

TO THE EDITOR:
Approximately 30 years after the start of the Human Genome Project, we sequenced the genome of an infant with encephalopathy in just over 11 hours. The results led to a clinical diagnosis of thiamine metabolism dysfunction syndrome 2 (THMD2) 16.5 hours after a blood sample was obtained and 13 hours after we initiated sequencing, which informed treatment of the infant, thereby illustrating the fulfillment of the promise of the Human Genome Project to transform health care.
A 5-week-old, previously healthy male infant was admitted after 2 hours of inconsolable, atypical crying and irritability (Fig. 1). Examination revealed downward eye deviation when he cried. Computed tomography of the head showed multiple large, bilateral hypodensities. Ten years earlier, his parents, who were first cousins, had had a child with a similar neurologic presentation that rapidly progressed to epileptic encephalopathy; the child died at 11 months of age without an etiologic diagnosis, despite extensive evaluation.
Figure 1. Clinical and Diagnostic Course in the Patient and His Sibling.
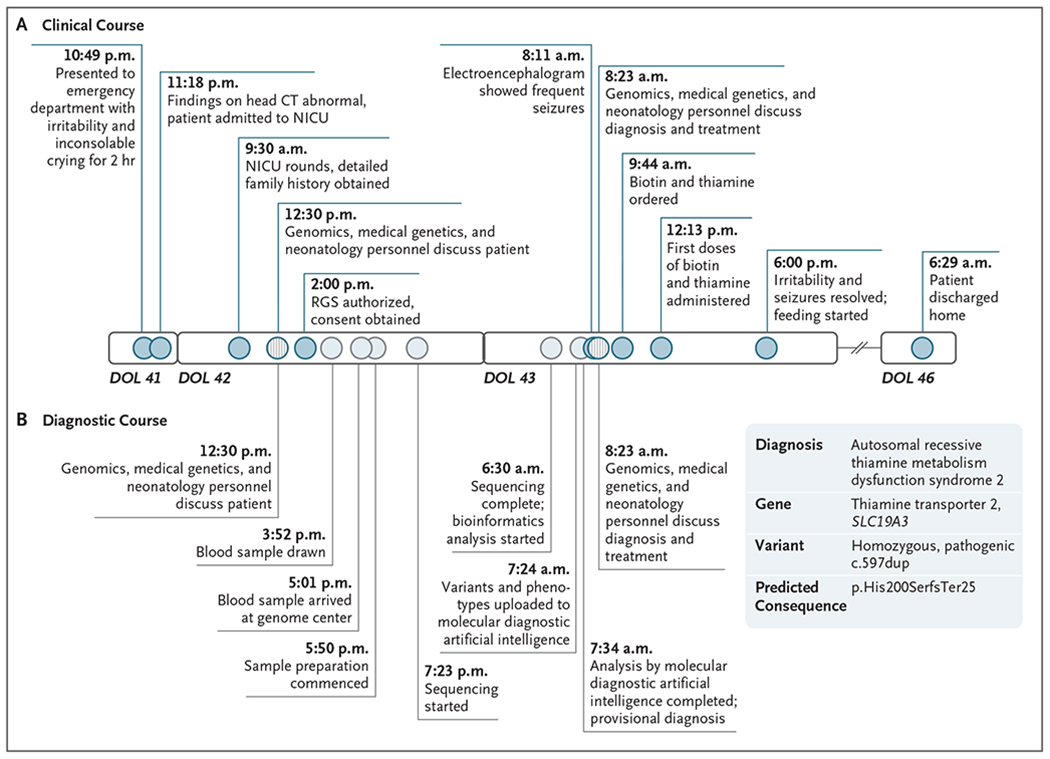
The homozygous frameshift variant in the thiamine transporter 2 gene SLC19A3 that was detected in the patient had previously been reported as pathogenic both in a child with a similar presentation1 and in the ClinVar database (accession number, VCV000533549.2). Circles along the timeline indicate events that occurred during the clinical course (darker blue) and the diagnostic course (lighter blue). Circles with vertical lines indicate points of interaction among neonatology, genomics, and medical genetics personnel. CT denotes computed tomography, DOL day of life, NICU neonatal intensive care unit, and RGS rapid genome sequencing.
Infantile encephalopathy is associated with approximately 1500 genetic diseases, many of which are clinically indistinguishable but have unique, effective treatments. Without prompt treatment, permanent neurologic injury or death occurs in many infants with these diseases. A resemblance to common conditions, such as hypoxic ischemic encephalopathy, can lead to inappropriate or delayed treatment. These considerations prompted us to seek a diagnosis through genome sequencing. With written informed consent from the parents, we obtained blood samples 17 hours after admission (Fig. 1) and performed genome sequencing, using standard and prototypic methods in parallel.2,3 We used the criteria of the American Society of Medical Genetics to evaluate the pathogenicity of a frameshift variant (c.597dup; p.His200fs) in SLC19A3 (ClinVar accession number, VCV000533549.2) and make a provisional diagnosis of THMD2 14 hours 33 minutes after his blood sample arrived at the genome center. (SLC19A3 encodes a thiamine transporter.) This provisional diagnosis was clinically confirmed 49 minutes later.
Video electroencephalography showed numerous seizures occurring in the interim. Thiamine and biotin administration was started 37.5 hours after admission, and phenobarbital administration was started 2 hours later. One 15-second seizure was recorded thereafter. Six hours later, the patient was alert, calm, and bottle feeding. Standard, trio genome sequencing confirmed the diagnosis. After a further 24 hours passed without seizures, the patient was discharged. He is now thriving at 7 months of age.
Early infantile, “Leigh-like” THMD2 is characterized by rapid neurologic deterioration and, if untreated, childhood death.4,5 We believe that the patient’s sibling died in infancy from THMD2, given the carrier status of both parents and the similarity of radiographic and neurologic findings (Fig. S1 in the Supplementary Appendix, available with the full text of this letter at NEJM.org).
This case illustrates the potential for decreased suffering and improved outcomes through the implementation of rapid genome sequencing in a multidisciplinary, integrated, precision medicine delivery system.1 Such a system includes identification of infants with suspected genetic diseases on the day of admission, rapid genome sequencing as a first-tier test, communication of results in a manner that facilitates prompt transition from empirical to etiologically informed treatment, and implementation within a learning health care system.1 Currently, rapid genome sequencing is being implemented in Australia, England, Germany, and Wales and in Medicaid pilot programs in California, Florida, and Michigan.
Supplementary Material
Acknowledgments
Supported by Alexion, Ernest and Evelyn Rady, the J. Willard and Alice S. Marriott Foundation, and a grant (UL1TR002550) from the National Institutes of Health.
Footnotes
Disclosure forms provided by the authors are available with the full text of this letter at NEJM.org.
Contributor Information
Mallory J. Owen, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Anna-Kaisa Niemi, Rady Children’s Hospital, San Diego, CA
David P. Dimmock, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Mark Speziale, Rady Children’s Hospital, San Diego, CA
Mark Nespeca, Rady Children’s Hospital, San Diego, CA
Kevin K. Chau, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Luca Van Der Kraan, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Meredith S. Wright, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Christian Hansen, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Narayanan Veeraraghavan, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Yan Ding, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Jerica Lenberg, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Shimul Chowdhury, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Charlotte A. Hobbs, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Sergey Batalov, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Zhanyang Zhu, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Shareef A. Nahas, Rady Children’s Institute for Genomic Medicine, San Diego, CA
Sheldon Gilmer, Rady Children’s Hospital, San Diego, CA.
Gail Knight, Rady Children’s Hospital, San Diego, CA
Sebastien Lefebvre, Alexion Pharmaceuticals, Boston, MA
John Reynders, Alexion Pharmaceuticals, Boston, MA
Thomas Defay, Alexion Pharmaceuticals, Boston, MA
Jacqueline Weir, Illumina, San Diego, CA
Vicki S. Thomson, Illumina, San Diego, CA
Louise Fraser, Illumina, San Diego, CA
Bryan R. Lajoie, Illumina, San Diego, CA
Tim K. McPhail, Illumina, San Diego, CA
Shyamal S. Mehtalia, Illumina, San Diego, CA
Chris M. Kunard, Illumina, San Diego, CA
Kevin P. Hall, Illumina, San Diego, CA
Stephen F. Kingsmore, Rady Children’s Institute for Genomic Medicine, San Diego, CA
References
- 1.Kingsmore SF, Ramchandar N, James K, et al. Mortality in a neonate with molybdenum cofactor deficiency illustrates the need for a comprehensive rapid precision medicine system. Cold Spring Harb Mol Case Stud 2020;6(1):a004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark MM, Hildreth A, Batalov S, et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med 2019;11(489):eaat6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimmock DP, Clark MM, Gaughran M, et al. An RCT of rapid genomic sequencing among seriously ill infants results in high clinical utility, changes in management, and low perceived harm. Am J Hum Genet 2020;107:942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabarki B, Al-Hashem A, Alfadhel M. Biotin-thiamine-responsive basal ganglia disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews [Internet]. Seattle: University of Washington, Seattle, 2013:1993–2020 (https://www.ncbi.nlm.nih.gov/books/NBK169615/). [PubMed] [Google Scholar]
- 5.Lee JS, Yoo T, Lee M, et al. Genetic heterogeneity in Leigh syndrome: highlighting treatable and novel genetic causes. Clin Genet 2020;97:586–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.