

Abstract
Mucosal sites, such as the lung, serve as crucial, yet vulnerable barriers to environmental insults such as pathogens, allergens, and toxins. Often, these exposures induce massive infiltration and death of short-lived immune cells in the lung, and efficient clearance of these cells is important for preventing hyperinflammation and resolving immunopathology. Herein, we review recent advances in our understanding of efferocytosis – a process whereby phagocytes clear dead cells in a non-inflammatory manner. We further discuss how efferocytosis impacts the onset and severity of asthma in humans and mammalian animal models of disease. Finally, we explore how recently identified genetic perturbations or biological pathway modulations affect pathogenesis and shed light on novel therapies aimed at treating or preventing asthma.
Efferocytosis
‘According to most studies, people’s number one fear is public speaking. Number two is death. Death is number two. Does that sound right? This means to the average person, if you go to a funeral, you’re better off in the casket than doing the eulogy.’ – Jerry Seinfeld
An integral component of maintaining homeostasis is the immunotolerant clearance of dead and dying cells, a process termed efferocytosis [1]. This occurs naturally on a daily basis, as billions of cells undergo genetically-programmed cell death, are sensed by tissue-resident and recruited phagocytic cells, and are engulfed and processed in a immunosilent manner [1]. Efferocytosis also promotes the return to homeostasis, once the imminent threat, such as exposure to a pathogen or allergen, is resolved [2]. Often, immune responses to such challenges cause as much tissue damage as the provoking agent itself, leaving large numbers of dying cells and cellular debris that must be removed [3]. In health, phagocytic cells sense the presence of these dying cells and facilitate their clearance without further inciting inflammation (Figure 1) [2, 4, 5]. However, it is becoming clear that failure to efficiently remove dying cells can lead to increased inflammatory responses that likely contribute to the severity of multiple diseases, including atherosclerosis, autoimmune disorders, and asthma. Improved mechanistic understanding of how efferocytosis occurs is already revealing exciting new molecular targets that might be therapeutically manipulated to effect changes in disease progression. Here, we review the molecular and cellular basis of efferocytosis, and discuss how failure to efficiently clear dead cells might contribute to the severity of asthma.
Figure 1: Efferocytosis is required for immunotolerance during homeostasis and inflammation.

Efferocytosis is required to maintain homeostasis during normal cellular turnover (left), as well as during inflammation in an evolutionarily conserved manner (right). During homeostasis, normal cell turnover via apoptosis is tolerated by the immunosilent clearance of cellular corpses by local and recruited phagocytes. During inflammation, innate immune cells, such as neutrophils, are recruited to sites of damage, including pathogen infection. Short-lived neutrophils, as well as infected host cells, undergo cell death, and cellular corpses are cleared by local and recruited phagocytes in a tolerant manner [1]. This figure was created using BioRender (https://biorender.com/).
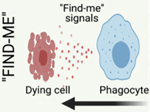
Efferocytosis (Latin: ‘to take to the grave’) requires active participation from both the dying cell and the phagocyte. Dying cells release molecules termed ‘find-me’ signals, to recruit phagocytic cells to sites of cellular death (Figure 2A, Key Figure). In mammals, these ‘find-me’ signals include nucleotides, such as ATP and UTP; chemokines, such as CX3CL1 (CX3C motif chemokine ligand 1; fractalkine); and lipid mediators, such as S1P (sphingosine-1-phosphate) and LPC (lysophosphatidylcholine) [6]. Sensing of these molecules through their cognate receptors on phagocytes results in their chemotaxis toward dying cells [7]. In addition, some molecules, such as S1P, have been shown to exert an additional priming effect on the phagocyte, wherein lipid sensing and engulfment receptors are upregulated for sustained efferocytosis [8].
Figure 2: Mechanisms of efferocytosis in vertebrates.

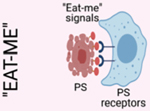
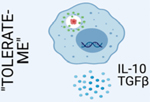
A. Efferocytosis is mediated by the recruitment of phagocytes to sites of cell death via “Find-me” signals, such as ATP and CXCL3, released by dying cells. Dying cells are distinguished from viable cells via active display of “Eat-me” signals, such as phosphatidylserine (PS). Phagocytes employ receptors that recognize “Eat-me” signals, such as PS receptors, which facilitate recognition and engulfment of dying cells. Efferocytosis is ultimately an immunotolerant process, and proper trafficking and processing of engulfed dying cells initiates “Tolerate-me” signaling via activation of LXR, RXR, and PPAR pathways [4]. B. Activation of nuclear receptors, such as LXRs, PPARs, and RXRs, largely mediates the immunotolerant response by inducing the transcription of genes such as Abca1, Il10, and Tgfb. C. A form of non-canonical autophagy called LC3-associated phagocytosis (LAP) is required for immunotolerant efferocytosis. Uptake of dying cells via engagement of PS receptors triggers the recruitment of the Class III PI3K complex, containing BECN1, VPS34, UVRAG, and RUBCN, to the dead cell-containing phagosome (or LAPosome). RUBCN binds to and stabilizes the NOX2 complex to promote production of ROS, which is required for recruitment of the downstream ATG5-ATG12 and LC3-PE conjugation systems. Decoration of the single-membraned LAPosome is required for fusion to the lysosomal network [7]. This figure was created using BioRender (https://biorender.com/).
Once phagocytes are recruited, they must distinguish between healthy cells and dying cells in need of removal (Figure 2B, Key Figure). To this end, dying cells display ‘eat-me’ signals that allow phagocytes to discriminate them from living cells [1, 3]. The best characterized ‘eat-me’ signal is phosphatidylserine (PS), a lipid located on the intracellular side of the plasma membrane of viable cells that is actively flipped extracellularly by caspase activity during apoptosis [9]. Phagocytes express a variety of receptors that specifically recognize PS, and hence discriminate between the living and the dead [6]. These receptors can be either membrane-bound, such as TIM1, TIM4, BAI1, RAGE, Stabilin1/2, and CD300f, or circulating, such as MFGE8 and GAS6/Protein S, which link PS-coated dying cells to phagocytes via interaction with αvβ5 integrin and TAM receptors (TYRO3, AXL, and MerTK), respectively [4].
Engagement of PS and other ‘eat me’ signals by specific receptors during efferocytosis triggers an immunosilent genetic program via ‘tolerate-me’ signaling pathways (Figure 2C, Key Figure) [3]. Dying cells are laden with lipids, and their uptake activates nuclear receptors, such as liver X receptors (LXRs) and peroxisome proliferator-activated receptors (PPARs), which control lipid metabolism, cholesterol efflux, and transport [10, 11]. Importantly, activation of these receptors, which heterodimerize with retinoid X receptors (RXRs), also upregulates efferocytosis receptors, such as Mertk and Mfge8, thus further promoting dead cell clearance [1]. The net result of this signaling is the efficient breakdown and shuttling of these surplus lipids [12, 13], the suppression of pro-inflammatory mediators such as IL-12 and IL-6, and the production of immunotolerant molecules such as IL-10, TGF β, and prostaglandin PGE2 [5, 14].
Trafficking of engulfed cellular corpses to the lysosomal pathway plays a crucial role in preventing inflammation, as proper degradation and processing of cargo requires the activity of the lysosomal acidic environment [15]. In the absence of efficient degradation and processing, phagosomal contents can leak into the cytosol, activating cytosolic receptors such as cGAS/STING, AIM2, and ZBP1 [16]. Activation of the receptors by nucleic acids triggers a robust type I interferon (IFN) response commonly associated with autoimmunity [17]. Recent studies have identified a form of non-canonical autophagy termed LC3-associated phagocytosis (LAP) that is pivotal for the efficient and anti-inflammatory removal of dead cells (Figure 2D) [18, 19]. In addition to engagement of toll-like receptors (TLRs) by pathogens, or Fc receptors by immune complexes, LAP can be triggered during efferocytosis via engagement of PS receptors, such as TIM4, on macrophages as well as non-professional phagocytes, such as epithelial cells [18, 20, 21]. This signaling during engulfment results in the recruitment of some, but not all, of the ATG family of proteins to the cargo-containing phagosome. This recruitment of LAP machinery, demonstrated in Caenorhabditis elegans and Drosophila melanogaster in addition to mammals, allows for efficient and proper processing of engulfed cargo [22]. Despite some molecular overlap between LAP and canonical, starvation-induced autophagy [23], LAP is molecularly distinct [24]. LAP proceeds independently of the most upstream complex of autophagy, the pre-initiation complex composed of ULK1, ATG13, and FIP200 [18, 19, 25]. LAP utilizes a Class III PI3K complex containing Beclin1, VPS34, and UVRAG and does not require ATG14 [25–27]. Proteomic analysis of the LAP-engaged phagosome revealed that Rubicon (RUBCN) forms a complex with the Class III PI3K complex and is uniquely and significantly associated with the cargo-containing phagosome [25]. During LAP, RUBCN supports the generation of the signaling molecule, PI(3)P, by VPS34, presumably via stabilization of the Class III PI3K complex, as opposed to RUBCN’s role during canonical autophagy [25]. Importantly, RUBCN binds p22phox, a component of the NADPH complex NOX2, to stabilize and promote the production of reactive oxygen species (ROS) [25, 28, 29]. NOX2 activity is also positively regulated by RUBCN-mediated PI(3)P, which binds the NOX2 component p40phox and stabilizes its association with the cargo-containing phagosome [25, 29]. Both PI(3)P and ROS are required for the association and activation of downstream conjugation systems, such as the ATG5–12 conjugation system and the LC3-PE conjugation [25, 30, 31]. The net result is the decoration of the single-membrane, cargo-containing phagosome with lipidated LC3-II to form a structure termed the LAPosome. Fusion of LAPosome with the lysosome allows the intralysosomal acidic environment to properly process and degrade the engulfed cargo [25].
The absence of LAP has a dramatic effect on the immunological outcome of efferocytosis. The normally immunosilent process of dead cell clearance becomes hyperinflammatory in LAP-deficient conditions, such as in the Rubcn−/− mouse [32]. Rubcn-deficiency results in age-associated autoinflammation [32], increased onset and severity of Alzheimer’s disease [30, 33], increased skin inflammation [34], and enhanced anti-tumor T cell immunity (LLC carcinoma mouse models) [31]. Despite these findings, it remains to be determined how LAP might impact type 2 immunopathologies, such as asthma.
Asthma
‘The sea was angry that day, my friends. Like an old man trying to return soup at a deli! I got about fifty feet out and then suddenly the great beast appeared before me. I tell you, he was ten stories high if he was a foot. As if sensing my presence, he let out a great bellow. I said, ‘Easy, big fella!’ And then, as I watched him struggling, I realized something was obstructing its breathing.’ – George Costanza
Asthma is a common disease affecting approximately 26 million patients in the U.S. In addition to its profound effects on the quality of life of affected individuals, asthma exacts an enormous economic burden in the form of costs associated with physician visits, hospital stays, and lost work and school hours [35]. The prevalence of this disease has remained high, and new approaches are needed to better manage the symptoms of asthma, and presumably, to eventually prevent its onset.
It is well-established that asthma can stem from inappropriate, or maladaptive, immune responses to otherwise harmless agents in the environment. These responses in turn give rise to inflammation of the airway, excessive mucus production, and bronchial airway hyperresponsiveness (AHR). Consequently, individuals with asthma suffer from periodic episodes of dyspnea, or shortness of breath [36]. For many years, asthma was regarded as a single disease because virtually all affected individuals displayed airway inflammation and shortness of breath. However, it has long been known that not all asthmatics are responsive to glucocorticoid administration, the gold standard treatment for asthma. Indeed, it has recently become clear that despite these overlapping pathologies, or phenotypes, asthma is in fact heterogeneous in nature (Figure 3). The relatively new concept of asthma ‘endotypes’ postulates that different forms of asthma arise from perturbations of distinct molecular and cellular pathways [37]. The most common and best-characterized form of this disease is allergic asthma, characterized by eosinophilic airway inflammation, increased production of type 2 cytokines IL-4, IL-5, and IL-13, and elevated allergen-specific IgE titers [37, 38]. Fortunately, this so-called ‘type 2’ (T2) form of asthma generally responds well to treatment with inhaled or oral glucocorticoids. However, approximately half of asthmatic patients display non-eosinophilic forms of disease [39]. These patients often display neutrophilic inflammation of the airway [40] and are notoriously resistant to inhaled corticosteroids [41]. An improved understanding of the molecular and cellular basis of non-eosinophilic asthma endotypes should reveal novel pathways that might be selectively targeted to treat these forms of asthma.
Figure 3: Expression of efferocytotic machinery in human lung resident cells.

The schematic depicts the relative RNA expression amounts of key efferocytotic machinery molecules in lung resident immune cells (A) and non-immune cells (B). Immune cell expression amounts were acquired from ImmGen (https://www.immgen.org/), and non-immune cell expression amounts were acquired from Human Lung Cell Atlas (https://hlca.ds.czbiohub.org/) [115]. n.d. denotes not determined. This figure was created using BioRender (https://biorender.com/).
While asthma is a debilitating disease, it is also associated with increased risk of other pulmonary diseases, including chronic bronchitis, emphysema, and chronic obstructive pulmonary disease (COPD) [42]. One might anticipate, therefore, that individuals with asthma might also be at increased risk of developing more severe Coronavirus disease 2019 (COVID-19), and some studies have indeed shown that. Indeed, studies have shown that severe asthmatics are at higher risk of developing more severe COVID-19 [43], or dying from it, than individuals who not presenting with severe asthma [44].
However, several other studies have failed to demonstrate such a relationship [45]. It is possible that these apparently discrepant findings arise from differences in patient groups. In support of this idea, a recent study found that the increased overall risk of asthmatics might be driven primarily by individuals with nonallergic asthma, whereas there has been no statistically significant association of more severe disease in individuals with allergic asthma [43]. This is an intriguing result, as it suggests that therapies targeting non-allergic asthma might be particularly effective in preventing COVID-19-related deaths. Additional, larger studies are needed to determine whether the risk for severe COVID-19 disease is dependent on asthma endotypes.
Given that non-eosinophilic forms of asthma are glucocorticoid-resistant and often associated with airway neutrophilia [41], it will be important to identify putative immune pathways whose perturbation might give rise to this form of inflammation. As in all forms of inflammation, the numbers of any particular leukocyte cell type are determined by the balance between recruitment of new cells to that tissue and the clearance of those cells. Chemokines such as IL-8, which promote neutrophil recruitment, are present at much higher concentrations in asthmatics than in individuals with healthy airways, and IL-8 concentrations correlate with neutrophil numbers [46]. Airway neutrophilia is also associated with the production of IL-17 by Th17 cells in both humans [47] and mouse models of asthma [48]. IL-17 indirectly recruits neutrophils by binding to its receptor IL17RA on epithelial cells, which in turn, produce neutrophil-recruiting chemokines [49]. A major source of IL-17 is Th17 cells, whose master transcription factor is RORγt. Inverse agonists of RORγt were recently shown to suppress IL-17 production, neutrophil recruitment, and AHR, in a mouse model of neutrophilic asthma in which lipopolysaccharide functioned as an adjuvant to promote immune responses to inhaled ovalbumin [50]. These experiments showed that inhibiting neutrophil recruitment could reduce asthma-like features in animals, further suggesting that augmented clearance of these cells through efferocytosis might also potentially be an effective therapeutic approach in asthma.
Efferocytosis and Asthma
‘Poor little bubble boy. He’s sitting there waiting for you in his bubble, or igloo thing or whatever.’ – Elaine Benes
While defects at any point of the efferocytosis pathway can result in inflammatory pathology in any tissue, the lung is especially susceptible, as it contains an inordinate number and variety of phagocytes -- specifically alveolar macrophages (AM) and dendritic cells, with varying expression of PS receptors and other core efferocytosis machinery molecules such as Rac1, Dock1, and Elmo1 (Figure 4A). In addition to the mere clean-up function of efferocytosis, CD103+ dendritic cells play an essential role in engulfing dying cells and cross-presenting antigens to CD8+ T cells in regional lymph nodes. Further, non-immune cells, such as airway epithelial cells and endothelial cells (Figure 4B), are crucial executioners of efferocytosis in the lung [51, 52]. Thus, in a murine model of asthma, efferocytotic defects in airway epithelial cells have resulted in increased disease severity [53].
Figure 4: General model of asthma in mammals.

A. During allergic sensitization, inhaled allergens and adjuvants are taken up by lung resident cells, such as dendritic cells (DCs), alveolar macrophages (AMs), and epithelial cells. Stressed epithelial cells produce alarmins, such as IL-33, IL-25, and TSLP, which are important for a type 2 immune response [116]. Alveolar macrophages and dendritic cells (DCs) also take up allergen and become activated [117]. B. Activated DCs traffic to draining lymph nodes where they present antigen to naïve CD4+ T cells and promote the differentiation of allergen-specific Th2 (GATA3+) and Th17 (ROR γt+) cells [118]. C. During challenge, Th2 and Th17 cells become activated and produce their signature cytokines. Th2 cell production of IL-5, and IL-13 promotes and synergizes with epithelial cell-derived eotaxin to drive eosinophil recruitment and activation [119]. Th17-derived IL-17 induces and synergizes with epithelial cell-derived CXCL2 to promote the recruitment and activation of neutrophils [106]. This figure was created using BioRender (https://biorender.com/).
The efficiency of pulmonary efferocytosis is underscored by the virtual absence of uncleared apoptotic cells in healthy lung tissue [54]. During acute inflammatory lung diseases such as pneumonia and acute respiratory distress syndrome, apoptotic cell presence remains relatively low, no greater than 2% [55]. Conversely, chronic inflammatory pulmonary pathologies such as COPD, cystic fibrosis, idiopathic pulmonary fibrosis, and asthma, often feature uncleared dead and dying cells, suggesting a crucial role for efferocytosis in the onset and severity of these diseases. The observations that apoptotic cells are increased in asthma and that patients with glucocorticoid-resistant disease have ‘M1’(inflammatory)-skewing of monocytes and AM raises the possibility that defective efferocytosis might contribute to asthma pathogenesis in at least some forms of that disease [55, 56].
Because asthma represents a spectrum of pathologies that differ in terms of triggers and immune responses, the role that efferocytosis plays mirrors the heterogeneity of the diseases themselves. Compared with macrophages from patients with eosinophilic asthma, macrophages from non-eosinophilic asthmatics displayed less ex vivo efferocytosis of epithelial cells rendered apoptotic by exposure to UV light [57, 58]. These studies suggest that defective efferocytosis by AM might at least partly explain persistent airway neutrophilia in non-eosinophilic asthma. Furthermore, both bronchoalveolar lavage fluid (BALF)- and induced sputum-derived macrophages from non-eosinophilic asthmatics have exhibited defective efferocytotic capacity, suggesting that the latter procedure might represent a rapid method for testing efferocytotic activity of macrophages [57]. However, efferocytosis of neutrophils, which comprise the bulk of cellular corpses in the lungs of non-eosinophilic asthmatics, was not directly assessed in that study, nor was the extent to which efferocytosis occurred in bronchial tissue, as opposed to the airway lumen [59]. A subsequent study did not reveal a significant difference in efferocytosis in macrophages derived from monocytes of eosinophilic and non-eosinophilic donors [60], but it is possible that such differences would only be seen in lung macrophages, which might have received different cues in these two different groups of asthmatics.
Disease severity can correlate with levels of efferocytosis. Thus, AM from patients with severe asthma demonstrated significantly lower levels of dead cell clearance, both in vivo and ex vivo, than AMs isolated from lungs of healthy human controls or from patients with mild-to-moderate asthma [61]. Furthermore, when cultured ex vivo, AMs from severe asthmatics produced higher amounts of the proinflammatory molecule, TNF-α and lower amounts of the immunotolerant molecules PGE2 and 15-hydroxyeicosataenoic acid [61]. Co-morbidities in asthmatic patients can also have a dramatic effect on efferocytotic capacity of airway cells in asthmatic patients. Obese patients with asthma, who typically have glucocorticoid-resistant disease, were also found to have reduced efferocytosis, as determined by counting sputum-derived macrophages that contained pyknotic nuclei [62]. Thus, a growing body of associative evidence now links efferocytosis to the severity of asthma.
‘Find-me’ signaling
‘If she can’t find me, she can’t break up with me.’ – George Costanza
Defects in ‘find-me’ signaling (see below) (Figure 2A) can contribute to asthma pathogenesis; asthmatic patients have shown upregulation of chemoattractants such as fractalkine in airway monocytes and S1P in BAL fluid [63, 64]. In line with these observations, recent studies have shown that the variants of the fractalkine receptor CX3CR1 (rs938203, rs2669849, rs1050592, T280M, and V249I) have a positive association with the development of asthma, whereas minor alleles (rs2669849 and V249I) of CX3CR1 confer protection [65].
Not all find-me signaling is created equally. Relative to wildtype mice, P2Y2−/− mice (deficient in the purinergic receptor P2Y(2)) have shown reduced allergic airway inflammation and pathology in an OVA-alum model of asthma, due to defective chemotaxis of DCs and eosinophils in response to ATP -- a key ‘find-me’ signal [66]. Indeed, P2Y2R expression has been reported to be significantly increased in DCs and eosinophils from human asthmatics compared to healthy controls [66]. However, HSP70, a ubiquitous, stress-induced chaperone with ATPase activity, was found to be increased in the sputum and plasma of severe asthmatics, compared to non-asthmatics and mild asthmatics [67].
‘Eat-me’ signaling
‘You dipped the chip. You took a bite. And you dipped again. That’s like putting your whole mouth right in the dip! From now on, when you take a chip — just take one dip and end it.’ — Timmy to George Costanza
Recognition and physical uptake of dying cells (Figure 2B) is also important for the regulation of airway inflammation. AXL is a member of the TAM (TYRO3, AXL, and MERTK) receptor tyrosine kinase family that recognizes PS-bound Gas6 or Protein S, and DNA methylation of AXL at birth is associated with increased risk for asthma in childhood [68]. Cleavage of AXL from the plasma membrane of airway macrophages to generate soluble AXL (sAXL) results in decreased efferocytosis, and sputum from patients with moderate-to-severe asthma contains increased sAXL relative to controls, suggesting diminished efferocytotic capacity [69]. Murine studies have been less clear. In a model of asthma in which mice were first sensitized with antigens from Aspergillus fumigatus and then challenged by oropharyngeal administration of live conidia from that organism, intraperitoneal delivery of monoclonal antibodies against AXL decreased plethysmograph-measured airway hyperresponsiveness, diminished concentrations of IL-4 and IL-13, and reduced numbers of mucus-secreting goblet cells, compared with controls [70]. Another group, however, demonstrated that Axl was exclusively expressed on murine AM, and that Axl−/− mice developed exacerbated lung inflammation during influenza virus infection (IAV), compared with controls [71]. Another member of the TAM family, MerTK, has also been implicated in allergic airway inflammation: Mer-deficient (MerKD) mice exhibited delayed resolution of allergic asthma and increased AHR levels and BALF protein amounts, compared to controls. Further, after intraperitoneal administration of the glucocorticoid dexamethasone, asthmatic Mer-deficient mice displayed defective efferocytosis of apoptotic eosinophils [72].
Milk Fat Globule-EGF factor 8 (MFGE8) is a bridging molecule that links PS-displaying dead cells to phagocytes via αvβ3–5 integrin, and studies have demonstrated that protein amounts of MFGE8 are decreased in endobronchial biopsies from human asthmatics compared to healthy patients [73]. Similarly, Mfge8−/− mice develop exacerbated immunopathology and AHR in experimental models of ovalbumin (OVA)-induced asthma relative to controls [73, 74].
Galectin-3 (encoded by LGALS3) is a β-galactoside-binding, endogenous lectin that mediates effects on immune cells intracellularly and extracellularly [75]. Upon stress or stimulation, macrophages secrete Galectin-3, which can act as a bridging molecule between dying cells and phagocytes to promote efferocytosis [76, 77]. Studies have demonstrated that LGALS3 expression in AM is lower in asthmatics compared with healthy controls, and that recombinant Galectin-3 improves efferocytosis in vitro by macrophages derived from patients with neutrophilic asthma [60]. This finding is consistent with previous studies showing that Galectin-3 amounts are particularly low in the sputum from individuals with neutrophilic asthma, and that the ratio of galectin-3 to galectin-3-binding protein is also reduced relative to controls [78]. Together, these data suggest that reduced galectin-3-mediated efferocytosis can contribute to the severity of neutrophilic asthma.
In terms of pulmonary disorders, the most well-characterized ‘eat-me’ receptor is RAGE (receptor for advanced glycation end products). Genome-wide association studies (GWAS) identified a variant (rs2070600) in AGER (encoding RAGE) that increases ligand binding affinity and is associated with increased pulmonary/asthmatic disease severity [79–81]. In addition, sputum samples of asthma patients contain higher expression of endogenous and soluble RAGE and correlate with disease severity [82, 83].
In addition to recognizing advanced glycation end products, RAGE binds numerous danger-associated molecular patterns (DAMPs), such as S100A8/A9 or HMGB1, which are associated with asthma [84]. Both of these ligands are elevated in the sputum of asthmatics, compared to healthy patients [83, 85]. Noteworthy, increased expression of HMGB1 correlates with increased asthma severity, immune activation, and inflammation in humans [86], while S100A8/A9 induces mast cell degranulation and IgE responses in the lung [85].
The generation of the Ager−/− mouse (RAGE-KO) has facilitated a better understanding of the molecular mechanisms by which RAGE mediates airway inflammation. RAGE-KO mice exhibited reduced asthma immunopathology in two models of experimental allergic airway inflammation. While IL-4 production was equivalent between RAGE-KO and controls, IL-5 and IL-13 (type 2 cytokines crucial for eosinophils and mucus production, respectively), were absent in RAGE-KO mice [87]. The alarmin IL-33 was also reduced in the lungs of RAGE-KO mice during allergic asthma, compared to controls; as a result, after induction of allergic asthma, RAGE-KO mice failed to accumulate another type 2 cell subset, group 2 innate lymphoid cells (ILC2s) in the lung, as evidenced by flow cytometry [88]. The cells in which RAGE primarily acts, however, are still being determined.
The engulfment machinery downstream of PS receptor engagement is also an important mediator of airway inflammation. Efferocytosis by bronchial epithelial cells is required to limit allergic airway inflammation and promote immunotolerant responses. Rac1 is a Rho-family GTPase that functions downstream of multiple signaling pathways to induce actin reorganization during phagocytosis [3]. Another member of Rho-family GTPases, RhoA, acts as an antagonist to Rac1 to inhibit uptake [89]. Mice harboring bronchial epithelial cells that were deficient in Rac1 (CCSP-Cre/Rac1flox/flox mice) failed to efficiently clear apoptotic cells in vivo, and when these mice were challenged with house dust mite (HDM), they exhibited significantly increased pulmonary inflammation, production of type 2 cytokines, and AHR, relative to controls. These CCSP-Cre/Rac1flox/flox mice failed to produce IL-10 during challenge with apoptotic cells and airway inflammation, and recombinant IL-10, delivered intranasally, dampened hyperinflammation in knockout mice [53]. This study highlights the role that non-professional phagocytes such as epithelial cells, play in efferocytosis. It is now clear that both immune and non-immune cells play important roles in clearing the inevitable cellular graveyard during allergic airway inflammation, and defects in this process can exacerbate inflammation and pathology.
‘Tolerate-me’ signaling
‘I’m lactose intolerant. I have no tolerance for lactose and I won’t stand for it!’ – Jerry Seinfeld
At the heart of efferocytosis is the question of how a phagocyte handles the burden of dramatically increasing its load of lipids and cholesterol when it engulfs a dying cell. LXRα/β are isoforms of LXR nuclear receptors that mediate cellular lipid and cholesterol metabolism. Consistent with the inverse correlation between circulating cholesterol concentrations and asthma [90], Lxrα−/−β−/− mice exhibit reduced AHR, type 2 cytokine production, and immunopathology in the HDM model of eosinophilic asthma, relative to controls [91]. Conversely, activation of the LXR pathway (with the LXR agonist GW3965, for example) has decreased the production of pro-inflammatory mediators such as IL-6 via LPS-stimulated human airway macrophages, and has reduced lung neutrophilia in LPS-challenged rodents [92].
The PPAR family of nuclear receptors are also crucial regulators of immunotolerance associated with efferocytosis. Cc10-Cre/Ppargflox/flox mice, which lack Pparg in airway epithelial cells, display heightened asthma-like responses, including increased AHR and elevated production of type 2 cytokines and alarmins [93]. Similarly, the PPARγ agonist rosiglitazone, improved lung function in mice in an OVA-induced asthma model [94] as well as in human asthmatic smokers [95]. In addition, mice with a ubiquitous Pparα deletion (Ppar α−/− mice) phenocopy Cc10-Cre/Ppargflox/flox mice and exhibit increased allergic airway responses relative to controls [96].
Given the contrasting role of LXRs and PPARs in asthma pathogenesis, researchers have examined the role of downstream cholesterol transporter, ATP-binding cassette (ABC), subfamily A, member 1 (ABCA1) in asthma. Mice that overexpress human ABCA1 in their vascular endothelial cells (Tie2-human Abca1 mice) are significantly protected against OVA-induced neutrophilic asthma, with reduced peribronchial inflammation, decreased titers of OVA-specific IgE, and reduced airway epithelial thickness, relative to controls [97].
How LAP and other mediators of dead cell processing affect allergic airway inflammation remains unknown. Collectively, these studies demonstrate a delicate balance in terms of efferocytosis, cholesterol metabolism, and airway inflammation, with more in-depth studies needed to explore the role that each of these molecules plays in differing types of asthma.
Concluding Remarks (and therapeutic options)
‘Mother Nature’s a mad scientist, Jerry.’ – Cosmo Kramer
The discovery that the efficient clearance of dying cells plays an important role in asthma pathogenesis has opened many therapeutic avenues. Glucocorticoids, the gold standard for the treatment of asthma, have been shown to enhance efferocytosis by upregulating key efferocytotic machinery, such as MERTK, LXRs, PPARs, and RXRs [98]. Macrolide antibiotics, such as azithromycin, have anti-inflammatory effects and have been reported to promote efferocytosis in lung macrophages [99]. Similarly, studies have demonstrated that the macrolide antibiotic, telithromycin, can significantly reduce asthmatic symptoms and improve lung function in patients with moderate-to-severe asthma [100].
Drugs that modulate phagocytosis are also possible candidates. One such candidate is lipoxin A4, which is an endogenous lipoxygenase-derived eicosanoid mediator that promotes cytoskeletal rearrangement and phagocytosis, and has been shown to display anti-inflammatory properties [101]. Indeed, pediatric patients with severe asthma present decreased sputum concentrations of lipoxin A4, compared to those with mild-to-moderate asthma [102]. Recent studies have demonstrated that lipoxin A4 can promote efferocytosis and reduce inflammation and pathology in murine models of acute lung inflammation [103, 104]. However, targeting an axis with such wide-ranging roles in fundamental biological processes could result in unwanted side effects.
Given the connection between cholesterol and asthma pathogenesis, statins have been explored as a potential therapeutic for airway inflammation. Widely used as cholesterol-lowering agents, statins also have broad, anti-inflammatory effects and have been demonstrated to increase efferocytotic capacity in vitro and ex vivo in a mouse model of lung inflammation [105]. Statin use is also associated with reduced asthma-related hospitalization or severe events [106]. However, clinical trials exploring the efficacy of statins as a treatment for asthmatics has yielded mixed results [106], suggesting further studies are needed.
Manipulating cholesterol efflux via PPAR agonism is also a potential treatment option. Synthetic PPAR agonists such as thiazolidinediones have been shown to enhance dead cell clearance and improve resolution in a mouse model of lung inflammation [107, 108].
These agonists have also decreased immunopathology in mouse models of asthma [109, 110]. While different PPAR isoforms retain specific activity in response to pharmacological PPAR agonists, an alternative approach would be to promote the production of broadly-acting natural PPAR ligands, such as eicosanoids, polyunsaturated fatty acids (PUFAs), and endocannabinoids [109].
Recent studies have highlighted the association of autophagic processes and allergic airway inflammation. Epithelial cells and airway smooth muscle from human asthmatics express increased BECN1 and ATG5, compared to non-asthmatic control tissues [111]. A SNP in ATG5 (rs510432) was found to be associated with childhood asthma, with the minor variant allele associated with increased asthma risk. [112]. Given this association, pharmacological modulation of autophagy has been explored in murine models of asthma. Intranasal administration of chloroquine, which inhibits lysosomal fusion [113], during HDM exposure resulted in decreased inflammation, AHR, and airway remodeling relative to controls [111]. However, chloroquine acts broadly on the lysosomal network and does not distinguish between autophagy and LAP. By specifically targeting RUBCN -- which is required for LAP but not autophagy, one might preserve vital autophagy quality control mechanisms while modulating the LAP response. One such candidate is Tat-N8, a cell-penetrating, RUBCN inhibitory peptide, comprised of the HIV protein Tat, conjugated to an N-terminal 8-amino acid sequence derived from the NOX2 subunit, p22phox. Tat-N8 can robustly block the RUBCN-p22phox interaction, which is required for LAP [114]. While studies have demonstrated the ability of Tat-N8’s to increase survival in a murine model of sepsis [114], this peptide (or its derivatives) have not yet been examined in the context of experimental asthma. It will be of great interest to determine whether selective targeting of LAP might represents an effective candidate therapy to treat asthma or other inflammatory diseases (see outstanding questions).
Outstanding Questions.
The lung contains an inordinate number of phagocytic cells - do professional phagocytes and non-phagocytes in the lung play distinct efferocytotic roles during asthma?
Do different endotypes of asthma exhibit different requirements for efferocytosis? Do these differences, whether in expression or function, represent possible therapeutic targets to treat asthma?
What role does LC3-associated phagocytosis (LAP) play in the onset and severity of asthma?
Would PS-receptor agonists or LAP modulators be an effective therapeutic against asthma?
Table 1.
|
“Find-me” signaling | ||
| Molecule | Phenotype | Ref | |
| Fractalkine | Increased upon induction of asthma | 63 | |
| S1P | Increased upon induction of asthma | 64 | |
| CX3CR1 | Variant associated with increased risk of asthma | 65 | |
| Possible protective effect of minor allele | 65 | ||
| PY(2)R | Py2r-deficient mice exhibit protection against experimental asthma | 66 | |
| HSP70 | Circulating and sputum concentrations are increased in asthmatics | 67 | |
|
“Eat-me” signaling | ||
| Molecule | Phenotype | Ref | |
| AXL | DNA methylation associated with increased risk of childhood asthma | 68 | |
| Sputum concentrations of soluble AXL (sAXL) are increased in asthmatics | 69 | ||
| Antibodies against AXL mitigate asthma-like features in mice | 70 | ||
| MerTK | Mer-deficient mice exhibit increased susceptibility to, and delayed resolution of, experimental asthma | 72 | |
| MFGE8 | Mfge8-deficient mice exhibit increased AHR and immunopathology in a model of experimental asthma | 73, 74 | |
| Galectin-3 | Galectin-3 expression decreased in alveolar macrophages from asthmatics | 60 | |
| Sputum concentrations of Galectin-3 and the ratio of Galectin-3 to Galectin-3 binding protein are particularly low in individuals with neutrophilic asthma | 78 | ||
| Recombinant Galectin-3 improves efferocytosis in macrophages derived from asthmatics with neutrophilic disease | 60 | ||
| RAGE | RAGE-KO mice are protected from asthma and exhibit reduced inflammation, AHR. And type 2 cytokine production | 87 | |
| Rac-1 | Mice with Rac1-deficient bronchial epithelial cells develop more severe allergic airway inflammation and fail to mount an immunotolerant response | 53 | |
|
“Tolerate-me” signaling | ||
| Molecule | Phenotype | ||
| LXRα/β | LXRα/β-deficient mice exhibit protection from asthma and reduced AHR | 91 | |
| PPARγ | Ppparg-deficient mice exhibit more severe asthma and increased AHR | 93 | |
| PPARα | Pppara-deficient mice exhibit more severe asthma and increased AHR | 96 | |
| ABCA1 | Overexpression of human ABCA1 in murine endothelial cells improved AHR and lung function | 97 | |
The table includes molecules within the efferocytosis pathway that affect asthma pathogenesis.
This table was created using BioRender (https://biorender.com/).
Abbreviations: AHR, airway hyperresponsiveness; KO, knockout.
Highlights.
Efferocytosis is a carefully orchestrated process that requires dying cells to release and display signals to facilitate recruitment of and recognition by phagocytes.
Efferocytosis is characterized by its immunotolerant response, and defects in the efferocytotic machinery are associated with autoimmune and inflammatory disorders in mice and humans.
Asthma affects over 200 million people worldwide and results from inappropriate immune responses to inhaled allergens leading to airway inflammation, excessive mucus production, and bronchial airway hyperresponsiveness (AHR).
Impaired efferocytosis has been observed in asthmatics, and defects in efferocytosis are associated with increased risk or severity of asthma.
Therapeutics that promote efferocytosis are promising candidates for the treatment of asthma, in which certain endotypes present unmet medical needs.
Acknowledgments
This work was supported by NIH Intramural Research Program 1ZIAES10328601 (to JM) and ZIA ES102025-13 (to DNC).
Glossary
- Absent in melanoma 2 (AIM2)
Cytosolic DNA sensor that forms the AIM2 inflammasome with ASC and Caspase-1 to mediate pro-inflammatory cytokine production and pyroptosis
- Airway hyperresponsiveness
Hallmark of asthma: defined as increased sensitivity and reactivity of the airways to normally innocuous stimuli; correlated to disease severity
- Autophagy
Evolutionarily conserved process, classically triggered by nutrient deprivation; cellular components are sequestered into de novo autophagosomal structures and trafficked to lysosomes for recycling and degradation
- Autophagy-related genes (ATG)
Evolutionarily conserved set of genes required for autophagy
- Cyclic GMP-AMP synthase (cGAS)
Cytosolic DNA sensor that regulates the synthesis of cGAMP, which subsequently activates the type I IFN response via STING-mediated signaling
- Damage-associated molecular patterns (DAMPs)
Small molecular motifs produced or released by damaged and dying cells that subsequently activate pathogen recognition receptors to stimulate innate immunity
- Efferocytosis
clearance of dying cells; recruitment of phagocytes via ‘find-me’ signals, the recognition and engulfment of dying cells via ‘eat-me’ signaling, and an immunotolerant response via ‘tolerate-me’ signaling
- Fc receptors (FcRs)
Family of surface receptors that recognize the Fc region of antibodies bound to opsonized particles, such infected cells or pathogens
- LAPosome
Cargo-containing, LC3-decorated phagosome generated during LAP
- LC3-associated phagocytosis (LAP)
Form of non-canonical autophagy triggered by receptor engagement during phagocytosis wherein RUBCN and components of the autophagy machinery are recruited to the cargo-containing phagosome to facilitate trafficking to the lysosomal network
- Liver X Receptor (LXR)
Member of the nuclear receptor family of transcription factors; regulators of cholesterol, lipid, and general metabolic homeostasis
- Peroxisome proliferator-activated receptors (PPARs)
Members (alpha, gamma, and delta (beta) of the nuclear receptor family of transcription factors; regulators of cellular differentiation, tumorigenesis, and metabolic homeostasis
- Pyknotic nuclei
Nuclei of cells undergoing apoptosis or necrosis, wherein chromatin is characterized as dense, compact, and possibly fragmented
- Retinoid X receptor (RXR)
Member of the steroid/thyroid hormone nuclear receptor superfamily of transcription factors; regulators of cellular differentiation, metabolism, and cell death
- Stimulator of interferon genes (STING)
Cytosolic five-transmembrane protein activated by cGAMP to mediate the type I IFN response
- Toll-like receptors (TLRs)
Family of membrane-bound pathogen recognition receptors that recognize and respond to PAMPs or DAMPs to mediate immune responses
- Z-DNA-binding protein 1 (ZBP1)
cytosolic sensor of Z-DNA and Z-RNA, present in numerous viruses; promotes the production of antiviral mediators, such as IFN β
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boada-Romero E. et al. (2020) The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 21 (7), 398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arandjelovic S. and Ravichandran KS (2015) Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16 (9), 907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez J. (2017) Prix Fixe: Efferocytosis as a Four-Course Meal. Curr Top Microbiol Immunol 403, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poon IK et al. (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14 (3), 166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagata S. et al. Autoimmunity and the clearance of dead cells. Cell 140 (5), 619–30. [DOI] [PubMed] [Google Scholar]
- 6.Ravichandran KS (2010) Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 207 (9), 1807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DR et al. (2016) The clearance of dying cells: table for two. Cell Death Differ 23 (6), 915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo B. et al. (2016) Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 44 (2), 287–302. [DOI] [PubMed] [Google Scholar]
- 9.Segawa K. et al. (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344 (6188), 1164–8. [DOI] [PubMed] [Google Scholar]
- 10.A.-G N. et al. (2009) Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31 (2), 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukundan L. et al. (2009) PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med 15 (11), 1266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiss RS et al. (2006) Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol 16 (22), 2252–8. [DOI] [PubMed] [Google Scholar]
- 13.Fond AM et al. (2015) Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest 125 (7), 2748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez J. et al. (2013) The Relationship between Metabolism and the Autophagy Machinery during the Innate Immune Response. Cell Metab 17 (6), 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Settembre C. et al. (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14 (5), 283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Underhill DM and Goodridge HS (2012) Information processing during phagocytosis. Nat Rev Immunol 12 (7), 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crow MK (2016) Autoimmunity: Interferon alpha or beta: which is the culprit in autoimmune disease? Nat Rev Rheumatol 12 (8), 439–40. [DOI] [PubMed] [Google Scholar]
- 18.Martinez J. et al. (2011) Microtubule-associated protein 1 light chain 3 alpha (LC3)associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States of America 108 (42), 17396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Florey O. et al. (2011) Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 13 (11), 1335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henault J. et al. (2012) Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 37 (6), 986–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanjuan MA et al. (2007) Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450 (7173), 1253–7. [DOI] [PubMed] [Google Scholar]
- 22.Birge RB et al. (2016) Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ 23 (6), 962–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N. (2007) Autophagy: process and function. Genes Dev 21 (22), 286173. [DOI] [PubMed] [Google Scholar]
- 24.Sil P. et al. (2018) A ravenous defense: canonical and non-canonical autophagy in immunity. Curr Opin Immunol 50, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez J. et al. (2015) Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17 (7), 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Zhong Y. et al. (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature cell biology 11 (4), 468–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsunaga K. et al. (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature cell biology 11 (4), 385–96. [DOI] [PubMed] [Google Scholar]
- 28.Yang CS et al. (2012) Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe 11 (3), 264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ueyama T. et al. (2011) Cooperation of p40(phox) with p47(phox) for Nox2-based NADPH oxidase activation during Fcgamma receptor (FcgammaR)-mediated phagocytosis: mechanism for acquisition of p40(phox) phosphatidylinositol 3-phosphate (PI(3)P) binding. J Biol Chem 286 (47), 40693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heckmann BL et al. (2019) LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 178 (3), 536–551 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunha LD et al. (2018) LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 175 (2), 429–441 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez J. et al. (2016) Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533 (7601), 115–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Heckmann BL et al. (2020) Noncanonical function of an autophagy protein prevents spontaneous Alzheimer’s disease. Sci Adv 6 (33), eabb9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sil P. et al. (2020) Noncanonical autophagy in dermal dendritic cells mediates immunosuppressive effects of UV exposure. J Allergy Clin Immunol 145 (5), 1389–1405. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Yaghoubi M. et al. (2019) The Projected Economic and Health Burden of Uncontrolled Asthma in the United States. Am J Respir Crit Care Med 200 (9), 11021112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez FD and Vercelli D. (2013) Asthma. Lancet 382 (9901), 1360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson GP (2008) Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 372 (9643), 1107–19. [DOI] [PubMed] [Google Scholar]
- 38.Woodruff PG et al. (2009) T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180 (5), 388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wenzel SE et al. (1999) Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 160 (3), 1001–8. [DOI] [PubMed] [Google Scholar]
- 40.Ray A. and Kolls JK (2017) Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol 38 (12), 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Green RH et al. (2002) Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax 57 (10), 875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silva GE et al. (2004) Asthma as a risk factor for COPD in a longitudinal study. Chest 126 (1), 59–65. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Z. et al. (2020) Association of asthma and its genetic predisposition with the risk of severe COVID-19. J Allergy Clin Immunol 146 (2), 327–329 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williamson EJ et al. (2020) Factors associated with COVID-19-related death using OpenSAFELY. Nature 584 (7821), 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang R. et al. (2020) Treating asthma in the COVID-19 pandemic. Thorax 75 (10), 822–823. [DOI] [PubMed] [Google Scholar]
- 46.Ordonez CL et al. (2000) Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: Clinical and biologic significance. Am J Respir Crit Care Med 161 (4 Pt 1), 1185–90. [DOI] [PubMed] [Google Scholar]
- 47.Zhou T. et al. (2017) Associations between Th17-related inflammatory cytokines and asthma in adults: A Case-Control Study. Sci Rep 7 (1), 15502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson RH et al. (2009) Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med 180 (8), 720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen K. et al. (2016) IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae. Cell Host Microbe 20 (5), 596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitehead GS et al. (2019) Therapeutic suppression of pulmonary neutrophilia and allergic airway hyperresponsiveness by a RORgammat inverse agonist. JCI Insight 5 (14), e125528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCubbrey AL and Curtis JL (2013) Efferocytosis and lung disease. Chest 143 (6), 1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grabiec AM and Hussell T. (2016) The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol 38 (4), 409–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juncadella IJ et al. (2013) Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493 (7433), 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kasahara Y. et al. (2001) Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 163 (3 Pt 1), 737–44. [DOI] [PubMed] [Google Scholar]
- 55.Vandivier RW et al. (2006) Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 129 (6), 1673–82. [DOI] [PubMed] [Google Scholar]
- 56.Goleva E. et al. (2008) Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J Allergy Clin Immunol 122 (3), 550–9 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simpson JL et al. (2013) Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin Exp Allergy 43 (1), 29–35. [DOI] [PubMed] [Google Scholar]
- 58.Yun JH et al. (2008) Phagocytic clearance of apoptotic cells: role in lung disease. Expert Rev Respir Med 2 (6), 753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Persson C. (2013) Airway, apoptosis, and asthma. Clin Exp Allergy 43 (9), 1083–5. [DOI] [PubMed] [Google Scholar]
- 60.Erriah M. et al. (2019) Galectin-3 enhances monocyte-derived macrophage efferocytosis of apoptotic granulocytes in asthma. Respir Res 20 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huynh ML et al. (2005) Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med 172 (8), 972–9. [DOI] [PubMed] [Google Scholar]
- 62.Fernandez-Boyanapalli R. et al. (2013) Obesity impairs apoptotic cell clearance in asthma. J Allergy Clin Immunol 131 (4), 1041–7, 1047 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Upton N. et al. (2017) Rhinovirus induction of fractalkine (CX3CL1) in airway and peripheral blood mononuclear cells in asthma. PLoS One 12 (8), e0183864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ammit AJ et al. (2001) Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J 15 (7), 1212–4. [DOI] [PubMed] [Google Scholar]
- 65.Tremblay K. et al. (2006) Association study between the CX3CR1 gene and asthma. Genes Immun 7 (8), 632–9. [DOI] [PubMed] [Google Scholar]
- 66.Muller T. et al. (2010) The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy 65 (12), 1545–53. [DOI] [PubMed] [Google Scholar]
- 67.Hou C. et al. (2011) Increased heat shock protein 70 levels in induced sputum and plasma correlate with severity of asthma patients. Cell Stress Chaperones 16 (6), 663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao L. et al. (2017) Epigenetic regulation of AXL and risk of childhood asthma symptoms. Clin Epigenetics 9, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grabiec AM et al. (2017) Diminished airway macrophage expression of the Axl receptor tyrosine kinase is associated with defective efferocytosis in asthma. J Allergy Clin Immunol 140 (4), 1144–1146 e4. [DOI] [PubMed] [Google Scholar]
- 70.Shibata T. et al. (2014) Axl receptor blockade ameliorates pulmonary pathology resulting from primary viral infection and viral exacerbation of asthma. J Immunol 192 (8), 3569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujimori T. et al. (2015) The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol 8 (5), 1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Felton JM et al. (2018) Mer-mediated eosinophil efferocytosis regulates resolution of allergic airway inflammation. J Allergy Clin Immunol 142 (6), 1884–1893 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kudo M. et al. (2013) Mfge8 suppresses airway hyperresponsiveness in asthma by regulating smooth muscle contraction. Proc Natl Acad Sci U S A 110 (2), 660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khalifeh-Soltani A. et al. (2018) The Mfge8-alpha8beta1-PTEN pathway regulates airway smooth muscle contraction in allergic inflammation. FASEB J, fj201800109R. [DOI] [PubMed]
- 75.Dong R. et al. (2018) Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int J Mol Med 41 (2), 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Diaz-Alvarez L. and Ortega E. (2017) The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediators Inflamm 2017, 9247574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sano H. et al. (2003) Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest 112 (3), 389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao P. et al. (2015) Anti-inflammatory deficiencies in neutrophilic asthma: reduced galectin-3 and IL-1RA/IL-1beta. Respir Res 16, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hancock DB et al. (2010) Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet 42 (1), 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Osawa M. et al. (2007) De-N-glycosylation or G82S mutation of RAGE sensitizes its interaction with advanced glycation endproducts. Biochim Biophys Acta 1770 (10), 1468–74. [DOI] [PubMed] [Google Scholar]
- 81.Repapi E. et al. (2010) Genome-wide association study identifies five loci associated with lung function. Nat Genet 42 (1), 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.El-Seify MY et al. (2014) Serum level of soluble receptor for advanced glycation end products in asthmatic children and its correlation to severity and pulmonary functions. Clin Lab 60 (6), 957–62. [DOI] [PubMed] [Google Scholar]
- 83.Watanabe T. et al. (2011) Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir Med 105 (4), 519–25. [DOI] [PubMed] [Google Scholar]
- 84.Brandt EB and Lewkowich IP (2019) RAGE-induced asthma: A role for the receptor for advanced glycation end-products in promoting allergic airway disease. J Allergy Clin Immunol 144 (3), 651–653. [DOI] [PubMed] [Google Scholar]
- 85.Yang Z. et al. (2007) S100A12 provokes mast cell activation: a potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol 119 (1), 106–14. [DOI] [PubMed] [Google Scholar]
- 86.Shim EJ et al. (2012) The role of high-mobility group box-1 (HMGB1) in the pathogenesis of asthma. Clin Exp Allergy 42 (6), 958–65. [DOI] [PubMed] [Google Scholar]
- 87.Milutinovic PS et al. (2012) The receptor for advanced glycation end products is a central mediator of asthma pathogenesis. Am J Pathol 181 (4), 1215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oczypok EA et al. (2015) Pulmonary receptor for advanced glycation end-products promotes asthma pathogenesis through IL-33 and accumulation of group 2 innate lymphoid cells. J Allergy Clin Immunol 136 (3), 747–756 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim SY et al. (2017) Coordinated balance of Rac1 and RhoA plays key roles in determining phagocytic appetite. PLoS One 12 (4), e0174603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fessler MB et al. (2009) Novel relationship of serum cholesterol with asthma and wheeze in the United States. J Allergy Clin Immunol 124 (5), 967–74 e1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smet M. et al. (2016) Cholesterol-sensing liver X receptors stimulate Th2-driven allergic eosinophilic asthma in mice. Immun Inflamm Dis 4 (3), 350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Birrell MA et al. (2008) Liver X receptor agonists increase airway reactivity in a model of asthma via increasing airway smooth muscle growth. J Immunol 181 (6), 4265–71. [DOI] [PubMed] [Google Scholar]
- 93.Lakshmi SP et al. (2018) Airway Epithelial Cell Peroxisome Proliferator-Activated Receptor gamma Regulates Inflammation and Mucin Expression in Allergic Airway Disease. J Immunol 201 (6), 1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.El-Naa MM et al. (2015) In-vivo antioxidant and anti-inflammatory activity of rosiglitazone, a peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists in animal model of bronchial asthma. J Pharm Pharmacol 67 (10), 1421–30. [DOI] [PubMed] [Google Scholar]
- 95.Spears M. et al. (2009) Bronchodilatory effect of the PPAR-gamma agonist rosiglitazone in smokers with asthma. Clin Pharmacol Ther 86 (1), 49–53. [DOI] [PubMed] [Google Scholar]
- 96.Woerly G. et al. (2003) Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med 198 (3), 411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dai C. et al. (2014) ATP-binding cassette transporter 1 attenuates ovalbumin-induced neutrophilic airway inflammation. Am J Respir Cell Mol Biol 51 (5), 626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Garabuczi E. et al. (2015) Glucocorticoids enhance prolonged clearance of apoptotic cells by upregulating liver X receptor, peroxisome proliferator-activated receptor-delta and UCP2. Biochim Biophys Acta 1853 (3), 573–82. [DOI] [PubMed] [Google Scholar]
- 99.Gao X. et al. (2010) Macrolide antibiotics improve chemotactic and phagocytic capacity as well as reduce inflammation in sulfur mustard-exposed monocytes. Pulm Pharmacol Ther 23 (2), 97–106. [DOI] [PubMed] [Google Scholar]
- 100.Johnston SL (2006) Macrolide antibiotics and asthma treatment. J Allergy Clin Immunol 117 (6), 1233–6. [DOI] [PubMed] [Google Scholar]
- 101.Liu X. et al. (2017) Lipoxin A4 and its analog suppress inflammation by modulating HMGB1 translocation and expression in psoriasis. Sci Rep 7 (1), 7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gagliardo R. et al. (2016) Airway lipoxin A4/formyl peptide receptor 2-lipoxin receptor levels in pediatric patients with severe asthma. J Allergy Clin Immunol 137 (6), 1796–1806. [DOI] [PubMed] [Google Scholar]
- 103.Sekheri M. et al. (2020) 15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc Natl Acad Sci U S A 117 (14), 7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Godson C. et al. (2000) Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol 164 (4), 1663–7. [DOI] [PubMed] [Google Scholar]
- 105.Morimoto K. et al. (2006) Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol 176 (12), 7657–65. [DOI] [PubMed] [Google Scholar]
- 106.Fricker M. and Gibson PG (2017) Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J 50 (3). [DOI] [PubMed] [Google Scholar]
- 107.Greenlee-Wacker MC (2016) Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 273 (1), 357–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yoon YS et al. (2015) PPARgamma activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol 8 (5), 1031–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kytikova OY et al. (2020) Peroxisome Proliferator-Activated Receptors as a Therapeutic Target in Asthma. PPAR Res 2020, 8906968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shafi S. et al. (2019) PPARgamma: Potential Therapeutic Target for Ailments Beyond Diabetes and its Natural Agonism. Curr Drug Targets 20 (12), 1281–1294. [DOI] [PubMed] [Google Scholar]
- 111.McAlinden KD et al. (2019) Autophagy Activation in Asthma Airways Remodeling. Am J Respir Cell Mol Biol 60 (5), 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martin LJ et al. (2012) Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One 7 (4), e33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mauthe M. et al. (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14 (8), 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim YR et al. (2016) Peptide inhibition of p22phox and Rubicon interaction as a therapeutic strategy for septic shock. Biomaterials 101, 47–59. [DOI] [PubMed] [Google Scholar]
- 115.Travaglini KJ et al. (2020) A molecular cell atlas of the human lung from singlecell RNA sequencing. Nature 587 (7835), 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mitchell PD and O’Byrne PM (2017) Epithelial-Derived Cytokines in Asthma. Chest 151 (6), 1338–1344. [DOI] [PubMed] [Google Scholar]
- 117.Thomas SY et al. (2018) MyD88-dependent dendritic and epithelial cell crosstalk orchestrates immune responses to allergens. Mucosal Immunol 11 (3), 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lynch JP et al. (2016) Th2/Th17 reciprocal regulation: twists and turns in the complexity of asthma phenotypes. Ann Transl Med 4 (Suppl 1), S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coleman JM et al. (2012) Epithelial eotaxin-2 and eotaxin-3 expression: relation to asthma severity, luminal eosinophilia and age at onset. Thorax 67 (12), 1061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]