

Abstract
Folliculogenesis is a tightly coordinated process essential for generating a fertilization-competent gamete while also producing gonadal hormones that sustain endocrine function. In vitro follicle growth systems have been critical to our understanding of key events in folliculogenesis, such as gonadotropin-independent and dependent growth, steroid hormone production, and oocyte growth and maturation (cytoplasmic and meiotic). Although there are several successful follicle culture strategies, the following protocol details an encapsulated in vitro follicle growth (eIVFG) system for use with mouse ovarian follicles. Encapsulated IVFG is performed with alginate hydrogels, which are biologically inert, maintains cell-to-cell interactions between granulosa cells and the oocyte, and preserves follicle architecture as found in the ovary. The system supports follicle growth, development, and differentiation from the early primary follicle to the antral follicle stage. Moreover, post-folliculogenesis events including meiotic maturation, ovulation, and luteinization are also supported. Importantly, the culture of secondary follicles has successfully resulted in viable pups after blastocyst transfer. This alginate-based eIVFG system is versatile and has broad applications as a tool for interrogating the fundamental biology of the ovarian follicle in a controlled manner, a screening platform for toxicity and bioactivity, and a potential fertility preservation method for endangered species as well as humans.
Keywords: Folliculogenesis, Alginate, Oogenesis, Ovulation
This is a detailed protocol for culture of mouse ovarian follicles utilizing an alginate hydrogel-based encapsulated in vitro growth system.
Introduction
The development of in vitro follicle growth systems has allowed for the controlled investigation of events vital to folliculogenesis and has thus increased our understanding of the fundamental biology of the developing follicle. Various successful follicle growth systems have been developed over the last four decades, and selecting one is highly dependent on the species being used and the experimental goal [1]. This protocol describes an alginate hydrogel-based encapsulated in vitro follicle growth (eIVFG) system originally developed by the research groups of Teresa K. Woodruff and Lonnie D. Shea [2–34]. Alginate is a polysaccharide derived from brown algae that has been extensively utilized in tissue support and growth systems due to several properties, including its ability to undergo physiological gelation to form dilute hydrogels that are nonadhesive and nondegradable [35, 36].
Alginate-based eIVFG systems allow for follicle growth and development in a manner that maintains proper spatial oocyte–somatic cell interactions and is able to recapitulate critical components of folliculogenesis such as follicle growth and cellular differentiation, steroid production, meiotic maturation, and ovulation [3]. Importantly, gametes derived from eIVFG in a mouse model are fertilization-competent, can support preimplantation embryo development, and can give rise to live offspring [6]. For these reasons, this method has been widely applied to multiple mammalian models including mouse [2–9, 11, 12, 14–22, 24, 25, 27, 29–32, 37, 38], rat [39], dog [40], goat [26, 41, 42], nonhuman primate [9, 13, 23, 34], and human [10, 28, 33]. Encapsulated IVFG has also been utilized in the context of fertility preservation [8, 10, 43, 44], screening compounds for reproductive toxicity and bioactivity [27, 31, 45, 46], and as a means for nonhormonal contraceptive discovery [38]. The following protocol focuses on standard eIVFG methods, but various modifications in culture conditions, alginate composition, and uses for eIVFG are well documented in the literature [2–34, 38, 46–49]. Therefore, this protocol should be used as a starting point for development and application to fit specific research needs.
Key experimental design considerations need to be assessed prior to the utilization of eIVFG, including:
Follicle stage: The developmental stage of the follicles used is highly dependent upon the study question, with the potential to utilize primary, transitioning primary to secondary, two-layer secondary, and multilayer secondary follicles. Considerations include what stage(s) of folliculogenesis or oogenesis are of interest. Multilayer secondary follicles only require 6–8 days of culture before ovulation can be induced but will not allow the assessment of early folliculogenesis events. Transitioning and two-layer early secondary follicles require 10–14 days [18] before ovulation induction but better encompass both gonadotropin-independent and dependent stages. Primary follicles have been used to a lesser extent in eIVFG and require additional support systems such as co-culture with other follicles [18], stromal cell populations [14], or embryonic fibroblasts [17] to sustain survival and growth. The follicle stage will dictate if individual or multiple follicle encapsulation is required. Multilayer secondary follicles are suitable for individual culture. However, murine primary and early two-layer secondary follicles must be cultured in cohorts of 10 (multiple follicle culture) because secreted paracrine factors are required for their survival and growth [18].
Mouse strain and age: Regarding animal age, prepubertal mice are most commonly used for eIVFG due to the high yield of primary and secondary follicles that can be collected [50], but it is important to note that these follicles would not normally have contributed to physiologic fertility. Follicles can also be isolated from adult mice and used for eIVFG, but the total yield of follicles significantly decreases with age [16]. Mouse strain will also dictate the quantity of follicles that can be obtained. CD-1 [2, 14, 16–22, 24, 25, 27, 29–32, 38], F1 hybrids C57BL/6jxCBA/Ca [3–8, 11, 12, 15, 48], and CB6F1 [37] have been previously utilized for eIVFG, with the follicle size of specific developmental stages and growth dynamics differing between strains. For this protocol, all representative images are taken from CD-1 mice. The number of follicles from CD-1 mice with suitable morphology for culture is approximately 5–10 per adult mouse or 10–20 per prepubertal mouse with mechanical isolation. The yield will be approximately 10–20 follicles per mouse when using enzymatic digestion for follicle isolation.
Alginate concentration and supplements: Alginate concentrations of 0.25–2% have been examined for their effect on various folliculogenesis and endpoint parameters [7, 20]. Alginate is used as a scaffold for follicle growth and can be adjusted to mimic different ovarian microenvironments, such as the cortex and medulla, which have higher and lower rigidness, respectively [34]. Furthermore, alginate concentrations can be adjusted to mimic various ovarian phenotypes such as the aging ovary and polycystic ovarian syndrome. The current protocol utilizes 0.5% alginate, but different concentrations and modifications have been recently reviewed [51, 52]. Alternatively, blended alginate hydrogels such as fibrin-alginate interpenetrating networks [48], alginate enriched with a synthetic arginine-glycine-aspartic acid (RGD) adhesion peptide [4], and non-alginate hydrogels such as polyethylene glycol [49] can also be utilized for eIVFG.
Endpoints: Encapsulated IVFG allows for various endpoint analyses and post-culture assays. Follicle growth and survival are the most commonly tracked parameters, but others include follicle histology, hormone and secreted factor analysis in conditioned media, ovulation and luteinization, meiotic progression, gamete morphology, and fertilization and preimplantation embryo development potential.
Methods
The use of animals to develop this method was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Northwestern University.
The major components covered in this protocol include:
Media preparation: All media should be prepared before initiating eIVFG. Preparation instructions and components are listed in Table 1.
Set up for isolation: Media is warmed and equilibrated to the environment of the incubator if needed, and a pipette for follicle manipulation is prepared (Stripper or mouth pipette. Check with institutional safety and/or biohazard committees to determine if mouth pipetting is allowed for manipulating of murine samples. If not, handheld pipettes (Stripper) can be utilized for micromanipulation.)
Ovarian tissue dissection: Ovaries are removed from the mouse and released from the bursa.
Follicle isolation: Follicles are isolated from the ovary mechanically or enzymatically.
First recovery incubation: Isolated follicles are allowed to recover in the incubator for 1 h.
Follicle selection: Follicles of high quality that are healthy enough to undergo long-term culture are selected.
Alginate encapsulation: High-quality follicles are encapsulated in an alginate bead that they will grow in throughout the duration of culture.
Second recovery incubation: Encapsulated follicles are allowed to recover for 45 min in the incubator.
Plating: Encapsulated follicles are placed into growth media (GM) within a 96-well plate and imaged.
Culture: Follicles are cultured at 37 °C, 5% CO2. Media is refreshed and follicles are imaged every other day until the desired culture endpoint.
Table 1.
List of media and solutions used throughout the procedure with preparation and storage instructions
| Media | Component | Catalog no | Supplier | Final concentration |
|---|---|---|---|---|
| Dissection media (DM) | Leibovitz’s (L15) | 11415-064 | Gibco | Base |
| Penicillin–streptomycin | 15140-122 | Gibco | 0.50% | |
| Fetal bovine serum (FBS) | PS-FB2 | Peak serum | 1% | |
| • Typical volume prepared: 30 mL | ||||
| • Shelf life: 2 weeks at 4 °C | ||||
| • Components are added to a conical tubed, mixed by inversion, and then sterile filtered into a new conical tube. | ||||
| Maintenance media (MM) | MEMα + GlutaMAX | 32561-037 | Gibco | Base |
| Pen-Strep | 15140-122 | Gibco | 0.50% | |
| FBS | PS-FB2 | Peak serum | 1% | |
| • Typical volume prepared: 30 mL | ||||
| • Shelf life: 2 weeks at 4 °C | ||||
| • Components are added to a conical tubed, mixed by inversion, and then sterile filtered into a new conical tube. | ||||
| Growth media (GM) | MEMα + GlutaMAX | 32561-037 | Gibco | 50% |
| F-12 + GlutaMAX | 31765-035 | Gibco | 50% | |
| Bovine serum albumin (BSA) | 103700 | MP Biomedicals | 3 mg/mL | |
| Fetuin (refer to Table 2) | Table 2 | Table 2 | Table 2 | |
| Insulin-transferrin-selenium (10X) (ITS) | 41400045 | ThermoFisher | 1 μL/mL | |
| Follicle-stimulating hormone (FSH) | Gonal-F Rx only | EMD Serono, Inc. | 10 mIU/mL | |
| • Typical volume prepared: 10–30 mL | ||||
| • Shelf life: 2 weeks at 4 °C | ||||
| • MEMα, BSA, and fetuin are added to a conical tubed, placed on a shaker for 5 min to mix, sterile filtered into a new conical tube, and then FSH and ITS are added. GM should not include Pen-Strep, which has been observed to be detrimental to follicles in culture. | ||||
| • To obtain Gonal-F you might need a physician prescription | ||||
| Enzymatic digestion media (EM) | Leibovitz’s (L15) | 11415-064 | Gibco | Base |
| Pen Strep | 15140-122 | Gibco | 1% | |
| Liberase | 5401119001 | Sigma Aldrich | 25 μg/mL | |
| DNase I | 79254 | Qiagen | 200 μg/mL | |
| FBS | PS-FB2 | Peak serum | 10% (quench) | |
| • Typical volume prepared: Prepared directly in 35 mm dishes with 2.5 mL starting volume of base media per dish | ||||
| • Shelf life: Prepare for immediate use | ||||
| • L-15 is added to a 35 mm dish (one dish per every four ovaries) and warmed on a heated surface to 37 °C. Pen Strep, Liberase, and DNase I are added directly to each dish. After digestion, FBS is added to quench the enzymes. | ||||
| Maturation media (MatM) | MEMα + GlutaMAX | 32561-037 | Gibco | Base |
| FBS | PS-FB2 | Peak serum | 10% | |
| Mouse epidermal growth factor (EGF) | 354010 | BD Biosciences | 10 ng/mL (PBS) | |
| Human chorionic gonadotropin (hCG) | C1063-1VL | Sigma Aldrich | 1.5 IU/mL (PBS) | |
| FSH | Gonal-F Rx only | EMD Serono, Inc. | 10 mIU/mL | |
| • Typical volume prepared: 10 mL | ||||
| • Shelf life: 2 weeks at 4 °C | ||||
| • MEMα and FBS are added to a conical tube, sterile filtered into a new conical tube, and then EGF, hCG, and FSH are added. | ||||
| Hyaluronidase solution (100X) | Sterile PBS (+/+) | 14040-133 | Gibco | Base |
| Hyaluronidase | C5670 | Sigma Aldrich | 3% | |
| • Typical volume prepared: 1 mL | ||||
| • Shelf life: 1 year at –20 °C | ||||
| • Mix 30 mg hyaluronidase/mL PBS and make 20 μL aliquots | ||||
| Alginate lyase | Leibovitz’s (L15) | 11415-064 | Gibco | Base |
| Alginate lyase (≥ 10 000 units/g) | A1603 | Sigma Aldrich | 10 IU/mL | |
| Pen-Strep | 15140-122 | Gibco | 0.50% | |
| FBS | PS-FB2 | Peak Serum | 1% | |
| • Typical volume prepared: 30 mL separated into 1 mL aliquots | ||||
| • Shelf life: 2 years at –20 °C or – 80 °C | ||||
| • Combine L15, Pen-Strep, and FBS with alginate lyase powder in a conical tube and vortex to dissolve. Sterile filter solution into a clean conical tube and aliquot. | ||||
| Alcian blue solution | Sterile MilliQ H2O | Base | ||
| Alcian Blue 8GX | 33864-99-2 | Sigma | 0.50% | |
| Acetic acid, glacial | A38S-212 | Fisher Scientific | 0.25% | |
| • Typical volume prepared: 25 mL | ||||
| • Shelf life: Indefinite at room temperature | ||||
| • Combine components and mix by inversion | ||||
An overview of the protocol’s steps listed above and possible endpoints including follicle growth and survival tracking, ovulation, meiotic stage analysis, and histology are presented schematically in Figure 1.
Figure 1.

Schematic of eIVFG procedure and examples of endpoint analyses. Dissected ovaries are broken down mechanically or enzymatically to isolate follicles. These follicles then undergo a recovery incubation prior to the selection of high-quality follicles. Follicles are encapsulated in alginate beads individually (multilayer secondary follicles) or in cohorts of 10 (two-layer early secondary follicles) and then undergo another recovery incubation. Alginate-encapsulated follicles are then plated in a 96-well plate and cultured. Imaging and media changes take place every other day throughout the duration of the culture, and various endpoint parameters can be assessed.
Media preparation
All media should be prepared in a sterilized, Class II biological safety cabinet. Preparation instructions and suppliers of components for all media, solutions, growth factors, and enzymes are presented in Table 1. Preparation instructions and supplier information for fetuin are listed in Table 2.
Table 2.
Instructions for the preparation of fetuin aliquots
| Materials | Catalog no. | Supplier | Final concentration |
|---|---|---|---|
| Sterile MilliQ H2O | Base | ||
| Fetal bovine fetuin | F-3385 | Sigma | 10 mg/mL |
| BCA protein assay reagent kit | 23227 | FisherScientific | |
| Spectra/Pro regenerated cellulose Dialysis membranes | MWCO 8000 1231166 | Spectra/Pro Repligen | |
| Procedure: | |||
| • Dissolve fetuin to a 15–20 mg/mL concentration in MilliQ H2O | |||
| • Dialyze the fetuin solution at 4 °C overnight in MilliQ H2O in a 1:50 to 1:100 ratio. Stir gently during all the process. Next day, replace with fresh MilliQ H2O, and dialyze 1–2 more times. Dialysis is important to increase the purity of the fetuin concentrate. | |||
| • Measure the fetuin concentration using a BCA protein Assay Reagent Kit or Nanodrop | |||
| • Dilute fetuin solution to 10 mg/mL with MilliQ H2O | |||
| • Aliquot fetuin into 1.5 mL microcentrifuge tubes (1 mL/tube) | |||
| • Close all the tubes, and puncture a hole in the lid of each tube | |||
| • Freeze until completely solid (several days at −20 °C, overnight at −80 °C or 10 min in liquid nitrogen) | |||
| • Lyophilize the fetuin to dryness at −80 °C. It will take approximately 1 day | |||
| • Seal the holes of the lids with stickers to keep the fetuin dry | |||
| • Fetuin can be stored for long term at −80 °C or at −20 °C for short term use | |||
| We usually prepare 25 g of fetuin to have stock for 3–4 years, but this will depend on how often follicle culture will be performed in your laboratory | |||
| Lyophilization at – 80 °C is a complex process and you need a special equipment | |||
The follicle isolation protocol utilizes three types of media, dissection media (DM; L15-based, buffered for ambient air exchange), maintenance media (MM; α-MEM-based, sodium bicarbonate buffered for air exchange at 5% CO2), and enzymatic media (EM; L15-based). Follicles should be kept in DM when manipulated outside the incubator for extended periods of time, and in MM while in the incubator. When using enzymatic media, ensure follicles are in ambient air conditions since it is L15-based. GM is α-MEM-based and used for the extended culture of follicles within an incubator. Critical for a successful in vitro follicle growth is the addition of fetal bovine fetuin to the GM. Fetuin is a glycoprotein present in fetal blood and in the follicle fluid. Follicles are grown in serum-free medium, which induces hardening of the zona pellucida. The addition of fetuin during the oocyte growing and maturation steps is important because it prevents the hardening of the zona pellucida, and therefore increases fertilization and embryo development rates [53–57].
Note: Media should not be warmed multiple times. Warm only the volume that is needed and dispose of any extra which has been warmed.
Set-up for follicle isolation
All supplies, consumables, and equipment needed for the protocol are listed in Table 3. An overview of the tools, dishes, and media needed for each step is listed in Table 4. Aseptic technique should be used throughout the protocol. Follicle handling should be performed in a laminar flow hood and preferably on a heated stage. Setting up for this protocol involves the preparation of a series of dishes and plates that will be used throughout the protocol. Follicles will be manipulated outside of the incubator in DM, while they will recover in the incubator in MM. Dishes containing MM are prepared at the onset of the protocol because they need to equilibrate in the incubator before use.
Table 3.
Materials, consumables, and equipment used for eIVFG
| Equipment | Example | |
|---|---|---|
| Class II biosafety cabinet | Baker SterilGARD® III Advance Safety Cabinets SKU: 8038-30-1044 | |
| CO2 incubator | Heracell VIOS 160i CO₂ Incubator, Thermo Scientific VWR # 76459-168 | |
| Dissection microscope | Leica; MZ9.5, S9 series | |
| Heat stage | Tokai-Hit; Thermoplate TPI series heated glass stage | |
| Laminar flow hood | Cole-Parmer; Vertical Laminar Flow Cabinet #UX-78902-40 | |
| IVF cabinet (Encompasses dissecting scope, heated stage, and laminar flow hood) | IVFtech; Classica workstation | |
| Inverted imaging platform for culture plates | EVOS; ThermoFisher Scientific, AMF5000 | |
| Material | Source | Catalog no |
| Watch glass (45 mm) | Electron Microscopy Sciences | 7054345 |
| 28 ½ G insulin syringe | Exelint International, Co. | 26027 |
| Nunc IVF dish | ThermoScientific | 150260 |
| 35 mm Petri dish | Corning | 351008 |
| Disposable scalpel | Fisher Scientific | 12-460-453 |
| Low retention pipette tips | MIDSCI | PR-10RK-NS |
| Stripper/mouth pipette | Origio/homemade | MXL3-STR |
| Stripper tips (125 μm) | Origio | MXL3-125 |
| Stripper tips (150 μm) | Origio | MXL3-150 |
| 96-well culture plates | Corning | 3595 |
Table 4.
List of tools, dishes, and media needed for each step of eIVFG
| Step | Materials | Timing |
|---|---|---|
| Ovary dissection | 35 mm Petri dish with DM Fine tweezers or insulin needles | ~5–10 min |
| Isolation | Stripper or mouth pipette with stripper tips | ≤45 min |
| Mechanical isolation | 45 mm watch glass with DM | |
| 2 insulin syringes | ||
| Or | IVF dish with DM | |
| Enzymatic isolation | Scalpel | |
| 35 mm Petri dish with EM | ||
| IVF dish with DM | ||
| Heated shaker | ||
| Recovery incubation | IVF dish with MM (equilibrated) | 1 h |
| Selection | Stripper or mouth pipette with stripper tips IVF dish with DM | 1 h |
| Encapsulation | Stripper or mouth pipette with stripper tips | 15–45 min |
| Curved forceps | Depends on number of follicles | |
| Drop method | 35 mm Petri dish lid | |
| 10 μL displacement pipette | ||
| P10 low retention pipette tips | ||
| Or | 35 mm Petri dish with calcium solution | |
| 35 mm Petri dish with DM | ||
| Parafilm method | 35 mm Petri dish lid | |
| Parafilm cut into 0.5–1 cm2 squares | ||
| 35 mm Petri dish with calcium solution | ||
| 35 mm Petri dish with DM | ||
| Recovery incubation | 35 mm Petri dish with MM (equilibrated) | 45 min |
| Plating | Flat bottom 96-well plate with GM (equilibrated) Curved forceps | <15 min |
Note: All tubes and culture plates used for follicles must be polystyrene as other materials such as polypropylene can be toxic to oocytes.
2.1 Warm DM to 37 °C in heated beads or on a heating stage for 15–30 min. To prevent contamination, do not use a water bath.
2.2 Clean the workspace that will be used for follicle isolation with 70% ethanol.
2.3 Add 1 mL of MM to the center well of an in vitro fertilization (IVF) dish and 3 mL to the outer well. Place in a humidified incubator (37 °C, 5% CO2) to equilibrate for at least 1 h. This dish will be used to hold follicles during a recovery incubation, described later in the protocol (Section 5).
2.4 Add 3 mL of MM to a 35 mm dish and place into the incubator to equilibrate for at least 1 h. This dish will be used to hold alginate beads during a recovery incubation, described later in the protocol (Section 8).
2.5 Prepare mouth pipette or stripper for follicle micromanipulation.
Note: Either a pulled glass pipette or stripper tips can be used. Ensure that the opening diameter is sufficient for the selected follicle class. Stripper tips of particular sizes can be purchased and used (i.e., 125 μm for early secondary, 150 μm for multilayer secondary) to aid in size selection.
Dissection of ovarian tissue
Note: Start with processing only one mouse (two ovaries) at a time. This number can be increased with experience as long as the isolation does not exceed a total of 45 min. If you need to isolate a large number of follicles, you can conduct isolations sequentially or have multiple people isolating follicles at the same time. Figure 2 shows the anatomy surrounding the ovary in a prepubertal CD-1 mouse. Video 1 shows the dissection of a prepubertal mouse ovary from the bursa.
Figure 2.
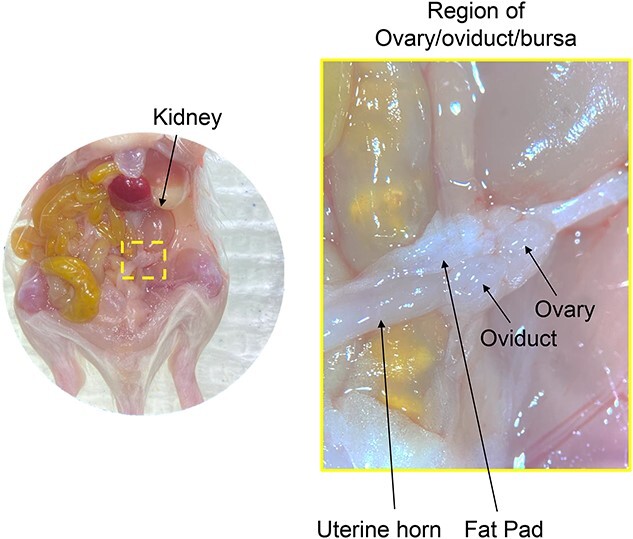
Identification of structures within the abdominal cavity of prepubertal mice aids in localization of the ovary due to its small size. When removing ovaries from prepubertal mice, it is often easier to dissect the ovary along with the fat pad, oviduct, and proximal part of the uterus as one unit and remove unnecessary tissue under a dissecting scope as a second step.
3.1 Prepare a 35 mm Petri dish with 3 mL of warmed DM. This will be used to remove the ovaries from the bursa under a dissecting scope. One dish can be used for all of the ovaries.
3.2 Euthanize a female mouse according to approved institutional regulations.
3.3 Dissect the mouse. The mouse is placed in dorsal recumbency, and the abdomen is lightly spayed with 70% ethanol to reduce fur getting into the dissection field. The abdomen cavity is opened with sharp scissors through the skin and body wall to expose the viscera. Move the viscera to the side to visualize the reproductive tract.
3.4 Locate the ovaries by finding the kidney and identifying two tube like structures connected to it: the thinner and more medial structure is the ureter and the wider and more lateral structure is one half of the uterus. For adults, follow the uterus up to just below the kidney and identify the ovary which is a small oval structure at the end connected to the uterus, the oviduct, and a small pad of fat. Remove ovaries from the bursa and place them in DM. For prepubertal mice, the ovary is not distinguishable by eye so a portion of the oviduct, uterus, and adipose tissues surrounding the ovaries are removed to avoid damaging the ovaries (Figure 2).
3.5 Under a dissection scope, remove any attached oviduct, uterus, and adipose tissue using fine forceps or insulin needles (Video 1).
Note: All following steps should be performed on a heated surface (37 °C) unless otherwise stated.
Follicle isolation
Follicle isolation can be performed either mechanically by breaking down the structural components of the ovary directly with insulin syringes or enzymatically by using several enzymes to degrade extracellular matrix components to assist in mechanical breakdown with a pipette. Mechanical isolation is more labor-intensive but preserves the basement membrane and some theca and stromal cells. Enzymatic isolation is less labor-intensive and typically results in higher yield, but the basement membrane is digested and follicles initially have decreased steroidogenic activity. In this protocol, enzymatic isolation is conducted using a combination of DNAse I and Liberase, though other enzymes such as collagenase have been used to successfully isolate follicles. The choice of isolation method should be carefully considered, but both methods work for most experimental designs. Isolation must be completed within 45 min to reduce the amount of time that follicles spend outside of the incubator. Though spending more than 45 min on isolation may allow for isolation of a larger number of follicles, the quality of follicles will suffer. Follicles become sticky and degenerate the longer they are kept outside of the incubator, so it is very important to complete isolation steps quickly to have the highest quality follicles for culture. As such, all follicles of the desired developmental stage should be collected initially without being selective for quality. Follicle stage can be determined by identifying the number of granulosa cell layers surrounding the oocyte and approximating follicle diameter with the end of a stripper tip (Figure 3). This will enable maximum yield while minimizing the time that follicles spend outside of the incubator. Selection for quality will take place after isolated follicles have been placed in the incubator to recover.
Figure 3.

Follicle size and morphology can be used to stage follicles during isolation. Size can be approximated using an eyepiece reticule attachment on the dissection microscope or by comparing the follicle diameter to the width of the opening of a stripper tip.
Note: After isolation, follicles are manipulated with stripper tips (or a pulled glass pipette) attached to a stripper or mouth pipette.
Mechanical isolation
Note: The method of mechanical isolation differs between ovaries from prepubertal mice and adult mice due to the difference in the amount of stromal tissue between these ages. Prepubertal ovaries have less stroma, resulting in softer, easier to disrupt ovaries, whereas the stroma is more prominent in adult ovaries. Furthermore, ovaries become fibrotic with advanced reproductive age [58]. Techniques for mechanical isolation of ovaries from different aged mice can be seen in Video 2.
4.2 Clean a 45 mm watch glass with 70% ethanol, dry completely with a Kimwipe, and add 2–3 mL of warm DM. Add the dissected ovaries to the dish.
Prepubertal mice
4.2.1 Isolate follicles from the ovaries using two 28 ½ G insulin syringes. In your nondominant hand, use one syringe to pin an ovary down and use the second syringe in your dominant hand to repeatedly “flick” the needle of the first syringe above the air-media interface (Video 2). This motion leads to vibrations in the ovary that expel intact follicles. Should the ovary become dislodged, re-stabilize it with the syringe and continue this process until the ovarian tissue is broken down into small fragments (Figure 4A). For two ovaries, this process should take ~20–30 min.
Figure 4.
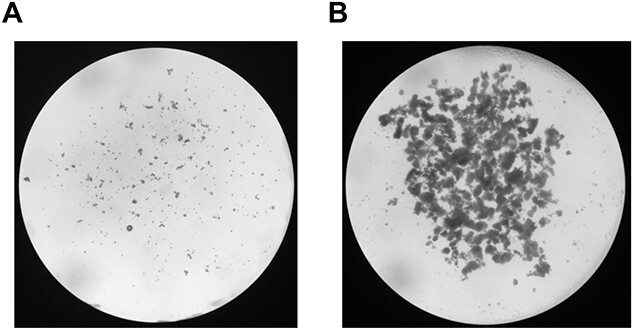
Ovarian tissue broken down after mechanical follicle isolation. (A) Prepubertal ovaries [4] broken down by the flicking method. (B) Adult ovaries [6] broken down by the poking method.
Adult mice
4.2.2 Isolate follicles from the ovaries using two 28 ½ G insulin syringes. In your nondominant hand, use one syringe to pin an ovary down and use the other insulin syringe (dominant hand) to puncture, or “poke”, the ovary repeatedly (Video 2). Follicular fluid and granulosa cells will be released, making the media cloudy. This process also releases various stages of follicles, oocytes, and cumulus oocyte complexes (COCs). Continue until poking fails to release material and the ovary is broken up into small pieces (Figure 4B). For two ovaries, this process should take ~10–20 min.
4.2.3 Follicles can either be collected after mechanical isolation or throughout the isolation process. Follicles of the correct size are moved to an IVF dish with warm DM. It is important to work quickly as follicles will become sticky and reaggregate.
Note: Cloudy DM can be moved to a larger Petri dish, diluted with fresh DM, and scanned for follicles. In addition, insulin needles can be used to tease apart the reaggregated tissue pieces.
4.2.4 Stop processing tissue at or before 45 min have elapsed. This is sufficient time to perform mechanical isolation by either method. With experience, more ovaries can be processed in this time. If the ovaries are not sufficiently processed (very few follicles isolated), the process needs to be stopped at 45 min, and additional mice should be used to obtain more follicles.
Enzymatic isolation
4.3.1 Prepare enzymatic media (EM) in a 35 mm Petri dish as outlined in Table 1 by adding 2.5 mL warmed L15, 25 μL Pen-Strep, 25 μL Liberase (working concentration 25 μg/mL), and 25 μL DNAse I (working concentration 200 μg/mL).
Note: Each dish of EM can be used to digest up to four ovaries, so plan for one dish for every two mice.
4.3.2 Place up to four ovaries in each dish of EM and use a scalpel or insulin syringes to cut each ovary into quarters. Use forceps to stabilize the ovary while pressing down with the scalpel to cut (Video 3).
4.3.3 Place the dish on a heated plate shaker at 37 °C for 15–20 min. Since this media is L-15 based, it should be incubated in an environment that is equivalent to ambient air.
4.3.4 Remove the dish from the shaker and quench the enzymatic reaction with 250 μL fetal bovine serum (FBS). Pipette up and down using a p1000 pipette set to 300–500 μL to mechanically break down the tissue until large pieces of ovaries are dispersed and individual follicles are released (50–100 times per plate) (Video 3).
Note: Be careful not to produce bubbles during this step as it will make selection more difficult.
4.3.5 After pipetting, collect follicles of the appropriate stage and transfer them to the center well of an IVF dish containing warm DM.
4.3.6 Stop isolating at or before the 45 min mark after enzyme quenching.
Recovery period
Following the initial isolation procedure, follicles are allowed to recover briefly at 37 °C, 5% CO2. Poor quality follicles and those damaged during the isolation process will begin to deteriorate during this time, making them easier to exclude during the selection process.
5.1 Collect and transfer follicles from the dish with DM to the center well of an IVF dish with equilibrated MM.
5.2 Return the MM dish containing the follicles to the incubator (37 °C, 5% CO2), and let the follicles recover for a minimum of 1 h but no longer than 3 h.
Follicle selection
6.1 The highest quality follicles are selected for encapsulation and downstream culture. Proper selection is key to a successful experiment because poor quality or damaged follicles will significantly compromise survival and growth trajectories. Follicles of high quality are those in which the oocyte borders are well defined, and the granulosa cell layers are translucent. Poor quality follicles will be more opaque, have oocyte irregularities, damage to the follicle wall, or are asymmetrical (oocyte not in the center of the follicle) (Figure 5).
6.2 Prepare an IVF dish with 1 mL warm DM in the center well and 3 mL in the outer ring.
6.3 Retrieve the IVF dish with follicles from the incubator and transfer the follicles to the dish with DM.
6.4 Examine follicles under the highest magnification possible. High-quality follicles are separated from those of poor quality and moved to designated areas of the IVF dish or a separate dish with DM.
Figure 5.
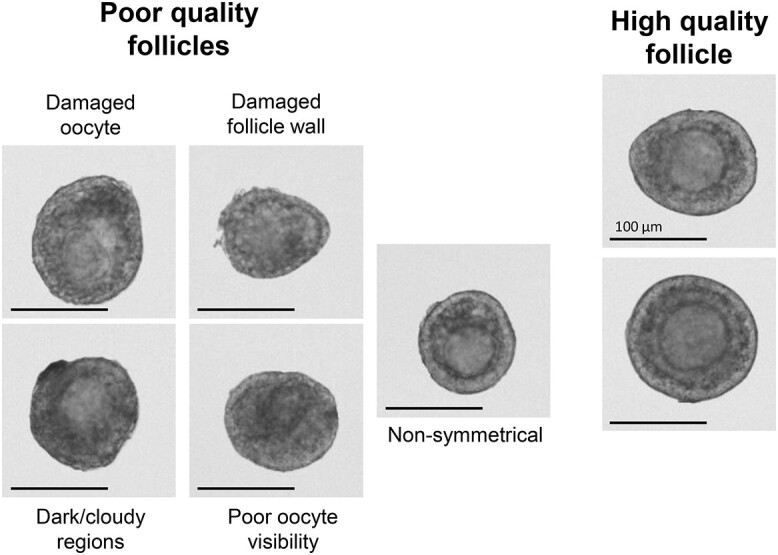
Representative images of poor and high-quality follicles. Only high-quality follicles should be encapsulated and cultured. Follicle features may appear different based on the viewing modality, but general criteria listed will be consistent.
Note: Brightfield and inclined transmitted illumination can accentuate different morphological features and defects of follicles which facilitates the selection process.
Note: After sorting all follicles, a second round of selection can be done on the pool of high-quality follicles to ensure rigor. Typically, approximately 1/3 of isolated follicles are of sufficient quality to be suitable for downstream culture.
Encapsulation
Multilayer secondary follicles are cultured individually (Figure 6A), while murine primary and two-layer early secondary follicles must be cultured in cohorts of 10 (multiple follicle culture) [18] (Figure 6B). Several methods can be used to encapsulate follicles into alginate hydrogels, and here we detail a droplet-based encapsulation method and a parafilm-based encapsulation method (Figure 6C and D). The choice between these methods is based on preference and both can be utilized for individual or multiple follicle culture. The components and preparation instructions for 0.5% alginate and the cross-linking calcium solution are listed in Table 5.
Figure 6.
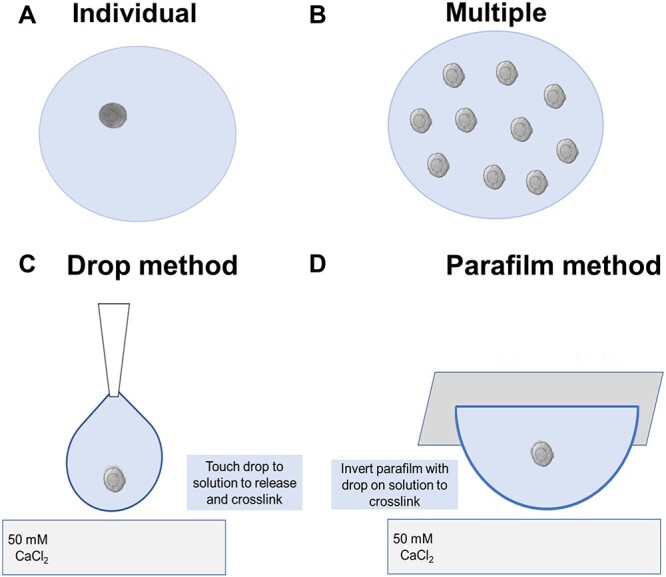
Schematic of encapsulation methods. (A) Individually encapsulating follicles is necessary for larger, multilayer secondary follicles that can grow in absence of paracrine factors from other follicles. (B) Multiple follicle encapsulation (10/bead) is necessary for two-layer secondary or earlier stage follicles that require secretion of paracrine factors from other follicles to grow. (C) The droplet method encapsulates follicles by expelling alginate directly from a pipette tip into the crosslinking solution. (B) The parafilm method encapsulates follicles by placing them in a drop of alginate on a piece of parafilm and inverting the drop onto the surface of the crosslinking solution.
Table 5.
Components and instructions for preparation of 0.5% alginate and calcium solution used for crosslinking
| Component | Catalog no. | Supplier | Final concentration | |
|---|---|---|---|---|
| 0.5% Alginate | Sterile PBS (−/−) Alginic acid sodium salt | 14190-144 71238 | Gibco Sigma Aldrich | Base 0.5% |
| • Typical volume prepared: 20 mL | ||||
| • Shelf life: 1 month at 4 °C | ||||
| • Sterile filter PBS into a 50 mL conical tube and add alginate to the top of the solution. Place on shaker at 4 °C overnight to dissolve. | ||||
| Calcium solution | Sterile MilliQ H2O | Base | ||
| CaCl2 | C5670 | Sigma Aldrich | 50 mM | |
| NaCl | S5886 | Sigma Aldrich | 140 mM | |
| • Typical volume prepared: 500 mL | ||||
| • Shelf life: Indefinite at room temperature | ||||
| • Combine components and mix on a stir plate until dissolved, then sterile filter. | ||||
Note: Encapsulation steps should be done at room temperature to avoid evaporation which will change the osmolarity and increase the concentration of alginate. Turn off any heated surface for this section of the protocol.
Note: Extended time in alginate prior to cross-linking can reduce follicle quality. Move quickly during the alginate wash steps.
Droplet encapsulation method
The droplet encapsulation method consists of a wash step followed by formation of the alginate bead by aspirating alginate and the follicle(s) directly into a low attachment P10 pipette tip. The bead is then crosslinked by touching the pre-formed drop gently to the alginate solution for crosslinking (Video 4).
7.1.1 Fill a 35 mm Petri dish with calcium solution. Take the lid of the 35 mm Petri dish and add two drops of 0.5% alginate to form one ~50 μL wash drop and one ~100 μL encapsulation drop (set up shown at the beginning of Video 4).
Note: A Petri dish lid is used due to the low lip which allows easier manipulation of the follicles through the wash and encapsulation drops.
Note: The volume of wash drops and encapsulation drop do not need to be an exact volume. Higher concentrations of alginate are more difficult to measure precisely with a displacement pipette.
7.1.2 Pick up follicles using a stripper tip and transfer them to the wash drop.
Note: Transfer follicles between IVF dish and alginate drops under the dissection scope to minimize the chance of losing follicles and avoid producing bubbles. When learning this technique, start with transferring just a few follicles at a time, but if using this technique for multiple follicle encapsulation, 10 follicles must be transferred for each encapsulation.
7.1.3 Position follicles in the top 1/3 of the wash drop in terms of depth and move quickly as the follicles will start to sink and will risk sticking to the surface of the plate (Video 4). If the follicle sinks to the plate lid, it will stick and will likely be damaged if removed. If the follicles are too close to the surface of the drop, the surface tension of the drop will be too high and the follicle will be damaged.
7.1.4 Under the dissection scope, use a P10 low retention pipette tip set to 5 μL to aspirate 5 μL containing the desired number of follicles.
Note: To try to position the follicle within the center of the bead, aspirate 1–2 μL of alginate prior to aspiration of a follicle (see schematic in Video 4). If encapsulating multiple follicles in one bead, try to spread them out as much as possible while aspirating by taking up a small amount of alginate between each follicle. If the follicles are too close together within the bead, they may merge together during culture.
7.1.5 Depress the pipette plunger while holding the pipette tip above the calcium solution at a 900 angle to form the drop of alginate, taking care not to expel the drop into the solution. Slowly lower the pre-formed drop and gently touch it to the calcium solution without having the pipette tip touch the liquid. If the tip comes in contact, change it before forming the next bead to prevent premature crosslinking of the alginate within the pipette tip (Video 4).
Note: If working with several follicles at once for individual encapsulation, work quickly to create several beads in sequence.
7.1.6 Allow the bead to crosslink in the alginate solution for 2 min. After 2 min, pick up the bead by scooping underneath it with curved forceps and transfer to warmed DM.
Note: Hold follicles in warmed DM after crosslinking and encapsulate as many follicles as time will allow within a 15 min period. After 15 min, transfer the beads to dish of equilibrated maintenance media in the incubator for recovery. Continue encapsulating with an additional set of follicles retrieved from the incubator.
Parafilm encapsulation method
This encapsulation method involves placing a 5 μL drop of alginate onto a piece of parafilm and then adding follicles to the drop. The parafilm and alginate drop is then flipped into the calcium solution for crosslinking (Figure 6D; Video 5).
7.2.1 Fill two 35 mm Petri dishes with 5 mL of 50 mM calcium solution and DM, respectively. Place one to three100 μL drops of alginate onto the lid of a Petri dish (35–45 mm) to use as wash drops, leaving space for the parafilm (setup shown at beginning of Video 5). The lower lip of the Petri dish lid allows for easier manipulation of the follicles.
Note: The number of wash drops can be increased if a large amount of follicles will be encapsulated since excess media transferred along with follicles to the wash drop can cause alginate crosslinking. Alternatively, the wash bead should be replaced as needed (i.e., when the alginate starts to thicken due to the introduction of media).
7.2.2 Cut parafilm into 0.5–1 cm2 squares. The paper backing can be left on the parafilm. Place one parafilm square, paraffin side up, onto the Petri dish lid and pipette a 5 μL drop of alginate onto the center. This will be the first encapsulation bead.
7.2.3 Pick up the follicle(s) (1 for multilayer secondary and 10 for two-layer early secondary) using a stripper tip and move them through the alginate wash drops. Place the follicle(s) in the top 1/3 of the wash bead in terms of depth to allow for the maximum amount of time before the follicle sinks to the bottom of the drop. Quickly, re-aspirate the follicle(s) before it reaches the bottom of the drop (Video 5).
7.2.4 Move the follicle(s) from the last wash drops to the encapsulation bead. The follicle(s) should be placed into the middle of the encapsulation drop to help ensure it is not too close to any edge.
Note: If encapsulating multiple follicles into a single bead, a slight stirring motion can be done when adding the follicles to the bead to help ensure they are spread out from each other (Video 5).
7.2.5 Pick up the parafilm square with curved forceps and quickly place it bead side down onto the calcium solution (Video 5). The parafilm will float on the solution but ensure that the parafilm is lying flat so that the bead is completely submerged. Allow the bead to crosslink for 2 min. Remove the paraffin square from the calcium solution with forceps and use the edge of a p100 pipette tip to gently move the alginate bead off the parafilm and into the 35 mm dish with DM (Video 5).
7.2.6 Continue this process until all follicles have been encapsulated.
Recovery incubation
To maintain the healthiest possible population of follicles, all follicles must be allowed to recover in the incubator between steps that require them to be outside of the incubator for an extended period of time. Thus, after encapsulation, follicles should be returned to the incubator for a period of 45–60 min before plating.
8.1 Retrieve the 35 mm Petri dish with 3 mL equilibrated MM from the incubator.
8.2 Gently scoop up the alginate beads with curved forceps and move to the MM dish.
8.3 Return the MM to the incubator, and let the encapsulated follicles recover for a minimum of 45 min.
Plating and imaging
While the beads are recovering, set up a 96-well plate for culture by filling central wells (one well/bead) with 100 μL of GM. Do not plate more than 20 beads per plate as a higher number of wells will increase the time it takes to perform imaging and media changes, resulting in temperature and pH fluctuations which will be harmful to the follicles. Add sterile Dulbecco’s phosphate-buffered saline (PBS) to the wells surrounding the media wells to help maintain a humid environment. Allow the media in the plate(s) to equilibrate for a minimum of 1 h in the incubator before adding encapsulated follicles.
9.1 Remove the equilibrated 96-well plate and Petri dish containing the encapsulated follicles from the incubator. Under a dissection scope, use curved forceps to pick up alginate beads and place one bead in each well.
9.2 Allow the plated encapsulated follicles to recover in the incubator for 1 h before imaging.
9.3 Image each follicle using an inverted brightfield microscope at 10–20× magnification. For multiple follicle beads, images at 4× magnification should also be taken. Ensure that each plate is out of the incubator for no more than 15 min at a time in order to prevent temperature and pH stress to the follicles.
Note: For multiple follicle beads, the bead can shift position and rotate during culture. 4× images can capture the whole alginate bead and are therefore useful in determining the spatial location of individual follicles over time.
Note: Keep an eye on the color of the pH indicator within the media (phenol red) during this time and return the plate to the incubator before 15 min if needed.
Culture
Transitioning primary to secondary and two-layer secondary follicles are cultured up to 12–14 days, while multilayer secondary follicles are cultured up to 6–8 days. At this point, follicles can then be used for downstream purposes such as in vitro maturation and ovulation. Follicles can also be cultured for shorter periods depending on the application. Half the media in each well needs to be replaced every 48 h of culture. Replacing only half of the volume ensures important paracrine and autocrine factors which are essential for growth are retained in sufficient amount [18]. To track growth and survival, imaging should be done on the same days as media changes.
Note: Aseptic technique is required.
10.1 Warm and equilibrate GM in the incubator for a minimum of 1 h prior to changing the media.
Note: You will need 50 μL of GM per well for each media change.
10.2 Image individual follicles at 10× magnification. In cultures with multiple follicles encapsulated together, image entire bead at 4× magnification to use as a reference to identify and track each individual follicle throughout culture.
10.3 After imaging, allow plates to recover for at least 45 min in the incubator before changing media.
10.4 Tilt the plate at a 45° angle and lightly tap against the bench so that the encapsulated follicle beads move to one side of the wells. This can be verified under a dissection scope. Aspirate 50 μL of GM per well, being careful to not disturb the alginate bead. Ensure that each plate is not out of the incubator for more than 15 min with the lid off at any given time, though the process of changing media should be able to be completed in under 5 min.
Note: If the media changes are taking longer than 5 min per plate, reduce the number of beads per plate in future cultures.
Note: Conditioned media can be saved and stored at –80 °C for future analysis of secreted factors such as steroid hormones, growth factors, and cytokines [3, 4, 33, 59].
Endpoints
Follicle growth and survival
Tracking follicle growth and survival is essential to assessing the health of cultured follicles. To use follicles for downstream applications, it is important to demonstrate that follicles are growing appropriately. Follicles cultured from the two-layer secondary stage or earlier should reach a terminal size of ~250 μm after 12–14 days. Multilayer secondary follicles should reach a terminal size of 350–400 μm after 6–8 days. Follicle survival should be tracked because low survival in a set of cultured follicles may indicate quality control issues in the process of isolating and culturing follicles. Image follicles every other day (same day as media changes) at 10× magnification. Individual follicles should be tracked to determine survival through the culture. In multiple follicle culture, this is done by using 4× magnification images to match up follicles based on their relative positions to each other. It is important to track individual follicle growth and survival.
Open the image file in ImageJ (https://imagej.nih.gov/ij/download.html), set the scale, and take two measurements of each follicle, one at the widest diameter and one perpendicular to the first measurement (Figure 7). The average of these measurements is designated as the follicle diameter. Typical growth curves for two-layer and multilayer secondary follicles are shown in Figure 7A and B, respectively.
Score each follicle as alive or dead based on morphology by looking for signs of deterioration (granular appearance or uneven borders; see Figure 8A for examples). Follicles that do not increase in size after 4 days should also be counted as dead even if they appear healthy. Representative images of a multiple follicle culture with high and low survival and the corresponding survival curves are shown in Figure 8B and C.
Figure 7.
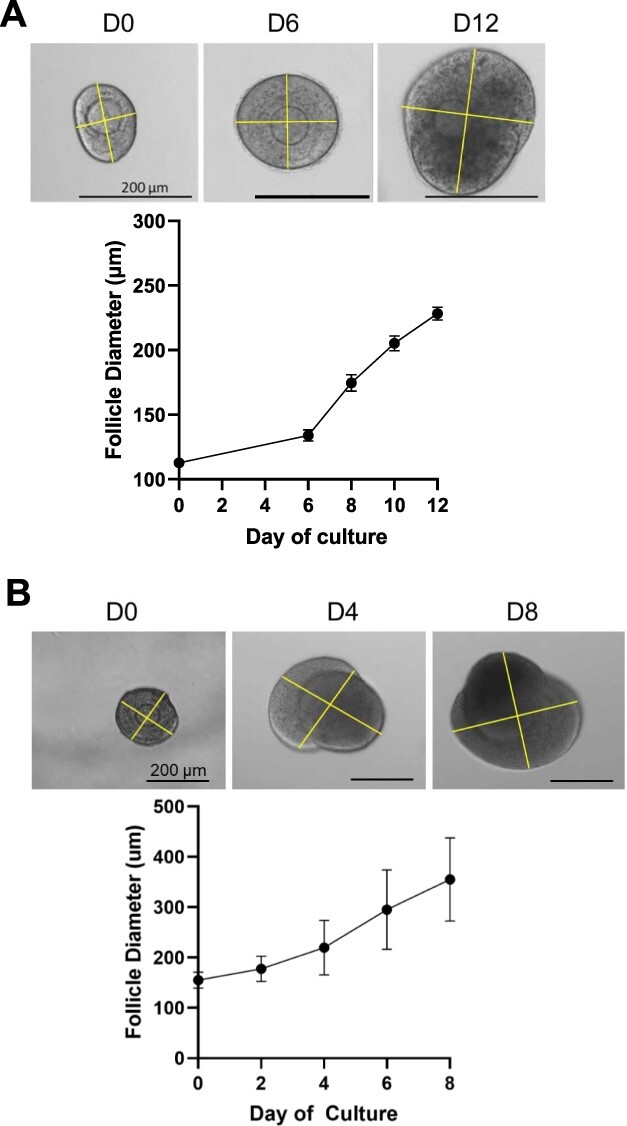
Example growth curves for (A) two-layer secondary follicles and (B) multilayer secondary follicles using perpendicular diameter measurements on ImageJ. Follicle size should be tracked every 48 h throughout the duration of culture.
Figure 8.

Assessment of follicle survival. (A) Examples of dead follicles. (B) Examples of two-layer secondary follicle cultures with high survival (top) and low survival (bottom). High survival follicles are characterized by an even border, continuous growth and translucent granulosa cell layers until the antrum is formed. Low survival follicles characteristics include granular appearance, opaque granulosa cells prior to antrum formation, uneven borders, and low growth rates. (C) Example of survival curves for cultures with high and low survival. (D) Examples of follicles that have a partially extruded oocyte and then recover later in culture. D0, culture day 0; D6, culture day 6; D8, culture day 8; D10, culture day 10.
Note: Some follicles may partially extrude their oocyte but are able to re-enclose it with granulosa cells shortly after to reform a normal follicle structure, continue a normal growth trajectory, and support the development of a high-quality egg. Oocyte extrusion occurs predominately in primary and early secondary follicles, but as long as normal follicle structure is recovered the follicle is considered alive [18]. An example of an extruded oocyte at day 6 (D6) that reforms by day 8 (D8) is shown in Figure 8D.
Note: While visual inspection of quality is routinely used for scoring survival, additional parameters can be assessed to confirm follicle and egg quality. Histology (see below) can be performed to confirm the presence of an antrum. For CD-1 follicles cultured in 0.5% alginate we observe antral formation in 80–100% of multilayer secondary and ~50–80% of early secondary follicles by the end of culture; however, this may differ depending on strain and culture conditions. Furthermore, ovulation (see below) and scoring of oocyte maturation scoring and can also be performed to assess oocyte quality [6].
Removing follicles from alginate
Follicles can be removed from alginate beads at any point during culture using alginate lyase, an enzyme that digests alginate. Several endpoints, including ovulation and histology, require follicles to be removed from alginate.
Prepare and thaw an appropriate volume of alginate lyase (Table 1). Pipette aliquot up and down a few times to mix and warm the vial prior to use.
To digest alginate from individual beads, remove all media from the well with the bead and add 70 μL of alginate lyase.
Alginate beads can also be digested in groups by removing 50% of the media from each well with a bead and taking a wide bore pipette tip to transfer the remaining 50% and the bead into the center well of an IVF dish. Once 10–12 beads are in the center well, carefully remove the media so that only beads remain and add 1 mL of warmed alginate lyase solution.
Place plate or dish with alginate lyase on a warming plate for 25–30 min.
Note: Removing follicles from alginate individually is only necessary if the desired endpoints require tracking growth and ovulation of a specific individual follicle or bead. If the beads are grouped together before treatment with alginate lyase, it will not be possible to track individual follicles from earlier stages in the culture.
Ovulation
Once encapsulated follicles are cultured to their terminal, antral stage they can be used for ovulation assays. Follicles can be removed from alginate and cultured with maturation media (MatM), which is supplemented with human chorionic gonadotropin (hCG—an analog of luteinizing hormone) to induce ovulation [33] as well as epidermal growth factor (EGF) and follicle-stimulating hormone (FSH). Ovulation in this in vitro system is characterized by rupture of the follicle wall and release of a COC. In addition, ovulated follicles can be cultured for an additional 48 h to allow for corpus luteum formation, where the remnants of the ovulated follicle wall luteinize and switch from predominant production of estradiol to production of progesterone.
Remove follicles from alginate as described above.
During the lyase treatment, prepare one IVF dish with 3 mL MatM in the outer ring and 1 mL MatM in the inner ring and warm in the incubator for at least 1 h.
Follicles can either be induced to ovulate individually, with each follicle in one well of a 96-well plate, or in groups of 10–12 in the center well of an IVF dish. If inducing follicles to ovulate individually, prepare a plate with 100 μL of MatM per bead and warm in the incubator. Place PBS in the wells surrounding the MatM to prevent evaporation.
Once the MatM is equilibrated and after the lyase treatment is complete, transfer isolated follicles from the alginate lyase solution one at a time to the outer ring of MatM in the IVF dish. If inducing follicles to ovulate individually, transfer them to an individual well of the 96-well plate with warmed MatM. If inducing ovulation in multiple follicles grouped together, transfer up to 12 follicles into the center well of the IVF dish containing MatM.
Incubate plates or dishes at 37 °C, 5% CO2 for 12–16 h.
Note: 12 h incubation will be the earliest time point of follicle rupture; longer incubations will allow for more complete COC expulsion from the follicle.
After incubation, image follicles and record the incidence of follicular rupture as assessed by the presence of a disrupted follicle wall, release of fluffy appearing cumulus cells, and/or visibility of a COC (Figure 9A and B). Follicles that have successfully ruptured can be cultured for an additional 48 h to assess for corpus luteum formation through measurement of hormone production and histologic analysis of cellular hypertrophy and luteinization (Figure 9C). Oocytes can be removed from COCs by incubation with 1X hyaluronidase solution in L15.
Figure 9.

Example images of an ovulation experiment. (A) Antral follicle prior to adding hCG, (B) follicle rupture after 12 h of culture with hCG, and (C) corpus luteum formation 48 h after ovulation.
Histological examination
Sectioning follicles for histology is useful for many downstream applications that are outside of the scope of this protocol, such as H&E staining, immunohistochemistry, or RNAscope assays. While the process of fixing and preparing follicles for sectioning shares similar elements to general tissue fixation and sectioning protocols, there are several unique steps including removing follicles from alginate before fixation, re-encapsulating follicles after fixation, staining alginate beads to aid in visualization, and encasing beads in biopsy wraps or filter paper before placing in cassettes to avoid losing the bead during processing. Follicles are removed from the alginate bead to facilitate their fixation. Alternatively, follicles can also be fixed in the alginate bead, but this is a complex and time-consuming approach [2]. The re-encapsulation of the follicles and the staining of the bead are necessary to keep the follicles close together and for easy manipulation of the follicles during the processing [3, 4]. As an alternative to alginate, HistoGel can be also used to prepare follicles for sectioning [60, 61].
Remove follicles from alginate beads as described above.
Prepare fixative with 3.8% paraformaldehyde (PFA) and 0.01% Triton X-100 in PBS and place 1 mL in the center well of an IVF dish.
Transfer follicles to the fixative solution, seal the dish with parafilm, and place the dish on a 37 °C heating stage for 1 h.
After fixation, wash follicles in PBS (3×, 5 min each) and re-encapsulate 5–10 follicles per bead (5 μL, 0.5% alginate) using the preferred encapsulation method.
Note: Antral follicles (>200 μm) should be encapsulated in groups of up to 5 follicles/bead. Earlier stage follicles can be grouped in groups of up to 10 follicles/bead.
Once the follicles are encapsulated, briefly stain the bead in an Alcian Blue solution for 15–30 s to allow the alginate but not the follicles to be stained light blue (Figure 10A). This is important for visualization of the bead within paraffin during later stages of processing.
Transfer the stained beads through increasing ethanol concentrations (50%, 60%, 70%; 15 min each) in 35 mm dishes.
Samples can be prepared for histology by folding each bead into biopsy wraps or filter paper before placing into a cassette to prevent the beads from slipping out of holes in the cassette. The cassette with the wrapped bead is then ready for clearing, embedding, and sectioning. An image of an embedded, stained bead is shown in Figure 10B.
Serial section the paraffin-embedded beads. Typically, every fourth section will be stained with hematoxylin and eosin (H&E) to determine follicle position (Figure 10C).
Figure 10.

Preparation of follicles for histology. (A) Follicles are re-encapsulated in alginate beads and stained with Alcian blue after fixing with 3.8% PFA. The stained beads can then be dehydrated and processed for embedding. (B) Blue-stained bead should be visible within the paraffin after embedding. (C) H&E-stained section containing follicles.
Note: Alternatively, sections can be examined under brightfield microscopy to help determine which sections contain follicles.
Troubleshooting
Low yield of high-quality follicles
Follicle yield varies based on mouse age and strain. For prepubertal (postnatal day 12–16) CD-1 mice, total follicle yield is generally 40–60 for both early and multilayer secondary follicles. For reproductively young (4–12 weeks) CD-1 mice, yield is ~20–30 early secondary follicles and 10–20 multilayer secondary follicles per mouse. It is essential to have a sufficient total yield, since only a fraction (~1/3) will be of high quality for culture. If yield is lower than this, first ensure mice are healthy and at an appropriate age for experimental goals and desired yield. Additionally, start by working with a smaller number of mice [1–2] to have more time to select follicles and process tissue without extending the time follicles are outside the incubator. Ensure that follicles are also not over digested should enzymatic isolation be performed (>30 min in enzymatic media before quenching) or damaged due to excessive mechanical isolation. If follicles appear to diminish in quality during recovery incubations, they may have been out of the incubator for too long during earlier stages of processing.
Low survival
Low viability is defined as survival <80% for each culture. To meet this benchmark, ensure that only the highest quality follicles are encapsulated and cultured. Additionally, minimize the time follicles spend out of the incubator during isolation, selection, encapsulation, imaging, and media changes. Ideally, follicles should be cultured in the back of an incubator designated for long-term follicle culture to reduce the number of times the incubator door is opened. If possible, a separate incubator should be used for media equilibration and recovery incubations throughout the isolation protocol. When follicles are taken out of the incubator for media changes and imaging, ensure there is time for recovery between steps. Keep an eye on the pH indicator in the media during these steps. If the media begins to turn pink, return the plate to the incubator for recovery. After ensuring the issue is not due to improper handling, check to make sure that the media components have appropriate activity to sustain follicle growth. Different suppliers and production lots of media components, particularly recombinant follicle-stimulating hormone (rFSH) and FBS, have different levels of bioactivity and may have variable efficacy in follicle culture. This may require troubleshooting between different suppliers. The reagents listed in Table 1 have been tested for efficacy in eIVFG.
Most of the published studies utilizing eIVFG have used rFSH sourced from Organon, which is no longer commercially available. Our lab has preliminarily tested a number of commercial and pharmacological-sourced rFSH for use in eIVFG and found that Gonal-F (follitropin alfa for injection) has a specific and working (10 mIU/mL [3–8, 11, 14–19, 21, 22, 24, 25, 27–31, 33, 34, 47–49] activity similar to the rFSH previously available through Organon). Gonal-F can be obtained in a reconstituted form (injection pen; 600 IU/mL) or as a lyophilized powder. If feasible, we recommend obtaining pharmacological grade rFSH (Gonal-F, Follistim) as these have been tested for their in vivo activity against the World Health Organization’s first International Standard for human rFSH. These formulations are stable for multiple years at 4 °C, but freezing results in loss of FSH activity. If purchasing commercial rFSH, we recommend testing it by eIVFG since the long-term nature of the culture is the most sensitive means of determining FSH in vitro activity.
Reduced ovulation
Ovulation rate should be >80% for each culture. If the rate is below 80%, first ensure that follicles are cultured for the appropriate number of days based on starting stage, to a terminal diameter of 240–260 μm for early secondary culture and 350–400 μm for multilayer secondary culture. Early secondary follicles typically require 12–14 days in culture, while multilayer secondary follicles require 6–8 days. If follicles are not cultured for long enough or if they are cultured for too long they will not ovulate as effectively. If the rate is still below 80%, ensure media components, particularly hCG and FBS, have appropriate activity to sustain follicle growth and ovulation. This may require troubleshooting between different suppliers. The reagents listed in Table 1 have been tested for efficacy in follicle culture.
Conclusions
Folliculogenesis and oogenesis are complex processes that are essential for fertility and endocrine function. Follicle activation, follicle growth, oocyte development, and ovulation are all necessary steps for fertilization to occur. In vitro follicle culture recapitulates these key stages of in vivo folliculogenesis and oogenesis and thus represents a powerful assay for the study of these processes in an isolated system.
Though there are several follicle culture strategies that have been used to recapitulate follicle growth, alginate is a nondegradable matrix that allows follicles to maintain an in vivo-like architecture. Specific elements of the protocol, from age of mice to encapsulation techniques, can be tailored based on the questions that are central to a particular study. The protocol described in this paper provides a versatile road map for designing experiments to answer basic questions about the biology of folliculogenesis and ovulation, screen compounds for ovotoxicity, or study the production of secreted factors from the ovarian follicle at different stages of development.
Data availability
All original data in this publication are available upon request to the corresponding author.
Authors’ contributions
AC and EJZ wrote the manuscript. FA assisted with writing and editing of the manuscript. FD took part in writing and editing of the manuscript. All authors approved the final submission.
Disclosures
The authors declare that they have no financial or other competing interests. Figure 1 was created using Biorender.
Supplementary Material
Acknowledgments
The authors would like to thank Dr Joan Jorgensen, Dr Jane Fenelon, and all other members of the SSR Virtual Education Committee for allowing this work to be presented as part of the SSR Experimental Methods and Techniques in Reproduction Webinar Series in a talk entitled “Recapitulating folliculogenesis and oogenesis outside the body: encapsulated in vitro follicle growth.” We would also like to thank Caroline Kratka and Teresa Chou for their assistance in testing the protocol for clarity. Finally, we would like to thank Dr. Teresa Woodruff and all past Woodruff lab members and collaborators who pioneered and optimized this methodology over the past two decades.
Footnotes
† Grant Support: This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD103384) and the Bill & Melinda Gates Foundation (INV-003385). Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission.
Contributor Information
Aubrey Converse, Department of Obstetrics and Gynecology, Feinberg School of Medicine, Northwestern University, Chicago, Illnois, USA.
Emily J Zaniker, Department of Obstetrics and Gynecology, Feinberg School of Medicine, Northwestern University, Chicago, Illnois, USA.
Farners Amargant, Department of Obstetrics and Gynecology, Feinberg School of Medicine, Northwestern University, Chicago, Illnois, USA.
Francesca E Duncan, Department of Obstetrics and Gynecology, Feinberg School of Medicine, Northwestern University, Chicago, Illnois, USA.
References
- 1. Simon LE, Kumar TR, Duncan FE. In vitro ovarian follicle growth: a comprehensive analysis of key protocol variables. Biol Reprod 2020; 103:455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pangas SA, Saudye H, Shea LD, Woodruff TK. Novel approach for the three-dimensional culture of granulosa cell-oocyte complexes. Tissue Eng 2003; 9:1013–1021. [DOI] [PubMed] [Google Scholar]
- 3. Kreeger PK, Fernandes NN, Woodruff TK, Shea LD. Regulation of mouse follicle development by follicle-stimulating hormone in a three-dimensional in vitro culture system is dependent on follicle stage and dose. Biol Reprod 2005; 73:942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kreeger PK, Deck JW, Woodruff TK, Shea LD. The in vitro regulation of ovarian follicle development using alginate-extracellular matrix gels. Biomaterials 2006; 27:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu M, West E, Shea LD, Woodruff TK. Identification of a stage-specific permissive in vitro culture environment for follicle growth and oocyte development. Biol Reprod 2006; 75:916–923. [DOI] [PubMed] [Google Scholar]
- 6. Xu M, Kreeger PK, Shea LD, Woodruff TK. Tissue-engineered follicles produce live. Fertile Offspring Tissue Eng 2006; 12:2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. West ER, Xu M, Woodruff TK, Shea LD. Physical properties of alginate hydrogels and their effects on in vitro follicle development. Biomaterials 2007; 28:4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu M, Banc A, Woodruff TK, Shea LD. Secondary follicle growth and oocyte maturation by culture in alginate hydrogel following cryopreservation of the ovary or individual follicles. Biotechnol Bioeng 2009; 103:378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu M, West-Farrell ER, Stouffer RL, Shea LD, Woodruff TK, Zelinski MB. Encapsulated three-dimensional culture supports development of nonhuman primate secondary follicles. Biol Reprod 2009; 81:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu M, Barrett SL, West-Farrell E, Kondapalli LA, Kiesewetter SE, Shea LD, Woodruff TK. In vitro grown human ovarian follicles from cancer patients support oocyte growth. Hum Reprod 2009; 24:2531–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin SY, Lei L, Shikanov A, Shea LD, Woodruff TK. A novel two-step strategy for in vitro culture of early-stage ovarian follicles in the mouse. Fertil Steril 2010; 93:2633–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barrett SL, Shea LD, Woodruff TK. Noninvasive index of cryorecovery and growth potential for human follicles in vitro. Biol Reprod 2010; 82:1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu M, Fazleabas AT, Shikanov A, Jackson E, Barrett SL, Hirshfeld-Cytron J, Kiesewetter SE, Shea LD, Woodruff TK. In vitro oocyte maturation and preantral follicle culture from the luteal-phase baboon ovary produce mature oocytes. Biol Reprod 2011; 84:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tingen CM, Kiesewetter SE, Jozefik J, Thomas C, Tagler D, Shea L, Woodruff TK. A macrophage and theca cell-enriched stromal cell population influences growth and survival of immature murine follicles in vitro. Reproduction 2011; 141:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parrish EM, Siletz A, Xu M, Woodruff TK, Shea LD. Gene expression in mouse ovarian follicle development in vivo versus an ex vivo alginate culture system. Reproduction 2011; 142:309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirshfeld-Cytron JE, Duncan FE, Xu M, Jozefik JK, Shea LD, Woodruff TK. Animal age, weight and estrus cycle stage impact the quality of in vitro grown follicles. Hum Reprod 2011; 26:2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tagler D, Tu T, Smith RM, Anderson NR, Tingen CM, Woodruff TK, Shea LD. Embryonic fibroblasts enable the culture of primary ovarian follicles within alginate hydrogels. Tissue Eng Pt A 2012; 18:1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornick JE, Duncan FE, Shea LD, Woodruff TK. Multiple follicle culture supports primary follicle growth through paracrine-acting signals. Reproduction 2013; 145:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skory RM, Bernabé BP, Galdones E, Broadbelt LJ, Shea LD, Woodruff TK. Microarray analysis identifies COMP as the most differentially regulated transcript throughout in vitro follicle growth. Mol Reprod Dev 2013; 80:132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiao ZX, Woodruff TK. Follicle microenvironment-associated alterations in gene expression in the mouse oocyte and its polar body. Fertil Steril 2013; 99:1453–1459.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahn RW, Barrett SL, Raja MR, Jozefik JK, Spaho L, Chen H, Bally MB, Mazar AP, Avram MJ, Winter JN, Gordon LI, Shea LD et al. Nano-encapsulation of arsenic trioxide enhances efficacy against murine lymphoma model while minimizing its impact on ovarian reserve in vitro and in vivo. Plos One 2013; 8:e58491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tagler D, Makanji Y, Anderson NR, Woodruff TK, Shea LD. Supplemented αMEM/F12-based medium enables the survival and growth of primary ovarian follicles encapsulated in alginate hydrogels. Biotechnol Bioeng 2013; 110:3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu J, Xu M, Bernuci MP, Fisher TE, Shea LD, Woodruff TK, Zelinski MB, Stouffer RL. Oocyte biology in fertility preservation. Adv Exp Med Biol 2013; 761:43–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tagler D, Makanji Y, Tu T, Bernabé BP, Lee R, Zhu J, Kniazeva E, Hornick JE, Woodruff TK, Shea LD. Promoting extracellular matrix remodeling via ascorbic acid enhances the survival of primary ovarian follicles encapsulated in alginate hydrogels. Biotechnol Bioeng 2014; 111:1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Makanji Y, Tagler D, Pahnke J, Shea LD, Woodruff TK. Hypoxia-mediated carbohydrate metabolism and transport promote early-stage murine follicle growth and survival. Am J Physiol Endocrinol Metab 2014; 306:E893–E903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silva GM, Rossetto R, Chaves RN, Duarte ABG, Araújo VR, Feltrin C, Bernuci MP, Anselmo-Franci JA, Xu M, Woodruff TK, Campello CC, Figueiredo JR. In vitro development of secondary follicles from pre-pubertal and adult goats cultured in two-dimensional or three-dimensional systems. Zygote 2015; 23:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu Y, Duncan FE, Xu M, Woodruff TK. Use of an organotypic mammalian in vitro follicle growth assay to facilitate female reproductive toxicity screening. Reprod Fertil Dev 2015; 28:1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiao S, Zhang J, Romero MM, Smith KN, Shea LD, Woodruff TK. In vitro follicle growth supports human oocyte meiotic maturation. Sci Rep 2015; 5:17323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao S, Duncan FE, Bai L, Nguyen CT, Shea LD, Woodruff TK. Size-specific follicle selection improves mouse oocyte reproductive outcomes. Reproduction 2015; 150:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lebbe M, Taylor AE, Visser JA, Kirkman-Brown JC, Woodruff TK, Arlt W. The steroid metabolome in the isolated ovarian follicle and its response to androgen exposure and antagonism. Endocrinology 2017; 158:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao S, Zhang J, Liu M, Iwahata H, Rogers HB, Woodruff TK. Doxorubicin has dose-dependent toxicity on mouse ovarian follicle development, hormone secretion, and oocyte maturation. Toxicol Sci 2017; 157:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu J, Wang Y, Kauffman AE, Zhang Y, Li Y, Zhu J, Maratea K, Fabre K, Zhang Q, Woodruff TK, Xiao S. A tiered female ovarian toxicity screening identifies toxic effects of checkpoint kinase 1 inhibitors on murine growing follicles. Toxicol Sci 2020; 177:405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Skory RM, Xu Y, Shea LD, Woodruff TK. Engineering the ovarian cycle using in vitro follicle culture. Hum Reprod 2015; 30:1386–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hornick JE, Duncan FE, Shea LD, Woodruff TK. Isolated primate primordial follicles require a rigid physical environment to survive and grow in vitro. Hum Reprod 2012; 27:1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Amsden B, Turner N. Diffusion characteristics of calcium alginate gels. Biotechnol Bioeng 1999; 65:605–610. [DOI] [PubMed] [Google Scholar]
- 36. Rowley JA, Madlambayan G, Mooney DJ. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 1999; 20:45–53. [DOI] [PubMed] [Google Scholar]
- 37. Rowley JE, Amargant F, Zhou LT, Galligos A, Simon LE, Pritchard MT, Duncan FE. Low molecular weight hyaluronan induces an inflammatory response in ovarian stromal cells and impairs gamete development in vitro. Int J Mol Sci 2020; 21:1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang K, Zhangb J, Huang Y, Wang Y, Xiao S, Hadden MK, Woodruff TK, Sun J. A platform utilizing Drosophila ovulation for nonhormonal contraceptive screening. Proc Natl Acad Sci USA 2021; 118:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heise M, Koepsel R, Russell AJ, McGee EA. Calcium alginate microencapsulation of ovarian follicles impacts FSH delivery and follicle morphology. Reprod Biol Endocrinol 2005; 3:47–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Songsasen N, Woodruff TK, Wildt DE. In vitro growth and steroidogenesis of dog follicles are influenced by the physical and hormonal microenvironment. Reproduction 2011; 142:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brito I, Silva G, Sales A, Lobo C, Rodrigues G, Sousa R, Moura A, Calderón C, Bertolini M, Campello C, Smitz J, Figueiredo J. Fibrin–alginate hydrogel supports steroidogenesis, in vitro maturation of oocytes and parthenotes production from caprine preantral follicles cultured in group. Reprod Domest Anim 2016; 51:997–1009. [DOI] [PubMed] [Google Scholar]
- 42. Brito IR, Silva CMG, Duarte ABG, Lima IMT, Rodrigues GQ, Rossetto R, Sales AD, Lobo CH, Bernuci MP, Rosa-e-Silva ACJS, Campello CC, Xu M et al. Alginate hydrogel matrix stiffness influences the in vitro development of caprine preantral follicles. Mol Reprod Dev 2014; 81:636–645. [DOI] [PubMed] [Google Scholar]
- 43. Rios PD, Kniazeva E, Lee HC, Xiao S, Oakes RS, Saito E, Jeruss JS, Shikanov A, Woodruff TK, Shea LD. Retrievable hydrogels for ovarian follicle transplantation and oocyte collection. Biotechnol Bioeng 2018; 115:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amorim CA, Langendonckt AV, David A, Dolmans MM, Donnez J. Survival of human pre-antral follicles after cryopreservation of ovarian tissue, follicular isolation and in vitro culture in a calcium alginate matrix. Hum Reprod 2009; 24:92–99. [DOI] [PubMed] [Google Scholar]
- 45. Wang Y, Liu M, Zhang J, Liu Y, Kopp M, Zheng W, Xiao S. Multidrug resistance protein 1 deficiency promotes doxorubicin-induced ovarian toxicity in female mice. Toxicol Sci 2018; 163:279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y, Xu J, Stanley JE, Xu M, Brooks BW, Scott GI, Chatterjee S, Zhang Q, Zelinski MB, Xiao S. A closed vitrification system enables a murine ovarian follicle bank for high-throughput ovotoxicity screening, which identifies endocrine disrupting activity of microcystins. Reprod Toxicol 2020; 93:118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shikanov A, Xu M, Woodruff TK, Shea LD. A method for ovarian follicle encapsulation and culture in a proteolytically degradable 3 dimensional system. J Vis Exp 2011; 49:2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shikanov A, Xu M, Woodruff TK, Shea LD. Interpenetrating fibrin–alginate matrices for in vitro ovarian follicle development. Biomaterials 2009; 30:5476–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shikanov A, Smith RM, Xu M, Woodruff TK, Shea LD. Hydrogel network design using multifunctional macromers to coordinate tissue maturation in ovarian follicle culture. Biomaterials 2011; 32:2524–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choi JH, Yoo CR, Ahn JY, Park JH, Lim JM. Growth of ovarian primary follicles retrieved from neonates of different ages and derivation of mature oocytes following in vitro-culture. Asian Austral J Anim 2012; 25:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vanacker J, Amorim CA. Alginate: a versatile biomaterial to encapsulate isolated ovarian follicles. Ann Biomed Eng 2017; 45:1633–1649. [DOI] [PubMed] [Google Scholar]
- 52. Brito IR, Lima IMT, Xu M, Shea LD, Woodruff TK, Figueiredo JR. Three-dimensional systems for in vitro follicular culture: overview of alginate-based matrices. Reprod Fertil Dev 2014; 26:915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kalab P, Kopf GS, Schultz RM. Modifications of the mouse zona pellucida during oocyte maturation and egg activation: effects of Newborn calf serum and Fetuin. Biol Reprod 1991; 45:783–787. [DOI] [PubMed] [Google Scholar]
- 54. Kalab P, Schultz RM, Kopf GS. Modifications of the mouse zona pellucida during oocyte maturation: inhibitory effects of follicular fluid, fetuin, and α2HS-glycoprotein. Biol Reprod 1993; 49:561–567. [DOI] [PubMed] [Google Scholar]
- 55. Schroeder AC, Schultz RM, Kopf GS, Taylor FR, Becker RB, Eppig JJ. Fetuin inhibits zona pellucida hardening and conversion of ZP2 to ZPZ during spontaneous mouse oocyte maturation in vitro in the absence of serum. Biol Reprod 1990; 43:891–897. [DOI] [PubMed] [Google Scholar]
- 56. Eppig JJ, O’Brien M, Wigglesworth K. Mammalian oocyte growth and development in vitro. Mol Reprod Dev 1996; 44:260–273. [DOI] [PubMed] [Google Scholar]
- 57. Eppig JJ, Wigglesworth K, O’Brien MJ. Comparison of embryonic developmental competence of mouse oocytes grown with and without serum. Mol Reprod Dev 1992; 32:33–40. [DOI] [PubMed] [Google Scholar]
- 58. Briley SM, Jasti S, McCracken JM, Hornick JE, Fegley B, Pritchard MT, Duncan FE. Reproductive age-associated fibrosis in the stroma of the mammalian ovary. Reproduction 2016; 152:245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou H, Decker JT, Lemke MM, Tomaszweski CE, Shea LD, Arnold KB, Shikanov A. Synergy of paracrine Signaling during early-stage mouse ovarian follicle development in vitro. Cell Mol Bioeng 2018; 11:435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bus A, van Hoech V, Langbeen A, Leroy JLRM, Bols PEJ. Effects of vitrification on the viability of alginate encapsulated isolated bovine pre-antral follicles. J Assist Reprod Genet 2018; 35:1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim J, Perez A, Claflin J, David A, Zhou H, Shikanov A. Synthetic hydrogel supports the function and regeneration of artificial ovarian tissue in mice. Npj. Regen Med 2016; 1:16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data in this publication are available upon request to the corresponding author.