsp5 mutants generally have reduced fitness of replication in cell culture compared to WT, and the growth attenuation is proportional to the reduction of enzymatic activity (Figures 8C, D). We also confirmed the nirmatrelvir resistance using these recombinant viruses. Overall, our results suggest that the five hotspot residues at the nirmatrelvir binding site can serve as makers for the monitoring of nirmatrelvir resistance in clinic. The X-ray crystal structures of Mpro mutants are also valuable in guiding the design of next generation of inhibitors with improved resistance profiles.
Figure 8.
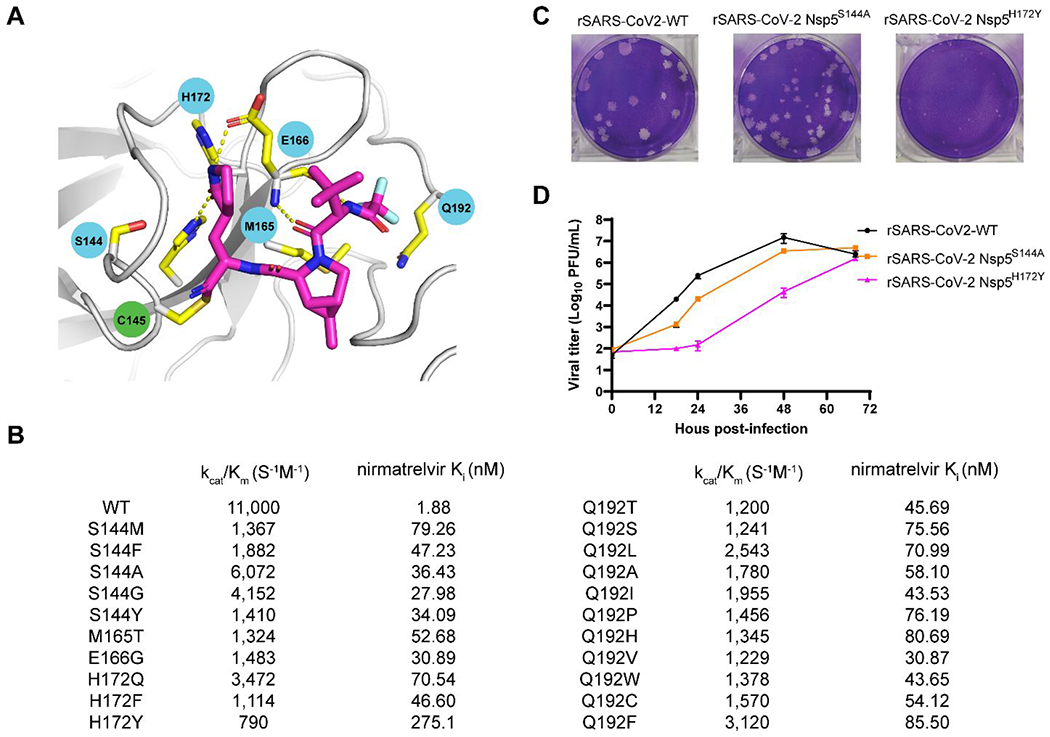
Naturally occurring SARS-CoV-2 Mpro mutants that are resistant to nirmatrelvir. (A) Mapping of the 5 drug resistant hot spot residues to the X-ray crystal structure of SARS-CoV-2 Mpro with nirmatrelvir (PDB: 7SI9). (B) The enzymatic activity and drug sensitivity to nirmatrelvir for the 20 identified naturally occurring SARS-CoV-2 Mpro mutants. (C) Plaque assay for the recombinant SARS-CoV-2 WT, nsp5S144A, and nsp5H172Y. (D) Growth curves for the recombinant SARS-CoV-2 WT, nsp5S144A, and nsp5H172Y.
CONCLUDING REMARKS
Cysteine proteases are typically low hanging drug targets in the preclinical development but face significant hurdles in translational development. Despite decades of research efforts and the numerous publications of cysteine protease inhibitors, nirmatrelvir represents the first example of FDA-approved cysteine protease inhibitor. The challenges facing cysteine protease inhibitors include but not limited to PK properties and target selectivity. Most of the cysteine protease inhibitors are covalent inhibitors, which require diligent design to achieve a fine balance between potency and selectivity. Gratifyingly, the malleability of the SARS-CoV-2 Mpro active site renders it feasible to accommodate structurally disparate covalent and non-covalent inhibitors.
Our current efforts have been devoted to optimizing the PK properties of the non-covalent inhibitor Jun8-76-3R and the covalent inhibitor Jun9-62-2R. We expect to obtain several lead compounds with comparable antiviral potency as nirmatrelvir and favorable oral bioavailability. In addition, leveraging the expertise gained from SARS-CoV-2 Mpro drug design, we are working on developing inhibitors targeting other viral cysteine proteases, including the SARS-CoV-2 PLpro and enterovirus D68 2A and 3C proteases.
Our discovery of di- and tri-haloacetamides novel cysteine reactive warheads further expand the toolbox in covalent drug design. It remains to be further validated whether these warheads can be generically applied to other cysteine proteases. Furthermore, we are working on determining the co-crystal structures of Mpro with di- and tri-haloacetamide-containing inhibitors to confirmatively delineate their mechanisms of action.
As antiviral drug resistance is inevitable, it is vital to predict Mpro drug resistant mutants before they become dominant in clinic. It is gratifying that our genome sequence mining approach identified similar drug resistant mutants (E166A, E166V, S144A) as the serial viral passage experiments.52 The clinic relevance of the high profile mutations identified from our genome mining approach need to be further characterized using recombinant SARS-CoV-2 viruses in cell culture and animal models. To avoid cross resistance, the Mpro inhibitors in development need to be tested against these nirmatrelvir resistant mutants.
We anticipate that several additional Mpro inhibitors will be approved in the coming years. The techniques, assays, and expertise accumulated in Mpro drug design is expected to catalyze the development of cysteine protease targeting antivirals.
KEY REFERENCES.
Ma, C.; Sacco, M. D.; Hurst, B.; Townsend, J. A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M. T.; Chen, Y.; Wang, J., Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678-692.1 This is one of the first papers reporting the discovery of GC376, boceprevir, and calpain inhibitors II and XII as SARS-CoV-2 Mpro inhibitors. The X-ray crystal structures laid the foundation for the subsequent drug design.
Sacco, M. D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J. A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; Hurst, B.; Tarbet, B.; Marty, M. T.; Kolocouris, A.; Xiang, Y.; Chen, Y.; Wang, J., Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020, 6, eabe0751.2 We solved the X-ray crystal structures of Mpro with several GC376 analogs and calpain inhibitors II and XII. The results suggest dual inhibitors targeting both Mpro and cathepsin L might be a novel approach in developing SARS-CoV-2 antivirals.
Kitamura, N.; Sacco, M. D.; Ma, C.; Hu, Y.; Townsend, J. A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; Kukuljac, A.; Marty, M. T.; Schultz, D.; Cherry, S.; Xiang, Y.; Chen, Y.; Wang, J., Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848-2865.3 Based on the X-ray crystal structures of Mpro with GC376 analogs and ML188, we designed hybrid non-covalent Mpro inhibitor using the Ugi four-component reaction. The lead compound Jun8-76-3R has potent antiviral activity and high target specificity, representing one of the most potent non-covalent Mpro inhibitors.
Ma, C.; Xia, Z.; Sacco, M. D.; Hu, Y.; Townsend, J. A.; Meng, X.; Choza, J.; Tan, H.; Jang, J.; Gongora, M. V.; Zhang, X.; Zhang, F.; Xiang, Y.; Marty, M. T.; Chen, Y.; Wang, J., Discovery of Di- and Trihaloacetamides as Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity. J. Am. Chem. Soc. 2021, 143, 20697-20709.4 Based on the same scaffold of Jun8-76-3R, we designed covalent Mpro inhibitors with novel reactive warheads including dichloroacetamide and tribromoacetamide. The lead compounds had potent enzymatic inhibition, antiviral activity, and high target specificity. Mass spectrometry and X-ray crystal structures revealed their novel mechanism of action.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases of Health (NIH-NIAID) grants AI147325, AI157046, and AI158775.
ABBREVIATIONS USED
- ACE2
angiotensin converting enzyme 2
- BSL-3
biological safety level 3
- COVID-19
coronavirus disease 2019
- 3CLpro
3-chymotrypsin-like protease
- DTT
dithiothreitol
- FRET
fluorescence resonance energy transfer
- GSH
glutathione
- i.v.
intravenous
- Mpro
main protease
- NSP
non-structural protein
- PK
pharmacokinetic
- PLpro
papain-like protease
- RdRp
RNA-dependent RNA polymerase
- TCCP
tris(2-carboxyethyl)phosphine)
Biographies
Jun Wang earned his Ph.D. degree from the University of Pennsylvania in 2010. He continued the postdoctoral training with Dr. William F. DeGrado at the University of California, San Francisco. In 2014 he started an independent career as an assistant professor at the college of pharmacy at the University of Arizona. He was promoted to associate professor in 2020. In 2022, he relocated his lab to the Ernest Mario School of Pharmacy at Rutgers University. His current research interests include antiviral drug discovery, assay development, drug resistance, and combination therapy. Dr. Wang serves as the associate editor and editorial board member for multiple journals including Journal of Medical Virology, Acta Pharmaceutica Sinica B, Medicinal Research Reviews, and European Journal of Pharmaceutical Sciences.
Bin Tan received his bachelor’s and master’s degrees from the China Pharmaceutical University in 2018 and 2020, respectively. After graduation, he worked for one year as a research assistant in Dr. Jing Xu’s group in Southern University of Science and Technology. He is now a second-year Ph.D. student in Dr. Jun Wang’s lab at Rutgers University and his research interests include drug discovery targeting SARS-CoV-2 Mpro and PLpro.
Ryan Joyce earned his BS degree from Salisbury University (2020, Salisbury, Maryland). In 2022, he joined Dr. Jun Wang Lab as a graduate student at the Ernest Mario School of Pharmacy at Rutgers University. He is currently a second-year Ph.D. student, and his research interests include drug discovery targeting enteroviruses, poxvirus, and coronavirus.
Haozhou Tan earned his BS degree from the Hunan Agricultural University (2016, Changsha, China) and the master’s degree from Northeastern University (2018, Boston, Massachusetts). In 2020, he joined Dr. Jun Wang Lab as graduate student at the College of Pharmacy at the University of Arizona. In 2022, he relocated to the Ernest Mario School of Pharmacy at the Rutgers University with his PI Dr. Wang. His current study focuses on antiviral drug research for Influenza and coronavirus.
Yanmei Hu earned her Ph.D. degree from the University of Arizona in August 2021. She continued the postdoctoral training with Dr. Jun Wang and relocated with the Wang laboratory to the Ernest Mario School of Pharmacy at the Rutgers University in 2022. Her current research interests include antiviral drug discovery, assay development, drug resistance mechanism study, and combination therapy targeting enteroviruses and coronaviruses.
REFERENCES
- 1.Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sacco MD; Ma C; Lagarias P; Gao A; Townsend JA; Meng X; Dube P; Zhang X; Hu Y; Kitamura N; Hurst B; Tarbet B; Marty MT; Kolocouris A; Xiang Y; Chen Y; Wang J Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv 2020, 6, eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitamura N; Sacco MD; Ma C; Hu Y; Townsend JA; Meng X; Zhang F; Zhang X; Ba M; Szeto T; Kukuljac A; Marty MT; Schultz D; Cherry S; Xiang Y; Chen Y; Wang J Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem 2022, 65, 2848–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma C; Xia Z; Sacco MD; Hu Y; Townsend JA; Meng X; Choza J; Tan H; Jang J; Gongora MV; Zhang X; Zhang F; Xiang Y; Marty MT; Chen Y; Wang J Discovery of Di- and Trihaloacetamides as Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity. J. Am. Chem. Soc 2021, 143, 20697–20709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu B; Guo H; Zhou P; Shi ZL Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol 2021, 19, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui J; Li F; Shi ZL Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol 2019, 17, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann M; Kleine-Weber H; Schroeder S; Kruger N; Herrler T; Erichsen S; Schiergens TS; Herrler G; Wu NH; Nitsche A; Muller MA; Drosten C; Pohlmann S SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shang J; Wan Y; Luo C; Ye G; Geng Q; Auerbach A; Li F Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh AK; Mishevich JL; Mesecar A; Mitsuya H Recent Drug Development and Medicinal Chemistry Approaches for the Treatment of SARS-CoV-2 and Covid-19. Chem Med Chem 2022, e202200440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [DOI] [PubMed] [Google Scholar]
- 11.Shyr ZA; Cheng Y-S; Lo DC; Zheng W Drug combination therapy for emerging viral diseases. Drug Discov. Today 2021, 26, 2367–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.V’Kovski P; Kratzel A; Steiner S; Stalder H; Thiel V Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol 2021, 19, 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musharrafieh R; Ma C; Zhang J; Hu Y; Diesing JM; Marty MT; Wang J Validating Enterovirus D68-2A(pro) as an Antiviral Drug Target and the Discovery of Telaprevir as a Potent D68-2A(pro) Inhibitor. J. Virol 2019, 93, e02221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y; Musharrafieh R; Zheng M; Wang J Enterovirus D68 Antivirals: Past, Present, and Future. ACS Infect. Dis 2020, 6, 1572–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia Z; Sacco M; Hu Y; Ma C; Meng X; Zhang F; Szeto T; Xiang Y; Chen Y; Wang J Rational Design of Hybrid SARS-CoV-2 Main Protease Inhibitors Guided by the Superimposed Cocrystal Structures with the Peptidomimetic Inhibitors GC-376, Telaprevir, and Boceprevir. ACS Pharmacol. Transl. Sci 2021, 4, 1408–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma C; Tan H; Choza J; Wang Y; Wang J Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and Protease-Glo luciferase assays. Acta Pharm. Sin. B 2022, 12, 1636–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Y; Lewandowski EM; Tan H; Zhang X; Morgan RT; Zhang X; Jacobs LMC; Butler SG; Gongora MV; Choy J; Deng X; Chen Y; Wang J Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. bioRxiv 2022, 2022.06.28.497978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim Y; Lovell S; Tiew KC; Mandadapu SR; Alliston KR; Battaile KP; Groutas WC; Chang KO Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol 2012, 86, 11754–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim Y; Liu H; Galasiti Kankanamalage AC; Weerasekara S; Hua DH; Groutas WC; Chang KO; Pedersen NC Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016, 12, e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morse JS; Lalonde T; Xu S; Liu WR Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. Chembiochem 2020, 21, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vuong W; Khan MB; Fischer C; Arutyunova E; Lamer T; Shields J; Saffran HA; McKay RT; van Belkum MJ; Joyce MA; Young HS; Tyrrell DL; Vederas JC; Lemieux MJ Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun 2020, 11, 4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu L; Ye F; Feng Y; Yu F; Wang Q; Wu Y; Zhao C; Sun H; Huang B; Niu P; Song H; Shi Y; Li X; Tan W; Qi J; Gao GF Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun 2020, 11, 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cáceres CJ; Cardenas-Garcia S; Carnaccini S; Seibert B; Rajao DS; Wang J; Perez DR Efficacy of GC-376 against SARS-CoV-2 virus infection in the K18 hACE2 transgenic mouse model. Sci. Rep 2021, 11, 9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y; Shuai L; Wen Z; Wang C; Yan Y; Jiao Z; Guo F; Fu ZF; Chen H; Bu Z; Peng G The preclinical inhibitor GS441524 in combination with GC376 efficaciously inhibited the proliferation of SARS-CoV-2 in the mouse respiratory tract. Emerg. Microbes Infect 2021, 10, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y; Ma C; Szeto T; Hurst B; Tarbet B; Wang J Boceprevir, Calpain Inhibitors II and XII, and GC-376 Have Broad-Spectrum Antiviral Activity against Coronaviruses. ACS Infect. Dis 2021, 7, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y; Ma C; Wang J Cytopathic Effect Assay and Plaque Assay to Evaluate in vitro Activity of Antiviral Compounds Against Human Coronaviruses 229e, OC43, and NL63. Bio Protoc. 2022, 12, e4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao M-M; Yang W-L; Yang F-Y; Zhang L; Huang W-J; Hou W; Fan C-F; Jin R-H; Feng Y-M; Wang Y-C; Yang J-K Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target Ther 2021, 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simmons G; Gosalia DN; Rennekamp AJ; Reeves JD; Diamond SL; Bates P Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 11876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ashhurst AS; Tang AH; Fajtová P; Yoon MC; Aggarwal A; Bedding MJ; Stoye A; Beretta L; Pwee D; Drelich A; Skinner D; Li L; Meek TD; McKerrow JH; Hook V; Tseng C-T; Larance M; Turville S; Gerwick WH; O’Donoghue AJ; Payne RJ Potent Anti-SARS-CoV-2 Activity by the Natural Product Gallinamide A and Analogues via Inhibition of Cathepsin L. J. Med. Chem 2022, 65, 2956–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma XR; Alugubelli YR; Ma Y; Vatansever EC; Scott DA; Qiao Y; Yu F; Xu S; Liu WR MPI8 is Potent against SARS-CoV-2 by Inhibiting Dually and Selectively the SARS-CoV-2 Main Protease and the Host Cathepsin L**. ChemMedChem 2022, 17, e202100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T; Luo S; Libby P; Shi GP Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther 2020, 213, 107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes CP; Fernandes DE; Casimiro F; da Mata GF; Passos MT; Varela P; Mastroianni-Kirsztajn G; Pesquero JB Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front Cell Infect. Microbiol 2020, 10, 589505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Unoh Y; Uehara S; Nakahara K; Nobori H; Yamatsu Y; Yamamoto S; Maruyama Y; Taoda Y; Kasamatsu K; Suto T; Kouki K; Nakahashi A; Kawashima S; Sanaki T; Toba S; Uemura K; Mizutare T; Ando S; Sasaki M; Orba Y; Sawa H; Sato A; Sato T; Kato T; Tachibana Y Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem 2022, 65, 6499–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng B; Abdullahi A; Ferreira IATM; Goonawardane N; Saito A; Kimura I; Yamasoba D; Gerber PP; Fatihi S; Rathore S; Zepeda SK; Papa G; Kemp SA; Ikeda T; Toyoda M; Tan TS; Kuramochi J; Mitsunaga S; Ueno T; Shirakawa K; Takaori-Kondo A; Brevini T; Mallery DL; Charles OJ; Baker S; Dougan G; Hess C; Kingston N; Lehner PJ; Lyons PA; Matheson NJ; Ouwehand WH; Saunders C; Summers C; Thaventhiran JED; Toshner M; Weekes MP; Maxwell P; Shaw A; Bucke A; Calder J; Canna L; Domingo J; Elmer A; Fuller S; Harris J; Hewitt S; Kennet J; Jose S; Kourampa J; Meadows A; O’Brien C; Price J; Publico C; Rastall R; Ribeiro C; Rowlands J; Ruffolo V; Tordesillas H; Bullman B; Dunmore BJ; Gräf S; Hodgson J; Huang C; Hunter K; Jones E; Legchenko E; Matara C; Martin J; Mescia F; O’Donnell C; Pointon L; Shih J; Sutcliffe R; Tilly T; Treacy C; Tong Z; Wood J; Wylot M; Betancourt A; Bower G; Cossetti C; De Sa A; Epping M; Fawke S; Gleadall N; Grenfell R; Hinch A; Jackson S; Jarvis I; Krishna B; Nice F; Omarjee O; Perera M; Potts M; Richoz N; Romashova V; Stefanucci L; Strezlecki M; Turner L; De Bie EMDD; Bunclark K; Josipovic M; Mackay M; Butcher H; Caputo D; Chandler M; Chinnery P; Clapham-Riley D; Dewhurst E; Fernandez C; Furlong A; Graves B; Gray J; Hein S; Ivers T; Le Gresley E; Linger R; Kasanicki M; King R; Kingston N; Meloy S; Moulton A; Muldoon F; Ovington N; Papadia S; Penkett CJ; Phelan I; Ranganath V; Paraschiv R; Sage A; Sambrook J; Scholtes I; Schon K; Stark H; Stirrups KE; Townsend P; Walker N; Webster J; Butlertanaka EP; Tanaka YL; Ito J; Uriu K; Kosugi Y; Suganami M; Oide A; Yokoyama M; Chiba M; Motozono C; Nasser H; Shimizu R; Kitazato K; Hasebe H; Irie T; Nakagawa S; Wu J; Takahashi M; Fukuhara T; Shimizu K; Tsushima K; Kubo H; Kazuma Y; Nomura R; Horisawa Y; Nagata K; Kawai Y; Yanagida Y; Tashiro Y; Tokunaga K; Ozono S; Kawabata R; Morizako N; Sadamasu K; Asakura H; Nagashima M; Yoshimura K; Cárdenas P; Muñoz E; Barragan V; Marquez S; Prado-Vivar B; Becerra-Wong M; Caravajal M; Trueba G; Rojas-Silva P; Grunauer M; Gutierrez B; Guadalupe JJ; Fernández-Cadena JC; Andrade-Molina D; Baldeon M; Pinos A; Bowen JE; Joshi A; Walls AC; Jackson L; Martin D; Smith KGC; Bradley J; Briggs JAG; Choi J; Madissoon E; Meyer KB; Mlcochova P; Ceron-Gutierrez L; Doffinger R; Teichmann SA; Fisher AJ; Pizzuto MS; de Marco A; Corti D; Hosmillo M; Lee JH; James LC; Thukral L; Veesler D; Sigal A; Sampaziotis F; Goodfellow HG; Matheson NJ; Sato K; Gupta RK; The C.-N. B. C.-C.; The Genotype to Phenotype Japan, C.; Ecuador, C. C. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pia L; Rowland-Jones S Omicron entry route. Nat. Rev. Immunol 2022, 22, 144–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boras B; Jones RM; Anson BJ; Arenson D; Aschenbrenner L; Bakowski MA; Beutler N; Binder J; Chen E; Eng H; Hammond H; Hammond J; Haupt RD; Hoffman R; Kadar EP; Kania R; Kimoto E; Kirkpatrick MG; Lanyon L; Lendy EK; Lillis JR; Logue J; Luthra SA; Ma C; Mason SW; McGrath ME; Noell S; Obach RS; MN OB; O’Connor R; Ogilvie K; Owen D; Pettersson M; Reese MR; Rogers TF; Rosales R; Rossulek MI; Sathish JG; Shirai N; Steppan C; Ticehurst M; Updyke LW; Weston S; Zhu Y; White KM; Garcia-Sastre A; Wang J; Chatterjee AK; Mesecar AD; Frieman MB; Anderson AS; Allerton C Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun 2021, 12, 6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kneller DW; Galanie S; Phillips G; O’Neill HM; Coates L; Kovalevsky A Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure 2020, 28, 1313–1320.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kneller DW; Li H; Phillips G; Weiss KL; Zhang Q; Arnould MA; Jonsson CB; Surendranathan S; Parvathareddy J; Blakeley MP; Coates L; Louis JM; Bonnesen PV; Kovalevsky A Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun 2022, 13, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiao J; Li YS; Zeng R; Liu FL; Luo RH; Huang C; Wang YF; Zhang H; Quan B; Shen C; Mao X; Liu X; Sun W; Yang W; Ni X; Wang K; Xu L; Duan ZL; Zou QC; Zhang HL; Qu W; Long YH; Li MH; Yang RC; Liu X; You J; Zhou Y; Yao R; Li WP; Liu JM; Chen P; Liu Y; Lin GF; Yang X; Zou J; Li L; Hu Y; Lu GW; Li WM; Wei YQ; Zheng YT; Lei J; Yang S SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobs J; Grum-Tokars V; Zhou Y; Turlington M; Saldanha SA; Chase P; Eggler A; Dawson ES; Baez-Santos YM; Tomar S; Mielech AM; Baker SC; Lindsley CW; Hodder P; Mesecar A; Stauffer SR Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem 2013, 56, 534–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lockbaum GJ; Reyes AC; Lee JM; Tilvawala R; Nalivaika EA; Ali A; Kurt Yilmaz N; Thompson PR; Schiffer CA Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quan B-X; Shuai H; Xia A-J; Hou Y; Zeng R; Liu X-L; Lin G-F; Qiao J-X; Li W-P; Wang F-L; Wang K; Zhou R-J; Yuen TT-T; Chen M-X; Yoon C; Wu M; Zhang S-Y; Huang C; Wang Y-F; Yang W; Tian C; Li W-M; Wei Y-Q; Yuen K-Y; Chan JF-W; Lei J; Chu H; Yang S An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol 2022, 7, 716–725. [DOI] [PubMed] [Google Scholar]
- 44.Julia S; Jevgenijs T; Guanyu W; Felipe AV; Christopher H; Andres Mauricio R; Caitlin E,M; Sharon P; Anne L; Jessica P; Mihai Burai P; Danielle V; Mitchell H; Anthony K,M; Nicolas M, Design, Synthesis and In Vitro Evaluation of Novel SARS-CoV-2 3CLpro Covalent Inhibitors. Eur. J. Med. Chem 2022, 229, 114046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markossian S; Grossman A; Brimacombe K; Arkin M; Auld D; Austin C; Baell J; Chung TDY; Coussens NP; Dahlin JL; Devanarayan V; Foley TL; Glicksman M; Gorshkov K; Haas JV; Hall MD; Hoare S; Inglese J; Iversen PW; Kales SC; Lal-Nag M; Li Z; McGee J; McManus O; Riss T; Saradjian P; Sittampalam GS; Tarselli M; Trask OJ; Wang Y; Weidner JR; Wildey MJ; Wilson K; Xia M; Xu X; editors.. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK53196/. [PubMed] [Google Scholar]
- 46.Ma C; Hu Y; Townsend JA; Lagarias PI; Marty MT; Kolocouris A; Wang J Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci 2020, 3, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma C; Wang J Dipyridamole, chloroquine, montelukast sodium, candesartan, oxytetracycline, and atazanavir are not SARS-CoV-2 main protease inhibitors. Proc. Natl. Acad. Sci. U.S.A 2021, 118, e2024420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Musharrafieh R; Lagarias P; Ma C; Hau R; Romano A; Lambrinidis G; Kolocouris A; Wang J Investigation of the Drug Resistance Mechanism of M2-S31N Channel Blockers through Biomolecular Simulations and Viral Passage Experiments. ACS Pharmacol. Transl. Sci 2020, 3, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sacco MD; Hu Y; Gongora MV; Meilleur F; Kemp MT; Zhang X; Wang J; Chen Y The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 2022, 32, 498–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greasley SE; Noell S; Plotnikova O; Ferre RA; Liu W; Bolanos B; Fennell K; Nicki J; Craig T; Zhu Y; Stewart AE; Steppan CM Structural basis for Nirmatrelvir in vitro efficacy against the Omicron variant of SARS-CoV-2. J. Biol. Chem 2022, 101972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ullrich S; Ekanayake KB; Otting G; Nitsche C Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett 2022, 62, 128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iketani S; Mohri H; Culbertson B; Hong SJ; Duan Y; Luck MI; Annavajhala MK; Guo Y; Sheng Z; Uhlemann A-C; Goff SP; Sabo Y; Yang H; Chavez A; Ho DD Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. bioRxiv 2022, 2022.08.07.499047. [DOI] [PMC free article] [PubMed] [Google Scholar]