

Abstract
Background
Constitutional mismatch repair deficiency (CMMRD) results from a biallelic germline pathogenic variant in a mismatch repair (MMR) gene. The most common CMMRD-associated malignancies are brain tumors; an accurate diagnosis is challenging when a malignant brain tumor is the only tumor at presentation. We describe two cases of glioblastoma as the initial CMMRD malignancy and discuss current diagnostic and therapeutic challenges.
Case presentation
Two children with brain tumors without remarkable family history had biallelic pathogenic germline variants in PMS2. Patient 1: A 6-year-old girl presented biallelic PMS2 germline pathogenic variants. Glioblastomas at the left frontal lobe and right temporal lobe were resistant to immune-checkpoint inhibitor, temozolomide, and bevacizumab. Patient 2: A 10-year-old boy presented biallelic PMS2 germline variants. His glioblastoma with primitive neuroectodermal tumor-like features responded to chemoradiotherapy, but he developed advanced colon cancer and acute lymphocytic leukemia. In both patients, only a monoallelic PMS2 germline variant was detected by conventional gene tests. PMS2 immunohistochemistry showed lack of staining at both the tumors and normal tissue as vascular endothelial cells. Further gene tests revealed large genomic deletion including the entire PMS2 gene, confirming biallelic PMS2 germline variants.
Conclusion
Conventional multi-gene panel tests are insufficient for detecting large deletions of MMR genes, resulting in misdiagnoses of CMMRD as Lynch syndrome. PMS2 variants have low cancer penetrance; family histories may thus be absent. Long-range gene analyses or immunohistochemical staining of MMR proteins in normal tissue should be considered for pediatric brain tumors with a single allele MMR variant when CMMRD is suspected.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-022-01403-9.
Keywords: Constitutional mismatch repair deficiency, CMMRD, Lynch syndrome, Glioblastoma, Colon cancer, Acute lymphocytic leukemia, PMS2, Genetic testing, Large deletion, Immune-checkpoint inhibitor
Background
Constitutional mismatch repair deficiency (CMMRD) syndrome, a rare condition that greatly increases the risk of cancers among children, adolescents, and young adults [1], is caused by biallelic (homozygous or compound heterozygous) germline pathogenic variants in one of the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2). Monoallelic germline variants in MMR genes cause Lynch syndrome (LS) [2], which predisposes individuals mainly to colorectal cancer, endometrial cancer, and other LS-related malignancies. Individuals whose father and mother both have LS have a one-quarter risk of inheriting biallelic pathogenic variants of MMR genes that cause CMMRD.
Patients with CMMRD have presented malignancies including colorectal cancer, brain tumor, LS-related malignancy, or hematological malignancy [3]. Malignant brain tumors such as glioblastoma are the most common type of CMMRD-related malignancy. In the Care for CMMRD (C4CMMRD) database, 53.4% of the patients with CMMRD developed brain tumors, and the mean age at the brain tumor diagnosis is 9 years [3]. Glioblastomas that are associated with CMMRD have different oncogeneses, and they may represent a distinct entity of pediatric glioblastoma. Physicians thus need to discriminate CMMRD-associated glioblastomas from other pediatric high-grade gliomas (HGGs). In addition, a precise diagnosis of CMMRD has important implications for treatment and for the surveillance of the patients' families.
CMMRD can be challenging to diagnose in patients without a family history. Genetic screening for MMR gene variants is not routinely performed for brain tumors, and thus some CMMRD-associated malignant brain tumors may have been overlooked. Moreover, patients with CMMRD could be misdiagnosed as having Lynch syndrome, because the conventional multi-gene analyses are insufficient for detecting long-range deletions of MMR genes. In this report, we describe two cases of glioblastoma as the initial malignancy of CMMRD. In light of the results of conventional multi-gene analyses in both cases, there was a risk of potentially underestimating the gene variants as indicating Lynch syndrome.
Case presentation
Patient 1
A six-year-old girl with no remarkable family history presented with headache and vomiting. She had hyperpigmented skin alternation, i.e., cafe au lait spots (Fig. 1). MRI showed a heterogeneous gadolinium-enhanced left frontal tumor (Fig. 2a, b). She underwent a left frontal craniotomy, and gross total removal of the tumor was achieved. Hematoxylin and eosin (HE) staining of the left frontal tumor showed glioblastoma with multinucleated giant cells (Fig. 2g). Molecular analyses by pyrosequencing showed no mutations in IDH1/2, H3F3A, HIST1H3B, or BRAF. The patient underwent standard local radiation therapy (RT): 60 Gy/30 fractions.
Fig. 1.
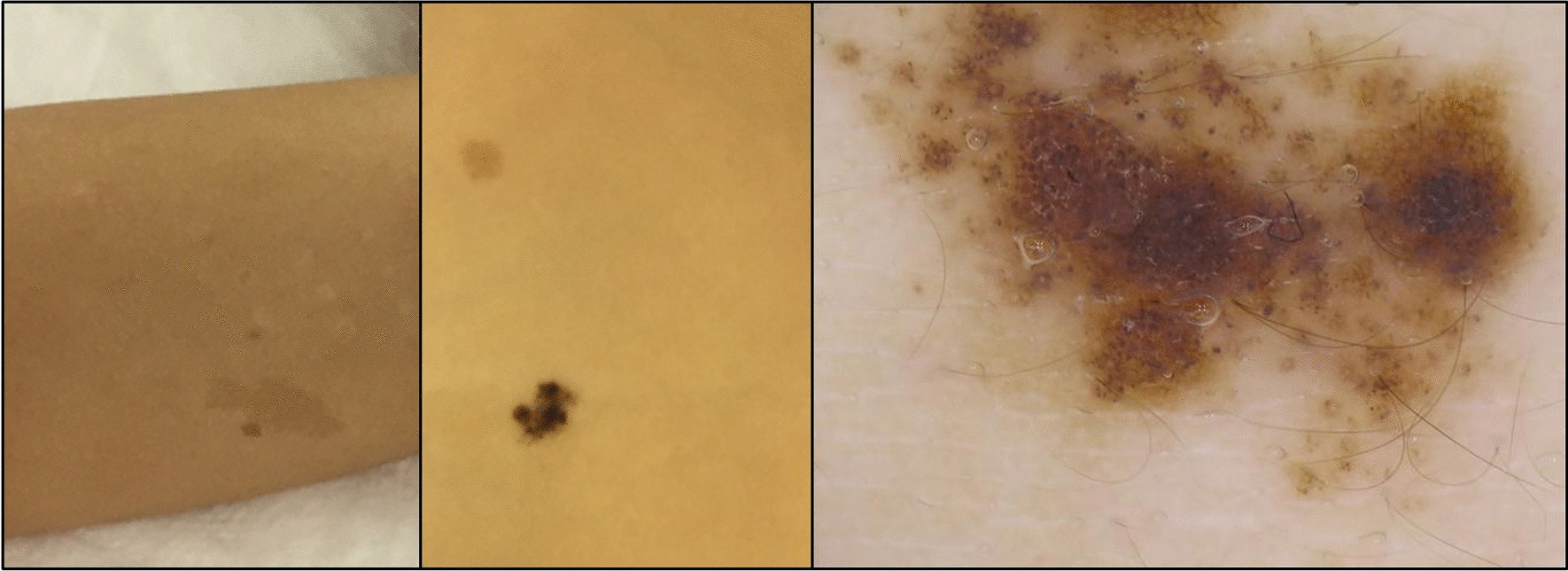
Multiple hyperpigmented skin alternations on the limbs and trunk of Patient 1, a 6-year-old girl
Fig. 2.
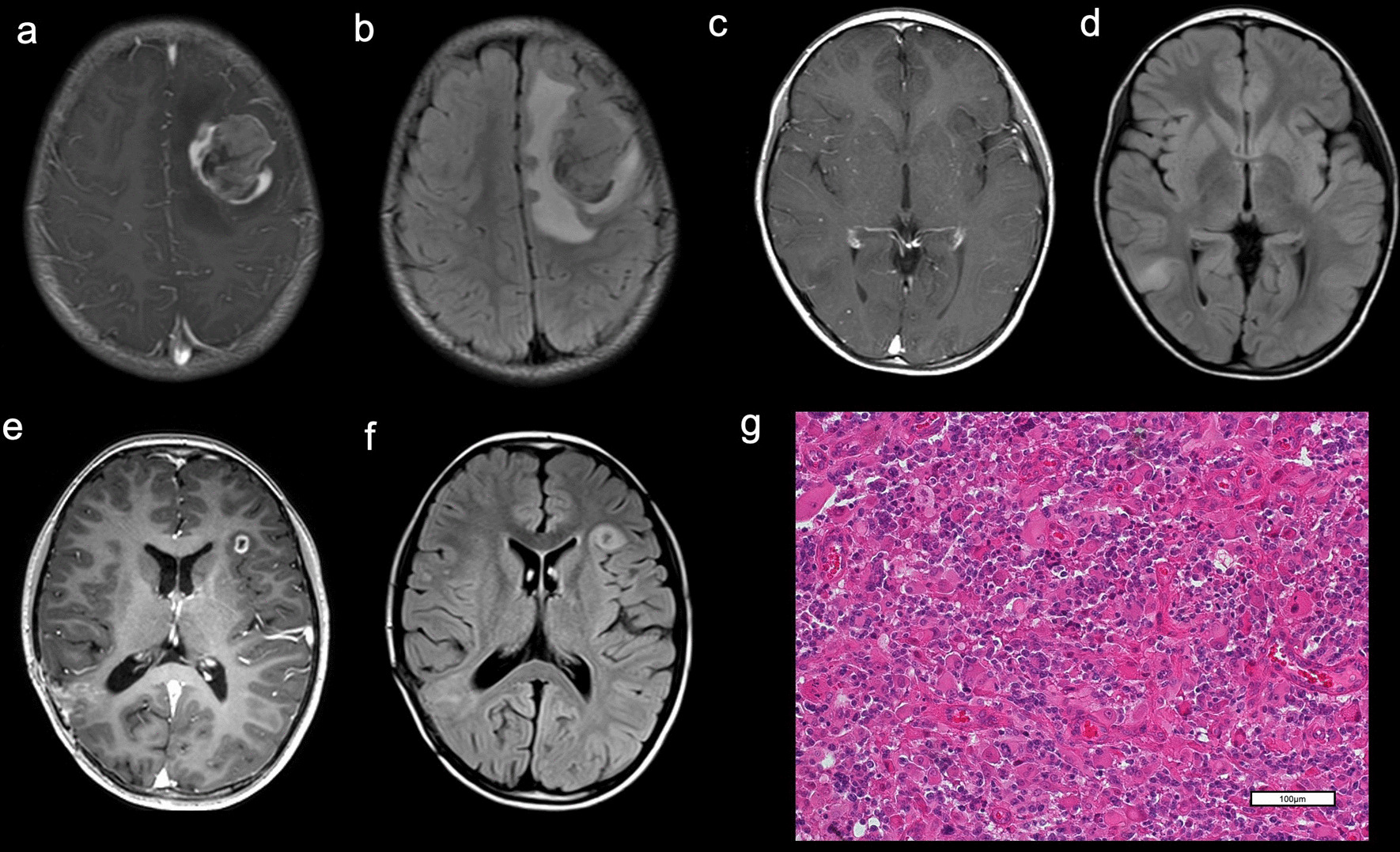
MRI of Patient 1 at onset showed a left frontal tumor. a The tumor was heterogeneously enhanced with gadolinium on T1WI. b FLAIR showed peritumoral edema. MRI c, d revealed that another non-enhancing tumor developed at the right temporal lobe. The tumor was hyper-intense on FLAIR d and was not enhancing with gadolinium on T1WI c. MRI e, f demonstrating a tumor at the left operculum. The tumor was enhanced with gadolinium on T1WI e and showed hyperintensity on FLAIR f. g Hematoxylin–eosin (HE) staining of a specimen from the 1st surgery for left frontal tumor showed a glioblastoma. (OLYMPUS BX43/ × 20 0.50FN 26.5, Nikon DIGITAL SIGHT DS-Fi2 Microscope C-mount Camera System, NIS ELEMENTS, resolution: 1280 × 960)
One year after the first surgery, another non-enhancing tumor developed at the right temporal lobe (Fig. 2c, d). This tumor was also completely resected. The pathological diagnosis was anaplastic astrocytoma, with the same molecular features as the initial glioblastoma. She underwent local RT: 50 Gy/25 fractions. The tumor later recurred at the right temporal tumor cavity, along with a new lesion at left operculum frontal lobe (Fig. 2e, f). These tumors were gross totally resected and carmustine wafers were implanted. The histological diagnosis of both tumors was glioblastoma. FoundationOne® CDx (F1CDx) (Foundation Medicine, Cambridge, MA, USA) identified pathogenic variants in 30 genes (POLE, ATM, HRAS, MET, NF1, PTEN, SMARCB1, STK11, ARID1A, RET, ATRX, CDKN1A, CIC, CSF1R, CTCF, DNMT3A, HSD3B1, KEL, KMT2A (MLL), MAP3K1, NOTCH1, NOTCH3, PBRM1, PMS2, PPP2R1A, RB1, RPTOR, STAG2, TP53, VHL) and 92 variants of uncertain significance (Additional file 1: Table S1). The F1CDx test also revealed a high tumor mutation burden (TMB-H) of 192 mutations per megabase (muts/Mb), though designated the tumor as microsatellite stable (MSS). By Promega Microsatellite Instability (MSI) Analysis System (Madison, WI, USA), the tumor was designated microsatellite instability-high (MSI-H).
Immunohistochemical (IHC) staining of a specimen from the 1st surgery for the left frontal tumor showed a loss of PMS2 expression in tumor cells and normal tissue as vascular endothelial cells and preserved expression of MLH1, MSH2, and MSH6 in both tumor cells and normal tissue (Additional file 2: Figure S1a-d). Genetic testing revealed biallelic variants, i.e., PMS2:c.[241G > T];[2276-125_2445 + 1584del]. PMS2:c.241G > T was identified by the initial cancer genomic profiling test with F1CDx. However, another pathogenic variant, PMS2:c.2276-125_2445 + 1584del (which indicates the deletion of 1,879 base pairs [bps] including Exon 14 of PMS2 gene, resulting in the stop codon in eight codons after codon 759 with frameshift mutation) was not detected by the F1CDx test, since the length of the deletion was too large to be detected by a next-generation sequencing (NGS) analysis in a cancer genomic profiling test. The large deletion was detected as described [4].
Parental genetic testing confirmed that the patient's father had one variant PMS2:c.241G > T, and the other variant, PMS2:c.2276-125_2445 + 1584del, was identified in the patient's mother. The interpretation of PMS2:c.241G > T in the ClinVar archive of the U.S. National Center for Biotechnology Information (ClinVar Variation ID: 439,243) is 'Pathogenic' and 'Likely pathogenic.' PMS2:c.2276-125_2445 + 1584del is registered as Class 5 (Pathogenic) in InSiGHT database of variants (https://insight-database.org/). A schematic image of the large deletion in PMS2 gene is provided as Fig. 7. From this genetic information, Patient 1 was diagnosed with CMMRD, indicating compound heterozygous variants in PMS2, inherited from each of her parents (Fig. 3).
Fig. 7.

Schematic image of the large deletions in chromosome 7 and PMS2 gene of Patients 1 and 2
Fig. 3.

Pedigree showing affected and unaffected members of Patient 1's family
The tumors at the patient's right temporal lobe and left operculum frontal lobe recurred at tumor cavity walls. She was administered pembrolizumab every 3 weeks for three cycles, and this regimen was terminated after three cycles because of progressive disease with an increasing size of the tumor and the development of a new lesion at the left temporal lobe. She was subsequently administered temozolomide and bevacizumab; however, the tumor did not respond to these treatments, and the patient died 4 years after the initial surgery.
Patient 2
A 10-year-old boy whose great-grandfather had colorectal cancer presented with a headache. His skin was not evaluated. MRI showed a gadolinium-enhanced left temporal tumor with peritumoral edema (Fig. 4a, b). The tumor was resected, and the histopathological diagnosis was glioblastoma with primitive neuroectodermal tumor-like features (Fig. 4c). The tumor was diagnosed with high-grade glioma, and the status of IDH1/2, H3F3A, HIST1H3B or BRAF was not analyzed.
Fig. 4.
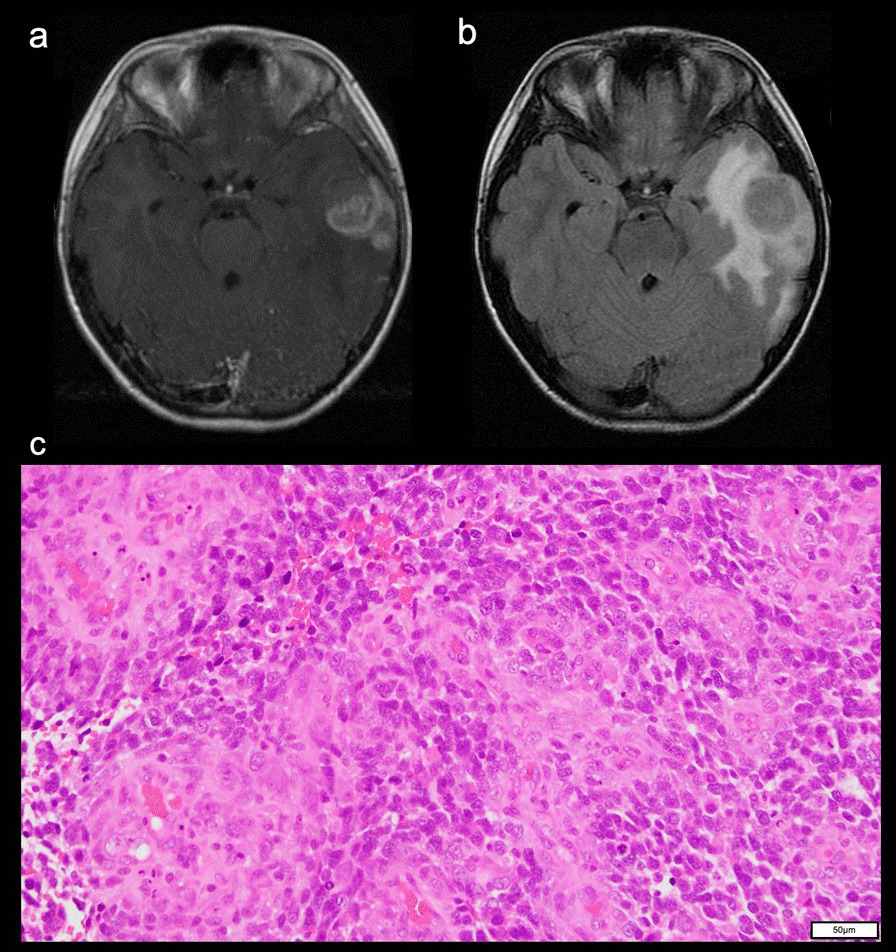
MRI of Patient 2, a 10-year-old boy at onset showing a left temporal tumor. a The tumor was heterogeneously enhanced with gadolinium on T1WI. b FLAIR showed peritumoral edema. cHE staining demonstrated a glioblastoma. (OLYMPUS BX53FZ/ × 20 0.50 FN 26.5, OLYMPUS DP 27, OLYMPUS Standard, resolution: 2448 × 1920)
Immunohistochemically, the tumor was positive for glial fibrillary acidic protein (GFAP). Immunohistochemical staining for MMR proteins showed a loss of PMS2 expression in tumor cells and vascular endothelial cells (Additional file 3: Figure S2a) and preserved expression of MLH1 (Additional file 3: Figure S2b), MSH2 (Additional file 3: Figure S2c) and MSH6 (Additional file 3: Figure S2d) in both tumor cells and vascular endothelial cells. The patient underwent craniospinal irradiation with local boost irradiation and eight cycles of platinum-based combination chemotherapy, and a complete response of the brain tumor was achieved.
Six years after the surgery, at the age of 16, the patient developed persistent abdominal pain. Colonoscopy revealed an adenocarcinoma of the cecum (Fig. 5a), and a right hemicolectomy with D3 lymph node dissection was performed. The diagnosis was moderately differentiated tubular adenocarcinoma (Fig. 5b), stage IIIb (pT3(A), ly3, v0, N2). After two cycles of chemotherapy with tegafur/uracil, the colon cancer recurred twice and the patient underwent local RT (60 Gy/20 fractions) each time to achieve the complete remission of the colon cancer.
Fig. 5.
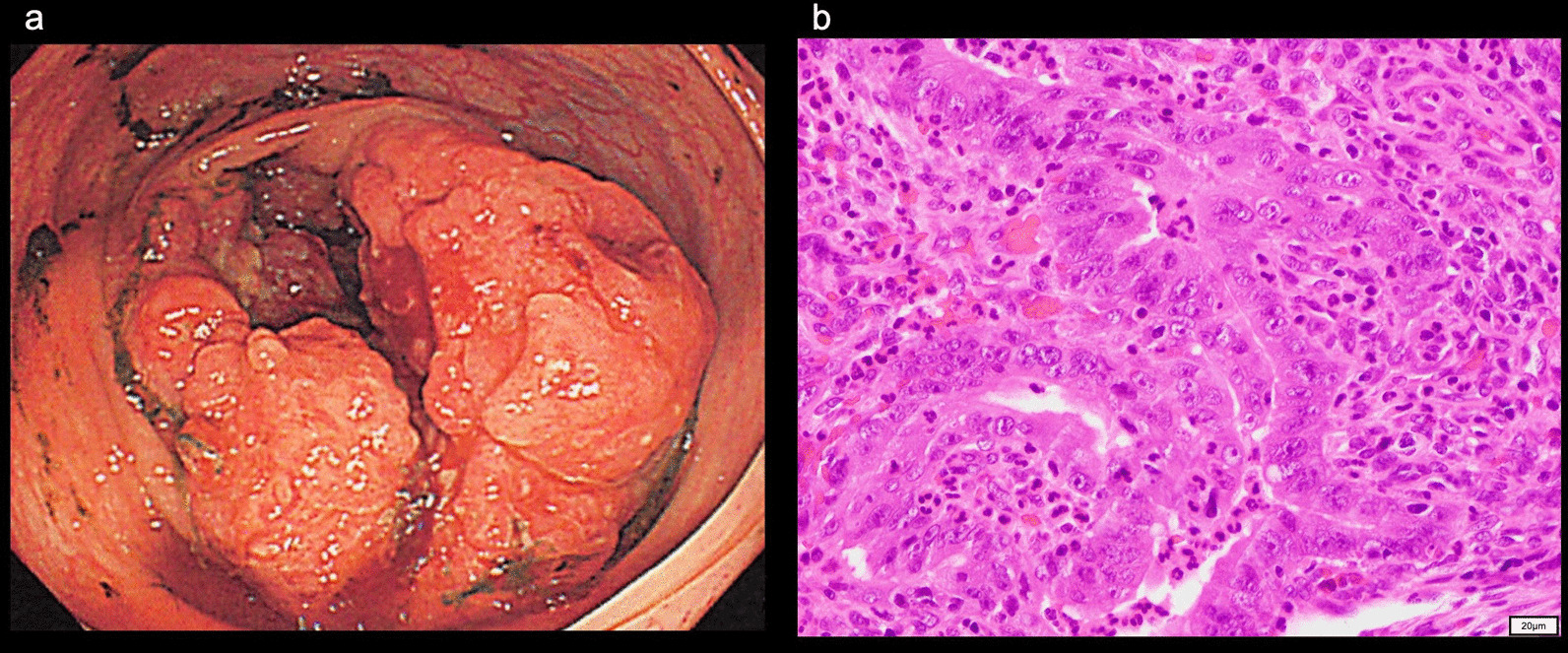
a Colonoscopy identified adenocarcinoma of the cecum. b HE staining showed a moderately differentiated tubular adenocarcinoma. (OLYMPUS BX53FZ / × 40 0.70FN 26.5, OLYMPUS DP 27, OLYMPUS Standard, resolution: 2448 × 1920)
Eight years after the brain tumor surgery, at 18 years old, the patient developed acute lymphocytic leukemia (ALL). He underwent chemotherapy based on the Japan Adult Leukemia Study Group (JALSG)-ALL202-U phase II multicenter study's schedule. Although a complete response of the ALL was obtained, colon cancer recurred, and mFOLFOX6 chemotherapy was administered.
IHC staining for MMR proteins of the patient's colon cancer revealed a loss of PMS2 in tumor cells and normal tissue as vascular endothelial cells and preserved expression of MLH1, MSH2, and MSH6 in both tumor cells and normal tissue (Additional file 4: Figure S3a-d). In addition, IHC staining of the initial brain tumor showed a loss of PMS2 expression and preserved expression of MLH1, MSH2, and MSH6 (Additional file 3: Figure S2a-d).
A germline analysis using DNA and RNA extracted from the patient's peripheral blood as described [4] revealed biallelic PMS2 variants, i.e., NC_000007.13 (chromosome 7): g.[5876369_612205del];[6043612C>A], which contains a large deletion including PMS2, and chromosome 7:g.5876369_612205del, which means PMS2:c.[−73408_*136661del], indicating the deletion of 245 kilo base pairs (kbps) including coding lesion of PMS2. Family genetic testing confirmed that the patient's paternal uncle had one variant, i.e., chromosome 7:g.6043612C>A, which means PMS2:c.241G>T, suggesting that the patient's father also carried the variant. The other variant, chromosome 7:g.5876369_6122058del, was identified in the patient's mother. The interpretation of PMS2:c.241G>T is 'Pathogenic' and 'Likely pathogenic' in ClinVar (ClinVar Variation ID: 439243). The other variant, chromosome 7: g.5876369_6122058del, caused the complete deletion of PMS2 and five other genes, but the deletion was not registered in any database. Schematic images of large deletions in chromosome 7 are shown in Figure 7. From this genetic information, the diagnosis was CMMRD, indicating compound heterozygous variants in PMS2, inherited from each parent (Fig. 6).
Fig. 6.

Pedigree showing affected and unaffected members of Patient 2's family
Subsequently, the ALL recurred again without the recurrence of brain tumor or colon cancer, and the patient died 8 years after his initial brain tumor surgery.
Discussion and conclusions
We have described the clinical pitfalls and diagnostic clues of CMMRD initially presenting as glioblastoma, and we presented two challenging cases of pediatric CMMRD-associated glioblastoma that were immune checkpoint inhibitor (ICI)-resistant, temozolomide-resistant, and bevacizumab-resistant. The precise diagnosis of CMMRD has important implications for the surveillance of patients' siblings and other family members. In Patient 1, metachronous HGG and cutaneous features were diagnostic clues. In a case series of CMMMRD-associated glioblastomas, 5 of 15 patients (33%) presented with metachronous brain lesions [5]. Skin alteration is also a characteristic of CMMRD. Most of the reported patients with CMMRD presented multiple café-au-lait maculae, which have a ragged edge and a slightly diffuse appearance [6]. The presence of HGG and multiple hyperpigmented skin alterations > 1 cm meet the indication criteria for CMMRD testing [3]. However, in the present Patient 2, CMMRD was diagnosed after he had developed three CMMRD-associated malignancies.
A comprehensive genomic profiling report of pediatric HGG revealed that 6% (9/157) of pediatric HGGs were hypermutated, and most of them harbored pathogenic variants in one of the MMR genes [7]. Among sporadic pediatric HGG patients evaluated in Jordan, MMRD was observed in 39% (17/44), and 82% (14/17) of these were biallelic MMRD [8]. These results implied that CMMRD may be overlooked in current clinical practice for pediatric HGG.
In the CMMRD database, PMS2 gene is also the most common causative gene of CMMRD-associated brain tumors, accounting for 60% of CMMRD patients [3]. However, PMS2 variants have low penetrance [9], and they may lack a family history of Lynch syndrome-spectrum malignancies. Therefore, the Amsterdam I/II and revised Bethesda guidelines could be insufficient for screening for Lynch syndrome [10], especially for the PMS2 variant. The estimated prevalence of Lynch syndrome in general populations is approx. one in 370 [11], and CMMRD could be underdiagnosed. Clinicians should consider the possibility of CMMRD when a brain tumor is detected, because a malignant brain tumor could develop as an initial presentation of CMMRD syndrome.
Biallelic MMR deficiency would be detected in a CMMRD-associated tumor; however, conventional multi-gene panel tests are not able to detect a large deletion of MMR genes. In Patient 1, the initial result of a multi-gene panel test (the F1CDx test) was a single monoallelic PMS2 variation, c.241G > T (p.Glu81*). The analysis pipeline of the F1CDx test can detect the short abnormal structure of the targeted gene, but it cannot detect a large deletion of the targeted gene. Another PMS2 variant, the deletion of 1,879 bps including Exon 14, was too large to be detected in conventional multi-gene panel tests. In Patient 2, chromosome 7:g.5876369_612205del indicates the deletion of 245 kbps caused all coding region of PMS2 with five other exons. The extremely large deletion including the entire PMS2 gene was undetectable in conventional multi-gene panel tests. Physicians may therefore misdiagnose CMMRD patients as having Lynch syndrome when only conventional multi-gene panel tests are used. Even if a pathogenic variant in one of the MMR genes is detected by a conventional analysis, the potential existence of another MMR pathogenic variant should be carefully examined by other methods such as a long-range analysis around MMR genes, as described [4].
Immunohistochemistry staining of MMR proteins in normal tissue could also be valuable for further investigations of MMR genes in clinical practice. IHC staining is a cost-effective strategy for screening MMR deficiency. MMR proteins are normally present in human cells. In LS patients, the expression of certain MMR proteins is lost in tumor cells and retained in normal cells such as endothelial cells. In CMMRD patients, the expression is lost in tumor and normal cells. The loss of one or more MMR proteins by IHC staining in 'normal tissues' thus suggests biallelic germline pathogenic variants of MMR gene [12]. The normal brain tissue surrounding a brain tumor should thus be carefully observed.
TMB-H and neoantigen loads have been suggested to be associated with the efficacy of ICIs [13], and based on these characteristics, ICIs are considered an effective therapy for CMMRD-associated glioblastoma. Several CMMRD-associated glioblastomas reportedly showed a significant response to ICI treatment [14, 15]. However, the experience of Patient 1 did not support the efficacy of ICI treatment for recurrent CMMRD-associated glioblastomas with TMB-H. There have been no clinical trials showing the benefit of an ICI in adjuvant therapy for glioblastomas [16], but the addition of an ICI as neoadjuvant treatment indicated more consistent immune activation and efficacy for recurrent glioblastoma [17]. To improve the treatment of CMMRD-associated glioblastoma, further evidence is necessary to establish predictive markers for the efficacy of ICIs and to identify the best treatment schedule including neoadjuvant settings.
The ICI therapies for MSI-H malignancies have shown promising results in some cancers. MSI testing with the conventional multi-gene panel tests (e.g., F1CDx) may lead to discrepancies in companion diagnostics using Bethesda and Promega panels. The advantage of a comprehensive genomic profiling (CGP) test is the simultaneous analysis of the MSI status, genomic aberrations, and the TMB in solid tumors. A cohort study that used the F1CDx test for diffuse glioma reported that most of the glioblastoma indicated MSS despite a high TMB score [18]. However, another study demonstrated that the accuracy of an NGS-based MSI analysis is not perfect [19]. The Promega MSI Analysis System is considered the gold-standard panel for MSI detection in cancers because of its higher sensitivity and specificity according to the revised Bethesda guidelines for colorectal cancers [20, 21]. In the present two patients, the results of the F1CDx microsatellite test were MSS, whereas the Promega MSI Analysis System revealed MSI-H in both patients. MSI testing should be performed by multiple methods if MSI-H is suspected based on other findings.
In conclusion, the accurate and early diagnosis of CMMRD has important implications for the management of the patients and their families. Our report highlights the shortcomings of conventional multi-gene panel tests for diagnosing CMMRD and microsatellite instability, which might have resulted in an underestimation of the incidence of CMMRD among pediatric patients with malignant brain tumors. Clinicians should consider conducting a long-range gene analysis and/or IHC staining of MMR proteins in normal tissue for pediatric patients with brain tumors and a single allele MMR variant for the diagnosis of CMMMRD.
Supplementary Information
Additional file 1: The list of pathogenic variants and variants of uncertain significance (VUS) detected in glioblastoma of patient 1 by FoundationOne® CDx test.
{kind=link}
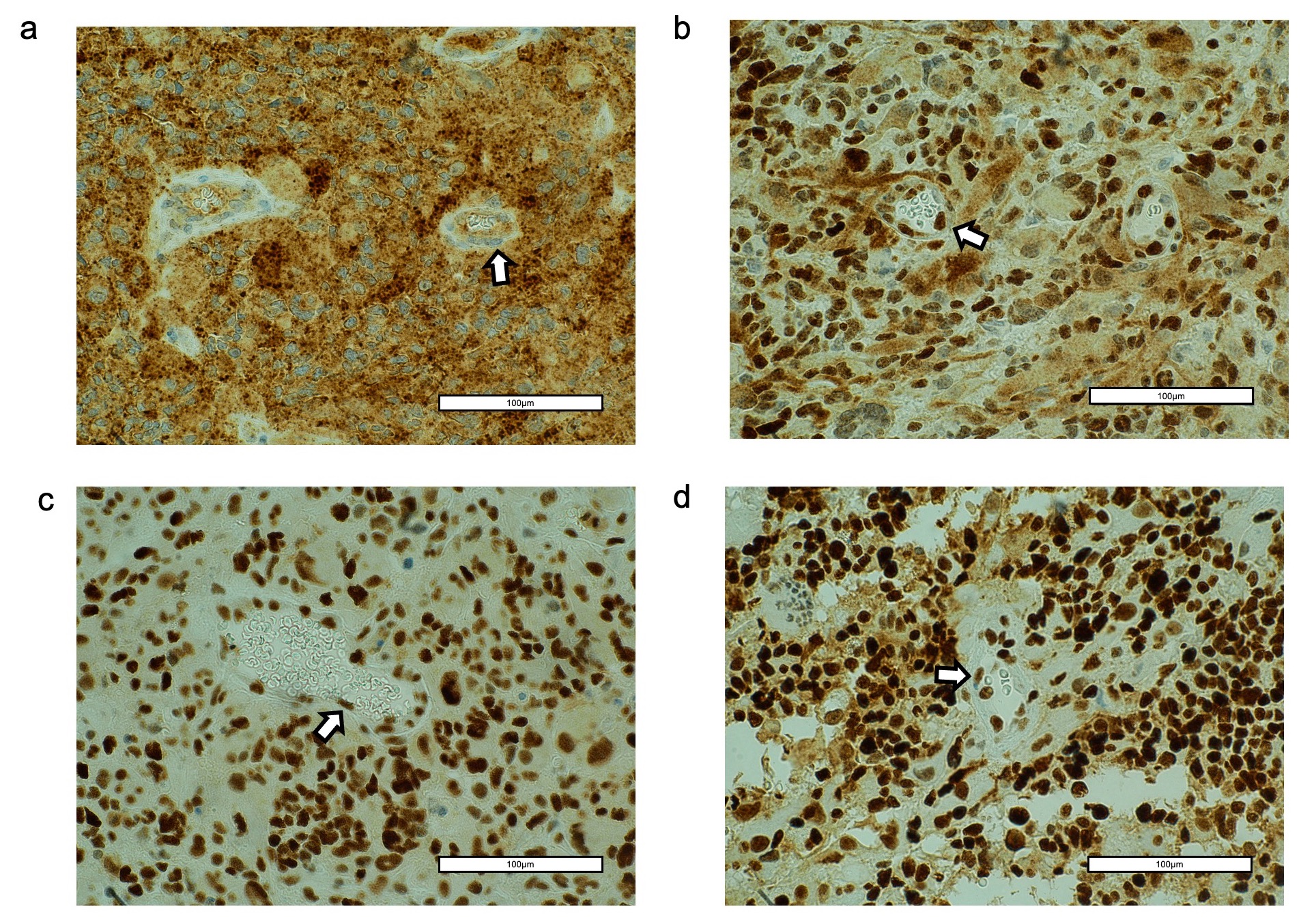
Additional file 2: Fig. S1. Immunohistochemical staining for MMR proteins showed a loss of PMS2 expression in tumor cells and normal tissue as vascular endothelial cells (a) and preserved expressions of MLH1 (b), MSH2 (c) and MSH6 (d) in both tumor cells and vascular endothelial cells. White arrows indicate the nucleus of the vascular endothelial cells (a-d). (OLYMPUS BX43/×40 0.70FN 26.5, Nikon DIGITAL SIGHT DS-Fi2 Microscope C-mount Camera System, NIS ELEMENTS, resolution: 1280 × 960)
{kind=link}
Additional file 3: Fig. S2 IHC staining for MMR proteins showed a loss of PMS2 expression in tumor cells and normal tissue as vascular endothelial cells (a) and preserved expressions of MLH1 (b), MSH2 (c) and MSH6 (d) in both tumor cells and vascular endothelial cells. (OLYMPUS BX53FZ/×20 0.50 FN 26.5, OLYMPUS DP 27, OLYMPUS Standard, resolution: 1224 x 960)
{kind=link}
Additional file 4: Fig. S3 IHC staining for MMR proteins showed a loss of PMS2 in tumor cells and normal tissue (c) and preserved expressions of MLH1 (d), MSH2 (e) and MSH6 (f) in both tumor cells and normal tissue. (OLYMPUS BX53FZ /×40 0.70FN 26.5, OLYMPUS DP 27, OLYMPUS Standard, resolution: 1224 x 960)
Acknowledgements
We thank Dr. Go Yamamoto for the expert technical assistance with analysis of germline variants.
Abbreviations
- bps
Base pairs
- CGP
Comprehensive genomic profiling
- CMMRD
Constitutional mismatch repair deficiency
- F1CDx
FoundationOne® CDx
- GFAP
Glial fibrillary acidic protein
- HGG
High-grade glioma
- ICI
Immune checkpoint inhibitor
- IHC
Immunohistochemical
- LS
Lynch syndrome
- MMR
Mismatch repair
- MSI
Microsatellite instability
- MSS
Microsatellite stable
- NGS
Next-generation sequencing
- TMB-H
High tumor mutation burden
Author contributions
SO, FY and TH contributed the conception of the study and wrote the manuscripts. KK performed the pathological analysis. SO, FM, AT and TT contributed to the acquisition of data. KA and TH analyzed and interpreted genomic data. All authors read and approved the final manuscript.
Funding
Data analysis of this research was supported by Japan Agency for Medical Research and Development (AMED) under grant JP18kk0205004(for K.A.) and JSPS KAKENHI Grant Numbers JP18K07339(for K.A.), JP22K07266(for K.A.). Data collection of this was supported by JP18K08694(for T.H.), and JP21H03002(for T.H.). The opinions, results, findings and/or interpretations are the sole responsibility of the authors and do not represent the opinions, interpretations or policy of the funders.
Availability of data and materials
The details of the variant analyzed in this study have been deposited into the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), under the accession number SCV002600096, SCV002600097 and SCV002600098.
Declarations
Ethics approval and consent to participate
This study was approved by Ethics Committee of Hiroshima University Hospital. Written informed consents were obtained from all of their parents.
Consent for publication
Written informed consents for publication of identifying images or other personal or clinical details were obtained from all of their parents.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Michaeli O, Tabori U. Pediatric high grade gliomas in the context of cancer predisposition syndromes. J Korean Neurosurg Soc. 2018;61:319–322. doi: 10.3340/jkns.2018.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aaltonen LA, Peltomäki P, Leach FS, Sistonen P, Pylkkänen L, Mecklin JP, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 3.Wimmer K, Kratz CP, Vasen HFA, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European Consortium "Care for CMMRD" (C4CMMRD) J Med Genet. 2014;51:355–365. doi: 10.1136/jmedgenet-2014-102284. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto G, Miyabe I, Tanaka K, Kakuta M, Watanabe M, Kawakami S, et al. SVA retrotransposon insertion in exon of MMR genes results in aberrant RNA splicing and causes Lynch syndrome. Eur J Hum Genet. 2021;29:680–686. doi: 10.1038/s41431-020-00779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrini-Rousseau L, Varlet P, Colas C, Andreiuolo F, Bourdeaut F, Dahan K, et al. Constitutional mismatch repair deficiency-associated brain tumors: report from the European C4CMMRD consortium. Neuro-Oncol Adv. 2019 doi: 10.1093/noajnl/vdz033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Vos M, Hayward BE, Charlton R, Taylor GR, Glaser AW, Picton S, et al. PMS2 mutations in childhood cancer. J Natl Cancer Inst. 2006;98:358–361. doi: 10.1093/jnci/djj073. [DOI] [PubMed] [Google Scholar]
- 7.Johnson A, Severson E, Gay L, Vergilio J, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 2017;22:1478–1490. doi: 10.1634/theoncologist.2017-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amayiri N, Tabori U, Campbell B, Bakry D, Aronson M, Durno C, et al. High frequency of mismatch repair deficiency among pediatric high grade gliomas in Jordan. Int J Cancer. 2016;138:380–385. doi: 10.1002/ijc.29724. [DOI] [PubMed] [Google Scholar]
- 9.Goodenberger ML, Thomas BC, Riegert-Johnson D, Boland CR, Plon SE, Clendenning M, et al. PMS2 monoallelic mutation carriers: the known unknown. Genet Med. 2016;18:13–19. doi: 10.1038/gim.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasen HFA, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62:812–823. doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hampel H, de la Chapelle A. The Search for unaffected individuals with Lynch syndrome: do the ends justify the means? Cancer Prev Res. 2011;4:1–5. doi: 10.1158/1940-6207.CAPR-10-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:1–12. doi: 10.1186/s13000-017-0613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galuppini F, Dal Pozzo CA, Deckert J, Loupakis F, Fassan M, Baffa R. Tumor mutation burden: from comprehensive mutational screening to the clinic. Cancer Cell Int. 2019;19:1–10. doi: 10.1186/s12935-019-0929-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larouche V, Atkinson J, Albrecht S, Laframboise R, Jabado N, Tabori U, et al. Sustained complete response of recurrent glioblastoma to combined checkpoint inhibition in a young patient with constitutional mismatch repair deficiency. Pediatr Blood Cancer. 2018;65:4–5. doi: 10.1002/pbc.27389. [DOI] [PubMed] [Google Scholar]
- 15.Bouffet E, Larouche V, Campbell BB, Merico D, De Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206–2211. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- 16.Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 Phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1003–1010. doi: 10.1001/jamaoncol.2020.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Omura T, Takahashi M, Ohno M, Miyakita Y, Yanagisawa S, Tamura Y, et al. Clinical application of comprehensive genomic profiling tests for diffuse gliomas. Cancers (Basel) 2022;14:2454. doi: 10.3390/cancers14102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int. 2020 doi: 10.1186/s12935-019-1091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. Revised bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baudrin LG, Deleuze JF, How-Kit A. Molecular and computational methods for the detection of microsatellite instability in cancer. Front Oncol. 2018;8:1–11. doi: 10.3389/fonc.2018.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: The list of pathogenic variants and variants of uncertain significance (VUS) detected in glioblastoma of patient 1 by FoundationOne® CDx test.
Additional file 2: Fig. S1. Immunohistochemical staining for MMR proteins showed a loss of PMS2 expression in tumor cells and normal tissue as vascular endothelial cells (a) and preserved expressions of MLH1 (b), MSH2 (c) and MSH6 (d) in both tumor cells and vascular endothelial cells. White arrows indicate the nucleus of the vascular endothelial cells (a-d). (OLYMPUS BX43/×40 0.70FN 26.5, Nikon DIGITAL SIGHT DS-Fi2 Microscope C-mount Camera System, NIS ELEMENTS, resolution: 1280 × 960)
Additional file 3: Fig. S2 IHC staining for MMR proteins showed a loss of PMS2 expression in tumor cells and normal tissue as vascular endothelial cells (a) and preserved expressions of MLH1 (b), MSH2 (c) and MSH6 (d) in both tumor cells and vascular endothelial cells. (OLYMPUS BX53FZ/×20 0.50 FN 26.5, OLYMPUS DP 27, OLYMPUS Standard, resolution: 1224 x 960)
Additional file 4: Fig. S3 IHC staining for MMR proteins showed a loss of PMS2 in tumor cells and normal tissue (c) and preserved expressions of MLH1 (d), MSH2 (e) and MSH6 (f) in both tumor cells and normal tissue. (OLYMPUS BX53FZ /×40 0.70FN 26.5, OLYMPUS DP 27, OLYMPUS Standard, resolution: 1224 x 960)
Data Availability Statement
The details of the variant analyzed in this study have been deposited into the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), under the accession number SCV002600096, SCV002600097 and SCV002600098.