

SUMMARY
Our comprehensive analysis of alternative splicing across 32 The Cancer Genome Atlas cancer types from 8,705 patients detects alternative splicing events and tumor variants by reanalyzing RNA and whole-exome sequencing data. Tumors have up to 30% more alternative splicing events than normal samples. Association analysis of somatic variants with alternative splicing events confirmed known trans associations with variants in SF3B1 and U2AF1 and identified additional trans-acting variants (e.g., TADA1, PPP2R1A). Many tumors have thousands of alternative splicing events not detectable in normal samples; on average, we identified ≈930 exon-exon junctions (“neojunctions”) in tumors not typically found in GTEx normals. From Clinical Proteomic Tumor Analysis Consortium data available for breast and ovarian tumor samples, we confirmed ≈1.7 neojunction- and ≈0.6 single nucleotide variant-derived peptides per tumor sample that are also predicted major histocompatibility complex-I binders (“putative neoantigens”).
Graphical Abstract
In Brief
A pan-cancer analysis by Kahles et al. shows increased alternative splicing events in tumors versus normal tissue and identifies trans-acting variants associated with alternative splicing events. Tumors contain neojunction-derived peptides absent in normal samples, including predicted MHC-I binders that are putative neoantigens.
INTRODUCTION
Analyses of cancer genomes have predominantly focused on the evaluation of somatic non-synonymous protein-altering mutations and the potentially pathogenic impact such mutations have on gene expression, protein function, and downstream pathways (Futreal et al., 2001; Greenman et al., 2007). The types of samples collected and the data generated by The Cancer Genome Atlas (TCGA) have been specifically chosen to support such analyses (Cancer Genome Atlas Research Network, 2008). However, the developed resources also provide an excellent opportunity for an in-depth analysis of the changes of the transcriptome in tumors, which has received much less attention so far.
Individual changes in regulatory binding sites or alterations to the protein coding sequences can have a strong functional impact, leading to selective growth advantages for tumor cells. Several cases have been reported where the physiological outcome of such alterations comes into functional effect through the alteration of splicing. A prominent example for cis-acting mutations is found in the splice junctions of MET leading to skipping of exon 14, resulting in activation of MET but also providing specific sensitivity to MET inhibitors (Frampton et al., 2015; Paik et al., 2015). In addition, trans-acting alterations have been described where a somatic variant in a splicing factor leads to many splicing changes across the genome. For instance, somatic alterations of the splicing factor U2AF1 lead to a widely altered landscape of splicing events in certain cancer types, such as lung adenocarcinomas (Brooks et al., 2014) or myelodysplastic syndromes (Graubert et al., 2012). Another well-characterized set of alterations are changes of the splicing factor SF3B1, which have been linked to changes in splicing patterns in various tumor types, such as uveal melanoma (Furney et al., 2013) or lymphocytic leukemia (Rossi et al., 2011), and are suggested to promote aberrant splicing patterns via alternative branchpoint usage (Alsafadi et al., 2016). More recently, the analysis of alternative splicing has also been shown to be of prognostic value for multiple cancer types, including non-small cell lung cancer (Li et al., 2017), ovarian cancer (Zhu et al., 2017), breast cancer (Bjørklund et al., 2017), uveal melanoma (Robertson et al., 2017), and glioblastoma (Marcelino Meliso et al., 2017).
RESULTS
Workflow for Integrated Pan-Cancer Analysis
We devised a versatile and comprehensive workflow to integrate analyses of RNA and whole-exome sequencing data from tumors from 8,705 donors, including 670 matched normal samples, spanning a range of 32 cancer types (Figure 1 left, middle). The main questions answered by the developed methodology are (1) the identification of underlying genetic changes leading to splicing variability in tumors (Figure 1 right top), (2) a comprehensive analysis of quantitative and qualitative changes of alternative splicing in tumors (Figure 1 right middle), and (3) determining the extent to which splicing aberrations can be exploited for immunotherapy (right bottom).
Figure 1. Project Overview.
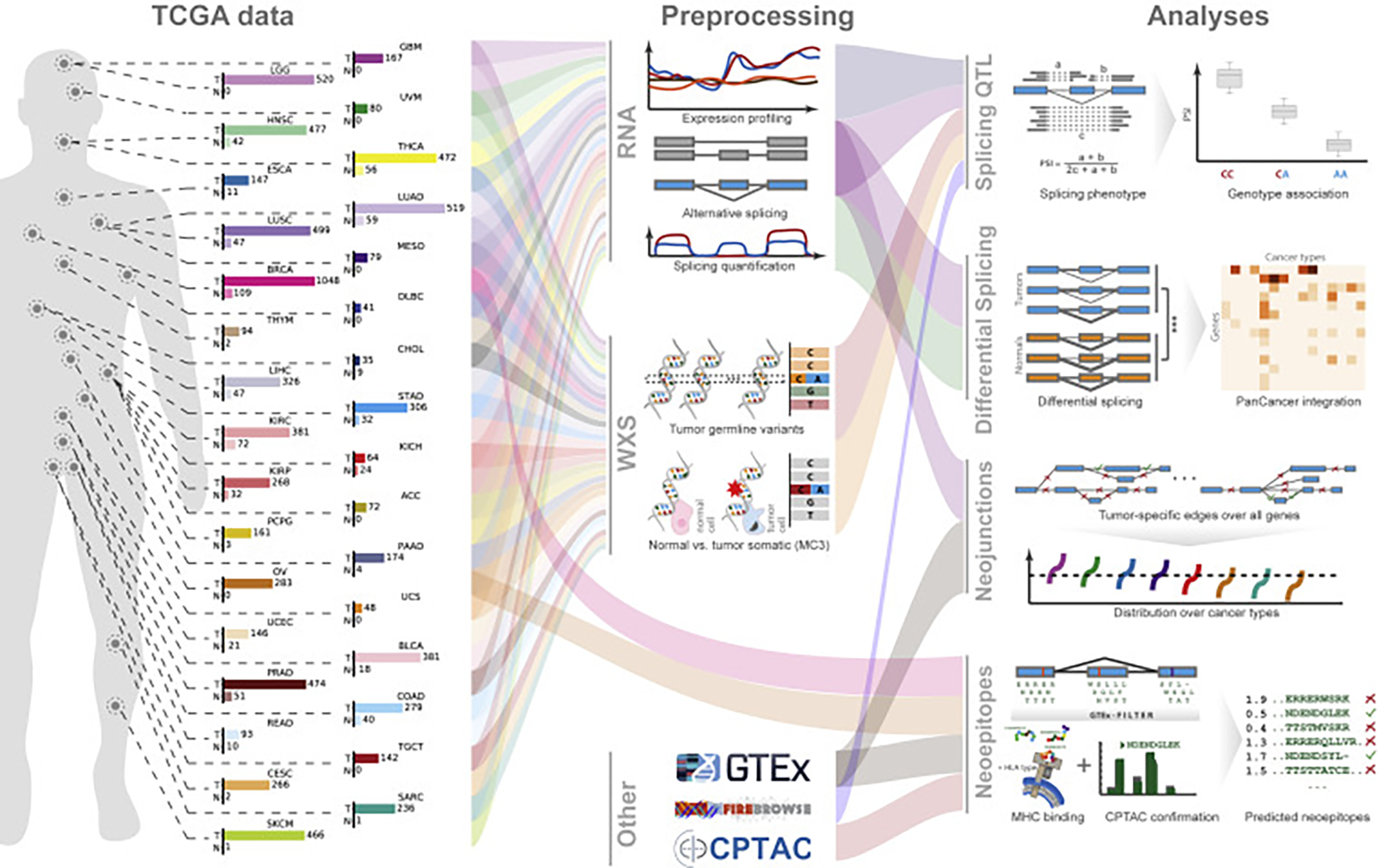
Flow diagram of data and analyses presented in this work. The left schema represents approximate body source sites for the samples of the 32 analyzed cancer types. Bar charts describe numbers of tumor and matched normal samples for each cancer. The numbers for tumor samples represent cases where both tumor RNA-seq as well as whole-exome sequencing (WXS) data are available. The numbers for normal represent matched normal RNA-seq. All samples underwent uniform preprocessing (middle, top), including sequence alignment, expression quantification, and alternative splicing analysis (middle, RNA). Furthermore, samples were used for tumor variant calling and somatic variant calling by the Multi-Center Variant Call (MC3) project (center). In addition, data from other sources, such as the GTEx project, the Broad Firebrowse, and Clinical Proteomic Tumor Analysis Consortium (CPTAC) were included (middle bottom). Different data types were then combined into four integrative analysis sections. For the identification of splicing quantitative trait loci (sQTL, right, top), we associated RNA-seq-derived splicing quantifications with WXS-derived genetic variants, to identify cis and trans effects. To highlight quantitative splicing differences between tumor and normal samples, we used the splicing quantifications to test for significant differences between tumor and normal (illustrated with ***) and ranked the results across all cancers (right, second). To discover neojunctions only present in cancer samples but unobserved in normals or a tissue-matched outgroup, we integrated TCGA RNA-seq data and GTEx RNA-seq data to determine the degree of splicing aberration per sample, marking stark splicing outliers (right, third). Lastly, we analyzed the neojunctions and tested the extent they are translated into proteins, utilizing CPTAC data, confirming a large number of peptides. Many confirmed peptides were also predicted to be MHC-I binders and are excellent neoantigen candidates, promising for immunotherapy (right, bottom). See also Figures S1–S5.
Landscape of Alternative Splicing Events in Cancer
Based on recently developed methodology to construct individual splicing graphs for large gene sets (Kahles et al., 2016), we have systematically quantified changes in splicing event usage across the full TCGA cohort. Throughout all cancer types we found a substantial number of high-confidence splicing events, confirmed by at least 20 RNA sequencing (RNA-seq) reads (Djebali et al., 2012; Nellore et al., 2016; Wang et al., 2008), that contain introns not annotated in GENCODE (Figures 2A, S1A, and S1B), increasing the total number of observed events at least 2-fold. Despite accounting for cohort size and read length effects, we still observed a high variability of additional splicing across individual cancer types. Compared with the alternative splicing events in the GENCODE annotation, we observed that exon skip and alternative 3′ site events represent the majority (27.1% and 27.5%, respectively) of the non-annotated events (Figures S1C and S1D).
Figure 2. Detection of Tumor Alternative Splicing and Splicing Landscape.
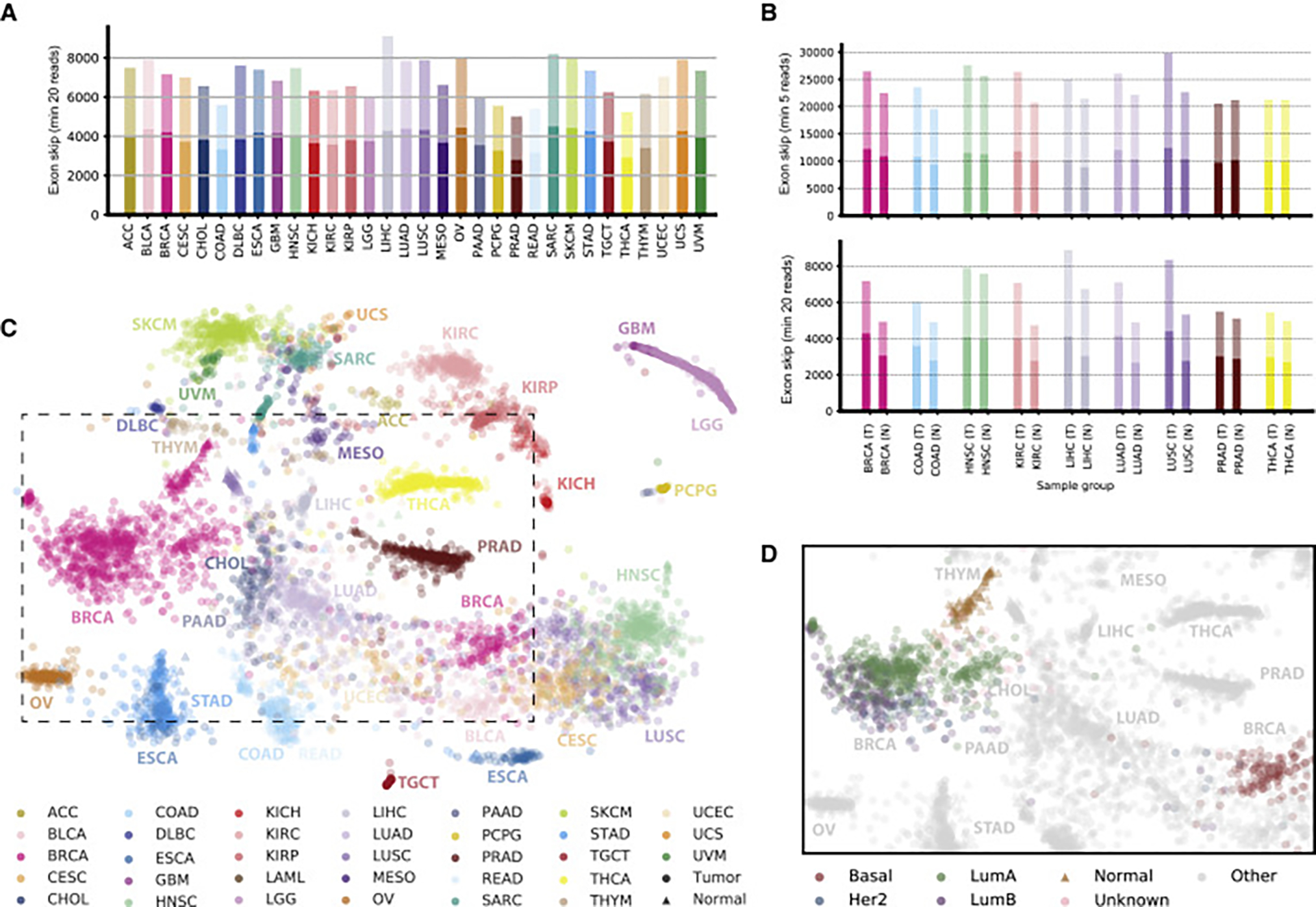
(A) Detection of alternative splicing events. For each cancer type, we considered 40 randomly chosen samples and jointly identified alternative splicing events (exon skipping events are shown) containing junctions that each can be confirmed with a minimum (min) of 20 spliced reads in at least one sample for the respective cancer type. The darker bar fractions correspond to known alternative splicing events and the lighter bar fractions to additional events that are not part of the GENCODE (v19) annotation.
(B) Comparison of the number of alternative splicing events on 40 matched tumor (T) and normal sample (N) pairs for TCGA cancer types with at least 40 normal samples, for events containing junctions confirmed with at least five reads (top) or 20 reads (bottom) in the respective cancer type.
(C) Landscape of alternative splicing for all considered TCGA samples computed on exon skipping PSI scores only. Each point represents a sample, colored according to its TCGA project code. The position of each sample is computed as a t-distributed stochastic neighbor embedding (t-SNE) representation of the higher-dimensional splice event PSI matrix. Tumor samples are shown as circles and normal samples as triangles. The dashed box represents an area detailed in (D).
(D) Samples in the splicing landscape highlighted for subtypes of BRCA. Normal samples are shown as triangles and tumor samples as circles colored according to subtype. Samples of all other cancer types are shown in gray.
When directly contrasted to matching normal tissue, we found a larger amount of alternative splicing events in tumor samples than in normal samples for the majority of the investigated cancers (Figures 2B and S1E–S1H; sample size of tumor and normal samples is 40 for all sets). This difference is especially pronounced for lung adenocarcinoma (LUAD), where we observed an over 30% increase in exon skip events in tumor samples. This effect became even stronger when only events with the strongest splicing changes (measured as an increased ΔPSI [percent spliced in]; Schafer et al., 2015) were used (Figures S1I and S1J).
We have visualized the splicing diversity across the full cohort utilizing a standard dimensionality reduction technique (t-distributed stochastic neighbor embedding [t-SNE]; Van der Maaten and Hinton, 2008; Figures 2C and S1K–S1N) highlighting both the tissue-specific nature of alternative splicing but also cancer-type-specific differences and commonalities. We observed that cancer types, such as colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ) or the group of squamous cell cancers, including lung squamous cell carcinoma (LUSC), cervical squamous cell carcinoma (CESC), and head and neck squamous cell carcinoma (HNSC), that are commonly ascribed with similar characteristics (Cancer Genome Atlas Network, 2012; Hoadley et al., 2014) clustered closely together, even overpowering the identity of the tissue of origin. Examples of the latter are LUAD and LUSC. The same pattern was observed based on a clustering of the median splicing profile (Figures S1O and S1P). Here, we also observed a cluster of uterine carcinosarcoma, uveal melanoma, mesothelioma, skin cutaneous melanoma, and sarcoma, which was less pronounced in a similar clustering based on gene expression profiles (Figures S1Q and S1R). Similarly, kidney chromophobe cancers (KICH) are clearly separated from kidney renal papillary cell carcinomas and kidney renal clear cell carcinomas in the t-SNE based on splicing profile as well as in the corresponding clustering (Figures S1K–S1N). We did not observe similarities in exon skip splicing patterns between breast basal-like and serous ovarian cancers as reported previously based on gene expression (Cancer Genome Atlas Network, 2012), suggesting that gene expression profiles did not drive the patterns in the same way as observed with alternative splicing. However, several breast basal-like cancers were located in the cluster of squamous cell cancers, including samples of LUSC, which had previously been reported as similar to basal-like breast cancer based on the analysis of transcriptional similarities (Chung et al., 2002). Interestingly, we found that, in breast cancer patients (BRCA), different cancer subtypes can be distinguished based on exon skip splicing features (Figures 2D and S1L), forming a notable trajectory across the four main subtypes with the luminal subtypes closely connected and the basal subtype clearly separated. For tumor-matched normal samples we found that, in almost all cases, they cluster clearly separated from the corresponding tumors (Figure S1M). With regard to possible confounding factors, such as library size, we did not observe clear associations to the clustering (Figure S1N). These observations were less pronounced for gene expression counts (Figures S1S and S1T).
Somatic trans Associations Drive Changes of Splicing Events
We performed an association study linking somatic single nucleotide variant (SNV) positions with alternative splicing changes in up to 8,255 donors. As phenotypes we considered a total of 94,749 exon skipping, 30,755 alternative 5′, and 48,365 alternative 3′ events. We considered recurrently called tumor sample population-level variant calls. For the pan-cancer association study we used a linear mixed model implemented in LIMIX (Lippert et al., 2014), correcting for population, tissue, and batch effects. We also checked trans-splicing quantitative trait loci (sQTL) for a potential bias toward purity and ploidy as well as a potential bias for patient gender and total mutational load (Figure S2A). We found that mutational load oftentimes strongly correlates with the genotype of individual variants (Figures S2B–S2D) and those variants also showed significant correlation among each other. This finding makes it difficult to determine whether individual variants themselves affect splicing event changes or are rather tagging higher mutational load, which in turn may have an effect on a wide range of splicing events. For this reason, we have excluded variants showing evidence of association with mutational burden (nominal p value <0.01) from further analysis. A subset of variants, including variants in SF3B1 and U2AF1, did not show this pattern (Figure S2E). In a joint analysis of cis and trans associations with 50% prior on each type, we identified 32 cis- and seven trans-sQTL (Bonferroni corrected p < 0.05).
The trans-sQTL genes included variants with known effect on splicing in SF3B1 (Alsafadi et al., 2016) (Figure 3A and 3B) and U2AF1 (Brooks et al., 2014) but also several candidates whose effects on splicing are less established. One such example is TADA1, where we observe that the distribution of splice event targets across the alternative event types shows a similar 3′ alternative splicing bias as the targets of the SF3B1 mutations (Figure 3C). TADA1 interacts with SF3B5, which itself interacts with various other splicing factors (including SF3B1) and suggests a possible mechanism (Figure 3D). We also found that mutations in the cancer driver gene PPP2R1A are associated with alternative splicing changes in SCRIB, which itself is a tumor suppressor gene and suggests a mechanism on how PPP2R1A may be driving tumorigenesis (Sayani et al., 2008). A further example is IDH1, where the same recurrent somatic missense variant had been associated with inhibiting the enzymatic functions of histones and demethylases. IDH1 variants have been shown to be most prevalent in brain lower grade glioma (LGG) and glioblastoma multiforme (GBM), which we also observe in the Pan-Cancer Atlas cohort (Figure S2F) (Yan et al., 2009). They often appear in patients with low-grade gliomas and have been associated with more favorable outcomes (Yen et al., 2010). Due to the prevalence pattern of IDH1 variants, we also tested for association within the glioma, glioblastoma, and pheochromocytoma and paraganglioma (GBM/LGG/PCPG) cohort to exclude the possibility of tissue-specific effects. In total, we observed broad splicing changes across 377 events (Figure 3B), which are also observed in 326 (243 for LGG only) events (Spearman correlation, Bonferroni corrected p value <0.05) within the GBM/LGG/PCPG cohort, excluding the possibility that this association was mainly driven by tissue identity. Here, we report a link between IDH1 variant and splicing, which is noteworthy since the importance of tumor-specific alternative splicing has already been established (Lefave et al., 2011; Venables et al., 2009).
Figure 3. Large-Scale Somatic cis- and trans-sQTL Analysis.
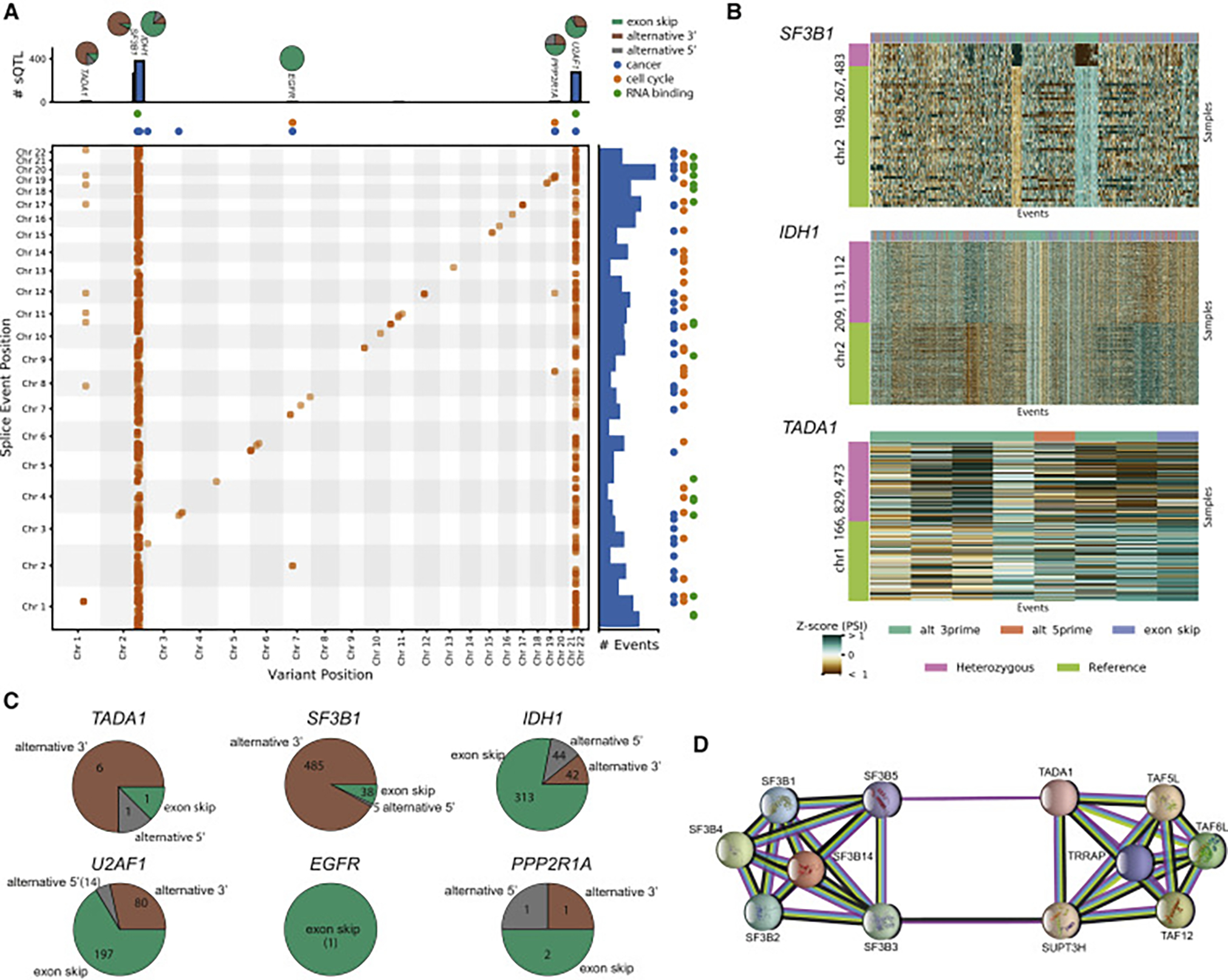
(A) Two-dimensional Manhattan plot with location of a variant (x axis) associated (p ≤ 0.05 after Bonferroni correction separately for cis and trans associations) with an alternative splicing event at a separate location (y axis). Points along the diagonal correspond to cis associations (window 1 Mb) and the remaining points correspond to trans associations. The marginal bar plots show the number of splicing events found to be associated with a single variant (top) and the number of associations found for each alternative splicing event (right). The colored points indicate whether an alternative splicing event or sQTL is within an RNA binding gene (green), cancer census gene (blue), or cell cycle gene (orange). The pie charts on top of the bar show the breakdown of splicing event type composition of the sQTL targets. Brown indicates alternative 3′ events, gray alternative 5′ events, and green exon skip events.
(B) Heatmaps of selected trans-sQTL: PSI z scores of alternative splicing events (columns) significantly associated in trans with the variant. The color bar on the left shows the mutation status for each sample (rows). For visualization purposes, the heatmaps are downsampled to highlight the differences.
(C) Pie charts from (A) detailing the distribution of splicing event targets across three categories (alternative 5′, alternative 3′, and exon skip events).
(D) Protein-protein interaction network of TADA1 and some selected partners (e.g., SF3B5).
See also Figure S2.
Tumor-Specific Splicing Patterns
While significant differences in splicing between tumor and normal samples have been described before (Sebestyén et al., 2015; Srebrow and Kornblihtt, 2006), our analysis strategy allowed us to draw a more complete picture of the splicing landscape over a large array of different tumor types and subtypes. Observations described in the previous sections have shown that a large fraction of the identified events are either quite rare in general or are observed across multiple cancer types but remain rare within the individual tissue, which complicates differential analysis. Also, tissue-specific splicing confounds the assessment of significant differences between tumors and normal samples across cancer types. Our strategy was therefore 2-fold: (1) uncovering rare splicing outliers in tumor samples that recur over multiple cancer types (PSI value deviates strongly from all other samples), and (2) differentially analyzing the broader changes in splicing within the cancer types where tissue-matched normal samples are available.
We identified a large set of 2,570 outlier events in 936 genes, 56 (6%) of which are included in the COSMIC (Catalogue of Somatic Mutations in Cancer) cancer gene census list. One prominent example is the tumor suppressor PTEN, which shows recurrent skipping of exon 3 in multiple cancer types (Figure 4A top) with a strong signal in COAD, LUSC, and uterine corpus endometrial carcinoma (UCEC), not correlating with sample size for the individual groups. Although alternative splicing of PTEN in the context of cancer has been described before (Agrawal and Eng, 2006; Okumura et al., 2011), the skipping of exon 3 has so far been mostly linked to predisposition for heritable disorders (Celebi et al., 2000; Chen et al., 2017). Another example not well linked to splicing is the metastasis suppressor gene NDRG1 (Kovacevic et al., 2011) (Figure 4A bottom). Although in each cancer type only very few outlier samples exist (with BRCA showing the strongest signal), a clear recurrence was apparent with 14 of the 32 cancer types showing at least one notable outlier. When comparing the splicing pattern with an outgroup set of more than 3,000 normal samples for 31 tissues from the GTEx study, we found none of the outliers to be present (Figure 4A).
Figure 4. Differential and Outlier Splicing.

(A) Strip plots showing outlier splicing for an exon skipping event in PTEN (top) and an alternative 3′ splice site event in NDRG1 (bottom). Each column represents a cancer type with its matched normal directly adjacent if available (left of dashed line) and GTEx normal samples (right of dashed line). Each dot corresponds to the PSI value of the selected splicing events in one sample. Outlier samples are emphasized through increased marker size with black outline.
(B) Result of differential splicing analysis between tumor and matched normals for 14 cancer types. Rows correspond to the 40 most significantly altered genes from the COSMIC cancer census set. Shading corresponds to −log10(p value). Columns represent cancer types.
(C) Number of neojunctions per sample for 32 cancer types. Each dot represents the number of tumor-specific introns of a single sample not observed in the annotation and not (or only very rarely) in tissue-matched GTEx samples. If at least five tumor-normal samples were available, the median of neojunctions is indicated by a horizontal dotted red line. Cancer types are sorted from left to right by the mean number of neojunctions.
(D) Overview of tumor introns exclusively detected in cancer samples but not in matched normals. The leftmost panel corresponds to TCGA tumor samples, the middle panel to TCGA matched normal samples, and the right panel to tissue-matched GTEx samples. Shading indicates the fraction of samples that have a tumor-specific intron confirmed with RNA-seq in the corresponding sample group. Rows are sorted according to a ranking that is the result of significance testing between tumor and matched normal samples. For multiple introns per gene, the most significant intron was chosen.
See also Figures S3 and S4.
In addition to rare outliers, we also analyzed broader shifts in splicing within the individual cancer types through a differential analysis of splice form usage between tumor and normal samples. We recovered a significant number of genes from the cancer gene census set as recurrently differentially spliced across tumor types (Figure 4B), partially showing pan-cancer properties (TPM3 in BRCA, HNSC, READ, and lung cancers). One of the genes we found most frequently differentially spliced across all tumor types is PKM. While alternative splicing of exon 9 exclusion giving rise to a change from PKM into PKM2 has been reported previously (Clower et al., 2010; David et al., 2010), suggesting a role not only in the alteration of metabolic function but also in tumor cell proliferation, we detect alternative 3′ site usage for exon 2. Another gene worth highlighting in the context of tumor-specific splicing is BCL2L1 (BCL-x), which produces two splice forms with opposite functions via differential 5′ splice site usage regulated by RBM4 expression, switching between anti-apoptotic or pro-apoptotic states (Wang et al., 2014). Among the top differentially spliced genes, we find a significant enrichment of cancer census genes (5 out of 50, p < 0.003, fold change 3.45, hypergeometric test). In addition, we also observed differential splicing in numerous other factors previously connected to cancer progression, such as NUMB, which encodes a negative regulator of NOTCH and has been previously linked to lung cancers (Pece et al., 2011). We found NUMB to be differentially spliced not only in lung cancers but also in UCEC (Figure S3). In summary, the joint ranking of differentially spliced genes provides a rich resource for the development of new hypotheses.
Increased Complexity of Splicing in Cancer
In addition to the differential usage of splice forms, we were also interested in the identification of exon-exon junctions (EEJs), predominantly observable in tumor samples. We call such tumor-specific EEJs “neojunctions”. Over all samples of the study, we identify ≈251,000 such neojunctions, with an average of 930 per sample (Figures 4C and S4A and S4B). Despite being similar in sample size, LUAD and UCEC had generally higher numbers of neojunctions than LUSC or prostate adenocarcinoma. We found the strongest outliers in bladder urothelial carcinoma (BLCA), UCEC, LUAD, BRCA, and COAD. We observed a marked distinction between tumors and normal samples, where normal samples had substantially lower levels of splicing burden than tumor samples (note that, according to our definition, normal samples can also have neojunctions). This difference appeared to vary across cancer types. Although BLCA, CESC, LUSC, and LUAD showed a very strong distinction, other cancer types, such as liver hepatocellular carcinoma or KICH, had no difference between tumor and normal samples. Notably, on the other end of the spectrum, cholangiocarcinoma seems to have an opposite pattern, with normal samples showing a consistently higher number of neojunctions. Further, different tumor types showed differences in their most extreme complexity values, which cannot be explained by library size or mutational load (Figures S4C and S4D).
To answer the question of which genes contribute most often to the set of neojunctions that could potentially be used as diagnostic or therapeutic markers, we derived a neojunctions-based ranking. Surprisingly, we observed EEJs that show RNA-seq support in over 50% of samples of specific tumor types but are virtually non-existent in TCGA normal samples or GTEx (Figure 4D). Further, we found a large degree of recurrence across cancer types but also observed tissue-specific patterns.
There is a large degree of variation among the cancer types with the largest numbers of neojunctions in BLCA, UCEC, LUAD, BRCA, and COAD that we cannot easily attribute to technical factors. We hypothesize that the large number of neojunctions in some samples can be attributed to a partial breakdown of the splicing machinery that may be the result of somatic mutations or dysregulation of splicing-related factors. In analogy to the term chromothripsis (Stephens et al., 2011), we call this effect syndeo mechanism thripsis, or syndeothripsis. We have identified 110 and 37 TCGA tumor samples with high and very high degree of splicing aberration (Figures S4E and S4F), respectively. The splicing burden in those samples goes far beyond what we observe in most normal samples and we therefore suggest that they are affected by syndeothripsis.
Neojunctions Lead to Potential Neoepitopes
The direct oncogenic effects of tumor-specific alternative splicing are only one of the many consequences splicing can have in a cancer context. We saw evidence indicating that a large fraction of the increased splicing diversity often seems to be a passenger rather than being the driving effect; in particular, we did not find an enrichment of neojunctions in the cancer census gene set (in contrast to the enrichment for differential exon usage). It is quite possible that the increased splicing complexity is due to a lower accuracy, or more “noise” (Pickrell et al., 2010), of splicing in cancer cells that may have a disrupted splicing machinery, although we did not find a direct correspondence between mutational load and detected junctions (Figure S4G). However, this additional transcriptomic complexity can potentially be used to inform cancer therapy. The classic argument is that a fraction of somatic alterations specific to the tumor is translated and can potentially lead to specific neoepitopes. Following this argument, we studied whether a similar effect can be observed for tumor-specific alternative splicing. This is motivated by our prior observation that such events are at least an order of magnitude more abundant than somatic variants. We will denote tumor-specific peptides generated through splicing and predicted to be major histocompatibility complex (MHC)-I binders as alternative splicing-derived putative neoepitopes (ASNs).
Due to the limited availability of proteomics data for TCGA samples, we have restricted the scope of this study to 63 donors for BRCA and ovarian serous cystadenocarcinoma (OV). Based on patient-specific splicing graphs, we derived all polypeptides generated by an EEJ. This resulted in a median of 539,925 EEJ-spanning polypeptides per donor (Figure 5A and Table 1). From these polypeptides, we extracted a list of candidate ASNs based on a pipeline of Clinical Proteomic Tumor Analysis Consortium (CPTAC) mass spectrometry (MS) data confirmation (Mertins et al., 2016; Zhang et al., 2016) and MHC-I binding affinity prediction (Andreatta and Nielsen, 2016) incorporating information on the human leukocyte antigen (HLA) type of each donor (Figure 5A). When considering only RNA-seq-confirmed EEJs, this resulted in, on average, 1.7 ASNs from 1.2 EEJs for each of the samples. For 43/63 (68%) of all considered samples we identified at least one ASN that was CPTAC confirmed and that was a predicted MHC-I binder (Figure 5B). If we do not require RNA-seq confirmation of the specific EEJ in a sample, the number of CPTAC-confirmed, MHC-I binding 9-mers increases significantly (on average ≈11 9-mers from eight EEJs per sample, Figure S5A). Generally, we expect the real number of ASNs to be higher as it would also include 9-mers not spanning an EEJ but completely residing inside a newly included exon or inside a retained intron (not counted in this analysis). Furthermore, a recent study showed that junction-spanning peptides resulting from alternative splicing are underrepresented in protein MS datasets due to the cleavage specificity of trypsin (Wang et al., 2017).
Figure 5. Alternative Splicing-derived Putative Neoepitopes (ASNs).
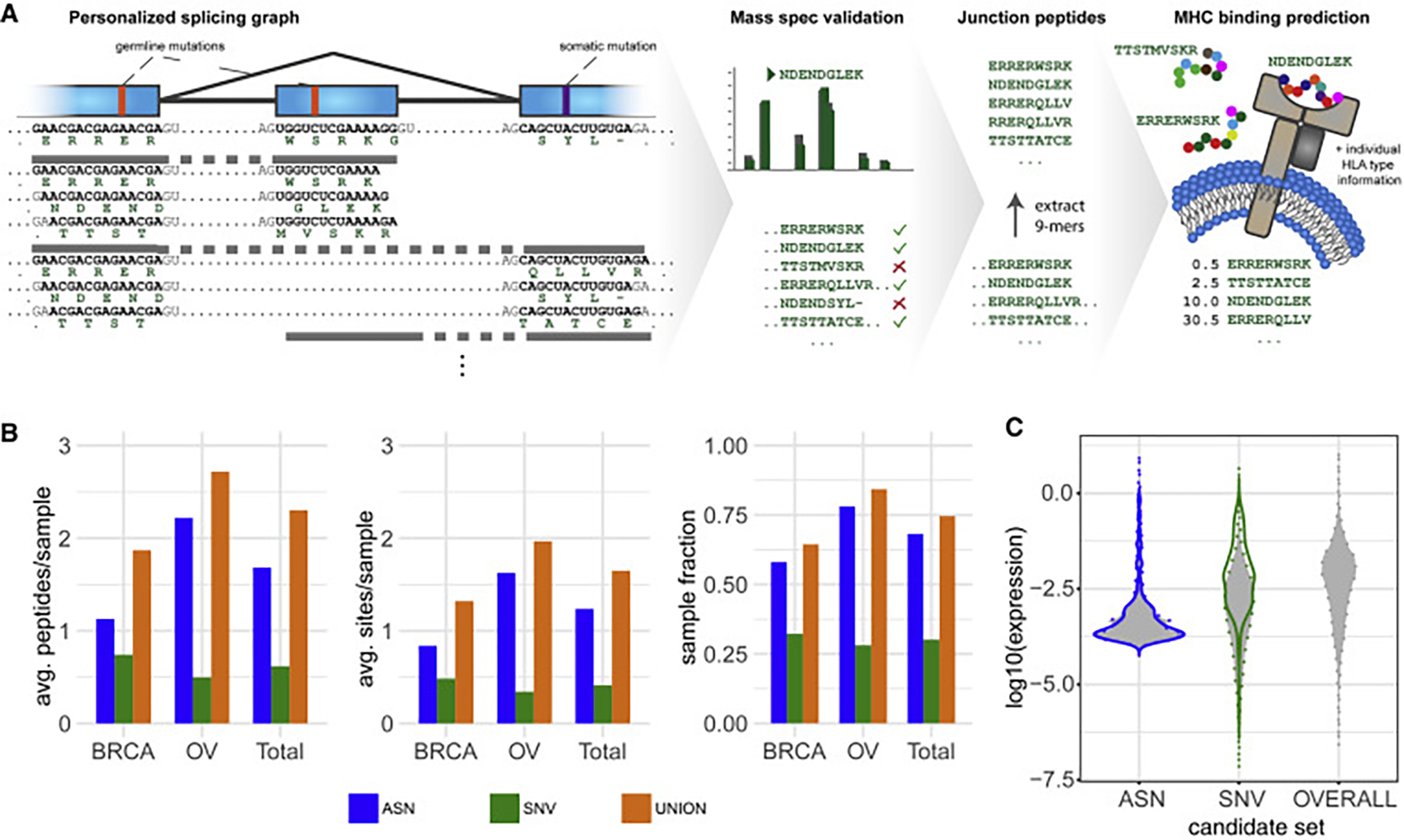
(A) Overview of the ASN detection and validation workflow. Starting from the personalized splicing graph including sample-specific germline and somatic SNVs and the GENCODE genome annotations, polypeptides are generated across the junctions of all introns (including neojunctions). Expression of the resulting polypeptides is validated using CPTAC mass spectra. From the expressed polypeptides, 9-mer substrings spanning junctions are enumerated and filtered based on their presence in a non-cancer background set. For the remaining 9-mers, MHC binding predictions (NetMHC) are obtained with respect to the individual’s HLA-I type. Predicted MHC-I binders (percentile rank <2.0) are considered ASNs. The analysis is repeated for somatic SNV-derived 9-mer peptides for comparison.
(B) Comparison of the contribution of alternative splicing and SNVs to the CPTAC-confirmed putative neoepitope landscape by cancer type. Average number of CPTAC-confirmed neojunction- and SNV-derived 9-mers per sample (left). Average number of CPTAC-confirmed alternative splicing and SNV sites generating putative neoepitopes per sample (center). Sample fractions with at least one CPTAC-confirmed alternative splicing- or SNV-derived putative neoepitope (right). “UNION” corresponds to the combination of both variant types. “Total” refers to the combination of both cancer types. Only neojunctions RNA-expressed in the respective sample or with a minimum RNA expression of 20 spliced reads in at least one of the samples are considered.
(C) Violin plot showing the RNA expression distribution over all expressed neojunction- and SNV-derived 9-mers as well as the overall 9-mer expression distribution. Expression of neojunctions is estimated using the library-size normalized read count confirming the neojunction. For SNV-derived peptides expression is determined by multiplying normalized segment read coverage by the SNV somatic variant allele fraction, and for overall 9-mer expression normalized segment read coverage of all 9-mers is used. The set of SNV-derived 9-mers is used as a representative peptide set for overall 9-mer expression. Filled violins with dotted margins represent the distribution over all 9-mers in the respective set; solid lines represent the distribution over the subset of CPTAC-confirmed 9-mers.
See also Figures S4 and S5 and Table S1.
Table 1.
Distribution of Intron-Spanning Polypeptide Sources
| Type | Median | Mean |
|---|---|---|
| Germline variant | 87,466.00 | 88,536.17 |
| Somatic variant | 172.00 | 610.94 |
| Germline + somatic variant | 42.00 | 208.63 |
| Reference | 518,831.00 | 518,831.00 |
| Total | 606,917.00 | 608,186.75 |
In order to compare ASNs with putative neoepitopes derived from SNVs, following an analogous protocol, we generated a list of all SNV-derived 9-mers that are observed in the respective tumor DNA, can be confirmed by CPTAC mass spectra, and are predicted MHC-I binders. On average we find 0.6 SNV-derived putative neoepitopes derived from 0.4 SNVs per sample. Overall, we found at least one SNV-derived putative neoepitope for 19/63 (30%) of all considered samples. Compared with other studies, these numbers appear relatively low. This can be explained by our requirement of MS validation, which retains only about 1% of otherwise viable peptides due to the low sensitivity of MS. For both cancer types, we found more ASNs than putative neoepitopes derived from SNVs (Figure 5B). Considering ASNs in addition to SNV-derived putative neoepitopes significantly increased the fraction for which at least one CPTAC-confirmed putative neoepitope can be confirmed from 30% to 75% (Figure 5B).
We used RNA-seq data to determine the expression of all neojunctions as a proxy for neojunction-derived 9-mer expression. Similarly, we used the product of RNA-seq-based expression estimates for an exon segment with an SNV and the respective variant allele frequency as a proxy for SNV-derived 9-mer peptide expression. For comparison, we also provide average exon fragment RNA expression as a proxy for overall 9-mer expression. The expression distribution for neojunctions is notably different from the SNV-derived and overall 9-mer expression distribution. Generally, neojunction-derived 9-mers show slightly lower expression than SNV-derived 9-mers (Figure 5C). CPTAC-confirmed SNV-derived putative neoepitopes show a higher overall associated RNA expression than ASNs, but there are fewer of them per sample.
Independent of the source of a neoepitope, potential therapeutic utility arises from recurrent observation across multiple patients. SNVs are typically rare, and we did not observe any recurring CPTAC-confirmed SNV-derived putative neoantigens. However, we did find that 15 ASNs in our study are observed across several samples within the same cancer type and five ASNs recur in both cancer types (Figure S5B and Table S1).
DISCUSSION
Alternative splicing events have previously been shown to contribute to cancer development and progression. Several examples of such mechanisms are known, but only a few comprehensive studies on transcript changes are available (Climente-González et al., 2017) and a complete picture of alternative splicing complexity and its potential to generate neoantigens is still missing.
In this work, we focus on five types of alternative splicing events, namely intron retention, exon skipping, mutually exclusive exons, and alternative 3′ and alternative 5′ splice site changes (Cartegni et al., 2002; Hatje et al., 2017; Roy et al., 2013; Wang et al., 2008). Our study builds on a previously published tool (Kahles et al., 2016) and analyzes specific splicing event types involving a small number of exons from RNA-seq data without the need to know complete transcripts. This study is a major contribution toward a comprehensive analysis of alternative splicing events across all suitable TCGA samples (another study without focus on cancer was performed in Nellore et al., 2016). Most previous studies considered isoform expression of known transcripts. For instance, a recent study analyzed the impact of isoform switches on gene function (Climente-González et al., 2017). We combine the splicing phenotypes with variants obtained from re-analysis of exome sequencing data for an sQTL association study. A previous study considered alternative splicing across 48 tissues from up to 620 donors (GTEx Consortium et al., 2017; Saha et al., 2017). Another work considered genetic determinants of alternative splicing in blood (Zhang et al., 2015). Both studies were restricted to cis associations of common germline variants with known isoform expression. Large QTL association studies of common variants with gene expression were reported on both TCGA (Gong et al., 2017; Li et al., 2013) and non-TCGA datasets (GTEx Consortium et al., 2017). In our study, we focus on variants that have been shown to occur as somatic variants in some individuals but may also occur in the germline genome in others. Those variants are typically substantially less frequent (between 0.1% and 5% across the cohort) than most common germline variants. The available data provide sufficient statistical power to detect trans-sQTL events that were difficult to detect previously (Fonseca et al., 2017; GTEx Consortium et al., 2017; Lehmann et al., 2015). Finally, our study comprehensively analyzes the extent to which alternative splicing in tumors leads to cancer-specific RNA transcripts that are translated into tumor-specific proteins and, hence, may be targeted by immunotherapy. This has been shown for specific genes for B cell lymphomas and ovarian cancers (Barrett et al., 2015; Vauchy et al., 2015). Here we use the data from TCGA and GTEx to identify alternative splicing events that are tumor specific and integrate them with re-analyzed CPTAC MS data (Mertins et al., 2016; Zhang et al., 2016) to show for two tumor types that the resulting mRNAs are indeed translated into tumor-specific proteins that contain peptides with the potential for MHC presentation.
We built a catalog of alternative splicing events found in these samples with hundreds of thousands of events of which ≈80% are not annotated in GENCODE. In addition, we show that in tumor samples we can observe on average ≈20% more alternative splicing than in matched normal samples. The analysis of RNA-seq data to extract splicing events is computationally demanding and we hope that the identified and quantified alternative splicing events for all Pan-Cancer Atlas donors can be used as a resource to simplify future analyses. One limitation of this study, however, is that we only analyze bulk RNA-seq and whole-exome sequencing data and we therefore have limited power to detect and understand subclonal effects.
To understand the impact of somatic variants on alternative splicing events, we performed a large-scale association study of tumor variants with alternative splicing variation across the genome. In order to characterize individual variants and to avoid a burden-type strategy, we based our analysis on tumor variant calls that overlap with recurrent highly confident somatic variant calls allowing us to leverage changes at the germline as well as the tumor levels. Association mapping in trans is technically challenging and requires large cohorts such as the one considered here. In particular, identifying and addressing confounders appropriately is often challenging. Here we have accounted for common confounders in the model and additionally checked our results against correlation with purity, ploidy, patient sex, as well as mutational load. Besides the aforementioned strong effect of mutational load, we did find that the variant in PPP2R1A is sex biased, which is expected as this gene is a known driver of ovarian/uterine cancer. We also observed a correlation between purity and one of the SF3B1 mutations. Eventually, this strategy allowed us to identify a small number of known (SF3B1, U2AF1) and a larger number of additional (TADA1, PPP2R1A, IDH1) distal sQTLs that affect multiple alternative splicing events. Overall, 385 genes have a splicing event that is the target of one of these sQTLs. This illustrates the power of the pan-cancer analyses of TCGA data to generate valuable hypotheses for further mechanistic studies; for instance, to understand how a somatic variant in IDH1 leads to widespread changes in alternative splicing across the genome. It is likely that splicing and expression patterns are changed as an indirect, downstream effect of altered histone and demethylase patterns. The link of TADA1 to alternative splicing events may be more direct, since TADA1 interacts with SF3B5 and also shows a similar distribution of affected AS types as the known mutations in SF3B1. PPP2R1A has previously been reported to affect nonsense-mediated decay (NMD) (Sayani et al., 2008). We hypothesize that the loss-of-function somatic mutation in PPP2R1A leads to a disruption of NMD function, which then leads to a detection of AS variants that would otherwise get degraded by NMD. This would explain why we find associations with alternative splicing. In summary, this sQTL analysis, utilizing a large sample set size, reveals promising additional long-range associations with changes in exon composition of multiple genes.
Our study of the alternative splicing landscape demonstrated that taking information on alternative splicing events into account is beneficial for characterizing cancer subtypes. A systematic analysis of splicing events in tumors enabled us to identify genes that are recurrently alternatively spliced across multiple cancer types. These events include well-understood examples of alternative splicing changes promoting tumor development (e.g., BCL2L1, PKM) but also alternative splicing in cancer genes for which the effect is not yet well understood (e.g., NUMB). However, in this context we would like to note that even though TCGA is a tremendous resource for cancer research, certain biases are inherent to the dataset (mostly related to the design of the study), which might not be representative in certain circumstances. For instance, TCGA tumors are treatment naive, consist predominantly of primary tumors, and are biased toward larger tumors with sufficient size to extract analysis material. Within this study we cannot directly address this sampling bias other than pointing it out and interpreting our results within its context.
One important element of this study was to determine the number of additional EEJs, which we called neojunctions, that appear predominantly in tumors. We found that some samples have a large degree of splicing aberration, where we can identify thousands of neojunctions. Overall, we identified ≈251,000 neojunctions with an average of ≈930 neojunctions per sample, and many of them are recurrent: ≈18,000 of those neojunctions appear in at least 100 samples. For comparison, there are only 13 somatic SNVs that were found in at least 100 tumors (the highly recurrent SNV BRAFV600E being one of them). The vast number of neojunctions and the high level of recurrence are very promising for future work.
To further develop the hypothesis of the importance of alternative splicing for the immune response to cancer, we have analyzed to what extent neojunctions contribute to the translation of potential neoepitopes. This required the development of an analysis pipeline to go from neojunctions to the predicted translations of peptides around the neojunctions to the MS confirmation, and the MHC-I binding prediction in order to determine which peptides are potential neoepitopes. Overall, by considering splicing-derived in addition to SNV-derived peptides, the fraction of samples with at least one CPTAC-confirmed putative neoepitope increases from 30% to 75% for BRCA and OV tumors. In addition, the splicing-derived putative neoepitopes have a high degree of recurrence, suggestive of potential use in immunotherapeutic intervention.
In addition to the already completed analyses, we are currently investigating further extensions and refinements. While the current work only focuses on MHC-I alleles for peptide binding predictions, incorporation of MHC-II alleles appears to be beneficial as well (Sun et al., 2017). Also, MHC binding is essential but not sufficient for a peptide to be capable of inducing an immune response. Both the actual expression and processing of the peptide as well as its immunogenicity need to be validated. The tandem MS-based proteomics analysis employed in this study aims at validating peptide expression. However, proteomics analysis alone cannot validate processing let alone presentation of a given peptide by MHC. An important improvement will be to replace this step; e.g., with MS-based immunopeptidomics (Bassani-Sternberg et al., 2016). Subsequently, immunogenicity of the detected naturally processed neoepitopes could be determined via CD8+ T cell killing (Vitiello and Zanetti, 2017). In addition, it may also be helpful to use more sensitive protein MS techniques; for instance, data-independent acquisition MS (Gillet et al., 2012). Lastly, our current choice of cancer types was mainly driven by availability within the TCGA and CPTAC cohorts. We are actively working on extending this work to other cancer types and into a more controlled experimental setup.
In summary, in this study we considered the many differences of alternative splicing in cancer compared with normal cells and suggest that these differences are characteristic for individual cancer types and could be used for the design of immunotherapeutic interventions, such as chimeric antigen receptor T cell therapy or personalized anti-cancer vaccines.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gunnar Rätsch (gunnar.ratsch@ratschlab.org).
METHOD DETAILS
Data Download
Raw RNA-sequencing samples in FASTQ format and whole-exome sequencing alignment files in BAM format were downloaded from the CancerGenomicsHub (CGHub) at UCSC (Wilks et al., 2013) using the cgtools software. CGHub has been decommissioned over the course of this project’s duration. All data is now available at the Genomic Data Commons (https://gdc.cancer.gov/, more information below). Proteomics data for TCGA breast and ovarian cancer samples were downloaded from the CPTAC data portal (Edwards et al., 2015).
RNA-Seq Alignment
All previously downloaded RNA-seq samples were individually aligned using a uniform processing pipeline based on the STAR aligner (Dobin et al., 2013). Due to the long duration of the whole project and the extensive analyses, we used two different alignment strategies to include further samples in a second run. While almost all analyses were performed with both strategies, the sQTL analysis was completed on strategy 1 only and the neoepitope analysis was completed on the junctions resulting from the intersection of strategies 1 and 2., For the remaining analysis, we compared all results and found no significant differences between the two alignment strategies.
Strategy 1
The STAR software (version 2.4.0i) was used in a 2-pass setup, where the first alignment pass was used to identify non-annotated junctions in the input data, allowing for the construction of a genome index containing non-annotetd junctions. The second pass alignment was then performed against the junction-aware index, allowing for a more sensitive recovery of non-annotated splice junction from the data. A complete set of command line parameters:
1st Pass.
STAR –genomeDir GENOME –readFilesIn READ1 READ2 –runThreadN 4 –outFilterMultimapScoreRange 1 –outFilter MultimapNmax 20 –outFilterMismatchNmax 10 –alignIntronMax 500000 –alignMatesGapMax 1000000 –sjdbScore 2 –align SJDBoverhangMin 1 –genomeLoad NoSharedMemory –readFilesCommand cat –outFilterMatchNminOverLread 0.33 –outFilter ScoreMinOverLread 0.33 –sjdbOverhang 100 –outSAMstrandField intronMotif –outSAMtype None –outSAMmode None.
Re-indexing.
STAR –runMode genomeGenerate –genomeDir GENOME_TMP –genomeFastaFiles GENOME_FASTA –sjdb Overhang 100 –runThreadN 4 –sjdbFileChrStartEnd SJ.out.tab (from 1st pass)
2nd Pass.
STAR –genomeDir GENOME_TMP –readFilesIn READ1 READ2 –runThreadN 4 –outFilterMultimapScoreRange 1 –out FilterMultimapNmax 20 –outFilterMismatchNmax 10 –alignIntronMax 500000 –alignMatesGapMax 1000000 –sjdbScore 2 –align SJDBoverhangMin 1 –genomeLoad NoSharedMemory –limitBAMsortRAM 70000000000 –readFilesCommand cat –outFilterMatch NminOverLread 0.33 –outFilterScoreMinOverLread 0.33 –sjdbOverhang 100 –outSAMstrandField intronMotif –outSAMattributes NH HI NM MD AS XS –outSAMunmapped Within –outSAMtype BAM SortedByCoordinate –outSAMheaderHD @HD VN:1.4 –out SAMattrRGline ID SM:
Strategy 2
Again, this strategy comprises a two-pass alignment approach. As a difference to strategy 1, a newer version of the STAR aligner was used (2.5.3a), that re-creates the index augmented with non-annotated junctions on the fly and does not require manual rebuild of the reference genome index. Hence only a single run per sample was necessary. The full list of command line parameters was as follows:
STAR –genomeDir GENOME –readFilesIn READ1 READ2 –runThreadN 4 –outFilterMultimapScoreRange 1 –outFilterMultimap Nmax 20 –outFilterMismatchNmax 10 –alignIntronMax 500000 –alignMatesGapMax 1000000 –sjdbScore 2 –alignSJDBoverhang Min 1 –genomeLoad NoSharedMemory–limitBAMsortRAM 70000000000–readFilesCommand cat –outFilterMatchNminOverLread 0.33 –outFilterScoreMinOverLread 0.33 –sjdbOverhang 100 –outSAMstrandField intronMotif –outSAMattributes NH HI NM MD AS XS –sjdbGTFfile GENCODE_ANNOTATION –limitSjdbInsertNsj 2000000 –outSAMunmapped None –outSAMtype BAM Sorted ByCoordinate –outSAMheaderHD @HD VN:1.4 –outSAMattrRGline ID::<ID> –twopassMode Basic –outSAMmultNmax 1
RNA-Seq Quality Control and Filtering
For each RNA-seq library we ran the FastQC analysis tool (version 0.11.6) and collected library statistics. Further we collected alignment statistics and computed a bias score between 3 and 5′ end of each gene to measure possible degradation. Based on these measurements, we developed a scoring scheme to exclude samples. A sample could be flagged as low-quality if at least 3 of key FastQC criteria were labeled as fail (criteria: per base quality, per sequence quality, gc content, N content, sequence overrepresentation), the degradation score was larger than Q3 + 1.5xIQR, the GC content was more than 1.5xIQR below Q1 or above Q3 or the number of reads was more than 1.5xIQR below Q1 or above Q3. A sample was excluded, if it was flagged for at least three low quality criteria, the degradation score was larger than Q3 + 3xIQR, the GC content was more than 3xIQR below Q1 or above Q3 or the number of reads was more than 3xIQR below Q1 or above Q3.
Tumor Variant Calling
We have used Picard (version 1.87) and the Genome Analysis Toolkit (GATK, version 3.4.46) (McKenna et al., 2010) for variant calling. We followed the good-practice guidelines for variant calling with GATK (Van der Auwera et al., 2013). We omitted a duplicate-marking of the input files as the alignment versions downloaded from CGHub already had duplicates marked. Each alignment file was then stripped of all unmapped reads and re-indexed using samtools (version 1.2).
Utilizing the capture-region information for the exome capture procedure of each file and dbSNP (version 138), the 1000 Genome Project Phase 1 and the Mills and 1000G gold standard set as compendium of known sites, we used GATK for base quality score recalibration.
Re-calibration Step 1
java -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R <genome.fasta> -I <alignment.bam> -knownSites <known_sites> -L <capture_region> -o <outfile1> -nct <threads>
Re-calibration Step 2
java -jar GenomeAnalysisTK.jar -T PrintReads -R <genome.fasta> -I <alignment.bam> -BQSR <outfile1> -o <outfile2> -nct <threads>
Variant calling was then performed using the GATK Haplotype Caller. The calling limit was defined as a +/− 1kb window around all genes in the GENCODE annotation (v19), including all intron regions.
Variant Calling
java -Xmx4g -Xms512m -Djava.io.tmpdir=<TMPDIR> -jar GenomeAnalysisTK.jar -T HaplotypeCaller -R <genome.fasta> -I <alignment.bam> –dbsnp <dbsnp_v138.vcf> -o <outfile> –output_mode EMIT_ALL_CONFIDENT_SITES -ERC GVCF –variant_ index_type LINEAR –variant_index_parameter 128000 -pairHMM VECTOR_LOGLESS_CACHING -mbq 15 –minPruning 5 -S STRICT –activeRegionOut <outfile_region> –activityProfileOut <outfile_profile> -L <calling_limit.bed> -nct <threads>
The gVCF files created in the previous step for each sample were then merged in an iterative process until less than 100 merged files remained:
java -jar GenomeAnalysisTK.jar -T CombineGVCFs -R <genome.fasta> –variant <s1> … –variant <sN> -o <outfile_merged1>
The merged gVCF files were then used for joint variant calling on each chromosome independently using the GATK:
java -Xmx16g -jar GenomeAnalysisTK.jar -T GenotypeGVCFs -L <chr> -nt <threads> –dbsnp <dbsnp_v138.vcf> -R <genome.fasta> -variant <outfile_merged1> … –variant <outfile_mergedN> -o <outfile_final>
Tumor Variant Filtering
Tumor variant calls have been filtered in the following way: Variants that have less than 100 samples with valid calls, quality of less than 100, are multi-allelic or indels have been removed from analysis. We further required more than 5 alternate alleles for each polymorphic position. All variants have been encoded into an additive scheme with 0 representing the homozygous reference state, 1 the heterozygous state and 2 the homozygous alternate allele. In this study, we ignore the existence of variants that appear sub-clonally. For somatic variant calls the unfiltered MC3 calls from PanCanAtlas have been used (version 0.2.8; Synapse ID: syn7834470). From that variant call set we extracted single nucleotide variants (SNVs) but excluded variants tagged by the following criteria:
StrandBias
contest
oxog
ndp
pcadontuse
nonpreferredpair
badseq
gapfiller
common_in_exac
PoN
We also required that at least three variant callers agree on a variant call and excluded variants which have a higher than 5% minor allele frequency in the 1,000 genomes cohort. Non-recurrent variant calls (variants which appear in only one sample) have also been excluded from further analysis. This filtering ensures a high-quality variant call set which includes intronic variants at exon boundaries.
The somatic and tumor variant calls have subsequently been intersected, resulting in a total of 4,041 variant calls considered in this analysis.
Gene Expression and Splicing Event Quantification
For expression counting we used a custom python script that counted a read towards a gene if at least one base of the read overlapped an exonic position of the gene. We did not count secondary alignments (as indicated in the BAM files with flag 256) and masked regions from the annotation where multiple genes overlapped. We also generated a second set of expression counts (non-alt) that excluded all genomic positions from counting that were annotated with both intron and exon.
Alternative splicing events were detected and quantified using the SplAdder toolkit (Kahles et al., 2016). Briefly, with the pipeline we generated a sample-specific splicing graph per sample and gene, integrating additional information based on RNA-seq alignment data. For each gene all graphs of all samples were then merged into a joint splicing graph. If a graph of a gene had more than 10,000 edges, we excluded it from further merging. Lastly, we pruned edges from the merged graphs if they were supported by less than 10 samples in the cohort. This procedure resulted in a single merged graph per gene for all samples.
Subsequently, we quantified nodes and edges of the merged graph for all samples based on the RNA-seq alignments. Edges were quantified as number of supporting spliced alignments and exons as mean read coverage over all exonic positions. From the quantified graphs we detected all alternative events of the following types: exon skipping, intron retention, alternative 3′ splice site and alternative 5′ splice site (Figure S1A). For each event we then computed percent spliced in (PSI) values based on the previously quantified splicing graphs.
Detection of Cancer-specific Introns
To account for cohort size and read length effects, this analysis was performed on a randomly selected subset of 40 tumor samples from each tumor type (for all types with sufficient number of samples) and the RNA-seq reads were trimmed to a uniform length of 50 nt if their length was exceeding this threshold. The detection and quantification of alternative splicing events was otherwise performed as described above.
Characterization of Neojunctions
Starting with the splicing graphs for all genes that we generated previously, we removed all intron edges that could be confirmed with at least 2 reads in at least 1% of samples (~30) from the GTEx cohort. Before thresholding, junction counts were normalized for library size differences. Further, samples with a library size (measured as the upper quartile expression of autosomal genes) of less than 2,500 were excluded from this analysis to exclude artifacts caused by low complexity libraries not caught by the global QC. We then computed splicing complexity (the number of neojunctions) as the sum of the total number of splice graph edges confirmed with at least 3 reads in a sample and at least 20 reads over the whole cohort as total sum over all genes of a sample.
For a ranking of neojunctions, we sorted all EEJs with an increased specificity towards tumor samples requiring a minimum number of spliced alignments across the EEJ per sample to count it as expressed (tumor: 10 spliced reads, normals: 3 spliced reads, GTEx: 2 spliced reads). Further, we removed all junctions that were present in more than 1% of GTEx or TCGA normal samples or had a higher mean expression in TCGA normals compared to TCGA tumor samples (within the same cancer type). We then ranked all EEJs by predominant occurrence in tumor samples based on Fisher’s exact test. To aggregate over multiple events in a gene, we show only the event with the strongest effect.
Identification of Rare Splicing Outliers
For outlier detection we applied a set of hard filter criteria on our full set of detected alternative splicing events. To allow for a stable and comparable analysis, we only checked for outliers in cancer types with at least 100 samples available. For each event we required that the maximum spread of PSI values in the GTEx cohort as well as within the TCGA normal sample is at most 0.3. We further excluded an event if it i) had less than 80 samples with sufficiently many reads (N = 10) to compute a PSI, ii) had a spread of PSI values in the respective cancer type of less than 0.4. We then computed the number of samples with a PSI value of at least 10 times the inter-quartile range above/below the upper/lower quartile and marked them as outliers. If we found less than 5 or more than 100 outliers for the event and cancer-type, there were no TCGA normal samples with sufficient read count (N = 10) available or the minimum PSI overall normal samples was lower than the smallest tumor sample PSI, we excluded the event. All remaining events were noted as outliers in the respective cancer type.
t-SNE
We have generated t-SNE figures for every event type (exon-skip, alternative events and intron retention) as well as for a list of concatenated events based on a matrix of sample by event matrix of percent spliced in values. All t-SNE figures have been produced using the package sklearn (Pedregosa et al., 2011). The aforementioned matrix has been filtered to remove events which had more than 30% samples missing values. A value is missing, if we were unable to compute a stable PSI value, which was the case when we had less than 10 spliced reads available in the denominator. Samples have been filtered if more than 10% of events had samples missing. Remaining missing values have been mean-imputed. Next, we performed a PCA based on a linear kernel of this data matrix. The first 100 principal components have been used for the t-SNE generation. t-SNE with learning rate 500 and perplexity 50 have been used for visualization throughout this work unless stated otherwise.
Differential Analysis of Splicing Events
The differential splicing analysis was run on all tumor types that had at least 50 tumor samples and 10 tissue-matched normal samples available. For each tumor type independently, we randomly subsampled the available groups to 50 tumor and 10 normal samples. We then used SplAdder to perform a differential test (based on a generalized linear model) between the two groups, utilizing the split-alignment counts acress the junctions of an event. To account for additional variability in the tumor samples, we repeated the testing 9 times, each time on a different random subset. For each event, the final p value was recorded as the median of the 9 results. If the same gene had more than one splicing event tested, we kept the one with the minimal p value. The results from all individual tissues were then aggregated into a common ranking using Fisher′s method for meta-analysis.
Filtering of Events and Variants for Somatic trans-Association
As phenotypes we considered a total of 94,749 exon skipping, 30,755 alternative 5′ and 48,365 alternative 3′ events for all samples that had a total of at least five reads across all junctions in the splicing event. We considered tumor sample population-level variant calls that are confirmed by at least three somatic variant callers as high-quality somatic variants in at least two donors in the MC3 variant calls, including intronic regions. For each of these positions, we re-analyzed the tumor whole exome sequencing data in order to determine the genotype in all samples. This strategy considered germline as well as somatic variants for the association analysis. Therefore, we leveraged the occurrence of single nucleotide variants on the germline genome in conjunction with somatic single nucleotide variants to determine functional effects of these variants.
Statistical Association of Genetic Variation and Alternative Splicing
A linear mixed model has been used (Lippert et al., 2014), accounting for population structure as a random effect and cancer type as fixed effect to account for cancer specific variation as well as batch effects. We also included gender and gene expression as fixed effects to account for potential detection bias. All splicing event quantifications have been quantile normalized to match a standard normal distribution. Depending on the amount of read support of individual splice events, we used up to 8,255 samples for the QTL-analysis. More specifically, we required that the sum of reads across all junction for every sample and splice event are covered by more or equal to 5 reads.
The splicing index is being used as a quantitative phenotype. In order to address some unwanted properties of this phenotype we have performed an inverse normal transform on all PSI’s estimated by SplAdder. To avoid ties, we have added a small amount of random pseudo-noise in the range of 10−5 to each estimate before transformation. Splicing events which exclusively exhibited ties, have been removed from analysis. We also excluded phenotypes in which less than 10% of the samples had any valid estimates.
We applied a Bonferroni multiple testing correction on cis-associations and trans-associations separately accounting for the total number of variants (cis-associations, p value < 6.19e-6) as well as the total number of events and variants tested (trans-associations, p value < 3.55e-11).
In the resulting set of sQTL, we have removed all events which showed over-inflation for the variants tested (more than 20 variants significantly associated). Further, we tested all variants for association with mutational load (Spearman Correlation) and removed all variants showing any evidence of correlation (nominal p value < 0.01). Mutational load has been calculated as total number of SNV based on MC3 calls from PanCanAtlas have been used (version 0.2.8 PUBLIC; Synapse ID: syn7834470).
Derivation of Splicing-Derived Peptides
Based on the splicing graphs, all intron-spanning polypeptides (encoding the translated amino acid sequence of a node pair) for a subset of 63 TCGA cancer samples (including BRCAand OV) were derived. For each gene, we generated a foreground splicing graph by collapsing the reference transcripts of each gene into a graph and augmenting it with patient-specific germline and somatic variants as well as additional junction information from RNA-seq across the TCGA cohort as follows.
The polypeptides were obtained by seeding the splicing graph traversal at the first CDS of the canonical transcripts and then following the splicing graph structure along any existing edges in read strand order. While traversing the graph, all possible read-frame shifts that could exist while translating an exon/CDS were taken into account. We define an intron-spanning polypeptide as the peptide generated by translating the pair of exons connected by the intron with respect to a certain reading-frame. The polypeptides were generated both for the reference DNA sequence and the personalized DNA sequences. Personalized DNA sequences are comprised of three subsets obtained by introducing variants into the reference genome as follows: (i) the germline variants of a particular donor only, (ii) the somatic variants of a particular donor only, and (iii) both the germline and the somatic variants. To obtain background sequences, we generated polypeptides that result from translating canonical transcripts annotated in the GENCODE reference annotation (version 19). Furthermore, we generate background peptides by using a splicing graph derived from GTEx control tissue samples. For each donor, we also generate a personalized GTEx background set by introducing germline variants.
MHC-I Binding Predictions
MHC class I binding predictions were performed using NetMHC-4.0 (Andreatta and Nielsen, 2016). Donor HLA-I types originate from a previous study on the same TCGA samples (Shukla et al., 2015) and were downloaded from the PanCanAtlas Jamboree server.
For each tumor sample, MHC-I binding affinity and corresponding ranks were determined for all 9-mers derived from background and personalized protein sequences with respect to all donor HLA-I alleles supported by NetMHC-4.0. (For seven donors only three HLA-I alleles were supported, for 17 donors all six were. Median number of supported alleles was 5.) For each 9-mer NetMHC-4.0 outputs a binding affinity rank per allele. This rank is based on a reference set of 400,000 random natural peptides. Peptides with a predicted binding affinity rank of better than 2% are considered binders. NetMHC-4.0 was used as follows:
MHC-I Binding Prediction
netMHC -a <donor_allele_string> -l 9 -f <proteins.fasta>
Identification of Expressed Peptides
For each of the 63 TCGA tumor samples under consideration, we generated individual polypeptide databases comprising reference-based and personalized versions of all sample-specific splicing-derived and reference annotation-derived protein sequences. Personalized versions of the reference annotation-derived protein sequences were generated analogously to those of the splicing-derived sequences.
OpenMS (Kohlbacher et al., 2007; Röst et al., 2016) was used to identify polypeptides from a sample′s polypeptide database as follows: In order to allow to control for false discovery rates, decoy sequences were added to the database. Subsequently, we used MS-GF+ to search the corresponding CPTAC data set for tryptic sequences from the database. A false discovery rate of 5% on the peptide-spectrum match level was used to filter the identified polypeptides. Any 9-mer contained in at least one of the identified polypeptides is considered CPTAC-confirmed. The following OpenMS commands were used to perform the polypeptide identification:
Add Decoy Sequences
DecoyDatabase -in <input.fasta> -out <decoy_db.fasta>
Search CPTAC Data Set
MSGFPlusAdapter -ini MSGF_iTRAQ.ini -in <cptac_spectra> -out <output.idXML> -database <decoy_db.fasta> -executable <path_to_msgfplus> -java_memory 80000 -threads 6
The file MSGF_iTRAQ.ini is available at https://github.com/ratschlab/pancanatlas_code_public.
Control for False Discovery Rate
PeptideIndexer -in <msgf_output.idXML> -fasta <decoy_db.fasta> -out <pi_output.idXML> -allow_unmatched -enzyme: specificity ‘semi’
FalseDiscoveryRate -in <pi_output.idXML> -out <fd_output.idXML>
IDFilter -in <fd_output.idXML> -out <fdr_filtered.idXML> -score:pep 0.05
Alternative Splicing-Derived Neoepitope Candidates
Starting from a sample’s splicing-derived polypeptide sequences we extracted all intron-spanning peptides of length 9. Due to the lack of normal RNA samples, tumor-specific splicing events cannot be accurately determined. In order to increase specificity, we consider all splicing events observed in GTEx as normal and exclude all GTEx 9-mers (including personalized peptides) from the list of alternative splicing-derived neoepitope candidates. Furthermore, all 9-mers also observed in the reference genome or the personalized reference genome, i.e., the reference genome after introduction of the respective donor′s germline variants and/or the somatic variants, were removed from the list. Furthermore, in order to increase specificity, we only considered 9-mers derived from EEJs also contained in the splicing graph generated on the new RNA-seq alignments (strategy 2).
SNV-Derived Neoepitope Candidates
Starting from a donor’s personalized reference genome representing either somatic variants only or both germline and somatic variants, all peptides of length 9 containing a somatic variant are extracted. All 9-mers also found in the reference genome or in the personalized genome containing germline variants only are removed from this list. Moreover, analogous to the identification of alternative splicing-derived neoepitope candidates, all GTEx 9-mers (including personalized peptides) are excluded.
Estimation of RNA Expression of 9-mer Peptides
RNA expression of 9-mers overall was determined by using the average RNA expression of the corresponding exon fragment as a proxy. For SNV-derived 9-mers this expression was multiplied by the respective variant allele frequency. We estimated the RNA expression of neojunction derived 9-mers by library size-normalizing the read counts confirming the respective junction.
Re-analysis on Representative Sample Subset
As a means to account for various sampling differences in the TCGA RNA-seq data set, we generated a representative sub-cohort with a reduced variability to repeat some of the key analysis. From the set of whitelisted samples passing our initial QC, we selected 10 tumor and 10 normal samples for all cancer types that had at least 10 tumor and 10 normal samples available. We pre-processed the fastq files of these samples and randomly subsampled each sample to contain 48,000,000 reads. All reads exceeding 50nt were trimmed down to 50nt. For alignment, we used strategy 2 as described above. All downstream analyses were analog as described above.
DATA AND SOFTWARE AVAILABILITY
Supplementary data accompanying this manuscript is available at the Genomic Data Commons (GDC) of the National Cancer Institute under the following URL: https://gdc.cancer.gov/about-data/publications/PanCanAtlas-Splicing-2018.
Research code that was used to implement methods described above along with further descriptions is publicly available on GitHub under the following address: https://github.com/ratschlab/pancanatlas_code_public.
Supplementary Material
KEY RESOURCES TABLE.
Highlights.
Systematic analysis of alternative splicing landscape across 8,705 cancer patients
Somatic trans-sQTL analysis identifies drivers of global splicing aberrations
Many tumors contain numerous neojunctions not typically found in normal samples
Neojunctions can be confirmed by MS and form a class of potential neoantigens
Significance.
Immunotherapy is currently a promising direction for treating cancer patients. Not all cancer types are suited for this type of approach. Among those that show potential benefit from immunotherapeutic treatment, deriving suitable antigens for a targeted vaccine is a considerable challenge. Tumor-specific splicing presents a large new class of splicing-associated potential neoantigens that may affect the immune response and could be exploited in immunotherapy; e.g., in personalized tumor vaccines. By considering neojunction-derived, in addition to SNV-derived, peptides as potential antigens, the fraction of samples for which at least one putative neoantigen can be identified and confirmed by mass spectrometry proteomics increases from 30% to 75%.
ACKNOWLEDGMENTS
We would like to thank the ICGC/PCAWG Transcriptome Working Group (in particular, Angela Brooks and Yuichi Shiraishi), Mitch Levesque, Mark Rubin, Ruedi Aebersold, Alessandra Curioni, Michal Bassani-Sternberg, George Coukos, and Nikolaus Schultz for fruitful discussions and feedback on project design, specific methods, and the manuscript. We also gratefully acknowledge the thorough review by the reviewers that has led to significant improvements of the manuscript. Data used in this publication were generated by the CPTAC (NCI/NIH). O.K. and T.S. acknowledge support from BMBF 031A535A. This work was funded by MSKCC core funding, ETH Zurich core funding to G.R., and SFA PHRT project grant PHRT #106 by the ETH Board to G.R.
Footnotes
DECLARATION OF INTERESTS
Michael Seiler, Peter G. Smith, Ping Zhu, Silvia Buonamici, and Lihua Yu are employees of H3 Biomedicine, Inc. Parts of this work are the subject of a patent application: WO2017040526 titled “Splice variants associated with neomorphic sf3b1 mutants.” Shouyoung Peng, Anant A. Agrawal, James Palacino, and Teng Teng are employees of H3 Biomedicine, Inc. Andrew D. Cherniack, Ashton C. Berger, and Galen F. Gao receive research support from Bayer Pharmaceuticals. Gordon B. Mills serves on the External Scientific Review Board of Astrazeneca. Anil Sood is on the Scientific Advisory Board for Kiyatec and is a shareholder in BioPath. Jonathan S. Serody receives funding from Merck, Inc. Kyle R. Covington is an employee of Castle Biosciences, Inc. Preethi H. Gunaratne is founder, CSO, and shareholder of NextmiRNA Therapeutics. Christina Yau is a part-time employee/consultant at NantOmics. Franz X. Schaub is an employee and shareholder of SEngine Precision Medicine, Inc. Carla Grandori is an employee, founder, and shareholder of SEngine Precision Medicine, Inc. Robert N. Eisenman is a member of the Scientific Advisory Boards and shareholder of Shenogen Pharma and Kronos Bio. Daniel J. Weisenberger is a consultant for Zymo Research Corporation. Joshua M. Stuart is the founder of Five3 Genomics and shareholder of NantOmics. Marc T. Goodman receives research support from Merck, Inc. Andrew J. Gentles is a consultant for Cibermed. Charles M. Perou is an equity stock holder, consultant, and Board of Directors member of BioClassifier and GeneCentric Diagnostics and is also listed as an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. Matthew Meyerson receives research support from Bayer Pharmaceuticals; is an equity holder in, consultant for, and Scientific Advisory Board chair for OrigiMed; and is an inventor of a patent for EGFR mutation diagnosis in lung cancer, licensed to LabCorp. Eduard Porta-Pardo is an inventor of a patent for domainXplorer. Han Liang is a shareholder and scientific advisor of Precision Scientific and Eagle Nebula. Da Yang is an inventor on a pending patent application describing the use of antisense oligonucleotides against specific lncRNA sequence as diagnostic and therapeutic tools. Yonghong Xiao was an employee and shareholder of TESARO, Inc. Bin Feng is an employee and shareholder of TESARO, Inc. Carter Van Waes received research funding for the study of IAP inhibitor ASTX660 through a Cooperative Agreement between NIDCD, NIH, and Astex Pharmaceuticals. Raunaq Malhotra is an employee and shareholder of Seven Bridges, Inc. Peter W. Laird serves on the Scientific Advisory Board for AnchorDx. Joel Tepper is a consultant at EMD Serono. Kenneth Wang serves on the Advisory Board for Boston Scientific, Microtech, and Olympus. Andrea Califano is a founder, shareholder, and advisory board member of Darwin-Health, Inc and a shareholder and advisory board member of Tempus. Toni K. Choueiri serves as needed on advisory boards for Bristol-Myers Squibb, Merck, and Roche. Lawrence Kwong receives research support from Array BioPharma. Sharon E. Plon is a member of the Scientific Advisory Board for Baylor Genetics Laboratory. Beth Y. Karlan serves on the Advisory Board of Invitae.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and one table and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.07.001.
REFERENCES
- Agrawal S, and Eng C (2006). Differential expression of novel naturally occurring splice variants of PTEN and their functional consequences in Cowden syndrome and sporadic breast cancer. Hum. Mol. Genet. 15, 777–787. [DOI] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. (2016). Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 7, 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M, and Nielsen M (2016). Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CL, DeBoever C, Jepsen K, Saenz CC, Carson DA, and Frazer KA (2015). Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc. Natl. Acad. Sci. USA 112, E3050–E3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, et al. (2016). Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 7, 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørklund SS, Panda A, Kumar S, Seiler M, Robinson D, Gheeya J, Yao M, Alnæs GIG, Toppmeyer D, Riis M, et al. (2017). Widespread alternative exon usage in clinically distinct subtypes of invasive ductal carcinoma. Sci. Rep. 7, 5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A, Wang P, and Zenklusen JC (2018). SnapShot: TCGA-analyzed tumors. Cell 173, 530. [DOI] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. (2014). A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9, e87361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, and Krainer AR (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 3, 285–298. [DOI] [PubMed] [Google Scholar]
- Celebi JT, Wanner M, Ping XL, Zhang H, and Peacocke M (2000). Association of splicing defects in PTEN leading to exon skipping or partial intron retention in Cowden syndrome. Hum. Genet. 107, 234–238. [DOI] [PubMed] [Google Scholar]
- Chen HJ, Romigh T, Sesock K, and Eng C (2017). Characterization of cryptic splicing in germline PTEN intronic variants in Cowden syndrome. Hum. Mutat. 38, 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CH, Bernard PS, and Perou CM (2002). Molecular portraits and the family tree of cancer. Nat. Genet. 32 Suppl, 533–540. [DOI] [PubMed] [Google Scholar]
- Climente-González H, Porta-Pardo E, Godzik A, and Eyras E (2017). The functional impact of alternative splicing in cancer. Cell Rep. 20, 2215–2226. [DOI] [PubMed] [Google Scholar]
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, and Krainer AR (2010). The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 107, 1894–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, and Manley JL (2010). HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards NJ, Oberti M, Thangudu RR, Cai S, McGarvey PB, Jacob S, Madhavan S, and Ketchum KA (2015). The CPTAC data portal: a resource for cancer proteomics research. J. Proteome Res. 14, 2707–2713. [DOI] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca NA, Kahles A, Lehmann K-V, Calabrese C, Chateigner A, Davidson NR, Demircioğlu D, He Y, Lamaze FC, Li S, et al. (2017). Pan-cancer study of heterogeneous RNA aberrations. bioRxiv. 10.1101/183889. [DOI] [Google Scholar]
- Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill JA, Lee C, Jentz D, et al. (2015). Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 5, 850–859. [DOI] [PubMed] [Google Scholar]
- Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. (2013). SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 3, 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Andrew Futreal P, Kasprzyk A, Birney E, Mullikin JC, Wooster R, and Stratton MR (2001). Cancer and genomics. Nature 409, 850–852. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Mei S, Liu C, Xiang Y, Ye Y, Zhang Z, Feng J, Liu R, Diao L, Guo A-Y, et al. (2017). PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 46, D971–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. (2012). Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 44, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium, Laboratory, Data Analysis & Coordinating Center (LDACC)-Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI, et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatje K, Rahman R-U, Vidal RO, Simm D, Hammesfahr B, Bansal V, Rajput A, Mickael ME, Sun T, Bonn S, et al. (2017). The landscape of human mutually exclusive splicing. Mol. Syst. Biol. 13, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, et al. (2014). Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahles A, Ong CS, and Rätsch G (2016). SplAdder: identification, quantification and testing of alternative splicing events from RNA-seq data. Bioinformatics 32, 1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlbacher O, Reinert K, Gröpl C, Lange E, Pfeifer N, Schulz-Trieglaff O, and Sturm M (2007). TOPP-the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197. [DOI] [PubMed] [Google Scholar]
- Kovacevic Z, Sivagurunathan S, Mangs H, Chikhani S, Zhang D, and Richardson DR (2011). The metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via p53-independent mechanisms. Carcinogenesis 32, 732–740. [DOI] [PubMed] [Google Scholar]
- Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan Y-X, and Cartegni L (2011). Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 30, 4084–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K-V, Kahles A, Kandoth C, Lee W, Schultz N, Stegle O, and Rätsch G (2015). Integrative genome-wide analysis of the determinants of RNA splicing in kidney renal clear cell carcinoma. Pac. Symp. Biocomput. 20, 44–55. [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Seo J-H, Stranger B, McKenna A, Pe’er I, Laframboise T, Brown M, Tyekucheva S, and Freedman ML (2013). Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 152, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun N, Lu Z, Sun S, Huang J, Chen Z, and He J (2017). Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 393, 40–51. [DOI] [PubMed] [Google Scholar]
- Lippert C, Casale F, Rakitsch B, and Stegle O (2014). LIMIX: genetic analysis of multiple traits. bioRxiv. 10.1101/003905. [DOI] [PubMed] [Google Scholar]
- Marcelino Meliso F, Hubert CG, Favoretto Galante PA, and Penalva LO (2017). RNA processing as an alternative route to attack glioblastoma. Hum. Genet. 136, 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellore A, Jaffe AE, Fortin J-P, Alquicira-Hernández J, Collado-Torres L, Wang S, Phillips RA III, Karbhari N, Hansen KD, Langmead B, et al. (2016). Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the sequence read archive. Genome Biol. 17, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Yoshida H, Kitagishi Y, Nishimura Y, and Matsuda S (2011). Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem. Biophys. Res. Commun. 413, 395–399. [DOI] [PubMed] [Google Scholar]
- Paik PK, Drilon A, Fan P-D, Yu H, Rekhtman N, Ginsberg MS, Borsu L, Schultz N, Berger MF, Rudin CM, et al. (2015). Responseto MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 5, 842–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S, Confalonieri S, Romano PR, and Di Fiore PP (2011). NUMB-ing down cancer by more than just a NOTCH. Biochim. Biophys. Acta 1815, 26–43. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, and Pritchard JK (2010). Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 6, e1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, Hess JM, Uzunangelov V, Walter V, Danilova L, et al. (2017). Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 33, 151. [DOI] [PubMed] [Google Scholar]
- Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, Fangazio M, Vaisitti T, Monti S, Chiaretti S, et al. (2011). Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 118, 6904–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rőst HL, Sachsenberg T, Aiche S, Bielow C, Weisser H, Aicheler F, Andreotti S, Ehrlich H-C, Gutenbrunner P, Kenar E, et al. (2016). OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748. [DOI] [PubMed] [Google Scholar]
- Roy B, Haupt LM, and Griffiths LR (2013). Review: alternative splicing (AS) of genes as an approach for generating protein complexity. Curr. Genomics 14, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Kim Y, Gewirtz ADH, Jo B, Gao C, McDowell IC, GTEx Consortium, Engelhardt BE, and Battle A (2017). Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 27, 1843–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayani S, Janis M, Lee CY, Toesca I, and Chanfreau GF (2008). Widespread impact of nonsense-mediated mRNA decay on the yeast intronome. Mol. Cell 31, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer S, Miao K, Benson CC, Heinig M, Cook SA, and Hubner N (2015). Alternative splicing signatures in RNA-seq data: percent spliced in (PSI). Curr. Protoc. Hum. Genet. 87, 11–16. [DOI] [PubMed] [Google Scholar]
- Sebestyén E, Zawisza M, and Eyras E (2015). Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 43, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, et al. (2015). Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 33, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srebrow A, and Kornblihtt AR (2006). The connection between splicing and cancer. J. Cell Sci. 119, 2635–2641. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Chen F, Meng F, Wei J, and Liu B (2017). MHC class II restricted neoantigen: a promising target in tumor immunotherapy. Cancer Lett. 392,17–25. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Maaten L, and Hinton G (2008). Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605. [Google Scholar]
- Vauchy C, Gamonet C, Ferrand C, Daguindau E, Galaine J, Beziaud L, Chauchet A, Henry Dunand CJ, Deschamps M, Rohrlich PS, et al. (2015). CD20 alternative splicing isoform generates immunogenic CD4 helper T epitopes. Int. J. Cancer 137, 116–126. [DOI] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, et al. (2009). Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 16, 670–676. [DOI] [PubMed] [Google Scholar]
- Vitiello A, and Zanetti M (2017). Neoantigen prediction and the need for validation. Nat. Biotechnol. 35, 815–817. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, and Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen D, Qian H, Tsai YS, Shao S, Liu Q, Dominguez D, and Wang Z (2014). The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 26, 374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Codreanu SG, Wen B, Li K, Chambers M, Liebler DC, and Zhang B (2017). Detection of proteome diversity resulted from alternative splicing is limited by trypsin cleavage specificity. Mol. Cell. Proteomics 17, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks C, Maltbie D, Diekhans M, and Haussler D (2013). CGHub: kick-starting the worldwide genome web. Proceedings of the Asia-Pacific Advanced Network 35, 10.7125/APAN.35.1. [DOI] [Google Scholar]
- Yan H, Williams Parsons D, Jin G, McLendon R, Ahmed Rasheed B, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. (2009). IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 350, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen KE, Bittinger MA, Su SM, and Fantin VR (2010). Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene 29, 6409–6417. [DOI] [PubMed] [Google Scholar]
- Zhang X, Joehanes R, Chen BH, Huan T, Ying S, Munson PJ, Johnson AD, Levy D, and O′Donnell CJ (2015). Identification of common genetic variants controlling transcript isoform variation in human whole blood. Nat. Genet. 47, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou J-Y, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 766, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chen Z, and Yong L (2017). Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol. Oncol. 748, 368–374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary data accompanying this manuscript is available at the Genomic Data Commons (GDC) of the National Cancer Institute under the following URL: https://gdc.cancer.gov/about-data/publications/PanCanAtlas-Splicing-2018.
Research code that was used to implement methods described above along with further descriptions is publicly available on GitHub under the following address: https://github.com/ratschlab/pancanatlas_code_public.