

Abstract
Membrane permeability plays an important role in oral drug absorption. Caco-2 and Madin-Darby Canine Kidney (MDCK) cell culture systems have been widely used for assessing intestinal permeability. Since most drugs are absorbed passively, Parallel Artificial Membrane Permeability Assay (PAMPA) has gained popularity as a low-cost and high-throughput method in early drug discovery when compared to high-cost, labor intensive cell-based assays. At the National Center for Advancing Translational Sciences (NCATS), PAMPA pH 5 is employed as one of the Tier I ADME assays. In this study, we have developed a quantitative structure activity relationship (QSAR) model using our ~6500 compound PAMPA pH 5 permeability dataset. Along with ensemble decision tree-based methods such as Random Forest and eXtreme Gradient Boosting, we employed deep neural network and a graph convolutional neural network to model PAMPA pH 5 permeability. The classification models trained on a balanced training set provided accuracies ranging from 71% to 78% on the external set. Additionally, an ~85% correlation was determined between PAMPA pH 5 permeability and in vivo oral bioavailability in mice and rats. These results suggest that data from this assay (experimental or predicted) can be used to rank-order compounds for preclinical in vivo testing with a high degree of confidence. Additionally, experimental data for 486 compounds (PubChem AID: 1645871) and the best models have been made publicly available (https://opendata.ncats.nih.gov/adme/).
Keywords: quantitative structure activity relationship, PAMPA, ADME, oral bioavailability, machine learning, in silico models
Graphical Abstract
Introduction
Absorption is a critical property for orally administrated drugs, as the drug must pass through the intestinal epithelium before reaching systemic circulation. Absorption is not only dependent upon the characteristics of the gastrointestinal (GI) tract, but also on the physicochemical properties of the drug1-3. Several studies have shown that absorption of drugs is regional 4,5 and the pH gradient in the intestinal tract (from acidic, i.e., pH 2-3 to basic i.e., pH 8-9) has been attributed as a major influencing factor6,7. For example, most intestinal absorption occurs in the small intestine (the duodenum, jejunum, and ileum specifically) where the pH ranges from 4-78. This phenomenon is explained by the pH-partition hypothesis which states that only uncharged compounds can permeate through the lipophilic membrane9. This suggests that the permeability of a compound would vary across different segments of the GI tract depending on the pH and permeability of a compound would be greatest at the pH where its least charged.
Two of the most popular cellular in vitro membrane permeability methods used in drug discovery include Caco-2 and Madin-Darby Canine Kidney (MDCK) monolayer assays. Caco-2 is a human epithelial colon adenocarcinoma cell line which presents both enterocytic and colonocytic10 characteristics. Additionally, data generated in these cells exhibit good correlation with in vivo bioavailability11 and permeability like that of the human jejunum12. MDCK cells, isolated from canine distal renal tissue, came in later as a faster and more cost-effective alternative to Caco-2 cells13. While these cell lines model active and passive transport, their use is often limited due to high costs, long membrane growth cycles (21-day and 5-day culture time for Caco- 2 and MDCK cells respectively), and lab-to-lab and batch-to-batch variation14-16. In addition to cell-based assays, Parallel Artificial Membrane Permeability Assay (PAMPA) is also popular as a cost effective, non-cell-based screening tool17-19. One of the major disadvantages of PAMPA (i.e., its inability to model active transport 20,18) is offset by the fact that more than 90% of drugs are absorbed via passive diffusion 21,2,22,17,20. PAMPA permeability at both pH 7.4 and pH 5.5 correlates well with Caco-2 permeability in small data sets 23. The adaptability of PAMPA to high-throughput in combination with its flexibility with experimental conditions (different lipid compositions/range of pH conditions) makes PAMPA an excellent screening method in early drug discovery. This technique has been extensively used in several published drug discovery projects with a great deal of success 24-30. At the National Center for Advancing Translational Sciences (NCATS), compounds are routinely screened for permeability using a high-throughput, double-sink PAMPA assay at pH 5 and pH 7.4, part of the ADME Tier I assays. On examining the correlation between PAMPA pH 5 permeability and preclinical oral bioavailability using in-house pharmacokinetic (PK) datasets, a correlation of ~85% was determined, further underlining the importance of this assay as a screening tool in drug discovery.
The current cost of bringing a new drug from drug discovery through to the market stands at $2.6 billion USD 31. This cost has risen steadily throughout the last few decades, making it critical to find alternatives to reduce costs in the drug discovery process. Quantitative structure activity relationships (QSAR) using machine learning approaches, a branch of artificial intelligence (AI), has been shown to improve the decision-making process across various steps in drug discovery, including its use in predicting PAMPA permeability. While a few PAMPA QSAR models do exist, they are primarily based on small datasets and in most cases, neither the data nor the models are made publicly available 1,2,32,33,19. In this study, we present an in silico model for predicting drug permeability at pH 5 based on experimental PAMPA data collected at NCATS, a complement to our previously published PAMPA pH 7.4 model 18. The PAMPA pH 5 model features a dataset of ~6500 compounds, representing a variety of small molecule drug discovery projects and chemotypes. We employed both classical and advanced machine learning techniques to develop the prediction models. The best model with both training and validation accuracies over 75% was made available on the publicly-accessible NCATS ADME portal (https://opendata.ncats.nih.gov/adme/). which can be useful in rank-ordering virtual compounds for their potential behavior in PAMPA pH 5 assay and identifying compounds with poor permeability profile.
Materials & Methods
Materials.
Dimethyl sulfoxide (DMSO, UPLC/MS grade), ammonium acetate, sodium hydroxide, ranitidine, dexamethasone, verapamil, and albendazole were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN, UPLC/MS grade) was purchased from Fisher Scientific (Hampton, NH). GIT-0 lipid (Catalog #110669), acceptor sink buffer (pH 7.4, Catalog #110139), PRISMA HT buffer (Catalog #110151), 96-well stirwell sandwich plates with stirrers (Catalog #120551-SUPP), and high sensitivity UV plates (Catalog #110286) were purchased from Pion Inc. (Billerica, MA).
Instrumentation.
Experiments were performed using a Freedom Evo 200 automated platform with a 96-channel (MCA96) head and 8-channel liquid handling (LiHa) system with EVOware software (version 3.2) (Tecan Inc., Männedorf, Switzerland). The system also includes a Gutbox (Pion Inc.) and a Nano Quant Infinite 200 Pro UV plate reader (Tecan Inc.). 200 μL pipette tips (MCA96: Catalog #14-223-552, Fisher Scientific, Hampton, NH; LiHa: Catalog #110126, Pion Inc., Billerica, MA) were used in the experiments.
PAMPA Permeability pH 5 Method:
Stirring double-sink PAMPA method (patented by Pion Inc.) was employed to determine the permeability of compounds in a high-throughput format 17 The GIT-0 lipid (proprietary Pion Inc. lipid, optimized to predict GI tract passive permeability) was immobilized on the plastic matrix of a 96-well “acceptor” filter plate placed atop a 96-well “donor” plate. pH 5 buffer (PRISMA HT buffer) was used in the donor wells and pH 7.4 buffer (acceptor sink buffer) was used in the acceptor wells. The test articles (in duplicates), stocked in 10 mM DMSO solutions, were diluted to 0.05 mM in aqueous buffer (pH 5), and the concentration of DMSO was 0.5% in the final solution. During the 30-minute incubation at room temperature, test samples in the donor compartment were stirred using Gutbox technology (Pion Inc.) to reduce the aqueous boundary layer. The test article concentrations in the donor and acceptor compartments were measured using a UV plate reader (Nano Quant, Infinite 200 PRO, Tecan Inc., Männedorf, Switzerland). Calculations were performed using Pion Inc. software and effective permeability (Peff) was expressed in units of 10−6cm/s. If the permeability could not be determined via UV, the samples were plated for analysis via UPLC-MS by plating 8 μL of the incubation solutions in 192 μL of Acetonitrile/Internal Standard (albendazole) solution in a 96-well plate (350 μL, Waters, Milford, MA). A previously published ultra-high-performance liquid chromatography- mass spectrometry method with minor modifications was used to analyze the samples 34
Compound Data Sets:
A total of 6500 measurements were available in the PAMPA pH 5 assay. These compounds were synthesized at NCATS and they represent a variety of small molecule drug discovery projects and chemotypes. Compounds were categorized with the following cutoffs: low permeability: <10 X 10−6cm/s and moderate/high permeability: >10 X 10−6cm/s. For the model, compound structures were standardized following best practices recommended in the literature 35. LyChI hash identifiers (https://github.com/ncats/lychi) were generated for all standardized structures to group them into unique compounds. For compounds with multiple measurements, the values were averaged if all values fell within the same category and compounds with conflicting experimental results were omitted. Finally, the processed data set comprised a total of 5227 unique compounds. The data set was randomly divided into a training set (80%; 4181 compounds labelled as Training set I) used to build the models and an external set (20%; 1046 compounds) used to validate the models. 486 out of 5277 compounds were identified as open access compounds and PAMPA pH 5 data for these compounds has been deposited in PubChem (AID: 1645871) as part of this study. The remaining 4741 compounds were identified as part of on-going projects at NCATS and data for these compounds will be released at some point in the future.
Due to imbalance in the distribution of training set compounds between the two classes (low permeability and moderate/high permeability), we decided to generate a balanced training set using the diversity under-sampling method 36. Retaining all minority class (i.e., low permeability) compounds, a structurally diverse set of majority class compounds that is double the size of minority class was obtained using RDKit Diversity Picker node in KNIME. Using this technique, a total of 1698 compounds were used as a balanced dataset (not to be confused with a perfectly balanced dataset; labelled as Training set II) to generate the same set of models. The same external set was employed for validating these models to mimic the imbalanced nature of the data in a realistic setting. An overview of the training and external data sets is provided in Table 1.
Table 1.
Overview of data sets employed for developing models in this study.
| Dataset | Total Compounds |
Class = 1 (Low Permeability) |
Class = 0 (Moderate to High Permeability) |
|---|---|---|---|
| Training Set I | 4181 | 566 | 3615 |
| Training Set II | 1698 | 566 | 1132 |
| External Set | 1046 | 141 | 905 |
Molecular Descriptors:
We used molecular descriptors available from the RDKit toolkit (https://www.rdkit.org/; version 2020.03.1) as one of the input features. Each compound in the data set is represented by a total of 119 RDKit descriptors. In addition, molecular fingerprints (bit vector representations of molecules) that encode substructural features were employed as input features. Morgan fingerprint 37, a circular molecular fingerprint that takes into account the neighborhood of individual atoms, was chosen for this study. Each fingerprint contains a total of 1024 bits, each bit set to either 1 or 0. On the other hand, molecular graphs as such also serve as input features for one of the modeling methods employed in this study.
Modeling Methods:
Random Forest.
A random forest (RF) 38 is an ensemble of several decision trees that are fitted on random subsets of input features of the data set. The outcome is decided via a majority vote on the outcomes from the individual trees in the forest. Using this averaging approach, RF is robust to overfitting and thereby improves prediction accuracy. We used the ‘RandomForestClassifier’ method implemented in Scikit-learn39, a Python library for machine learning. In this study we employed a total of 100 estimators (i.e., individual trees) per model. The ‘random state’ parameter was set to an integer (random state = 42). The remaining parameters were set to default. We built RF models based on both RDKit descriptors and Morgan fingerprints.
XGBoost.
eXtreme Gradient Boosting (XGBoost) is another method evaluated in this study. While RF builds a set of independent trees of unlimited depth, the gradient boosting technique builds a series of smaller trees where each tree corrects for the residuals in previous tree’s predictions. First implemented as Generalized Boosted Models (GBM), the method was considered to perform similarly to RF although the high number of adjustable parameters has limited its applicability on large datasets. Later, Chen and Guestrin implemented XGBoost 40 (https://github.com/dmlc/xgboost) that is based on the same idea behind GBM but uses an additive strategy to generate the prediction output. Furthermore, XGBoost uses a split-finding approach that can efficiently train on sparse data. This approach is particularly best suited when using sparse molecular representations such as fingerprints that contain many zeros. Due to its speed and widely recognized performance, we employed the XGBoost method using the same number of trees as RF. Both RDKit descriptors and Morgan fingerprints were employed.
Deep Neural Network.
Artificial neural networks (ANNs) have been applied to a wide range of QSAR tasks. More recently, the ANNs have evolved into deep neural networks (DNN). Unlike an ANN, a DNN consists of multiple fully connected layers with two or more hidden layers between the input and output layers. In a feedforward neural network (referred to simply as DNN in the rest of the study), the information passed through the input layer flows in forward direction through the hidden layers to the output layer. DNN models were implemented in Keras (https://keras.io) using the TensorFlow (https://tensorflow.org) backend. The number of hidden layers was adjusted based on the size of input descriptors (i.e., 119 for RDKit descriptors and 1024 for Morgan fingerprint).
Graph Convolutional Neural Network.
Graphs are natural ways to represent chemical structures where nodes represent atoms and edges represent bonds between them. We recently showed that graph convolutional neural network (GCNN) provided superior performance in modeling Tier I ADME endpoints (rat liver microsomal stability, PAMPA permeability, and kinetic aqueous solubility)41,42. A message passing variant of GCCN implemented in the ChemProp43 Python package (https://github.com/chemprop/chemprop/) was employed to build GCNN models. The algorithm generates graph features when chemical structures (as line notations) and associated target values (i.e., PAMPA pH 5) are provided as input. The model parameters were set to default.
Modeling and Validation:
The training sets (I & II) were used to build models that were validated on the external set. Each training set was randomly divided into internal training and internal test sets (at an 80:20 ratio) for a total of five times. Each time, the model developed using the internal training set was validated on the internal test set. This procedure, widely known as k-fold (k = 5) cross-validation 44, was employed to identify the best performing methods and the descriptors. The best models identified from the cross-validation were further validated on the external set.
The model performance was assessed using different statistical measures. A receiver operating characteristic (ROC) curve, that plots true positive rate against the false positive rate, was used to estimate the predictive power of the classification models. The area under the ROC curve (i.e., AUC) is a numerical value between 0 and 1. The higher the value, the better the predictive power. Sensitivity indicates the proportion of true positives correctly predicted as positive. Specificity is the ability of the model to correctly predict true negatives as negative. Balanced accuracy (BACC) is an average of the Sensitivity and Specificity. It is a useful alternative to accuracy when the datasets in hand have a large degree of class imbalance. Cohen’s Kappa is another metric used in this study that measures the agreement between the actual classes and the classes predicted by the classifier.
Here, TP = true positives, FN = false negatives, TN = true negatives, and FP = false positives. In the case of Kappa, pa is the proportion of observations in agreement and p∈ is the proportion in agreement due to chance.
Results
Assay Performance:
Three control compounds; ranitidine (low permeability), dexamethasone (moderate permeability), and verapamil (high permeability) were run with each plate to ensure assay quality. Table 2 shows the assay reproducibility data for these compounds, spanning 194 plates over 4 years. The minimum significant ratio (MSR)45 for all compounds was around 2.0, which indicates excellent assay reproducibility over time. Standard deviation and MSR values were not calculated for ranitidine as Peff values are always below the limit of quantification.
Table 2.
Reproducibility data for control compounds. Mean and S.D. permeability (Peff) values were calculated across 194 plates.
| Compound | Peff (10−6 cm/s) | MSR (102√2*S.D.) |
|---|---|---|
| Ranitidine | <1 | N/A |
| Dexamethasone | 61 ± 16 | 2.3 |
| Verapamil | 208 ± 52 | 2.1 |
Distribution of Molecular Properties:
The greatest number of compounds in our dataset were found in the moderate/high permeability category (~72%) followed by the low permeability category (28%) (Fig. 1). Molecular properties, sLogP, total polar surface area (TPSA), and molecular weight (MW), were calculated using an in-house compound dataset annotation tool, known as NCATS Find 46. A large proportion of compounds from both Peff categories fell within the 300-500 MW range, had Log P values between 2-6 and TPSA values less than 100. No significant differences were found between both categories based on the distribution of these molecular properties (Fig. 2).
Figure 1.

Number of compounds categorized into low permeability (black), and moderate/high permeability (gray).
Figure 2.

Distribution of dataset based on A) Molecular Weight, B) Log P, and C) TPSA. Dataset is divided into compounds with low permeability (black) and compounds with moderate to high permeability (gray).
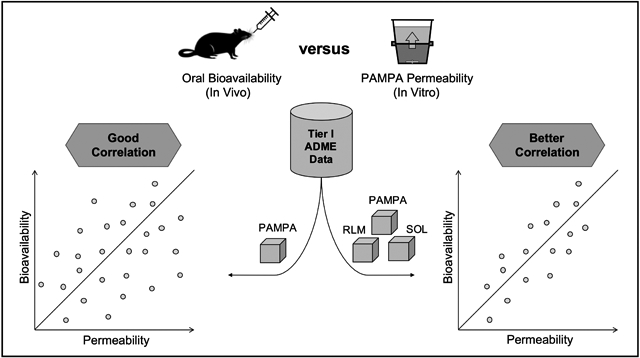
Correlating PAMPA pH 5 Permeability with Oral Bioavailability (%F):
Oral bioavailability (%F) is the fraction of an orally administered drug that reaches systemic circulation. To illustrate the application of our PAMPA pH 5 assay, we attempted to correlate log Peff values with oral bioavailability (%F) obtained from in-house pharmacokinetic (PK) studies (128 compounds). This in-house PK database was built with studies in mice (90%) and rats (10%), from a variety of projects with intravenous (IV) doses ranging from 1-5 mg/kg and oral (PO) doses ranging from 3-50 mg/kg, the median dosages being 3 mg/kg. We set the %F cut-off values at 20% as it represents an acceptable criteria for screening compounds in drug discovery 47,48.
The cut-off value for the PAMPA assay was set at 10 x 10− cm/sec since it is value differentiating compounds between low and moderate/high permeability. While we did not achieve a linear correlation, a categorical correlation of 74% was observed (Fig. 3A). %F is a complex property dependent on several factors such as GI physiology, physicochemical characteristics of the compound, drug metabolism, food, formulation, disease state, etc. Solubility and microsomal stability, two properties that affect %F, are also routinely tested for every compound synthesized at NCATS as part of Tier I ADME screening 49,41. To understand if a better correlation with %F could be obtained, we filtered our 128-compound dataset and eliminated compounds with poor solubility (<10 μg/mL)49 and poor microsomal stability (t1/2 < 30 min)41 (Fig. 3B). Peff values for the remaining 62-compounds were correlated with %F and an improved correlation of ~85% was observed (Fig. 3C).
Figure 3.

Correlating log Peff at pH 5 with %F (A) %F vs Log Peff with the 128-compound dataset. (B) Eliminating compounds with poor solubility and poor microsomal stability (C) %F vs Log Peff with the 62-compound dataset. Blue boxes in A and C show the categorical binning.
Cross-validation Results:
Training sets I and II were employed for 5-fold cross-validation (5-CV). DNN and GCNN models were compared with the baseline models based on RF and XGBoost. The baseline models RF and XGBoost provided similar performance on the training set I. Due to the high number of majority class examples in the dataset, the specificity values were high compared to sensitivity values. In case of both RF and XGBoost, RDKit descriptors provided slightly better sensitivity values compared to Morgan fingerprints. In contrast, the DNN models provided better sensitivity values with Morgan fingerprints. The performance of GCNN was found to be similar to DNN.
When evaluated using training set II, the overall performance of all models improved on account of enhanced sensitivity due to lower degree of class imbalance. The performance of RF and XGBoost models remained the same while DNN showed slightly better performance using RDKit descriptors compared to Morgan fingerprints. DNN and GCNN models provided a better balance between sensitivity and specificity. Figures 4 and 5 provide a comparison of the performance of models based on different methods and descriptors in terms of balanced accuracy, sensitivity, and specificity. The complete 5-CV results are provided in the supplementary information.
Figure 4.

Comparison of performances of models in 5-fold cross-validation measured as balanced accuracies. Each error bar represents the standard deviation of the average of the performance in five folds.
Figure 5.

Comparison of Sensitivity and Specificity values for training sets I and II. The error bars represent the standard deviations of average values for five folds.
External Validation Results:
Since training set II provided better performance in cross-validation, we only used the models based on this dataset to predict the external set. Once again, RF and XGBoost provided higher specificity values compared to sensitivity and RDKit descriptors provided superior performance over Morgan fingerprints. DNN model based on Morgan fingerprints provided better balanced accuracy due to improved sensitivity. GCNN model based on molecular graphs provided the best performance on the external set (Table 3).
Table 3.
External validation performance of models built using training set II.
| Method | Descriptor | AUC | BACC | Sensitivity | Specificity | Kappa |
|---|---|---|---|---|---|---|
| RF | RDKit | 0.83 | 0.74 | 0.62 | 0.87 | 0.41 |
| RF | Morgan FP | 0.78 | 0.71 | 0.60 | 0.83 | 0.32 |
| XGBoost | RDKit | 0.83 | 0.77 | 0.69 | 0.86 | 0.44 |
| XGBoost | Morgan FP | 0.80 | 0.71 | 0.57 | 0.84 | 0.33 |
| DNN | RDKit | 0.80 | 0.70 | 0.50 | 0.90 | 0.37 |
| DNN | Morgan FP | 0.77 | 0.73 | 0.64 | 0.82 | 0.34 |
| GCNN | Graph | 0.84 | 0.78 | 0.74 | 0.82 | 0.40 |
Feature Importance:
We analyzed our data further to understand structural features and important properties that are indicative of poor PAMPA permeability. The GCNN architecture, as implemented in the ChemProp package, provides a mechanism for identifying substructural features that explain the molecular property. For each compound, the interpretation module provides a predicted property value along with a substructural feature and an associated rationale score. The rationale score is the predicted property value for the substructure. We identified 11 features that were present in at least 30 compounds (log frequency > 1.5) in the training set and were predicted to have a rationale score > 2.5 (Fig. 6). These substructures are overrepresented in compounds with moderate to high PAMPA permeability (Peff > 2.5) from the training data. We also identified substructures that were predicted to have a rational score < = 1 and satisfy the frequency criterion (log frequency > 1.5). These substructures (Table 4) are overrepresented in compounds with low PAMPA permeability (Peff < 1) from our training data and can be of interest to medicinal chemists when dealing with liabilities due to PAMPA permeability. To the best of our knowledge, this is the first study to present analysis of substructural features relevant to PAMPA permeability.
Figure 6.

Features interpreted by the GCNN model. X-axis stands for the rationale score and Y-axis stands for the frequency of the feature in logarithmic scale. The top 11 features are shown on the plot.
Table 4.
Substructural features from GCNN model that represent low permeability compounds.
| Substructure | Rationale Score | Frequency |
|---|---|---|
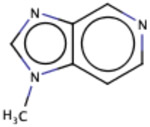
|
0.68 | 57 |
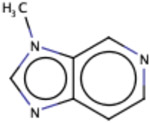
|
0.75 | 125 |
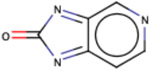
|
0.82 | 54 |
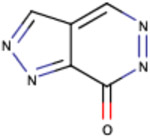
|
1.01 | 51 |
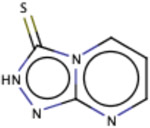
|
0.98 | 44 |
Similarly, we investigated the RF model to identify the RDKit descriptors that were scored higher in terms of importance of the RDKit features. The complete dataset was used for this purpose and out of the 119 RDKit descriptors, a total of 48 descriptors were identified to have importance of at least 0.01. Out of the 48 descriptors, we closely examined 17 descriptors (Fig. 7) with an importance > = 0.015. As anticipated, Log P (calculated by RDKit as SlogP) turned out to be the most important feature (average feature importance of 0.03) for the classification model followed by peoe_VSA8, smr_VSA3, peoe_VSA2 and topological polar surface area (TPSA). Log P, Log D and polar surface area have been previously discussed to be important descriptors for PAMPA permeability in former studies that reported for QSAR models 50-52. PEOE descriptors are those based on the partial charges of each atom in a molecule, calculated using Partial Equalization of Orbital Electronegativities (PEOE) method of calculating atomic partial charges, and depend only on the connectivity (i.e., elements, formal charges, and bond orders).
Figure 7.

RDKit descriptors identified as important features by RF model based on 5-CV using the complete dataset. For each descriptor, the feature importance score from the five folds is plotted.
Validation Using Molecular Weight and Time Split:
We previously demonstrated using the rat liver microsomal stability data that a time-based splitting of data provides an alternative view of model performance. A recent study 53 proposed molecular weight split combined with time-based splitting as a cross-validation strategy to validate ADME prediction models. The authors removed compounds with molecular weight higher than 500 g/mol from the training set and retained only those compounds with molecular weight higher than 600 g/mol in the external set. In the current study, we have data spanning 2016, 2017, 2018 and 2019 with approximately 25% of the data coming from the year 2019. Therefore, using the data from the years 2016, 2017 and 2018 as the training set and the rest as the external set closely resembles a random split of the data set at 80:20 ratio that resulted in the original external set. Additionally, we removed compounds with molecular weight > 500 g/mol from the training set and slightly adjusted the original criterion to retain compounds with weight > 550 g/mol in the external set. The reason behind adjusting this criterion is to accommodate higher number of compounds in the external set. After temporal and molecular weight split, the training and external sets comprised of 3,334 (Class 1: 407; Class 0: 2927) compounds and 119 (Class 1: 19; Class 0: 100) compounds, respectively. This model provided a balanced accuracy of 65% (Sensitivity: 58% and Specificity: 73%). While the performance is inferior compared to the performance of models on the other external set, this could be explained by the presence of unusually large molecules present in this external set (average molecular weight = 885 Daltons and average number of rings per molecule = 6) in comparison to the corresponding training set (average molecular weight = 400 Daltons and average number of rings per molecule = 4). It would be worth investigating this strategy on a larger dataset that spans multiple years and a wider chemical space.
Discussion
Oral bioavailability is a complex process dependent on many physiological, physicochemical, and pharmacological parameters including membrane permeability, solubility, metabolic stability, particle size, pH, surface area of the GI tract, activities of uptake and efflux transporters, etc. For an orally administered drug to reach systemic circulation, it must pass through the intestinal membrane by passive diffusion, carrier mediated uptake or active transporter mechanisms. Cell-based assays, such as Caco-2 and MDCK cell culture systems, have been used to model membrane permeability and these assays have become the standard in the pharmaceutical industry. However, since 80-95% of commercially available drugs are absorbed via passive diffusion 2,17,19,20,18, PAMPA is as a popular alternative approach. PAMPA has several advantages including low cost, amenability to high-throughput, shorter lead times as well as comparable prediction accuracy to the Caco-2 assay for prediction of intestinal permeability 54 Additionally, the good day-to-day reproducibility and lower data variability 54 make datasets generated through PAMPA assays highly sought after for in silico QSAR modeling. In this study, we used our 6500 compound PAMPA pH 5 dataset and built classification models to predict intestinal permeability of test compounds. While a few PAMPA QSAR models exist in literature, they are built using relatively small datasets and neither the model nor the datasets have been made publicly available. While we cannot make our entire dataset public due to its proprietary nature, a small subset (486 compounds; PubChem AID: 1645871) of our data and the best predictive models have been made public. Our PAMPA models (pH 5 and pH 7.4; published previously18) are to the best of our knowledge, the only open-access PAMPA models built using high-quality data, generated at a single laboratory.
A recent study by Oja and Maran highlighted the importance of understanding the pH-permeability relationship, especially for ionizable compounds 2. They also emphasized the fact that most PAMPA studies in literature have been performed at neutral or near-neutral pH and thus, there is a dearth of PAMPA permeability data and QSAR models at different pH values. To this end, Oja and Maran have published datasets and QSAR models (238 compounds) for PAMPA permeability at pH 3, 5, 7.4 and 9 22. Our PAMPA datasets go a long way towards filling this data gap. Although the openly accessible compounds represent a relatively small percentage of our total dataset, these represent by far, the largest datasets in literature (Table 5).
Table 5.
Summary of NCATS ADME Models and Datasets
| Assay | Type | Number of Compounds |
Location |
|---|---|---|---|
| PAMPA pH 5 | Dataset | 486 | PubChem- AID: 1645871 |
| PAMPA pH 5 | Model | 6,500 | https://opendata.ncats.nih.gov/adme/ |
| PAMPA pH 7.4 | Dataset | 2,532 | PubChem- AID: 1508612 |
| PAMPA pH 7.4 42 | Model | 22,000 | https://opendata.ncats.nih.gov/adme/ |
Multiple studies reported QSAR models to predict PAMPA permeability 55-57,23,58-61,19,18,22. However, as emphasized earlier by Chi et al 62, PAMPA permeability depends on several factors other than the pH of the assay. Therefore, creating a good QSAR model using data available in the public domain has been considered impractical. Most previously reported models were based on linear regression techniques such as partial least squares (PLS) and multiple linear regression. Later, having understood that there exists a bilinear relationship between Log D and PAMPA permeability, it became clear that the linear regression models were unable to capture the complex nonlinearity 63. Machine learning methods such as support vector machines, random forests and gradient boosting have been employed 64,62,65 using both public and proprietary datasets. Though most studies reported regression models, a few studies reported categorical models using classification criteria similar to those employed in the current study 55,66,67. While most studies relied on a handful of physicochemical properties, some of them employed large number of 2D or 3D descriptors from commercial software. Considering that type of assay used in our study (double sink PAMPA) is different from other published studies and the fact that there are very few PAMPA studies conducted at pH 5, we could not directly compare our models with those in literature.
Additionally, we correlated the PAMPA pH 5 permeability values with pre-clinical oral bioavailability and observed an accuracy of 74%. Considering that oral bioavailability is an extremely complex and multi-factorial property, this correlation was encouraging. Moreover, after accounting for solubility and metabolic stability, two parameters that affect oral bioavailability, this correlation increased to 85%. The corresponding correlation for the PAMPA pH 7.4 dataset was found to be 80% (unpublished data). This suggests that the proper use of our data and models could help minimize the risk of compounds failing in pre-clinical in vivo studies due to poor bioavailability.
Conclusion
In summary, we developed a robust QSAR model using our PAMPA pH 5 dataset and identified structural features and descriptors relevant for PAMPA pH 5 permeability. This model along with our previously published models 42 (https://opendata.ncats.nih.gov/adme/home) can be used to rank-order compounds for synthesis and thus, project teams can get to their lead compounds in fewer iterations. Implementing in silico tools in early drug discovery may ultimately prove to be game changing in the time-intensive, costly, and high-attrition drug discovery and development process.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Cordelle Tanega at NCATS for helping with the deposition of data in the PubChem bioassay database.
Funding
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Center for Advancing Translational Sciences.
Abbreviations
- 5-CV
5-fold cross-validation
- ANN
Artificial Neural Network
- AUC
Area Under the Curve
- BACC
Balanced Accuracy
- DNN
Deep Neural Network
- GI
Gastrointestinal
- QSAR
Quantitative Structure-Activity Relationship
- GCNN
Graph Convolutional Neural Network
- MSR
Minimum Significant Ratio
- ML
Machine Learning
- PAMPA
Parallel Artificial Membrane Permeability Assay
- PK
Pharmacokinetic
- RF
Random Forest
- XGBoost
eXtreme Gradient Boosting
Footnotes
Availability of data and materials
The dataset supporting the conclusions of this article is available in the PubChem repository, Assay ID 1645871. The models supporting the conclusions of this article are available in the Open Data NCATS ADME portal (ADME@NCATS), https://opendata.ncats.nih.gov/adme/. An Excel sheet containing the complete 5-fold cross-validation results for Training Set I & II is submitted as supplementary information.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Oja M, Sild S, Maran U (2019) Logistic Classification Models for pH-Permeability Profile: Predicting Permeability Classes for the Biopharmaceutical Classification System. J Chem Inf Model 59 (5):2442–2455. doi: 10.1021/acs.jcim.8b00833 [DOI] [PubMed] [Google Scholar]
- 2.Oja M, Maran U (2015) The Permeability of an Artificial Membrane for Wide Range of pH in Human Gastrointestinal Tract: Experimental Measurements and Quantitative StructureActivity Relationship. Mol Inf 34 (6-7):493–506. doi: 10.1002/minf.201400147 [DOI] [PubMed] [Google Scholar]
- 3.Volpe DA (2011) Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med Chem 3 (16):2063–2077. doi: 10.4155/fmc.11.149 [DOI] [PubMed] [Google Scholar]
- 4.Lennernas H (2014) Regional intestinal drug permeation: biopharmaceutics and drug development. Eur J Pharm Sci 57:333–341. doi: 10.1016/j.ejps.2013.08.025 [DOI] [PubMed] [Google Scholar]
- 5.Dahlgren D, Roos C, Lundqvist A, Abrahamsson B, Tannergren C, Hellstrom PM, Sjogren E, Lennernas H (2016) Regional Intestinal Permeability of Three Model Drugs in Human. Mol Pharmaceutics 13 (9):3013–3021. doi: 10.1021/acs.molpharmaceut.6b00514 [DOI] [PubMed] [Google Scholar]
- 6.Charman WN, Porter CJ, Mithani S, Dressman JB (1997) Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci 86 (3):269–282. doi: 10.1021/js960085v [DOI] [PubMed] [Google Scholar]
- 7.Avdeef A (2012) Absorption and Drug Development. Solubility, Permeability, and Charge State. 2nd edn. John Wiley & Sons, Hoboken, New Jersey [Google Scholar]
- 8.Vertzoni M, Augustijns P, Grimm M, Koziolek M, Lemmens G, Parrott N, Pentafragka C, Reppas C, Rubbens J, Van Den Alphabeele J, Vanuytsel T, Weitschies W, Wilson CG (2019) Impact of regional differences along the gastrointestinal tract of healthy adults on oral drug absorption: An UNGAP review. Eur J Pharm Sci 134:153–175. doi: 10.1016/j.ejps.2019.04.013 [DOI] [PubMed] [Google Scholar]
- 9.Shore PA, Brodie BB, Hogben CA (1957) The gastric secretion of drugs: a pH partition hypothesis. J Pharmacol Exp Ther 119:361–369 [PubMed] [Google Scholar]
- 10.Grasset E, Pinto M, Dussaulx E, Zweibaum A, Desjeux JF (1984) Epithelial properties of human colonic carcinoma cell line Caco-2: electrical parameters. Am J Physiol 247 (3):C260–C267. doi: 10.1152/ajpcell.1984.247.3.C260 [DOI] [PubMed] [Google Scholar]
- 11.Artursson P, Karlsson J (1991) Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 175 (3):880–885. doi: 10.1016/0006-291X(91)91647-U [DOI] [PubMed] [Google Scholar]
- 12.Lennernäs H, Palm K, Fagerholm U, Artursson P (1996) Comparison between active and passive drug transport in human intestinal epithelial (caco-2) cells in vitro and human jejunum in vivo. Int J Pharm 127 (1):103–107. doi: 10.1016/0378-5173(95)04204-0 [DOI] [Google Scholar]
- 13.Putnam WS, Pan L, Tsutsui K, Takahashi L, Benet LZ (2002) Comparison of Bidirectional Cephalexin Transport Across MDCK and Caco-2 Cell Monolayers: Interactions with Peptide Transporters. Pharm Res 19 (1):27–33. doi: 10.1023/A:1013647114152 [DOI] [PubMed] [Google Scholar]
- 14.D'Souza VM, Shertzer HG, Menon AG, Pauletti GM (2003) High Glucose Concentration in Isotonic Media Alters Caco-2 Cell Permeability. AAPS PharmSci 5 (3). doi: 10.1208/ps050324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bravo SA, Nielsen CU, Amstrup J, Frokjaer S, Brodin B (2004) In-depth evaluation of Gly-Sar transport parameters as a function of culture time in the Caco-2 cell model. Eur J Phram Sci 21 (1):77–86. doi: 10.1016/s0928-0987(03)00205-7 [DOI] [PubMed] [Google Scholar]
- 16.Anderle P, Niederer E, Rubas W, Hilgendorf C, Spahn-Langguth H, Wunderli-Allenspach H, Merkle HP, Langguth P (1998) P-Glycoprotein (P-gp) mediated efflux in Caco-2 cell monolayers: the influence of culturing conditions and drug exposure on P-gp expression levels. J Pharm Sci 87 (6):757–762. doi: 10.1021/js970372e [DOI] [PubMed] [Google Scholar]
- 17.Avdeef A (2005) The rise of PAMPA. Expert Opin Drug Metab Toxicol 1 (2):325–342. doi: 10.1517/17425255.1.2.325 [DOI] [PubMed] [Google Scholar]
- 18.Sun H, Nguyen K, Kerns E, Yan Z, Yu KR, Shah P, Jadhav A, Xu X (2017) Highly predictive and interpretable models for PAMPA permeability. Bioorg Med Chem 25 (3):1266–1276. doi: 10.1016/j.bmc.2016.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oja M, Maran U (2015) Quantitative structure-permeability relationships at various pH values for acidic and basic drugs and drug-like compounds. SAR QSAR Environ Res 26 (7-9):701–719. doi: 10.1080/1062936X.2015.1085896 [DOI] [PubMed] [Google Scholar]
- 20.Kerns E, Di L, Petusky S, Farris M, Ley R, Jupp P (2004) Combined Application of Parallel Artificial Membrane Permeability Assay and Caco-2 Permeability Assay in Drug Discovery. J Pharm Sci 93 (6):1440–1453. doi: 10.1002/jps.20075 [DOI] [PubMed] [Google Scholar]
- 21.Brennan MB (2000) Drug Discovery - filtering out failures early in the game. Chem. Eng. News, vol 78. American Chemical Society, [Google Scholar]
- 22.Oja M, Maran U (2018) pH-permeability profiles for drug substances: Experimental detection, comparison with human intestinal absorption and modelling. Eur J Pharm Sci 123:429–440. doi: 10.1016/j.ejps.2018.07.014 [DOI] [PubMed] [Google Scholar]
- 23.Verma RP, Hansch C, Selassie CD (2007) Comparative QSAR studies on PAMPA/modified PAMPA for high throughput profiling of drug absorption potential with respect to Caco-2 cells and human intestinal absorption. J Comput Aided Mol Des 21 (1-3):3–22. doi: 10.1007/s10822-006-9101-z [DOI] [PubMed] [Google Scholar]
- 24.Frings M, Bolm C, Blum A, Gnamm C (2017) Sulfoximines from a Medicinal Chemist's Perspective: Physicochemical and in vitro Parameters Relevant for Drug Discovery. Eur J Med Chem 126:225–245. doi: 10.1016/j.ejmech.2016.09.091 [DOI] [PubMed] [Google Scholar]
- 25.Yamaki S, Suzuki D, Fujiyasu J, Neya M, Nagashima A, Kondo M, Akabane T, Kadono K, Moritomo A, Yoshihara K (2017) Synthesis and structure activity relationships of glycine amide derivatives as novel Vascular Adhesion Protein-1 inhibitors. Bioorg Med Chem 25 (1):187–201. doi: 10.1016/j.bmc.2016.10.025 [DOI] [PubMed] [Google Scholar]
- 26.Furukawa A, Townsend CE, Schwochert J, Pye CR, Bednarek MA, Lokey RS (2016) Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J Med Chem 59 (20):9503–9512. doi: 10.1021/acs.jmedchem.6b01246 [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Glunz PW, Johnson JA, Jiang W, Jacutin-Porte S, Ladziata V, Zou Y, Phillips MS, Wurtz NR, Parkhurst B, Rendina AR, Harper TM, Cheney DL, Luettgen JM, Wong PC, Seiffert D, Wexler RR, Priestley ES (2016) Discovery of a Highly Potent, Selective, and Orally Bioavailable Macrocyclic Inhibitor of Blood Coagulation Factor VIIa-Tissue Factor Complex. J Med Chem 59 (15):7125–7137. doi: 10.1021/acs.jmedchem.6b00469 [DOI] [PubMed] [Google Scholar]
- 28.Grosche P, Sirockin F, Mac Sweeney A, Ramage P, Erbel P, Melkko S, Bernardi A, Hughes N, Ellis D, Combrink KD, Jarousse N, Altmann E (2015) Structure-based design and optimization of potent inhibitors of the adenoviral protease. Bioorg Med Chem Lett 25 (3):438–443. doi: 10.1016/j.bmcl.2014.12.057 [DOI] [PubMed] [Google Scholar]
- 29.Flanders Y, Dumas S, Caserta J, Nicewonger R, Baldino M, Lee C, Baldino CM (2015) A versatile synthesis of novel pan-PIM kinase inhibitors with initial SAR study. Tetrahedron Lett 56 (23):3186–3190. doi: 10.1016/j.tetlet.2015.01.119 [DOI] [Google Scholar]
- 30.Kim J, Chin J, Im CY, Yoo EK, Woo S, Hwang HJ, Cho JH, Seo KA, Song J, Hwang H, Kim KH, Kim ND, Yoon SK, Jeon JH, Yoon SY, Jeon YH, Choi HS, Lee IK, Kim SH, Cho SJ (2016) Synthesis and biological evaluation of novel 4-hydroxytamoxifen analogs as estrogen-related receptor gamma inverse agonists. Eur J Med Chem 120:338–352. doi: 10.1016/j.ejmech.2016.04.076 [DOI] [PubMed] [Google Scholar]
- 31.Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S (2019) Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci 40 (8):592–604. doi: 10.1016/j.tips.2019.06.004 [DOI] [PubMed] [Google Scholar]
- 32.Avdeef A, Artursson P, Neuhoff S, Lazorova L, Grasjo J, Tavelin S (2005) Caco-2 permeability of weakly basic drugs predicted with the double-sink PAMPA pKa(flux) method. Eur J Pharm Sci 24 (3):333–349. doi: 10.1016/j.ejps.2004.11.011 [DOI] [PubMed] [Google Scholar]
- 33.Bennion BJ, Be NA, McNerney MW, Lao V, Carlson EM, Valdez CA, Malfatti MA, Enright HA, Nguyen TH, Lightstone FC, Carpenter TS (2017) Predicting a Drug's Membrane Permeability: A Computational Model Validated With in Vitro Permeability Assay Data. J Phys Chem B 121 (20):5228–5237. doi: 10.1021/acs.jpcb.7b02914 [DOI] [PubMed] [Google Scholar]
- 34.Shah P, Kerns E, Nguyen DT, Obach RS, Wang AQ, Zakharov A, McKew J, Simeonov A, Hop CE, Xu X (2016) An Automated High-Throughput Metabolic Stability Assay Using an Integrated High-Resolution Accurate Mass Method and Automated Data Analysis Software. Drug Metab Dispos 44 (10):1653–1661. doi: 10.1124/dmd.116.072017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fourches D, Muratov E, Tropsha A (2016) Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J Chem Inf Model 56 (7):1243–1252. doi: 10.1021/acs.jcim.6b00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah P, Siramshetty VB, Zakharov AV, Southall NT, Xu X, Nguyen DT (2020) Predicting liver cytosol stability of small molecules. J Cheminf 12 (1):21. doi: 10.1186/s13321-020-00426-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan HL (1965) The Generation of a Unique Machine Description for Chemical Structures - A Technique Developed at Chemical Abstracts Service. J Chem Doc 5 (2):107–113. doi: 10.1021/c160017a018 [DOI] [Google Scholar]
- 38.Breiman L (2001) Random Forests. Machine Learning 45:5–32. doi: 10.1023/A:1010933404324 [DOI] [Google Scholar]
- 39.Pedregosa F, Varoquaux G, Gramfort A, al. e (2011) Scikit-learn: Machine Learning in {P}ython. J Mach Learn Res 12:2825–2830 [Google Scholar]
- 40.Chen T, Guestrin C (2016) XGBoost: A Scalable Tree Boosting System. Paper presented at the Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, California, USA, [Google Scholar]
- 41.Siramshetty VB, Shah P, Kerns E, Nguyen K, Yu KR, Kabir M, Williams J, Neyra J, Southall N, Nguyen T, Xu X (2020) Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR models. Sci Rep 10 (1):20713. doi: 10.1038/s41598-020-77327-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siramshetty VB, Williams J, Nguyen D, Neyra J, Southall N, Mathe E, Xu X, Shah P (2021) Validating ADME QSAR Models Using Marketed Drugs. SLAS Discovery: Advancing the Science of Drug Discovery. doi: 10.1177/24725552211017520 [DOI] [PubMed] [Google Scholar]
- 43.chemprop/chemprop Message Passing Neural Networks for Molecule Property Prediction.
- 44.Refaeilzadeh P, Tang L, Liu H (2009) Cross-Validation. In: Liu L, ÖZsu MT (eds) Encyclopedia of Database Systems. Springer US, Boston, MA, pp 532–538. doi: 10.1007/978-0-387-39940-9_565 [DOI] [Google Scholar]
- 45.Assay Guidance Manual (2004). Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD: [PubMed] [Google Scholar]
- 46.(NCATS) NCfATS Resolver.
- 47.Hou T, Wang J, Zhang W, Xu X (2007) ADME Evaluation in Drug Discovery. 6. Can Oral Bioavailability in Humans Be Effectively Predicted by Simple Molecular Property-Based Rules? J Chem Inf Model 47 (2):460–463. doi: 10.1021/ci6003515 [DOI] [PubMed] [Google Scholar]
- 48.Veber DF, Johnson SR, Chen H-Y, Smith BR, Ward KW, Kopple KD (2002) Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem 45 (12):2615–2623. doi: 10.1021/jm020017n [DOI] [PubMed] [Google Scholar]
- 49.Sun H, Shah P, Nguyen K, Yu KR, Kerns E, Kabir M, Wang Y, Xu X (2019) Predictive models of aqueous solubility of organic compounds built on a large dataset of high integrity. Bioorg Med Chem 27 (14):3110–3114. doi: 10.1016/j.bmc.2019.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Jiang L, Chen T, Hwang K (2002) A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur J Med Chem 37 (5):399–407. doi: 10.1016/s0223-5234(02)01360-0 [DOI] [PubMed] [Google Scholar]
- 51.Flaten GE, Dhanikula AB, Luthman K, Brandl M (2006) Drug permeability across a phospholipid vesicle based barrier: a novel approach for studying passive diffusion. Eur J Pharm Sci 27 (1):80–90. doi: 10.1016/j.ejps.2005.08.007 [DOI] [PubMed] [Google Scholar]
- 52.Over B, Matsson P, Tyrchan C, Artursson P, Doak BC, Foley MA, Hilgendorf C, Johnston SE, Lee IV MD, Lewis RJ, McCarren P, Muncipinto G, Norinder U, Perry MWD, Duvall JR, Kihlberg J (2016) Structural and conformational determinants of macrocycle cell permeability. Nat Chem Biol 12:1065–1074. doi: 10.1038/nchembio.2203 [DOI] [PubMed] [Google Scholar]
- 53.Feinberg EN, Joshi E, Pande VS, Cheng AC (2020) Improvement in ADMET Prediction with Multitask Deep Featurization. J Med Chem 63 (16):8835–8848. doi: 10.1021/acs.jmedchem.9b02187 [DOI] [PubMed] [Google Scholar]
- 54.Sugano K, Okazaki A, Sugimoto S, Tavornvipas S, Omura A, Mano T (2007) Solubility and Dissolution Profile Assessment in Drug Discovery. Drug Metab Pharmacokinet 22 (4):225–254. doi: 10.2133/dmpk.22.225 [DOI] [PubMed] [Google Scholar]
- 55.Chaturvedi PR, Decker CJ, Odinecs A (2001) Prediction of pharmacokinetic properties using experimental approaches during early drug discovery. Curr Opin Chem Biol 5 (4):452–463. doi: 10.1016/s1367-5931(00)00228-3 [DOI] [PubMed] [Google Scholar]
- 56.Ano R, Kimura Y, Shima M, Matsuno R, Ueno T, Akamatsu M (2004) Relationships between structure and high-throughput screening permeability of peptide derivatives and related compounds with artificial membranes: application to prediction of Caco-2 cell permeability. Bioorg Med Chem 12 (1):257–264. doi: 10.1016/j.bmc.2003.10.002 [DOI] [PubMed] [Google Scholar]
- 57.Dibbell DG, Cohn R (1966) Perforation of the colon during hydrostatic reduction. Am J Surg 115 (5):715–717. doi: 10.1016/0002-9610(66)90048-1 [DOI] [PubMed] [Google Scholar]
- 58.Fujikawa M, Nakao K, Shimizu R, Akamatsu M (2007) QSAR study on permeability of hydrophobic compounds with artificial membranes. Bioorg Med Chem 15 (11):3756–3767. doi: 10.1016/j.bmc.2007.03.040 [DOI] [PubMed] [Google Scholar]
- 59.Kalyanaraman C, Jacobson MP (2007) An atomistic model of passive membrane permeability: application to a series of FDA approved drugs. J Comput Aided Mol Des 21 (12):675–679. doi: 10.1007/s10822-007-9141-z [DOI] [PubMed] [Google Scholar]
- 60.Nakao K, Fujikawa M, Shimizu R, Akamatsu M (2009) QSAR application for the prediction of compound permeability with in silico descriptors in practical use. J Comput Aided Mol Des 23 (5):309–319. doi: 10.1007/s10822-009-9261-8 [DOI] [PubMed] [Google Scholar]
- 61.Akamatsu M, Fujikawa M, Nakao K, Shimizu R (2009) In silico prediction of human oral absorption based on QSAR analyses of PAMPA permeability. Chem Biodivers 6 (11):1845–1866. doi: 10.1002/cbdv.200900112 [DOI] [PubMed] [Google Scholar]
- 62.Chi CT, Lee MH, Weng CF, Leong MK (2019) In Silico Prediction of PAMPA Effective Permeability Using a Two-QSAR Approach. Int J Mol Sci 20 (13). doi: 10.3390/ijms20133170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kansy M, Fischer H, Kratzat K, Senner F, Wagner B, Parrilla I (2001) Pharmacokinetic Optimization in Drug Research. In: Testa B, van de Waterbeemd H, Folkers G, Guy R (eds) Pharmacokinetic Optimization in Drug Research. pp 447–464. doi: 10.1002/9783906390437.ch24 [DOI] [Google Scholar]
- 64.Sherer EC, Verras A, Madeira M, Hagmann WK, Sheridan RP, Roberts D, Bleasby K, Cornell WD (2012) QSAR Prediction of Passive Permeability in the LLC-PK1 Cell Line: Trends in Molecular Properties and Cross-Prediction of Caco-2 Permeabilities. Mol Inf 31 (3-4):231–245. doi: 10.1002/minf.201100157 [DOI] [PubMed] [Google Scholar]
- 65.Gousiadou C, Doganis P, Sarimveis H (2021) Development of QSAR ensemble models for predicting the PAMPA Effective Permeability of new, non-peptidic leads with potential antiviral activity against the coronavirus SARS-CoV-2. doi: 10.3762/bxiv.2021.22.v1 [DOI] [Google Scholar]
- 66.Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW (2002) Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther 303 (3):1029–1037. doi: 10.1124/jpet.102.039255 [DOI] [PubMed] [Google Scholar]
- 67.Wager TT, Hou X, Verhoest PR, Villalobos A (2010) Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci 1 (6):435–449. doi: 10.1021/cn100008c [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.