

Abstract
Acetaminophen (APAP) hepatotoxicity, a leading cause of acute liver failure in western countries, is characterized by mitochondrial superoxide and peroxynitrite formation. However, the role of iron, especially as facilitator of lipid peroxidation (LPO), has been controversial. Our aim was to determine the mechanism by which iron promotes cell death in this context. Fasted male C57BL/6J mice were treated with the iron chelator deferoxamine, minocycline (inhibitor of the mitochondrial calcium uniporter) or vehicle 1 h before 300 mg/kg APAP. Deferoxamine and minocycline significantly attenuated APAP-induced elevations in serum alanine amino transferase levels and hepatic necrosis at 6 h. This protection correlated with reduced 3-nitro-tyrosine protein adducts; LPO (malondialdehyde, 4-hydroxynonenal) was not detected. Activation of c-jun N-terminal kinase (JNK) was not affected but mitochondrial release of intermembrane proteins was reduced suggesting that the effect of iron was at the level of mitochondria. Co-treatment of APAP with FeSO4 exacerbated liver injury and protein nitration and triggered significant LPO; all effects were reversed by deferoxamine. Thus, after APAP overdose, iron imported into mitochondria facilitates protein nitration by peroxynitrite triggering mitochondrial dysfunction and cell death. Under these conditions, endogenous defense mechanisms largely prevent LPO. However, after iron overload, protein nitration and LPO contribute to APAP hepatotoxicity.
Keywords: Acetaminophen Hepatotoxicity, Ferrous iron, Lipid Peroxidation, Peroxynitrite, Deferoxamine, Minocycline
1. Introduction
Acetaminophen (APAP) is a widely used analgesic and antipyretic drug, which can induce liver injury and even acute liver failure after a substantial overdose in patients (Bernal and Wendon, 2013; Jaeschke, 2015). Investigations of the mechanisms of cell death identified key events which include cytochrome P450-dependent formation of the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI), hepatic GSH depletion, mitochondrial protein adducts formation and superoxide release. This causes c-jun N-terminal kinase (JNK) activation in the cytosol and its translocation to mitochondria, mitochondrial dysfunction and oxidative/nitrosative stress resulting in the mitochondrial permeability transition pore (MPTP) opening and the release of intermembrane proteins like endonuclease G, which translocate to the nucleus and cause DNA fragmentation (Jaeschke et al., 2012; Nelson, 1990; Ramachandran and Jaeschke, 2019). Based on the biochemical signalling events and the morphological characteristic of cell and organelle swelling and extensive cell contents release, the mode of cell death is generally considered to be oncotic necrosis (Gujral et al., 2002) or programmed necrosis (Jaeschke et al., 2019). Although there is some overlap in signalling events with what was traditionally considered apoptotic mechanisms, e.g., mitochondrial Bax translocation, mitochondrial release of cytochrome c, apoptosis inducing factor (AIF) and endonuclease G, nuclear DNA fragmentation, etc., neither caspase activation nor morphological characteristics of apoptosis are detectable after APAP overdose (Jaeschke et al., 2018; Jaeschke and Ramachandran, 2020).
Mitochondrial oxidant stress is considered a key feature of APAP-induced cell death (Du et al., 2016). Binding of NAPQI to mitochondrial proteins is the initial event that triggers superoxide release by complex III of the electron transport chain, which is responsible for the early activation of JNK in the cytosol (Nguyen et al., 2021). Activated JNK (P-JNK) translocates to the mitochondria (Hanawa et al., 2008) and binds to the anchor protein Sab on the outer mitochondrial membrane (Win et al., 2011), which causes further impairment of the electron transport chain through inactivation of Src (Win et al., 2016) and thus an amplification of the mitochondrial oxidant stress (Saito et al., 2010a). Interestingly, despite this oxidant stress inside the mitochondria, lipid peroxidation (LPO) is very limited and loading liver cells with an excessive amount of vitamin E does not protect against APAP toxicity (Knight et al., 2003). In contrast, APAP-induced liver injury can be dramatically amplified by partial deficiency of the mitochondria-specific MnSOD (Fujimoto et al., 2009; Ramachandran et al., 2011) and almost completely prevented by treatment with a mitochondria-targeted SOD mimetic (Mito-Tempo) (Du et al., 2017, 2019). Because there is evidence for formation of peroxynitrite, a reaction product of superoxide and nitric oxide, within mitochondria (Cover et al., 2005) and the fact that Mito-Tempo eliminates nitrotyrosine staining, an imprint of peroxynitrite modification of proteins (Du et al., 2017), these data strongly suggest that the critical oxidant responsible for APAP toxicity is peroxynitrite in the mitochondrial matrix where it triggers the MPTP opening (Kon et al., 2004; Masubuchi et al., 2005).
The role of iron in APAP hepatotoxicity has been controversial, with reports indicating that chelation of iron with deferoxamine has no effect on the injury (Younes and Siegers, 1985; Younes et al., 1988), causes significant protection (Sakaida et al., 1995; Yamada et al., 2020), or causes only some delay of the injury (Schnellmann et al., 1999). More recently, it was suggested an APAP overdose causes lysosomal instability (Woolbright et al., 2012), which releases lysosomal ferrous iron into the cytosol (Kon et al., 2010); this iron is then taken up into the mitochondria through the mitochondrial Ca2+, Fe2+ uniporter (Hu et al., 2016). Chelation of lysosomal ferrous iron or blocking its uptake into the mitochondria attenuated the MPTP opening and cell necrosis both in vitro (Hu et al., 2016; Kon et al., 2010) and in vivo (Hu and Lemasters, 2020). Because ferrous iron is a major catalyst of the Fenton reaction to generate hydroxyl radicals for the initiation of LPO and alkoxy radicals to propagate LPO, these data seem to directly contradict the limited lipid peroxidation seen after APAP overdose (Knight et al., 2003) and the hypothesis that peroxynitrite is the central oxidant in APAP hepatotoxicity (Knight et al., 2002). Thus, the objective of this study was to investigate whether iron can have alternate mechanisms of action that could explain and reconcile these contradictory findings in APAP-induced liver injury.
2. Materials and methods
2.1. Animals
All experiments were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Centre and followed the criteria of the National Research Council for the care and use of laboratory animals. Male C57BL/6J mice (8–10 weeks old) weighing 20–24 g were obtained from Jackson Laboratories (Bar Harbor, Maine) and kept in an environmentally controlled room under a 12-h light/dark cycle and allowed free access to food and water.
2.2. Experimental design
All reagents and compounds were purchased from Sigma-Aldrich (St. Louis, Missouri) unless otherwise stated. Mice were given intra-peritoneal (i.p.) injections of 300 mg/kg APAP (dissolved in warmed saline) or vehicle (saline) alone after a 16-h overnight fast. Groups of mice were injected i.p. with 200 mg/kg deferoxamine mesylate, 10 mg/kg minocycline hydrochloride or vehicle (saline) alone 1 h before APAP. Another group of mice received i.p. co-treatment with 300 mg/kg APAP and 0.15 mmol/kg FeSO4 1 h after administration of 200 mg/kg deferoxamine mesylate or saline (i.p.). Mice were then sacrificed under isoflurane anesthesia 6 h post APAP. Blood was collected from the vena cava into a heparinized syringe and centrifuged at 18,000g for 2 min to collect plasma. Livers were collected, rinsed in saline and cut into pieces, which were either snap frozen in liquid nitrogen or fixed overnight in 10% phosphate buffered formalin or used for mitochondrial isolation as described below.
2.3. Biochemical assays
Plasma alanine aminotransferase (ALT) activities were measured using an ALT test kit (MedTest, Canton, Michigan) according to the manufacturer’s instructions. Hepatic GSH levels were measured in snap-frozen tissue with a modified Tietze assay as described in detail (McGill and Jaeschke, 2015). To determine hepatic malondialdehyde (MDA) content, 100 mg of liver tissue was homogenized in 1 mL lysis buffer (25 mM HEPES, 5 mM EDTA, 0.19% CHAPS) containing protease inhibitors. The BCA assay (Thermofisher Scientific, Waltham, Massachusetts) was performed on the lysate to determine protein concentration. To 150 μL lysate, 300 μL ice cold trichloroacetic acid was added to precipitate protein. After centrifugation at 10,000g for 10 min, the supernatant was collected and incubated with equal volume 0.67%w/v thiobarbituric acid in a boiling water bath for 10 min. The samples were allowed to cool, and absorbance was measured at 532 nm to determine MDA concentration. MDA concentration was then normalized to protein concentration for each sample.
2.3.1. Histology and immunohistochemistry
Tissue was fixed in 10% phosphate buffered formalin and embedded in paraffin before being cut into 5 μm thick sections. Hematoxylin and eosin (H&E) staining was performed on sections to determine the areas of necrosis. For immunohistochemical staining, rehydrated tissue sections underwent heat mediated antigen retrieval using citrate buffer (pH = 6.0). The sections were blocked in 3% BSA in serum before being incubated overnight with either rabbit anti-4-hydroxynonenal (Abcam, ab46545) or rabbit anti-nitrotyrosine antibodies (Invitrogen, A-21285). Hydrogen peroxide (3%) was used to block endogenous peroxidase activity and an avidin-biotin conjugate detection system (Vector Laboratories, PK-4001) and diaminobenzidine substrate kit (Cell Signalling Technology, 8059) was used to develop and detect the signal. For immunofluorescent staining, goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 (Invitrogen, A11031) were used. Slides were then imaged using a Zeiss Axiovert inverted fluorescence microscope (Carl Zeiss AG, Jena, Germany) and intensity of staining quantified using QuPath software.
2.4. Subcellular fractionation and western blotting
Freshly harvested aliquots of liver were homogenized in ice cold mitochondrial isolation buffer (220 mM mannitol, 70 mM sucrose, 2.5 mM HEPES, 10 mM EDTA, 1 mM EGTA, 0.1% bovine serum albumin, pH = 7.4). The homogenate was centrifugated at 2500 g for 10 min to remove debris and nuclear material. The supernatant was then centrifugated at 20,000 g for 10 min to pellet mitochondria. The supernatant, which constitutes the cytosolic fraction (including microsomes), was collected, and the pellets were washed twice in isolation buffer and then lysed using lysis buffer. Protein quantification was performed on the cytosolic and mitochondrial fractions using the BCA assay (Thermofisher Scientific, Waltham, Massachusetts). To perform the western blots on the mitochondrial and cytosolic fractions, proteins were separated via gel electrophoresis on polyacrylamide gels followed by transfer to a nitrocellulose membrane. The membranes were blocked in 5% BSA and incubated overnight with the following primary antibodies at 1:1000 dilutions: rabbit anti-AIF (CST, 4642S), rabbit anti-β-actin (CST, 4967S), mouse anti-cytochrome c (Santa Cruz, sc-13,156), rabbit anti-JNK (CST, 9252), and p-JNK (CST, 9251). The membranes were then probed with HRP-conjugated secondary antibodies: goat anti-rabbit HRP, 1:5000 (Invitrogen, 32,460) and goat anti-mouse HRP, 1:3000 (Santa Cruz, sc-2302) and visualized using chemiluminescence detecting reagents (Cytiva, Malborough, Massachusetts) on a LI-COR Odyssey Imager (LI-COR Biosciences, Lincoln, Nebraska). Densitometric analysis was performed on LI-COR Image Studio (LI-COR Biosciences) to quantify the difference in signal intensity of the bands.
2.5. Statistical analysis
Comparisons between two groups were made using the Student’s t-test. Comparisons between more than two groups were made using one way analysis of variance (ANOVA) followed by a post-hoc Tukey test. P < 0.05 was considered to be statistically significant. Statistical analysis was performed on GraphPad Prism version 8.0.1 for Windows (GraphPad Software, San Diego, California).
3. Results
3.1. Effect of iron chelation in APAP hepatotoxicity
Treatment of overnight fasted mice with 300 mg/kg APAP caused extensive liver injury by 6 h as indicated by the high levels of plasma ALT activities (Fig. 1A) and the extensive area of centrilobular necrosis (Fig. 1B, C). Pre-treatment for 60 min with 200 mg/kg of the iron chelator deferoxamine (DFO) before APAP resulted in significant attenuation of liver injury as shown by an 80% decrease in ALT release and 65% smaller necrotic areas (Fig. 1A–C). This protection was not caused by changes in the baseline GSH content as the levels in DFO-treated animals (4.01 ± 0.18 μmol GSH/g liver, n = 3) were similar to controls (3.79 ± 0.23 μmol GSH/g liver) before injection of APAP. Interestingly, the baseline levels of MDA (32.0 ± 6.5 nmol/g protein) did not significantly increase after APAP treatment (30.0 ± 4.3) and were not affected by additional DFO administration (27.8 ± 3.7) reiterating that APAP did not induce relevant LPO. In contrast, APAP induced substantial nitro-tyrosine staining as an indicator of peroxynitrite formation, which was significantly attenuated by DFO (Fig. 2A). JNK activation and translocation of P-JNK to mitochondria are critical early events in APAP toxicity (Hanawa et al., 2008), which could be confirmed in these animals (Fig. 2B). However, DFO had no effect on these parameters (Fig. 2B) suggesting that the protective effect of iron chelation had to be at the level of preserving mitochondrial function. Permeabilization of the outer mitochondrial membrane with release of intermembrane proteins such as cytochrome c and AIF into the cytosol are hallmarks of APAP-induced necrotic cell death (Jaeschke et al., 2019). Consistent with these previous observations, there was a significant increase in cytosolic levels of both cytochrome c and AIF 6 h after APAP (Fig. 2C). DFO significantly attenuated mitochondrial cytochrome c release and caused a trend to lower AIF levels (Fig. 2C). Together, these data suggest that the protective effect of DFO correlates with reduced nitro-tyrosine protein adducts formation and reduced mitochondrial dysfunction but not LPO.
Fig. 1.
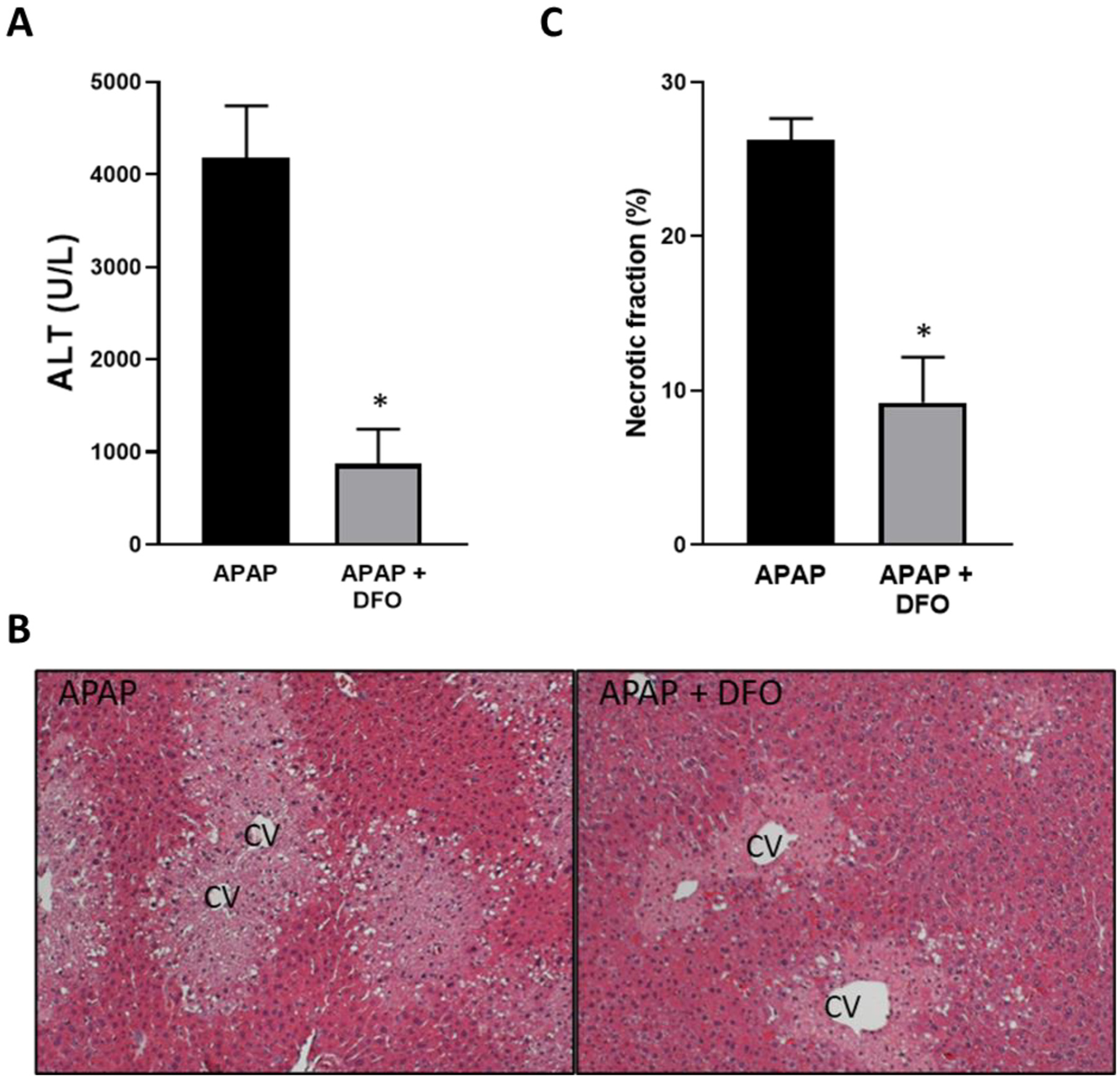
Effect of pre-treatment with deferoxamine (DFO) on APAP-induced liver injury. Mice were injected with 200 mg/kg DFO 1 h before 300 mg/kg APAP (i.p.). Blood and liver tissue were collected at 6 h after APAP. (A) Plasma alanine amino transferase (ALT) activities. (B) H&E-stained liver sections. (C) Quantification of areas of necrosis. Bars represent mean ± SEM for n = 5 mice. *p < 0.05 compared to APAP.
Fig. 2.
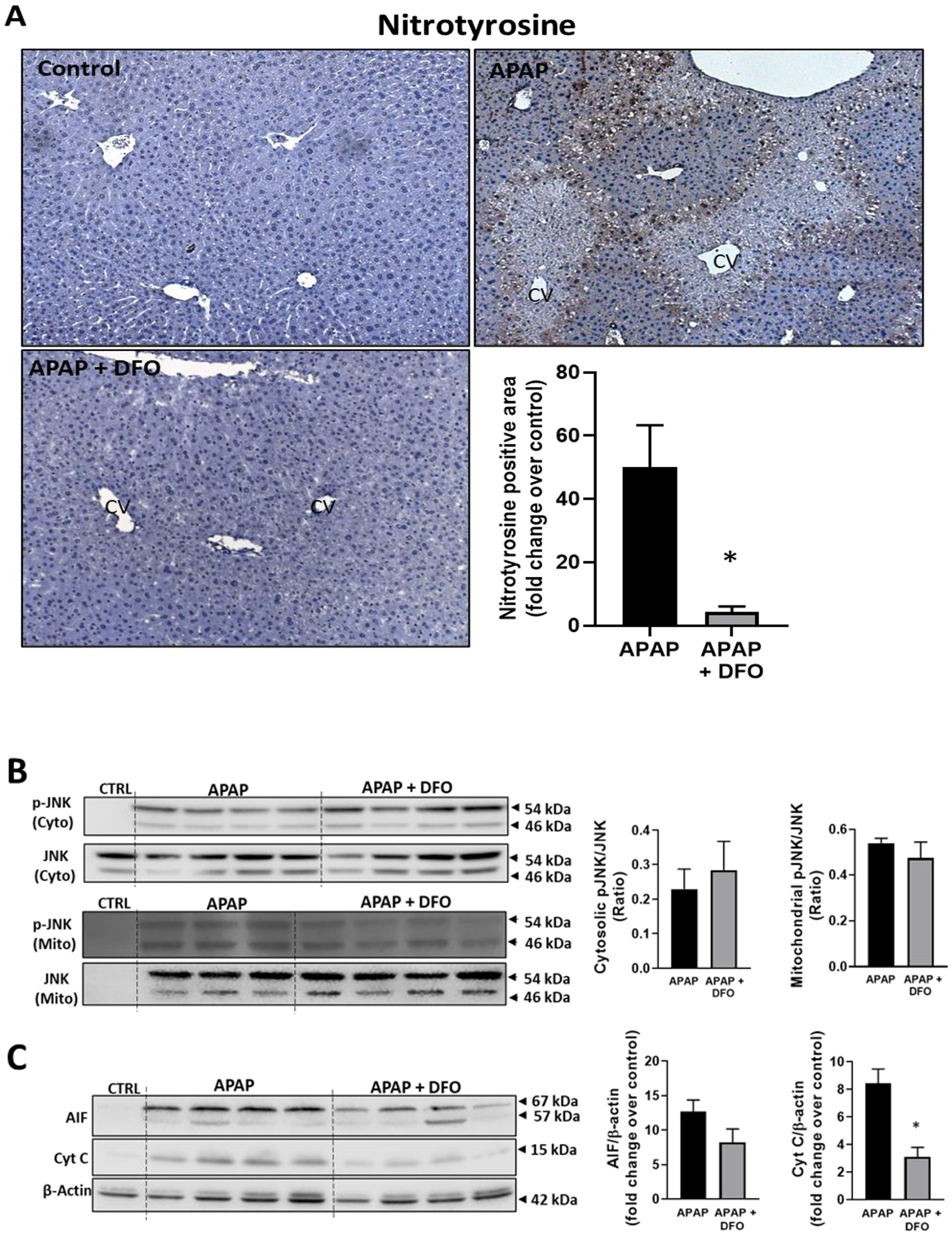
Effect of pre-treatment with DFO on protein nitration, JNK activation and MPTP opening. Mice were injected with 200 mg/kg DFO 1 h before 300 mg/kg APAP (i.p.). Blood and liver tissue were collected at 6 h after APAP. (A) Immunostaining for nitro-tyrosine protein adducts and quantification of positive stained areas. (B) Western blots and densitometric quantification of mitochondrial and cytosolic JNK and p-JNK. (C) Western blots and densitometric quantification of cytosolic AIF and cytochrome c. Bars represent mean ± SEM for n = 3–4 mice. *p < 0.05 compared to APAP.
3.2. Effect of minocycline on APAP hepatotoxicity
Minocycline is an inhibitor of the mitochondrial Ca2+, Fe2+ uniporter (Schwartz et al., 2013) and was shown to protect against APAP toxicity in vitro (Hu et al., 2016) and in vivo (Hu and Lemasters, 2020). Treatment with 10 mg/kg of minocycline 60 min before 300 mg/kg APAP attenuated APAP-induced ALT release by 59% and the area of necrosis by 65%, confirming the protective effect of minocycline in this model (Fig. 3A–C). Again, this protection was not caused by changes in the hepatic GSH content as the levels in minocycline-treated animals (3.64 ± 0.14 μmol GSH/g liver, n = 3) were similar to controls (3.79 ± 0.23 μmol GSH/g liver) before injection of APAP. Baseline MDA levels (29.5 ± 3.4 nmol/g protein) were not significantly increased after an APAP overdose (35.9 ± 2.0); minocycline had no effect on these results (34.1 ± 6.3). In contrast, minocycline extensively reduced APAP-induced nitro-tyrosine staining (Fig. 4A). Like DFO, minocycline did not affect JNK activation and P-JNK translocation to mitochondria (Fig. 4B) but significantly attenuated the increased cytochrome c and AIF release from mitochondria (Fig. 4C).
Fig. 3.
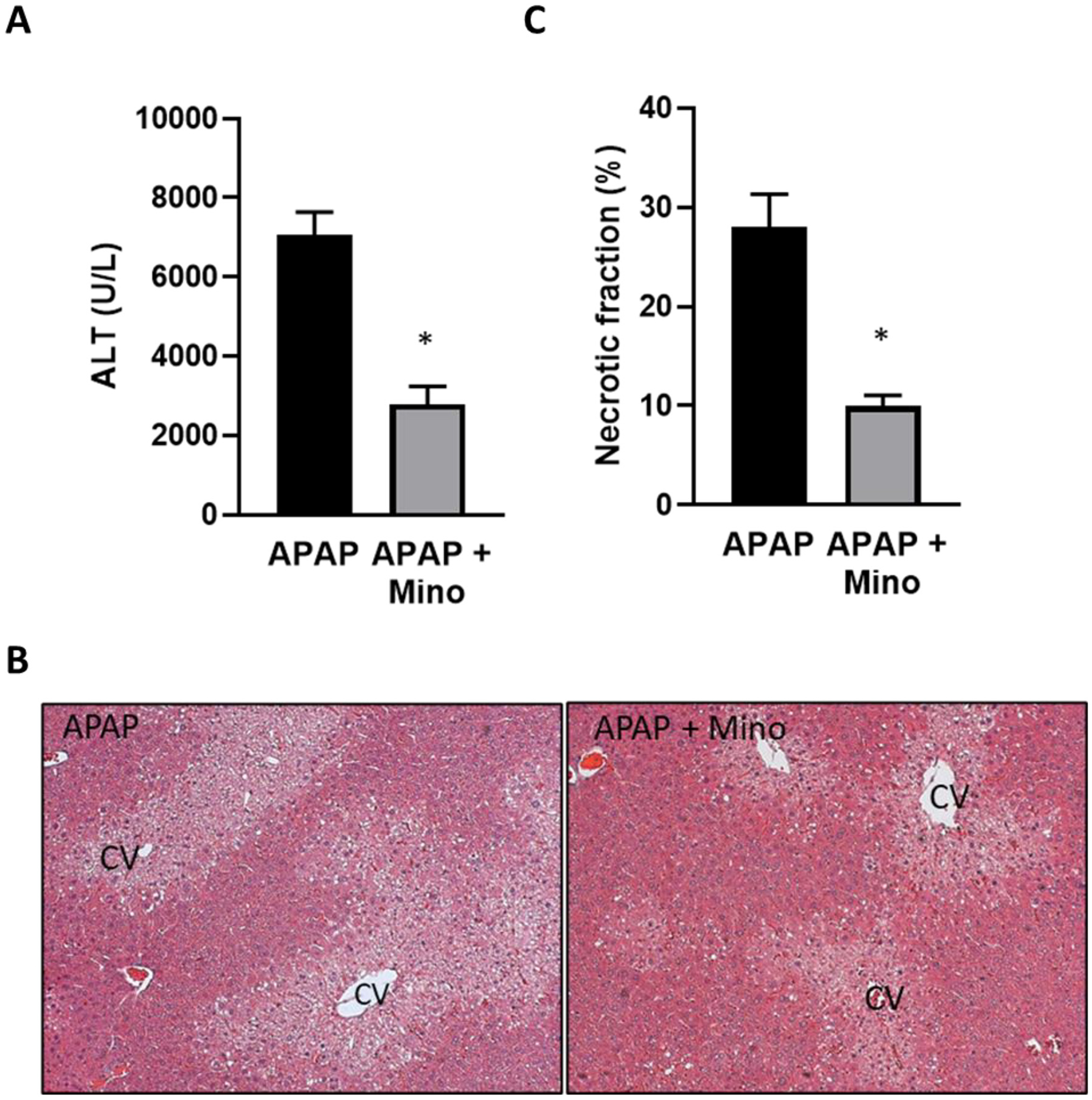
Effect of pre-treatment with minocycline on APAP-induced liver injury. Mice were injected with 10 mg/kg minocycline 1 h before 300 mg/kg APAP (i.p.). Blood and liver tissue were collected at 6 h after APAP. (A) Plasma alanine amino transferase (ALT) activities. (B) H&E-stained liver sections. (C) Quantification of areas of necrosis. Bars represent mean ± SEM for n = 5 mice. *p < 0.05 compared to APAP.
Fig. 4.
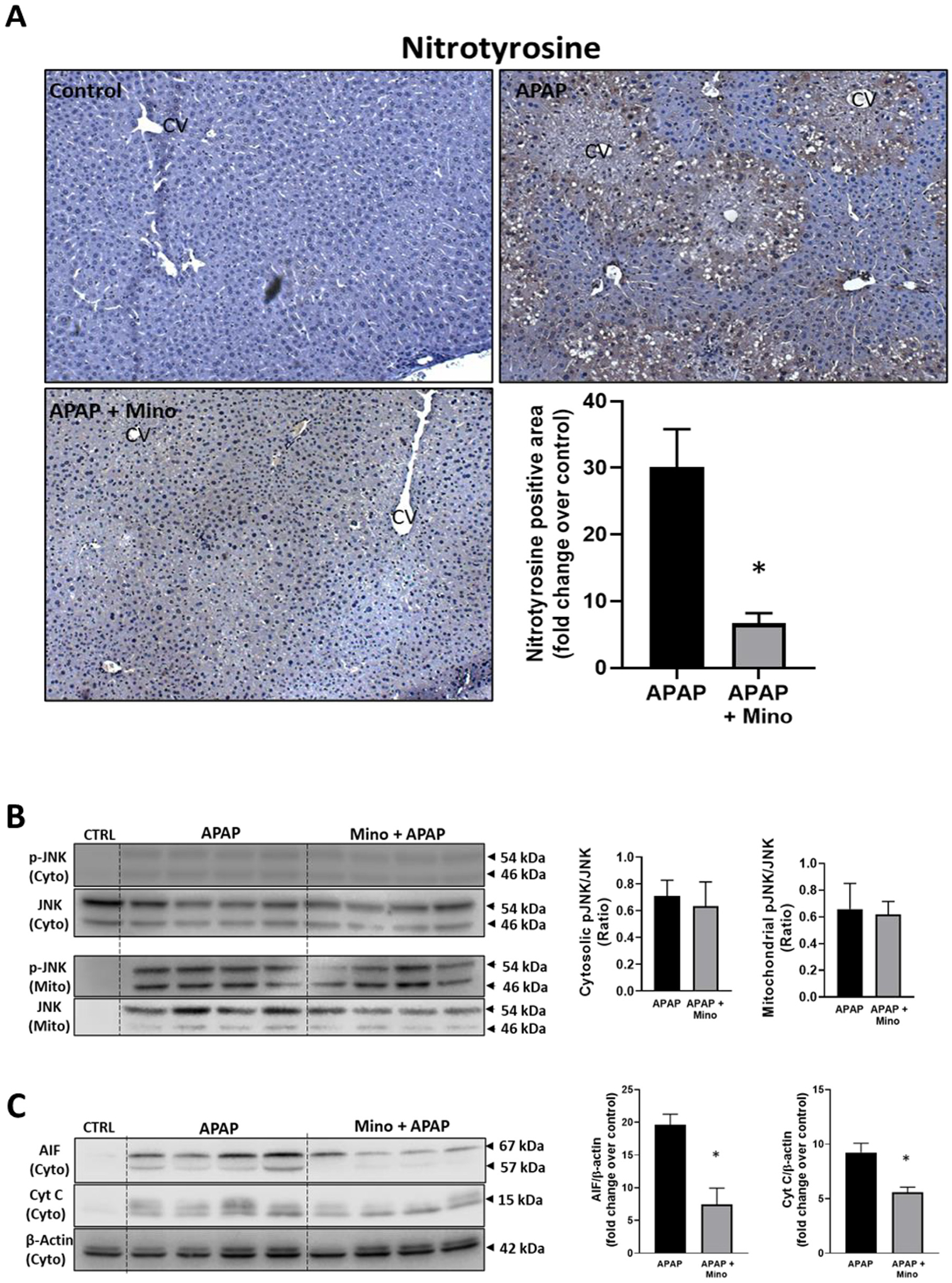
Effect of pre-treatment with minocycline on protein nitration, JNK activation and MPTP opening. Mice were injected with 10 mg/kg minocycline 1 h before 300 mg/kg APAP (i.p.). Blood and liver tissue were collected at 6 h after APAP. (A) Immunostaining for nitro-tyrosine protein adducts and quantification of positive stained areas. (B) Western blots and densitometric quantification of mitochondrial and cytosolic JNK and p-JNK. (C) Western blots and densitometric quantification of cytosolic AIF and cytochrome c. Bars represent mean ± SEM for n = 3–4 mice. *p < 0.05 compared to APAP.
3.3. Aggravation of APAP hepatotoxicity by iron supplementation
To evaluate the effect of iron loading on APAP toxicity, animals were co-treated with 0.15 mmol/kg FeSO4 and 300 mg/kg APAP. Iron treatment dramatically increased APAP-induced liver injury at 6 h as indicated by the 385% increase of plasma ALT activities in the APAP+Fe2+ group compared to APAP alone and the 75% increase of the area of necrosis (Fig. 5). Interestingly, pre-treatment of the FeSO4 group with DFO did not only prevent the aggravation of liver injury but caused further protection like the DFO effect in APAP-treated animals alone (Fig. 1). Consistent with the other data, FeSO4 did not affect JNK activation or mitochondrial translocation (Fig. 6B) but enhanced nitro-tyrosine staining and the release of AIF and cytochrome c from mitochondria into the cytosol (Fig. 6C). DFO pre-treatment almost completely prevented nitro-tyrosine staining and the mitochondrial release of AIF and cytochrome c (Fig. 6). However, FeSO4 treatment also caused a 656% increase of MDA levels over baseline and more extensive 4-hydroxy-nonenal (4-HNE) staining suggesting significant LPO in these animals (Fig. 7). These effects were eliminated with DFO pre-treatment (Fig. 7).
Fig. 5.
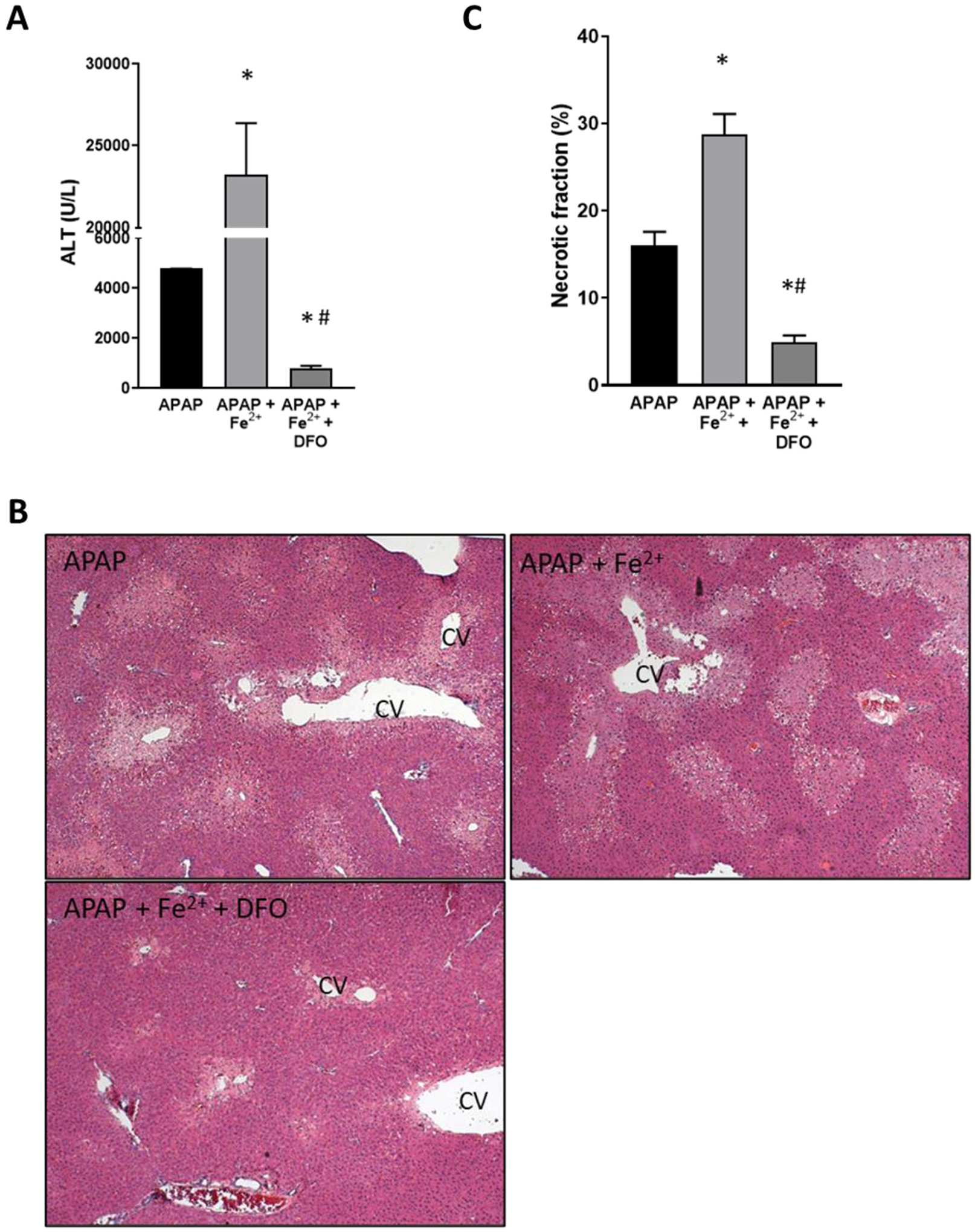
Effect of co-treatment of FeSO4 with APAP on liver injury. Mice were injected with DFO 200 mg/kg or saline 1 h before receiving i.p. injections of 300 mg/kg APAP with or without 0.15 mmol/kg FeSO4. Blood and liver tissue were collected at 6 h after APAP. (A) Plasma alanine amino transferase (ALT) activities. (B) H&E-stained liver sections. (C) Quantification of areas of necrosis. Bars represent mean ± SEM for n = 5 mice. *p < 0.05 compared to APAP, #p < 0.05 compared to APAP + FeSO4.
Fig. 6.
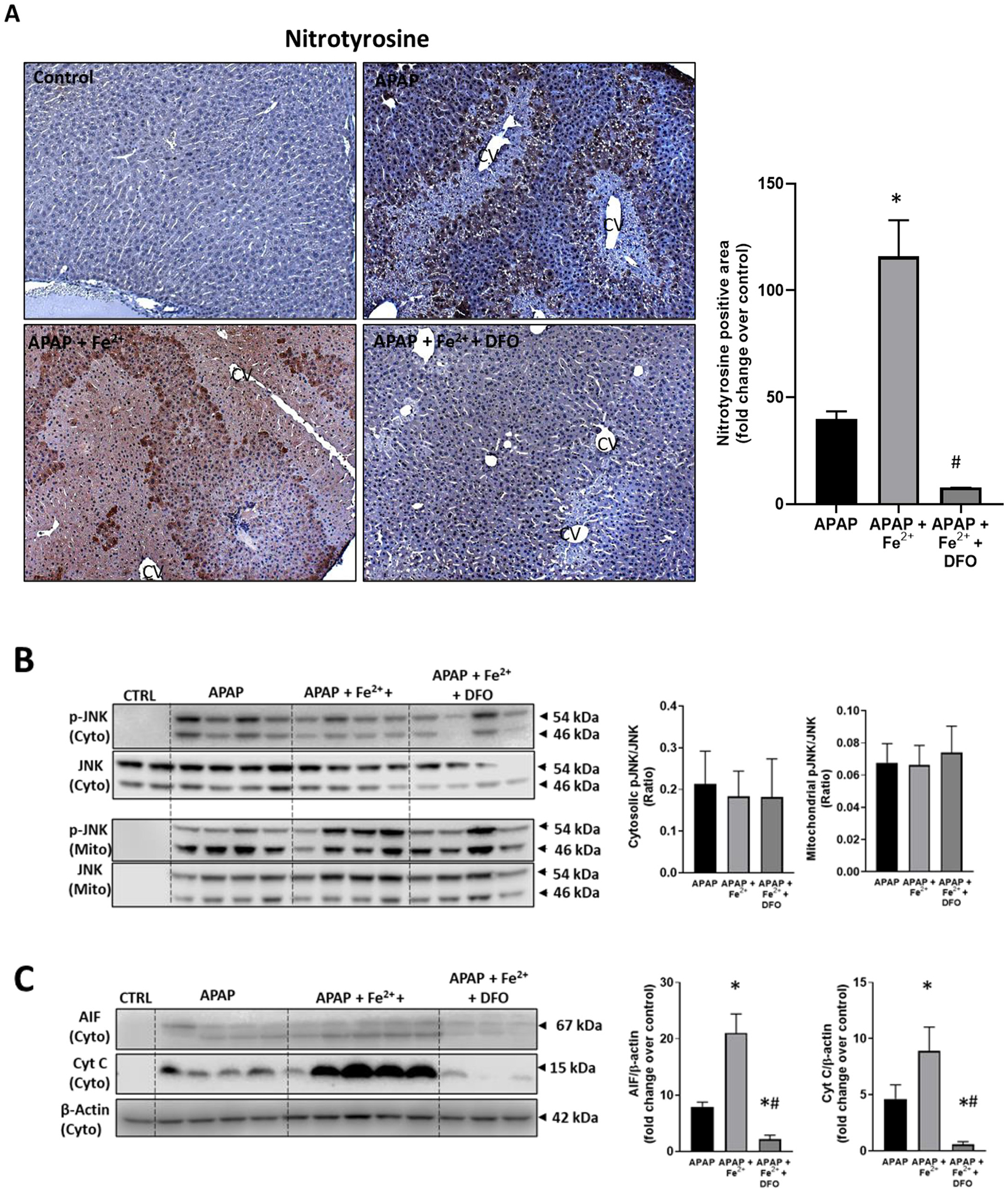
Effect of co-treatment of FeSO4 with APAP on protein nitration, JNK activation and MPTP opening. Mice were injected with DFO 200 mg/kg or saline 1 h before receiving i.p. injections of 300 mg/kg APAP with or without 0.15 mmol/kg FeSO4. Blood and liver tissue were collected at 6 h after APAP. (A) Immunostaining for nitro-tyrosine protein adducts and quantification of positive stained areas. (B) Western blots and densitometric quantification of mitochondrial and cytosolic JNK and p-JNK. (C) Western blots and densitometric quantification of cytosolic AIF and cytochrome c. Bars represent mean ± SEM for n = 3–4 mice. *p < 0.05 compared to APAP, #p < 0.05 compared to APAP + FeSO4.
Fig. 7.
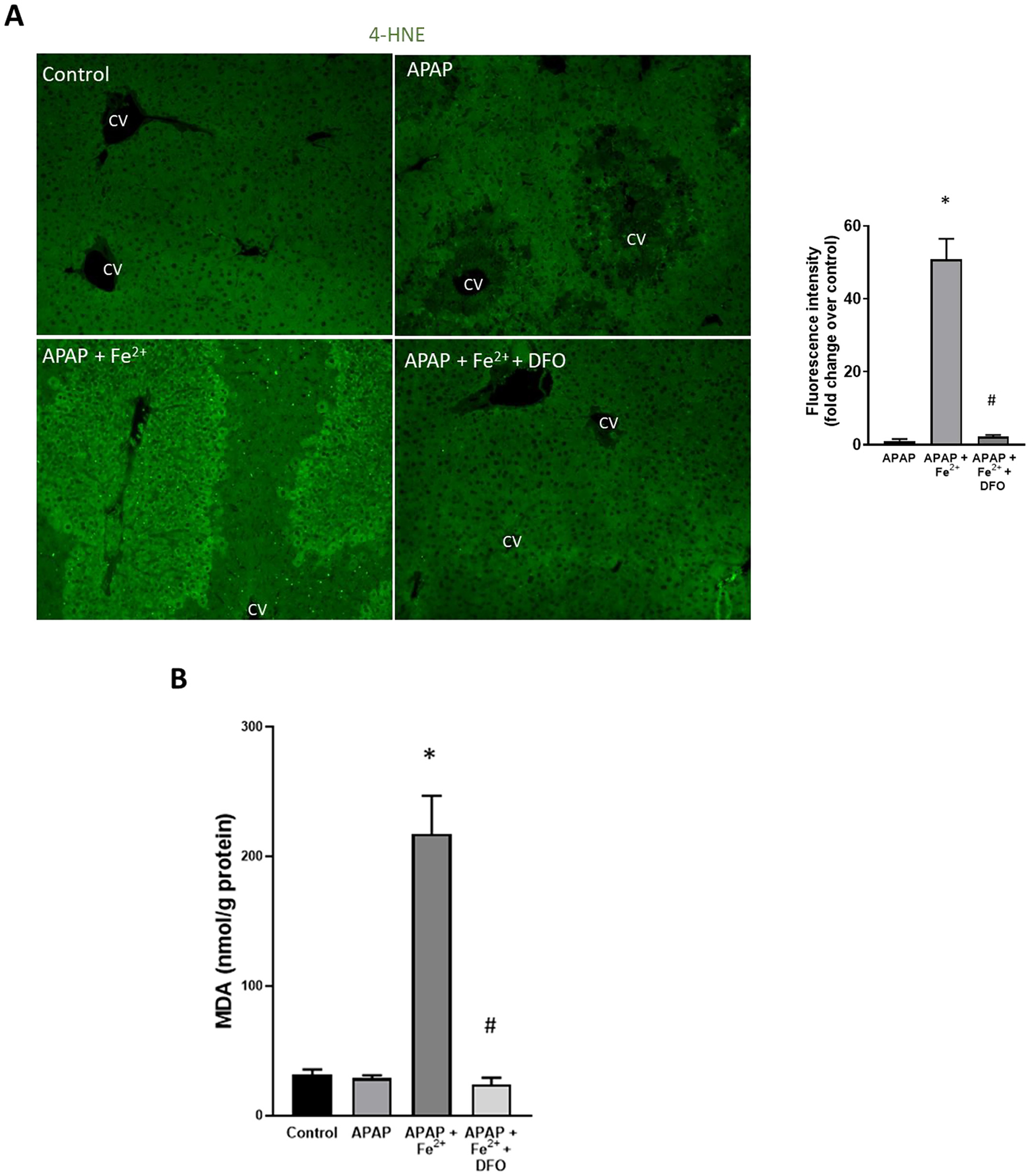
Effect of co-treatment of FeSO4 with APAP on lipid peroxidation. Mice were injected with DFO 200 mg/kg or saline 1 h before receiving i.p. injections of 300 mg/kg APAP with or without 0.15 mmol/kg FeSO4. Blood and liver tissue were collected at 6 h after APAP. (A) Immunofluorescence staining for 4-hydroxynonenal (4-HNE) and quantification of fluorescence intensity. (B) Hepatic malondialdehyde (MDA) content. Bars represent mean ± SEM for n = 5 mice. *p < 0.05 compared to APAP, # < 0.05 compared to APAP + FeSO4.
4. Discussion
4.1. Role of iron in APAP hepatotoxicity
The objective of this investigation was to assess a potential role of iron in APAP-induced liver injury. Previous studies reported conflicting results including that chelation of iron with deferoxamine had no effect on the injury (Younes and Siegers, 1985; Younes et al., 1988), caused significant protection (Sakaida et al., 1995; Yamada et al., 2020), or only delayed the injury (Schnellmann et al., 1999). More recent studies indicated that ferrous iron is released from lysosomes after an APAP overdose in mice (Kon et al., 2010) and is transported into mitochondria through the mitochondrial Ca2+, Fe2+ uniporter (Hu et al., 2016); both the specific chelation of lysosomal iron and the inhibition of iron uptake into mitochondria by minocycline protected against APAP hepatotoxicity (Hu et al., 2016; Hu and Lemasters, 2020; Kon et al., 2010). Our data confirmed these findings by showing the extensive protection of the iron chelator deferoxamine and the mitochondrial Ca2+, Fe2+ uniporter inhibitor minocycline. In addition, we could show that co-treatment of APAP with FeSO4 dramatically aggravated APAP-induced liver injury, which could be prevented by iron chelation with DFO. However, important for the proper interpretation of these data is the question if any of the interventions affected the metabolic activation of APAP. In fact, it was previously reported that DFO did not affect protein adduct formation (Schnellmann et al., 1999) and that minocycline did not prevent the initial GSH depletion (Hu and Lemasters, 2020) after APAP overdose. In addition, we showed here that neither DFO, minocycline nor Fe2+ treatment affected JNK activation in the cytosol or impacted its translocation to mitochondria (Figs. 2,4,6). Because NAPQI formation and mitochondrial protein adducts induce the initial mitochondria-derived oxidant stress in the cytosol that ultimately triggers JNK activation (Nguyen et al., 2021), P-JNK levels in the cytosol and mitochondria are sensitive markers of the metabolic activation of APAP. Thus, based on direct assessment of protein adducts, GSH depletion kinetics and of JNK activation, it can be concluded that DFO, minocycline and Fe2+ did not affect the early events in the toxicity of APAP but acted at the level of the mitochondria. This hypothesis was further confirmed by the observation that AIF and cytochrome c release from the intermembrane space of the mitochondria were reduced with iron chelation. Although these proteins can be released by a Bax pore (Bajt et al., 2008), the bulk of the release of intermembrane proteins is triggered by the rupture of the outer mitochondrial membrane due to MPTP-induced matrix swelling (Ramachandran and Jaeschke, 2019). In addition, the reduction of nitro-tyrosine staining by DFO indicated reduced peroxynitrite formation, which takes place predominantly inside the mitochondria (Cover et al., 2005), also points towards an iron effect within the mitochondria. This conclusion is further supported by previous in vitro experiments where calcein and mitoferrofluor fluorescence quenching was used as indicator of cytosolic and mitochondrial chelatable Fe2+, respectively. In these studies, it was shown that the lysosomal targeted iron chelator starch-desferal reduces cytosolic and mitochondrial Fe2+ while protecting against APAP-induced cell death (Hu et al., 2016). However, the inhibitors of the mitochondrial electrogenic Ca2+, Fe2+ uniporter Ru360 and minocycline only reduced mitochondrial iron while protecting against APAP-induced injury (Hu et al., 2016). This suggests that there is still lysosomal iron release with minocycline, which seems to have limited pathophysiological consequences if Fe2+ is not taken up into mitochondria. Together, our findings are consistent with previous reports of lysosomal iron being taking up into mitochondria and being involved in triggering the MPTP opening and APAP-induced cell death (Hu et al., 2016; Hu and Lemasters, 2020; Kon et al., 2010).
4.2. Role of iron in lipid peroxidation and protein nitration
Free iron is generally considered an integral part of the Fenton reaction to initiate and propagate LPO, which is widely considered an important pathophysiological event. However, the role of LPO has been questioned in APAP toxicity (Du et al., 2016; Ramachandran and Jaeschke, 2021). One central argument against the relevance of LPO is that this process in APAP hepatotoxicity is quantitatively insufficient to cause cell death (Jaeschke and Ramachandran, 2018). All studies in the literature that measure parameters of LPO in APAP toxicity report no evidence of LPO or increases of these parameters of at most 200% of baseline levels (Jaeschke and Ramachandran, 2018). In contrast, LPO-mediated cell death in vitamin E-deficient mice correlates with increases of LPO parameters of 1000–3000% above baseline (Wendel and Feuerstein, 1981; Jaeschke et al., 1987). This is consistent with our finding with iron co-treatment, where MDA levels increased by 656% (Fig. 7). Thus, under normal conditions, i.e., in the absence of iron overload or serious deficiency of vitamin E, the endogenous antioxidant defense mechanisms are sufficient to prevent excessive LPO that could directly cause cell death. This conclusion is also supported by the fact that increasing vitamin E levels in the liver by 7-fold did not protect against APAP toxicity but reduced liver injury in animals pre-treated with FeSO4 and exposed to allyl alcohol as a positive control for LPO-mediated hepatotoxicity (Knight et al., 2003).
Several decades ago, it was recognized that the APAP-induced centrilobular necrosis correlated with nitro-tyrosine staining in the same areas suggesting peroxynitrite formation (Hinson et al., 1998). Peroxynitrite, a spontaneous reaction product between two radicals, i.e., superoxide and nitric oxide, is a potent oxidant and a nitrating species (Ferrer-Sueta and Radi, 2009). Whereas the mitochondrial electron transport chain as source of superoxide in this context is undisputed (Knight et al., 2001; Nguyen et al., 2021), the specific nitric oxide synthase (NOS) generating nitric oxide remained unclear for some time (Hinson et al., 2002). More recently evidence was provided that neuronal NOS (NOS1) may be the main source of nitric oxide during APAP toxicity (Agarwal et al., 2012; Banerjee et al., 2015). In addition, scavenging of peroxynitrite with endogenous GSH and preventing its formation with mitochondria-targeted SOD mimetics effectively protected against APAP-induced liver injury (Du et al., 2017; James et al., 2003; Knight et al., 2002), suggesting that peroxynitrite is the dominant cytotoxic mediator in the pathophysiology.
Based on the experimental data that dispute LPO as a relevant injury mechanism but support peroxynitrite as the critical toxicant in APAP toxicity, the question remained how iron can affect peroxynitrite toxicity. Based on work by Radi and coworkers, peroxynitrite can spontaneously decompose or react with various chemical entities in biological systems (Campolo et al., 2014). One direct reaction could be with thiols, which would oxidize the thiol to sulfenic acid and reduce peroxynitrite to nitrite () (Campolo et al., 2014). In case of protein thiols, this may cause protein dysfunction and contribute to cell toxicity; in case of interaction with hepatic GSH, this represents a detoxification reaction that consumes GSH (Knight et al., 2002). However, this detoxification reaction is limited after an APAP overdose due to the extensive and prolonged depletion of hepatic GSH (McGill et al., 2013). Nevertheless, this reaction becomes important when animals are treated with GSH precursors (N-acetylcysteine) that facilitate GSH synthesis. Under these conditions, peroxynitrite is scavenged and nitro-tyrosine formation is prevented, and the animals are effectively protected (James et al., 2003; Knight et al., 2002; Saito et al., 2010b). Thus, in the presence of sufficient thiol reagents such as GSH, protein nitration and toxicity are prevented. However, in the absence of thiol reagents, peroxynitrite can react with transition metals such as Fe, Cu or Mn, which can reduce peroxynitrite by one electron thereby producing nitrogen dioxide (•NO2) and a high oxidation state oxo-metal complex (O = Me(n + 1)+). This oxo-metal complex can oxidize tyrosine to a tyrosyl radical, which then reacts with •NO2 to form nitro-tyrosine protein adducts (Campolo et al., 2014). Thus, it is likely that during the early phase after APAP administration when hepatic and mitochondrial GSH levels are maximally depleted, the lack of thiols causes peroxynitrite generated inside the mitochondrial matrix to react with transition metals, e.g., Fe2+ imported into mitochondria through the Ca2+, Fe2+ uniporter, to cause extensive protein nitration leading to the MPTP opening and mitochondrial dysfunction. The dramatic reduction of protein nitration with DFO (iron chelation) and minocycline (inhibition of mitochondrial iron uptake) is consistent with this mechanism and would reconcile most findings regarding the role of peroxynitrite and iron in APAP toxicity.
Despite the lack of evidence of relevant LPO, a recent paper showed a beneficial effect of an inducer of the mitochondrial aldehyde dehydrogenase 2 (Wimborne et al., 2020) suggesting that a very localized and limited LPO within mitochondria cannot be completely ruled out. This local LPO may not be sufficient to cause the MPTP opening alone but may act in combination with the detrimental effects of peroxynitrite to cause mitochondrial dysfunction. Thus, cell death may be the result of multiple simultaneous insults at the level of mitochondria, which could explain why the injury is attenuated by highly diverse interventions (Jaeschke et al., 2021). This potential co-operative effect between two iron-dependent reactions within mitochondria, i.e., peroxynitrite-mediated protein nitration and localized LPO, requires further investigation.
4.3. Iron overload and acetaminophen hepatotoxicity
In contrast to normal conditions, animals co-treated with APAP and FeSO4, showed not only a further increase in nitro-tyrosine staining, consistent with the role of iron in protein nitration, but also substantial LPO, consistent with iron-mediated Fenton-type reactions. This is similar to effects of iron in allyl alcohol toxicity, where the alcohol itself does not cause injury but GSH depletion and iron treatment in combination with allyl alcohol dramatically induced LPO and liver injury (Knight et al., 2003). In addition, both APAP and allyl alcohol induced dramatic LPO in animals fed a diet high in polyunsaturated fatty acids and deficient of vitamin E (Wendel and Feuerstein, 1981; Jaeschke et al., 1987). Based on these previous data and the current findings, it can be concluded that under normal conditions APAP overdose induces peroxynitrite formation in the mitochondrial matrix and its reaction with imported iron facilitates protein nitration and toxicity largely independent of LPO. On the one hand, this would suggest that under these conditions endogenous defense mechanisms (iron chelation, vitamin E) are sufficient to prevent excessive LPO that could cause cell death. On the other hand, due to excessive GSH depletion, thiols are lacking that could effectively scavenge peroxynitrite, and the imported iron is sufficient to promote protein nitration but is insufficient to induce significant LPO. However, if one of these defense systems that keep LPO in check is overwhelmed (excessive iron) or weakened (not enough chain-breaking vitamin E), sufficient LPO can be induced to significantly contribute to the overall injury process. Given the ubiquitous nature of vitamin E in human diet, it would be difficult to achieve sufficiently low levels of vitamin E to increase the susceptibility to APAP overdose in humans. However, the availability of iron supplements raises the possibility that excessive iron can be ingested together with an APAP overdose in a suicide attempt (Audimoolam et al., 2011; Nye and Singh, 2022). Under these specific conditions, it may be possible that the mechanism of cell death could involve LPO in patients. As our data showed, use of iron chelators might be beneficial by limiting both protein nitration and LPO.
4.4. Summary and conclusions
Our data demonstrated that both iron chelation with DFO and preventing iron uptake into the mitochondria by minocycline effectively protected against APAP overdose at the level of mitochondria. Importantly, the intervention did not affect LPO, but attenuated nitro-tyrosine protein adduct formation. Given the known biochemical mechanisms of protein nitration requiring transition metals (Campolo et al., 2014), our data are consistent with a role of iron predominantly in protein nitration (Fig. 8). However, despite the critical role of peroxynitrite and protein nitration in APAP hepatotoxicity under normal circumstances, a contribution of a very localized and limited LPO in mitochondria cannot be ruled out at this time (Fig. 8). Another important aspect of our study is the fact that co-treatment with FeSO4 increased protein nitration but also caused extensive LPO and dramatically aggravated liver injury. These data are consistent with the paradigm that LPO is not part of the regular pathophysiology but if some of the defense systems are impaired (excessive free iron, lack of vitamin E) significant LPO can develop and then may be part of the injury process (Fig. 8). This scenario, although rare, could play a role when patients ingest not only an overdose of APAP but also of iron supplements as shown in several case reports (Audimoolam et al., 2011; Nye and Singh, 2022). Under these conditions, iron chelation therapy might be beneficial by limiting both protein nitration and LPO.
Fig. 8.
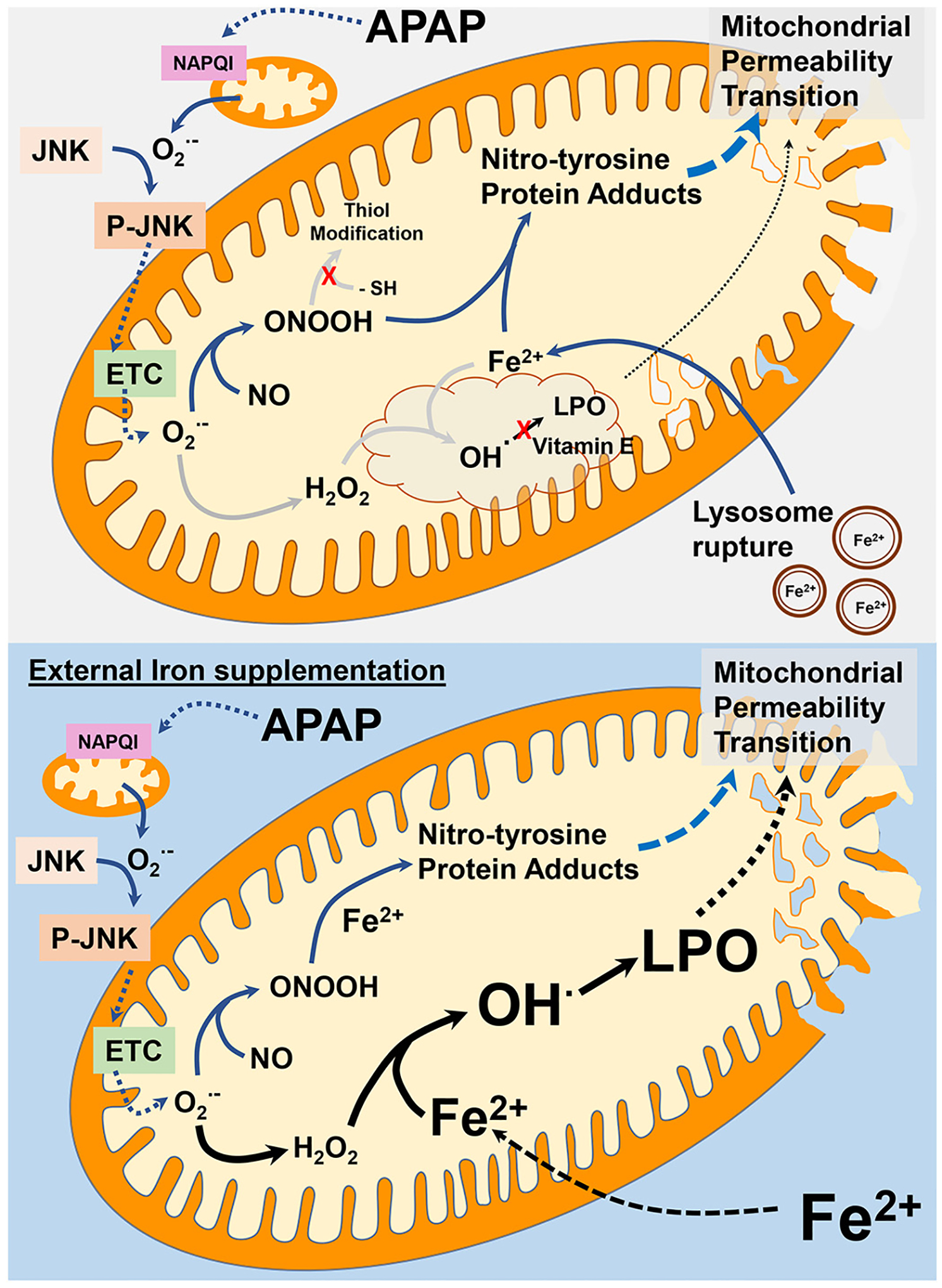
Role of iron in APAP-induced mitochondrial damage. Under typical APAP overdose conditions, iron mediated lipid peroxidation (LPO) does not play a significant role in APAP hepatotoxicity. Formation of mitochondrial NAPQI protein adducts after APAP metabolism induces superoxide-mediated JNK activation and its mitochondrial translocation. This amplifies mitochondrial superoxide generation through electron transport chain (ETC) dysfunction, which reacts with nitric oxide to produce peroxynitrite. Though peroxynitrite can be scavenged by thiol groups of GSH, this does not occur because of the significant GSH depletion due to NAPQI detoxification. The influx of iron into mitochondria from lysosomal instability instead facilitates peroxynitrite reaction with iron to produce nitro-tyrosine protein adducts, ultimately inducing the mitochondrial permeability transition. Since vitamin E levels are typically maintained after APAP, any hydroxyl and alkoxyl radicals generated by reaction of superoxide derived hydrogen peroxide or fatty acid hydroperoxide with iron are rapidly scavenged, preventing relevant lipid peroxidation. In a situation of significant iron loading, however, these reactions could be overwhelmed by the excess iron which causes extensive lipid peroxidation along with nitro-tyrosine formation to induce the mitochondrial permeability transition.
Acknowledgements
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01 DK102142 (HJ), R01 DK125465 (AR) and R01 DK119523 (JJL), National Institute of General Medicine (NIGMS)-funded Liver Disease COBRE grants P20 GM103549 (HJ) and P30 GM118247 (HJ); and National Institute of Environmental Health Sciences (NIEHS) grant R21 ES031335 (JJL).
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests.
Hartmut Jaeschke reports financial support was provided by National Institutes of Health. Anup Ramachandran reports financial support was provided by National Institutes of Health. John Lemasters reports financial support was provided by National Institutes of Health. Hartmut Jaeschke reports a relationship with Johnson & Johnson Consumer Companies Inc. that includes: consulting or advisory and funding grants.
Abbreviations:
- AIF
apoptosis-inducing factor
- ALT
alanine aminotransferase
- APAP
acetaminophen
- DFO
deferoxamine
- GSH
glutathione
- H&E
hematoxylin & eosin
- HNE
4-hydroxy-nonenal
- JNK
c-Jun N-terminal kinase
- LPO
lipid peroxidation
- MDA
malondialdehyde
- MPTP
mitochondrial permeability transition pore
- NAPQI
N-acetyl-p-benzoquinone imine
- SOD
superoxide dismutase
Footnotes
CRediT authorship contribution statement
Olamide B. Adelusi: Formal analysis, Investigation, Writing – original draft. Anup Ramachandran: Formal analysis, Investigation, Writing – review & editing, Visualization. John J. Lemasters: Conceptualization, Writing – review & editing. Hartmut Jaeschke: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
References
- Agarwal R, Hennings L, Rafferty TM, Letzig LG, McCullough S, James LP, MacMillan-Crow LA, Hinson JA, 2012. Acetaminophen-induced hepatotoxicity and protein nitration in neuronal nitric-oxide synthase knockout mice. J. Pharmacol. Exp. Ther 340, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audimoolam VK, Wendon J, Bernal W, Heaton N, O’Grady J, Auzinger G, 2011. Iron and acetaminophen a fatal combination? Transpl. Int 24, e85–e88. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Farhood A, Lemasters JJ, Jaeschke H, 2008. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther 324, 8–14. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Melnyk SB, Krager KJ, Aykin-Burns N, Letzig LG, James LP, Hinson JA, 2015. The neuronal nitric oxide synthase inhibitor NANT blocks acetaminophen toxicity and protein nitration in freshly isolated hepatocytes. Free Radic. Biol. Med 89, 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal W, Wendon J, 2013. Acute liver failure. N. Engl. J. Med 369, 2525–2534. [DOI] [PubMed] [Google Scholar]
- Campolo N, Bartesaghi S, Radi R, 2014. Metal-catalyzed protein tyrosine nitration in biological systems. Redox Rep 19, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H, 2005. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther 315, 879–887. [DOI] [PubMed] [Google Scholar]
- Du K, Ramachandran A, Jaeschke H, 2016. Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol 10, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Farhood A, Jaeschke H, 2017. Mitochondria-targeted antioxidant Mito-tempo protects against acetaminophen hepatotoxicity. Arch. Toxicol 91, 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Ramachandran A, Weemhoff JL, Woolbright BL, Jaeschke AH, Chao X, Ding WX, Jaeschke H, 2019. Mito-tempo protects against acute liver injury but induces limited secondary apoptosis during the late phase of acetaminophen hepatotoxicity. Arch. Toxicol 93, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Radi R, 2009. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem. Biol 4, 161–177. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Kumagai K, Ito K, Arakawa S, Ando Y, Oda S, Yamoto T, Manabe S, 2009. Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. Toxicol. Pathol 37, 193–200. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H, 2002. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol. Sci 67, 322–328. [DOI] [PubMed] [Google Scholar]
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N, 2008. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem 283, 13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JA, Pike SL, Pumford NR, Mayeux PR, 1998. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem. Res. Toxicol 11, 604–607. [DOI] [PubMed] [Google Scholar]
- Hinson JA, Bucci TJ, Irwin LK, Michael SL, Mayeux PR, 2002. Effect of inhibitors of nitric oxide synthase on acetaminophen-induced hepatotoxicity in mice. Nitric Oxide 6, 160–167. [DOI] [PubMed] [Google Scholar]
- Hu J, Lemasters JJ, 2020. Suppression of iron mobilization from lysosomes to mitochondria attenuates liver injury after acetaminophen overdose in vivo in mice: protection by minocycline. Toxicol. Appl. Pharmacol 392, 114930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Kholmukhamedov A, Lindsey CC, Beeson CC, Jaeschke H, Lemasters JJ, 2016. Translocation of iron from lysosomes to mitochondria during acetaminophen-induced hepatocellular injury: protection by starch-desferal and minocycline. Free Radic. Biol. Med 97, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, 2015. Acetaminophen: dose-dependent drug hepatotoxicity and acute liver failure in patients. Dig. Dis 33, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Ramachandran A, 2018. Oxidant stress and lipid peroxidation in acetaminophen hepatotoxicity. React. Oxyg. Species (Apex) 5, 145–158. [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Ramachandran A, 2020. Acetaminophen-induced apoptosis: facts versus fiction. J. Clin. Transl. Res 6, 36–47. [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Kleinwaechter C, Wendel A, 1987. The role of acrolein in allyl alcohol-induced lipid peroxidation and liver cell damage in mice. Biochem. Pharmacol 36, 51–57. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A, 2012. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev 44, 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Duan L, Akakpo JY, Farhood A, Ramachandran A, 2018. The role of apoptosis in acetaminophen hepatotoxicity. Food Chem. Toxicol 118, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Ramachandran A, Chao X, Ding WX, 2019. Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch. Toxicol 93, 3491–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Adelusi OB, Ramachandran A, 2021. Ferroptosis and acetaminophen hepatotoxicity: are we going down another rabbit hole? Gene Expr 20, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LP, McCullough SS, Lamps LW, Hinson JA, 2003. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol. Sci 75, 458–467. [DOI] [PubMed] [Google Scholar]
- Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H, 2001. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol. Sci 62, 212–220. [DOI] [PubMed] [Google Scholar]
- Knight TR, Ho YS, Farhood A, Jaeschke H, 2002. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J. Pharmacol. Exp. Ther 303, 468–475. [DOI] [PubMed] [Google Scholar]
- Knight TR, Fariss MW, Farhood A, Jaeschke H, 2003. Role of lipid peroxidation as a mechanism of liver injury after acetaminophen overdose in mice. Toxicol. Sci 76, 229–236. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Jaeschke H, Lemasters JJ, 2004. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40, 1170–1179. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ, 2010. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol. Sci 117, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masubuchi Y, Suda C, Horie T, 2005. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol 42, 110–116. [DOI] [PubMed] [Google Scholar]
- McGill MR, Jaeschke H, 2015. A direct comparison of methods used to measure oxidized glutathione in biological samples: 2-vinylpyridine and N-ethylmaleimide. Toxicol. Mech. Methods 25, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, Williams CD, Wilkins DG, Rollins DE, Jaeschke H, 2013. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol. Appl. Pharmacol 269, 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SD, 1990. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis 10, 267–278. [DOI] [PubMed] [Google Scholar]
- Nguyen NT, Du K, Akakpo JY, Umbaugh DS, Jaeschke H, Ramachandran A, 2021. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-Jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol. Lett 338, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye R, Singh T, 2022. Use of CRRT and Plasmapheresis to treat simultaneous Iron and acetaminophen overdose. Blood Purif 51, 292–295. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H, 2019. Acetaminophen hepatotoxicity. Semin. Liver Dis 39, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H, 2021. Oxidant stress and acetaminophen hepatotoxicity: mechanism-based drug development. Antioxid. Redox Signal 35, 718–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H, 2011. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol 251, 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito C, Lemasters JJ, Jaeschke H, 2010a. C-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol 246, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito C, Zwingmann C, Jaeschke H, 2010b. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 51, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaida I, Kayano K, Wasaki S, Nagatomi A, Matsumura Y, Okita K, 1995. Protection against acetaminophen-induced liver injury in vivo by an iron chelator, deferoxamine. Scand. J. Gastroenterol 30, 61–67. [DOI] [PubMed] [Google Scholar]
- Schnellmann JG, Pumford NR, Kusewitt DF, Bucci TJ, Hinson JA, 1999. Deferoxamine delays the development of the hepatotoxicity of acetaminophen in mice. Toxicol. Lett 106, 79–88. [DOI] [PubMed] [Google Scholar]
- Schwartz J, Holmuhamedov E, Zhang X, Lovelace GL, Smith CD, Lemasters JJ, 2013. Minocycline and doxycycline, but not other tetracycline-derived compounds, protect liver cells from chemical hypoxia and ischemia/reperfusion injury by inhibition of the mitochondrial calcium uniporter. Toxicol. Appl. Pharmacol 273, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel A, Feuerstein S, 1981. Drug-induced lipid peroxidation in mice–I. Modulation by monooxygenase activity, glutathione and selenium status. Biochem. Pharmacol 30, 2513–2520. [DOI] [PubMed] [Google Scholar]
- Wimborne HJ, Hu J, Takemoto K, Nguyen NT, Jaeschke H, Lemasters JJ, Zhong Z, 2020. Aldehyde dehydrogenase-2 activation decreases acetaminophen hepatotoxicity by prevention of mitochondrial depolarization. Toxicol. Appl. Pharmacol 396, 114982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win S, Than TA, Han D, Petrovic LM, Kaplowitz N, 2011. C-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial sab protein expression in mice. J. Biol. Chem 286, 35071–35078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win S, Than TA, Min RW, Aghajan M, Kaplowitz N, 2016. C-Jun N-terminal kinase mediates mouse liver injury through a novel sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 63, 1987–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright BL, Ramachandran A, McGill MR, Yan HM, Bajt ML, Sharpe MR, Lemasters JJ, Jaeschke H, 2012. Lysosomal instability and cathepsin B release during acetaminophen hepatotoxicity. Basic Clin. Pharmacol. Toxicol 111, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada N, Karasawa T, Kimura H, Watanabe S, Komada T, Kamata R, Sampilvanjil A, Ito J, Nakagawa K, Kuwata H, Hara S, Mizuta K, Sakuma Y, Sata N, Takahashi M, 2020. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis 11, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Siegers CP, 1985. The role of iron in the paracetamol- and CCl4-induced lipid peroxidation and hepatotoxicity. Chem. Biol. Interact 55, 327–334. [DOI] [PubMed] [Google Scholar]
- Younes M, Sause C, Siegers CP, Lemoine R, 1988. Effect of deferrioxamine and diethyldithiocarbamate on paracetamol-induced hepato- and nephrotoxicity. The role of lipid peroxidation. J. Appl. Toxicol 8, 261–265. [DOI] [PubMed] [Google Scholar]