

Abstract
Ageing is a complex, multifaceted process leading to widespread functional decline affecting every organ and tissue. Remarkably, it is still unknown if ageing has a unifying causal mechanism or is grounded in multiple sources. Phenotypically, the ageing process is associated with a wide variety of features at the molecular, cellular and physiological level, e.g., genomic and epigenomic alterations, loss of proteostasis, declining overall cellular and sub-cellular function, deregulation of signaling systems. However, the relative importance, mechanistic interrelationships and hierarchical order of those ageing features have not been clarified. Here, we synthesize accumulating evidence that DNA damage affects most if not all aspects of the ageing phenotype making it a most likely unifying cause of ageing. Hence, targeting DNA damage and its mechanistic links with the ageing phenotype will provide a logical rationale for developing interventions to counteract age-related dysfunction and disease in concert.
The ultimate cause of ageing
There is wide agreement that ageing in metazoa is ultimately caused by the declining force of natural selection, once genes have been passed on to the next generation1. Hence, mutations that only have adverse effects late in life, are not eliminated by purifying selection and therefore allowed to accumulate in the germline2. Pleiotropic mutations with beneficial effects before, but adverse effects after reproduction, are even positively selected3. The consequences of accumulation of such germline mutations only become evident when lifespan is no longer curtailed by extrinsic sources of early mortality, as with modern humans or animals kept in protective environments, explaining the steep rise in multimorbidity at advanced age.
While the evolutionary logic of ageing is clear, surprisingly little is known about its proximate causes, even though ageing is the source of most chronic diseases and the main burden for healthcare in advanced societies world-wide. Does ageing have a sheer infinite number of origins, as predicted by evolutionary theory, or could there be one ancestral cause present from the beginning that with increasing complexity of life was later joined by many secondary causes? In an attempt to better understand ageing, a number of processes that causally contribute to pathologies occurring at old age have been identied4. In this perspective we show how the main features of the ageing phenotype, causally and mechanistically, converge onto one factor: DNA damage (Figure 1), rendering this a strong candidate as the primary cause of ageing.
Figure 1. DNA damage is the driver of ageing.
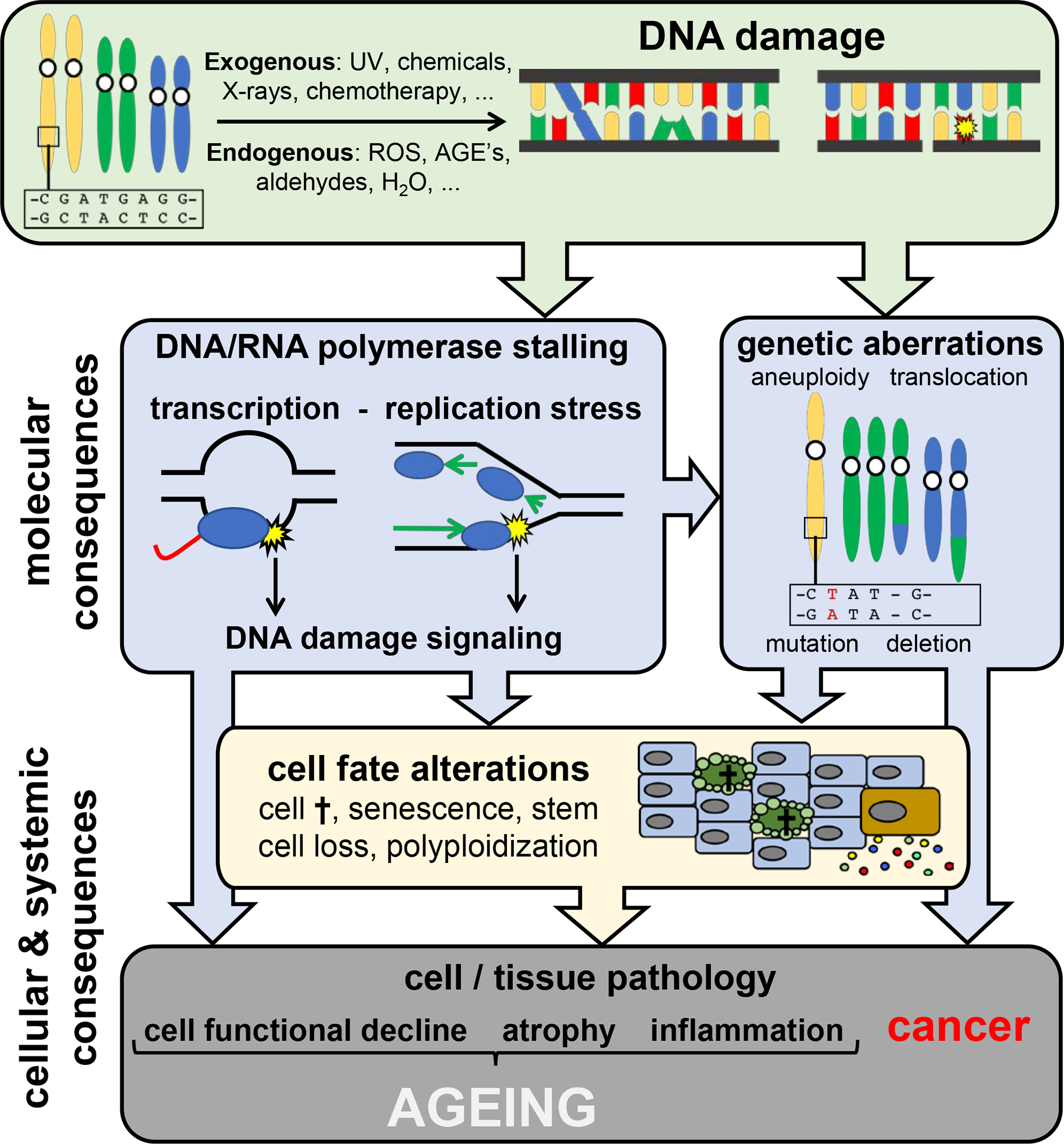
The nuclear and mitochondrial genomes are continuously damaged by exogenous agents (UV, X-rays, chemical compounds in food, water, air), endogenous sources such as reactive oxygen species (ROS), aldehydes and advanced glycation endproducts (AGEs) and spontaneous reactions (hydrolysis). Molecular consequences of time-dependent accumulating DNA damage are: i) genetic aberrations, such as mutations and chromosomal instability, and ii) stalling of RNA and DNA polymerases by DNA lesions, which provokes DNA damage signaling and interferes with primary DNA functioning. Cellular and tissue consequences of DNA damage include cell fate decisions such as cell death and senescence leading to functional loss of cells and organs, cancer, atrophy and inflammation.
Effects of DNA damage at the molecular level
The inherently instable genome
As the primary template encoding all genetic information, DNA is surprisingly instable. Genome instability can be defined as the tendency of the genome to undergo mutation, i.e., any permanent, transmittable DNA sequence alteration in the genome such as a base substitution, a deletion or insertion, copy number variation, chromosomal aberration or retrotransposition. Mutations generally adversely affect function and are a major cause of cancer and genetic disease. However, in the germ line they are also the substrate of evolution.
Mutations are an inherent characteristic of both nuclear and mitochondrial genomes and a consequence of erroneous replication or repair often starting from DNA damage. In a broader sense, genome instability can refer to the inherent characteristic of DNA to undergo chemical modification, generally termed DNA damage, that alters its structure and functional properties5. DNA damage has been a problem from the onset of DNA-based life, given the ubiquitous abundance of DNA-damaging agents, such as UV-rays from the sun, causing lesions that block transcription and replication. DNA damage ranges from spontaneous deamination and hydrolysis to a plethora of chemical alterations including different types of breaks, nicks, gaps, abasic sites, adducts, inter-, intra-strand and DNA-protein crosslinks, subtle chemical modifications, etc. Also, aberrant DNA structures, such as R-loops, G-quadruplexes and persistent single-strand regions or arrested intermediates in DNA transactions such as stalled transcription, replication and recombination complexes should be considered as DNA damage, as they compromise DNA functionality and trigger the same responses. DNA injuries hamper accurate replication, controlled transcription and secure storage of the genetic information. At the apex of the informational hierarchy, nuclear DNA is usually present in only two (distinct) copies and, in contrast to all other biomolecules that can be remade based on instructions carried by the corresponding genes, DNA integrity can only be maintained by constant repair. An elaborate network of highly sophisticated DNA repair and DNA damage response (DDR) systems counteract the time- and exposure-dependent erosion of the genetic information. Inherited defects in these maintenance systems not only predispose to cancer but also underlie numerous, segmental forms of premature ageing in humans, indicating a tight link between genome integrity, cancer and ageing6 (Box 1).
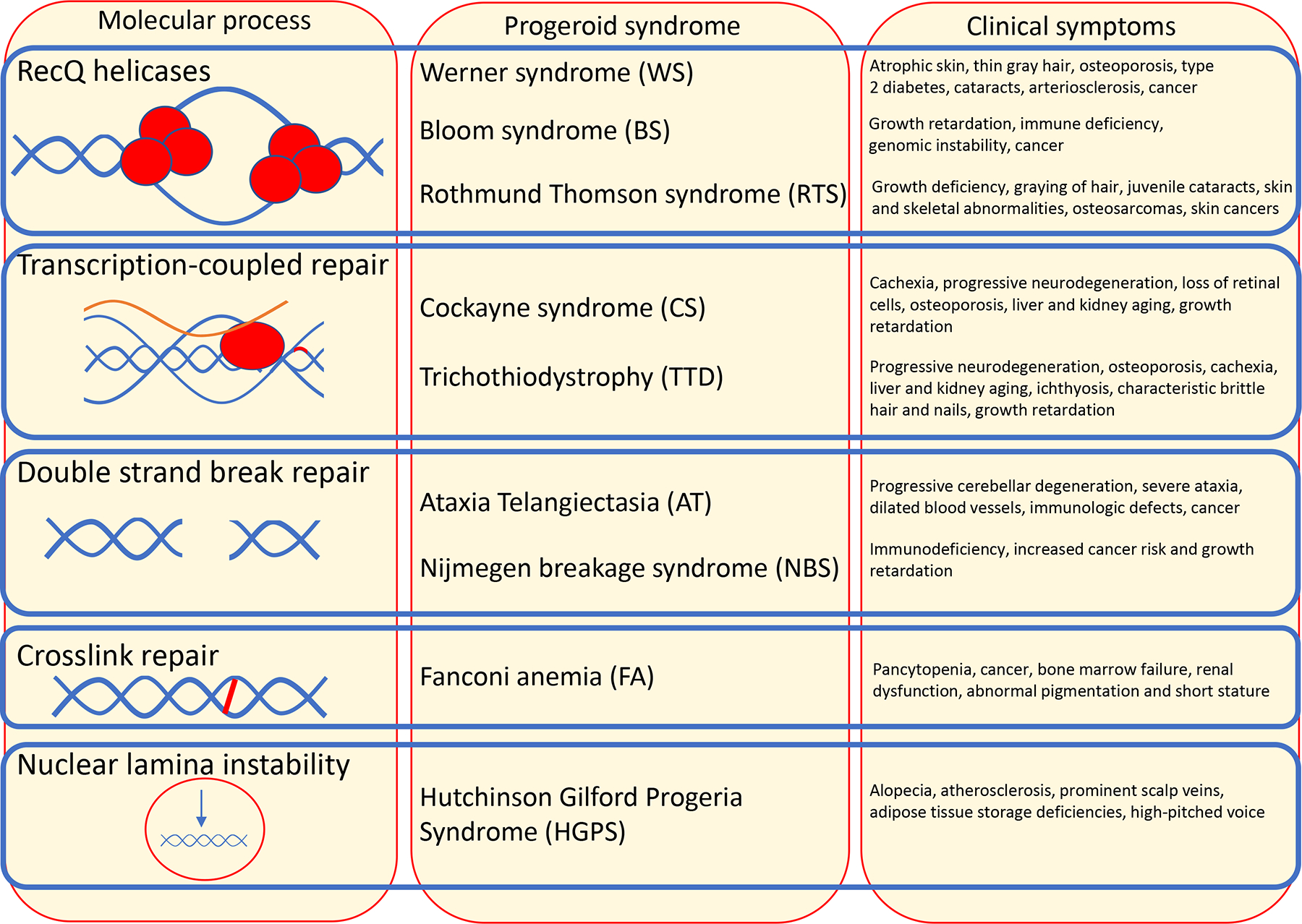
Text Box 1. DNA repair defects accelerate human ageing.
Most progeroid (“premature ageing-like”) syndromes are caused by mutations in genes involved in maintaining genome stability. Werner syndrome patients display many overt signs of ageing such as hair greying, type 2 diabetes, osteoporosis and cataracts, which often manifest prior to the age of 30. Werner as well as Bloom and Rothmund-Thomson syndromes are caused by mutations in RecQ helicases that function in DNA recombination, replication, repair, and telomere maintenance. Typical ageing-associated pathologies such as neurodegeneration, atherosclerosis and osteoporosis occur in Cockayne syndrome (CS) and trichothiodystrophy (TTD) before the age of 10, caused by impaired transcription-coupled repair. Global-genome nucleotide excision repair defects cause several-thousand-fold increased sun-induced skin cancer susceptibility in xeroderma pigmentosum patients, some of whom also suffer from accelerated neurodegeneration. Defects in DSB repair result in the progeroid conditions Ataxia telangiectasia (AT) and Nijmegen breakage syndrome (NBS), while DNA crosslink repair deficiencies cause Fanconi anemia (FA). Also nuclear lamina dysfunction that underlies Hutchinson Gilford progeria has been linked to nuclear genome instability6.
Progeroid syndromes are segmental as a specific DNA repair defect predominantly affects specific tissues, such as hematopoiesis in AT or FA. Neurodegenerative phenotypes occur widespread throughout those progeroid syndromes suggesting that neurons might be particularly sensitive to multiple defects in DNA repair120. Premature ageing is also found in long-term cancer survivors that suffer from the long-lasting consequences of genotoxic chemo- and radiotherapy7. An additional category of progressive progeroid disorders affecting multiple organs is due to mitochondrial defects50, which likely involve DNA damage as well (see main text).
Text Box 1 Figure. Examples of progeroid syndromes caused by DNA repair defects.
During normal ageing, DNA damage occurs continuously on a massive scale, due to numerous exogenous and endogenous genotoxins. The pro-ageing effects of genotoxins are visible during photoaging of the skin but also DNA-damaging chemotherapy accelerates ageing features7. Even mechanical stress to tissues can cause genome instability and may contribute to the accelerated ageing in Hutchinson-Gilford Progeria, where mechanical resilience of the nucleus is compromised by a mutation affecting the scaffold protein lamin A8. It is estimated that up to 105 DNA lesions occur in an active mammalian cell on a daily basis, with spontaneous hydrolysis alone causing ~104 abasic (mostly apurinic) sites5. Even though most of these lesions are efficiently removed, some escape detection, are irreparable, repaired too late, or repaired in an erroneous way. In time, DNA injuries inevitably accumulate9 making genome instability a true hallmark of ageing (Figure 2).
Figure 2. Molecular, cellular and systemic consequences of DNA damage.
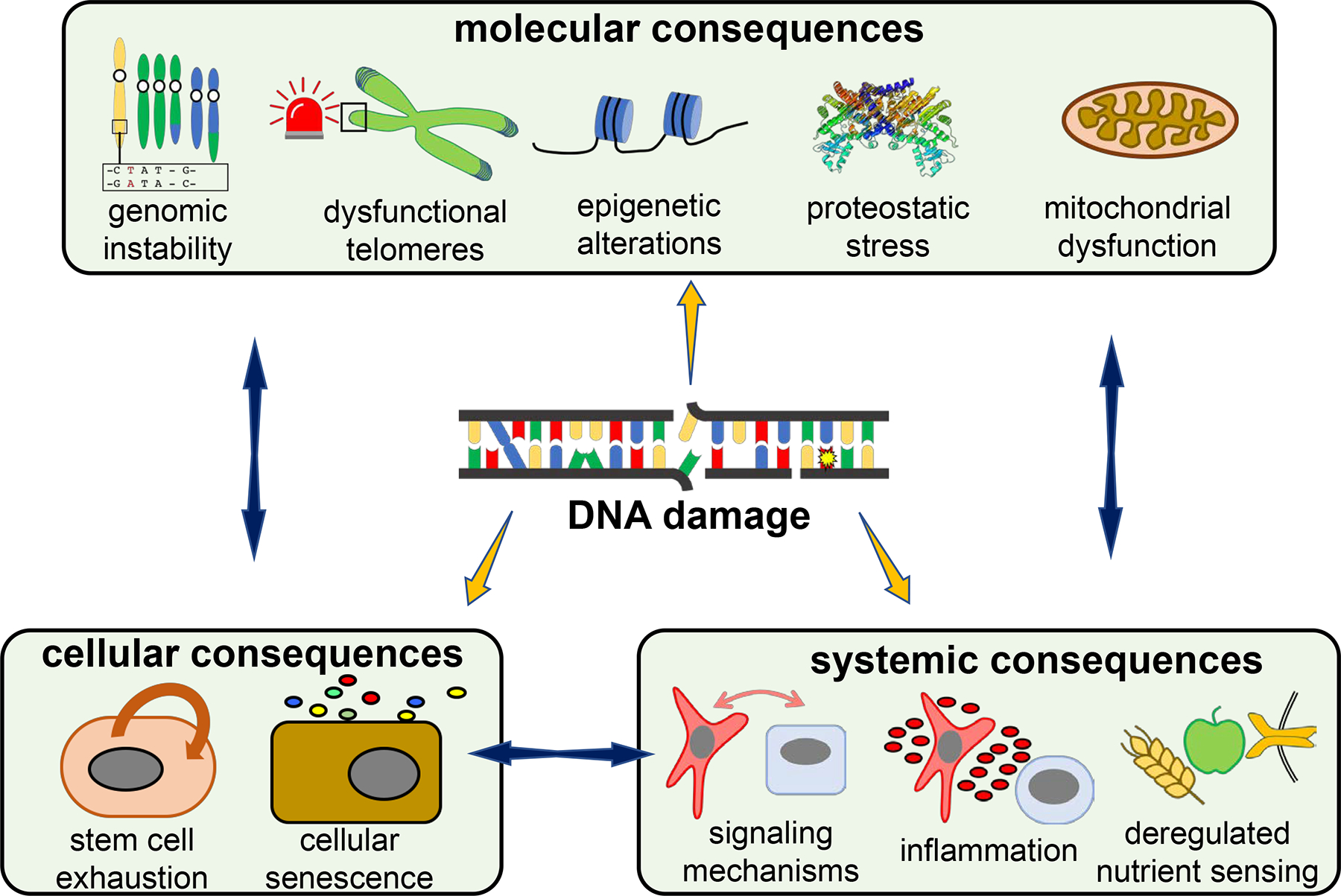
DNA damage and the cellular DNA damage response (DDR) can impinge on molecular processes, alter cell fate and deregulate intercellular communication. DNA damage leads to mutations or chromosomal aberrations thus triggering genome instability. Critically shortened telomeres activate the DDR triggering cellular senescence. DNA repair leads to chromatin-remodeling, while the chromatin structure affects DNA damage susceptibility and repair access. The DDR affects autophagy, the UPRER and leads to a loss of protein complex stoichiometry. Mitochondrial dysfunction is driven by NAD+ deprivation by nuclear DNA repair, DNA damage-induced mitophagy defects, and altered mtDNA polymerase expression that affects mtDNA replication. DNA damage induces dampening of nutrient sensing pathways, which in turn affect DNA damage repair and signaling. Cellular senescence is induced in response to DNA damage. DNA damage causes exhaustion of stem cell pools through DDR-induced apoptosis, senescence, premature differentiation and alterations of the stem cell niche. The DDR impacts intercellular communication through inflammatory cytokines and dampened growth signaling.
Genome instability at dysfunctional telomeres
The discovery in the late 1980s that S. cerevisiae “ever shorter telomeres” (EST1) mutants undergo replicative senescence10 has popularized the concept that progressive telomere shortening drives the ageing process. In mammals, telomeres consist of thousands of TTAGGG repeats covered by the shelterin complex that facilitates formation of a lariat-like T-loop, and thereby hides the telomeric end preventing activation of the DDR sensors11. Due to incomplete lagging strand synthesis during DNA replication, the number of repeats decreases with each cell division. In the germline and in some somatic stem cells this loss is compensated by telomerase, which is silenced in most somatic cells during early development, restricting the number of cell divisions until telomeres become critically short. An unprotected telomere resembles a persistent DNA double strand break (DSB) triggering chronic DDR activation resulting in replicative senescence12. Even a single DSB suffices to cause full-blown cell cycle blockade13. The pathogenicity of telomere shortening in ageing is an antagonistic pleiotropic effect of a trait that must have been selected for its early benefits such as limiting unrestrainted proliferation and hence tumor formation14.
Genetic defects in telomere maintenance cause human telomeropathies, including dyskeratosis congenita, aplastic anemia, and pulmonary and liver disease exhibiting multiple progeroid features15. In mice, segmental premature ageing only manifest in telomerase mutants after several generations, likely because their particularly long telomeric repeats take several generations to become critically shortened and thus dysfunctional16. The estimated telomere length in bulk human tissues does not suggest that on average telomeres become critically short in normal ageing, even at old age17. However, progressive telomere shortening might alter expression of specific subtelomeric genes18, the in vivo relevance of which during ageing is yet to be determined.
DNA damage-induced epigenetic alterations
The epigenome is comprised of DNA methylation and many histone modifications and is unstable over the lifetime of somatic cells. Some changes are similar between cells in a tissue and are likely adaptive or programmed, others are progressive and/or stochastic, similar to DNA damage and mutations, contributing to intercellular heterogeneity, possibly with important functional consequences.
Chromatin modifications include phosphorylation, methylation, acetylation, ubiquitination, sumoylation, citrullination, and polyADPribosylation (PAR), most of which are also part of the DDR19. Age-dependent chromatin modifications include loss of histones20 and increased “fuzziness” of nucleosomes21, linked with local and global chromatin remodeling, an imbalance of activating and repressive histone modifications, and transcriptional changes. In humans and experimental animals, diverse sets of age-related alterations in DNA methylation in various tissues have been found to strongly correlate with chronological age and are now used as epigenetic clocks. Because such clocks tick similarly from cell to cell the underlying CpG methylation statuses likely reflecting adaptive changes22.
Increasing evidence suggests that DNA damage is a major driver of age-associated epigenetic changes. The DNA methyltransferase, Dnmt1, localizes to sites of DNA repair23 and many chromatin remodelers regulate the assembly of distinct repair machineries, lesion removal, and restoration of the original chromatin state, which may leave epigenetic marks. For example, after the repair of transcription-blocking lesions in C. elegans, H3K4me2 deposition facilitates the resumption of transcription of genes regulating protein biosynthesis and homeostasis and consequently promotes longevity24. The DDR in human cells leads to loss of H3K27me3, promoting cellular senescence25. The phosphorylated histone variant γH2AX forms foci at the site of DSBs. Such foci accumulate in various mouse tissues with ageing26 indicative of persistent chromatin alterations resulting from DNA damage. ‘DNA segments with chromatin alterations reinforcing senescence’ (DNA-SCARS) have been found enriched in senescent cells. Such DNA-SCARS exemplify persistent local chromatin changes due to irreparable DNA lesions27. In cell lines it has been demonstrated that DNA methylation patterns are altered during homologous recombination (HR) repair, followed by further modification weeks later by base excision repair-mediated transcription-associated demethylation28. Poly-ADP-ribosylation of histones and the Poly-ADP-Ribose polymerase 1 (PARP1) itself facilitates repair of single-strand breaks serving as a landing platform for proteins in base excision repair. PARylation severely reduces cellular NAD+ pools which may trigger apoptosis or may indirectly inhibit Sirtuin proteins, which in turn affect genome-wide chromatin acetylation, ageing and DNA repair29 and trigger gene expression changes that resemble those observed in ageing mouse brain30.
It is thus plausible that continuous DNA damage induction and repair for tens of thousands of lesions daily leave epigenetic marks and thereby contribute to intercellular epigenetic heterogeneity in ageing, particularly since somatic cells do not have to function forever and epigenetic memory is erased in the germline at the start of the next generation. Consistent with these ideas, transcription in aged cells appears far more variable than in young cells31. Hence, the DDR likely is a primary cause of epigenetic changes that lead to deterioration of control of gene expression, which in turn contributes to somatic heterogeneity and time-dependent overall functional decline.
DNA damage-induced proteostatic stress
Proteostatic pathways control the synthesis, folding and degradation of proteins. Several age-related diseases are associated with protein misfolding and aggregation such as Alzheimer (AD) and Parkinson disease (PD). Misfolded proteins can arise when structural alterations affect solubility, thus causing protein aggregates, e.g. upon oxidative, heat, or endoplasmic reticulum stress. Multiple lines of evidence link DNA damage to proteostatic stress. Children with the premature ageing condition Cockayne syndrome (CS), which is caused by a defect in transcription-coupled repair (TCR), show neurofibrillary tangles in the cerebellar cortex32 occurring decades earlier than in familial early-onset AD. Defective TCR accelerates neurodegeneration in a C. elegans model for CS thus further underlining the ancestral role of DNA damage in driving age-related neuronal pathology33. DNA damage and altered expression and activity of DNA repair genes have been implicated in the pathogenesis of AD and other dementias34–38, such as reduced nucleotide excision repair (NER) efficiency in human PD39. Several DNA repair mechanisms, particularly mismatch repair, are involved in the repeat expansion underlying Huntington disease40 and vice versa mutant huntingtin has been linked to defects in repairing transcription-associated DNA strand breaks41.
DNA damage could trigger proteostatic stress, for example, through increased stalling of transcription (transcriptional stress) or (epi)mutation-mediated transcriptional noise. This likely affects assembly, stoichiometry, proper folding and functioning of protein(complexe)s, triggering proteostatic stress and aggregation. Single cell sequencing of human neurons has confirmed that somatic mutations increase during ageing and do so at a higher rate in cells from patients with neurodegenerative diseases42. Stochastic transcription-blocking DNA lesions accumulating in post-mitotic tissues such as neurons, which do not dilute DNA damage by replication, likely cause the genome-wide reduced expression preferentially of large genes observed during natural ageing and in an accelerated fashion in progeroid NER/TCR-deficient mice43. These DNA-damage-driven mechanisms would explain the decoupling of transcription and protein expression44 and loss of stoichiometry of protein complexes noted during ageing in different species45, thus creating proteotoxic stress and protein aggregates.
Defects in chaperones, the ubiquitin proteasome system and autophagy can result in accumulation of misfolded proteins. The DDR itself can strain the proteostatic machineries46 and IRE1α and transcription factor XBP1 –both key regulators of the endoplasmic reticulum unfolded protein response (UPRER)– are induced in DNA repair defective progeroid mice43. Also autophagy is induced by DNA damage signaling and is indeed required for survival amid persistent DNA damage46. When unrepaired DNA lesions drive cells into senescence they exert a chronic senescence-associated secretory phenotype47; which is thought to strain the UPRER 48. In contrast, calorie restriction reduces transcription stress and simultaneously alleviates the UPRER 43, providing a direct link between DNA-damage-driven transcription stress and proteostatic stress.
Taken together, these observations support a central role of DNA damage and (epi)mutations as major causes of proteotoxic stress with age.
Mitochondrial dysfunction
As the organelles that regulate energy and metabolic homeostasis, mitochondria have since long been associated with ageing, mostly as main source of ROS49 and linked with ageing diseases, such as PD and sarcopenia50. The primary cause of mitochondrial dysfunction has often been sought in ROS-induced damage to mitochondria’s own genome, which measuring less than 17 Kb is infinitely smaller than the 3 billion bp of its nuclear counterpart but is present in multiple copies in each of the thousands of organelles in a typical mammalian cell50.
The most popular hypothesis to explain age-related mitochondrial dysfunction is accumulation of somatic mutations in the mitochondrial genome, as a consequence of errors during replication and the lack of most of the sophisticated repair pathways active in the nucleus. Mice expressing a proofreading-deficient mitochondrial DNA polymerase (POLG) have greatly elevated mtDNA mutations and display multiple symptoms of premature ageing51,52. Increased mtDNA mutations have been correlated to loss of Cytochrome C oxidase (COX) activity in aged human skeletal muscle fibers53,54, substantia nigra and hippocampus of normally aged human brain55 and various other tissues56. However, it is unclear if the frequency of such mtDNA mutations reaches functionally important levels with natural age to ever cause phenotypic effects57. More advanced methods, such as digital PCR, indicated fairly low frequencies of mtDNA deletions58 and ultra-deep sequencing did not show an age-dependent increase of mutations in wild type mice and instead suggested that most somatic mtDNA mutations originate from replication errors during development59.
An important connection between nuclear DNA damage and mitochondrial dysfunction implicates mitophagy, the selective degradation of mitochondria by autophagy. High levels of nuclear DNA damage, e.g. in cells from aged organisms or DNA repair mutants, lead to prolonged activation of PARP1, a DNA break sensor that upon activation consumes large amounts of NAD+60. Inhibition of PARP or supplementation of NAD+ was reported to alleviate some premature ageing phenotypes associated with defects in DNA repair by restoring mitochondrial function and mitophagy29.
Hence, while the role of mtDNA mutations remains subject to debate, aspects that are not yet well explored are the effect of DNA damage itself (as opposed to mutations) on mitochondrial DNA replication and transcription and damage to the over 1000 mitochondrial genes in the nuclear genome.
DNA damage-driven Cell Fate Decisions
Cellular senescence
Cellular senescence permanently arrests cell proliferation in response to various stresses, most of which DNA-damage-related. Senescence was discovered as a mechanism that limits the number of population doublings in cultured human fibroblasts due to telomere attrition, triggering DNA-damage-signaled cell cycle arrest61,62. Senescence has likely evolved as a mechanism contributing to embryogenesis, regeneration (e.g. wound healing)63 and cellular defense against overproliferation and thereby cancer. However, senescent cells acquire a “senescence-associated secretory phenotype” (SASP), secreting many pro-inflammatory cytokines, proteases, and growth and angiogenesis factors that can disrupt microenvironments and compromise tissue structure and function thereby contributing to local and systemic ageing-associated pathologies47 and promote cancer64. The proinflammatory mediators can promote sterile inflammation, in this context often referred to as ‘inflammaging’. Recently, attention has focused on the effect of senescence in vivo where purging p16-positive senescent cells in transgenic mice increased mean lifespan as well as aspects of healthspan65. Application of ‘senolytic’ agents that selectively eliminate senescent cells confirm that they contribute to ageing e.g. in atherosclerotic plaques66 and osteoarthritic lesions67.
Cells are driven into senescence by clastogenic compounds, such as bleomycin, doxorubicin, or cisplatin often causing irreparable DNA damage resulting in DNA SCARs27. DNA damage is also responsible for oncogene-induced senescence, which involves replication stress and subsequent DSBs as the consequence of hyper-replication associated with activated oncogenes68. DDR pathways, including ATR, ATM, and p53 that converge on activation of the cyclin-dependent kinase inhibitors p16, p21, and p27 and hyperphosphorylation of the retinoblastoma protein, trigger the withdrawal from the cell cycle69. In addition, cellular senescence can also arise as a consequence of chromosomal aneuploidy70.
Even the only non-genotoxin related “mitochondrial-dysfunction-associated senescence” (MiDAS)71 type is most likely also driven by DNA damage given the above described links to mitochondrial dysfunction. Hence, cellular senescence appears a bona fide part of the DDR or, as in MiDAS, can be attributed indirectly to DNA damage.
Stem cell exhaustion
Somatic stem cell exhaustion has two components, decline of stem cell number and reduced functional capacity. Different stem cells utilize distinct DDR mechanisms72: for instance, quiescent hematopoietic stem cells (HSCs) and hair follicle stem cells (HFSCs) employ fast but less accurate non-homologous end-joining (NHEJ), while cycling HSCs and intestinal stem cells prefer accurate HR or in case of too extensive damage opt for apoptosis, as do embryonic stem cells. In contrast, irreparable damage drives melanocyte stem cells and aged HFSCs into premature differentiation thereby clearing the stem cell pool73. Accumulation of DNA damage has been observed in human and mouse HSCs as well as in muscle, intestinal, mesenchymal, neural, skin, and germ stem cells72. Various DNA repair deficiencies trigger stem cell exhaustion. Muscle-forming satellite cells in progeroid Ercc1 repair mutant mice were incapable of following the regular proliferation and differentiation programs74 and third-generation telomerase-deficient mouse mutants display stem cell insufficiencies in the hematopoietic system, gut, skin and testis15.
The underlying role of DNA damage has been particularly well documented in HSCs. During ageing, HSCs expand in number but decline in pluripotency, skewing towards the myeloid lineage75. DNA damage increases in aged HSCs76 likely from replication stress77. As most adult stem cells, HSCs reside predominantly in a quiescent state, which offers some protection from endogenous genotoxic stress such as metabolic ROS, but their extended time for accumulating DNA lesions and use of error-prone NHEJ increase mutagenesis78. Defective DNA repair limits HSC functionality in ageing and progeroid mice79. Thus, time-dependent accumulation of stochastic DNA damage severely hampers stem cell functionality, increasing mutations during human HSC ageing80, impairing functional properties, promoting clonal expansion of positively selected somatic mutations resulting in loss of clonal diversity81 or raising the potential for oncogenic transformation. Age-dependent accumulation of somatic mutations has indeed been observed in various cells types82, such as satellite cells in humans that acquire on average 13 somatic mutations per year83.
Also the non-cell-autonomous DDR can compromise the stem cell niche and promote stem cell exhaustion. Genome instability amid dysfunctional telomere maintenance or Sirt6 deficiency results in niche-dependent defects in hematopoietic stem cells84,85. Notch signaling by the niche regulates the level of p53 in muscle stem cells via Mdm2 repression86. With increasing age, fading niche support drives these cells into cell death via mitotic catastrophe upon activation. In C. elegans somatic niche cells regulate the DDR in germ stem cells via FGF-like signaling and a similar niche regulation of the p53-mediated DDR was observed in mouse HFSCs87.
In conclusion, accumulating DNA damage is increasingly recognized to drive stem cell exhaustion during ageing through a combination of apoptotis, premature differentiation, cytostatic DNA damage checkpoint signaling, accumulation of mutations, and DNA damage-driven alterations in intercellular communication affecting stem cell niches.
Systemic effects of DNA damage
Signaling mechanisms impact the ageing phenotype
The importance of signal transduction mechanisms in ageing has become evident since the paradigm-shifting discovery of lifespan-extending mutations in insulin-like signaling (IIS) in C. elegans88. Consequently, several signaling systems have been shown to regulate longevity in species ranging from yeast to mammals. Interventions such as calorie restriction (CR) at least in part exert their anti-ageing effects by inhibiting signaling cascades such as IIS and the mTOR pathways89. In contrast, inflammatory signaling is thought to promote a range of age-related pathologies.
The DDR is a potent activator of inflammatory responses. This is literally obvious in the response to UV-induced DNA damage in the skin where inflammation is counteracted by systemic immunosuppression triggered by Langerhans cells migrating from the skin to the lymph nodes to activate regulatory T cells90. As mentioned, DNA-damage-induced senescent cells exert complex non-cell-autonomous effects63,64, which senolytics aim to curb66,67. DNA damage triggers innate immune responses that in C. elegans regulate systemic stress signaling91. Inflammatory responses have also been observed in DNA-repair-deficient progeroid mice92, which at the same time attenuate the somatotrophic (including IIS), thyrotrophic, and lactotrophic hormonal axes, as an anti-aging response, which resembles CR and IGR-1R and other dwarf mutant mice that are long-lived85,92,93. Unrepaired transcription-blocking lesions suppress IGF-1 signaling in mouse and human cells resulting in elevated stress resistance94. In C. elegans IIS attenuation enhanced tissue maintenance amid DNA damage accumulation through the activation of the FOXO transcription factor DAF-1695. The paradoxical similarity between responses triggered by DNA damage and interventions delaying ageing suggested that a systemic DDR triggers a ‘survival response’ to counteract the detrimental consequences of DNA damage.
Taken together, the DDR exerts multiple effects on age-related alterations in local and systemic communication mechanisms by affecting inflammatory and key endocrine signaling components that impact the ageing process.
Anti-ageing responses to nutritional interventions are impacting genome stability
Nutritional interventions impact ageing and lifespan throughout the animal kingdom. Initially observed in the 1930s in rats96, CR –reduced calorie intake without malnutrition– is the most robust universal health- and lifespan-promoting intervention in species ranging from yeast to mammals. It is thought that CR exerts its lifespan-extending effects through specific nutrient sensing pathways, including IIS, Sirtuins, and the AMP-activated protein kinase (AMPK) regulated mammalian target of rapamycin (mTOR) pathway97. In addition to the IIS attenuation in DNA-repair-deficient progeroid mice and worms discussed above, the DDR kinase ATM phosphorylates several key proteins of the IIS–mTOR pathways after DNA damage98.
CR dramatically delays premature ageing in DNA repair mutant mice likely by decreased levels of ROS and other reactive compounds leading to reduced DNA damage levels43. Longevity-promoting changes in nutrient sensing pathways can also stimulate DNA repair itself, suggesting that some of the observed health benefits in normal ageing could be due to improved genome maintenance. mTOR inhibition by rapamycin in vivo, which extends lifespan, increases levels of the DNA repair protein O-6-methylguanine-DNA methyltransferase (MGMT)99. CR also activates Sirt1 and AMPK4, promoting DNA damage repair and signaling as an epigenetic regulator100 and increasing NER capacity101, respectively. The protein kinase AKT, a central positive regulator of various nutrient sensing pathways, negatively regulates DNA repair and inhibits key DDR factors including Chk1, Topbp1, and p53102. FOXO3a, which is activated by reduced IIS, promotes the binding of TIP60 with ATM, optimizing ATM activation after DNA damage103.
In summary, abundant evidence indicates that DNA damage affects key signaling mechanisms –by impinging on IIS, Sirtuins, AMPK and mTOR– that regulate lifespan and elicit anti-ageing effects of CR in model organisms.
Is DNA damage the primary cause of ageing?
Spontaneous DNA damage thus impinges on all major aspects of the ageing phenotype. Some of the physiological alterations in turn boost genome instability thus amplifying the deterioration of homeostasis during ageing. The strong mechanistic link of DNA damage with ageing, and the role of DNA as the primary template for all cellular functions, make it a major candidate as the primary cause of ageing. However, at least three important arguments against this conclusion should be addressed.
First, if DNA damage is central to the ageing process, one would expect that improving DNA repair extends lifespan and evidence for this is scarce104–106. However, it is important to realize that DNA damage is comprised of a plethora of distinct chemical alterations, the repair of which does not depend on one gene and not even on one pathway. Instead DNA repair involves at least 7 well-balanced multi-enzyme core pathways and many more accessory processes that encompass hundreds of genes, many of which have other roles as well. Hence, the function of DNA repair as a longevity assurance system cannot be generally improved by simply upregulating the activity of one or few genes. It took evolution millions of years improving DNA repair in long-lived species, such as primates. DNA repair capacities have evolved under specific selection conditions largely driven by environmental genotoxins, such as high fluxes of UV or natural compounds. Moreover, apart from DNA repair per sé, cellular systems affecting DNA damage generation and outcome, such as metabolism, anti-oxidant defense, cell death, senescence, and mutagenesis are relevant as well.
Second, reliable quantification of spontaneous DNA damage in animal or human tissues appears technically extremely difficult hampering efforts to show an age-related increase to levels that likely impair cellular function and explain age-related pathologies (Text Box 2). However, DNA mutations, a consequence of erroneous DNA repair, can now be accurately determined and have been shown to accumulate with age in humans and mice in a tissue-specific manner42,107–110. Nevertheless, while there is no doubt that accumulating mutations cause cancer and, possibly, increased cancer risk with age, it is –as yet– unknown if their frequency is high enough to account for the loss of tissue function and increased disease risk at old age. However, besides causing mutations, accumulating DNA damage also interferes with gene expression and replication causing replication and transcription stress, senescence, functional decline and cell death, all main drivers of ageing (Figure 1).
Text Box 2. Methods to detect DNA damage.
A serious challenge for linking DNA lesions to ageing has remained the methodological difficulty of accurately measuring the plethora of chemical alterations in DNA. Key problems are the limited sensitivity and/or specificity of technologies to detect physiological levels of DNA damage and the occurrence of artifacts (e.g. oxidation) during DNA isolation and handling or due to interrupted DNA-metabolizing transactions (e.g. topoisomerases) when cells are lysed. Most lesions can only be determined at semi-quantitative or relative manners or after exposure to unphysiologically high levels of genotoxic agents. Only some lesion types can be directly identified (but not quantified in absolute terms) through lesion-specific antibodies towards CPD, 6–4PP or 8-oxo-dG structures or rough overall DSBs and SSBs assessment through the (variable) COMET assay. HPLC combined with advanced mass spectrometric methodologies can detect specific chemical alterations of nucleosides121. There are only few examples of highly sensitive assays reporting reliable quantitation of spontaneous oxidative DNA damage, most notably 8-oxo-dG and cyclopurine lesions. Cyclopurines are endogenous transcription-blocking DNA lesions that were shown to increase from a density of 2 to 4 in young mice to 10–20 per million base pairs in old mice122. Indirectly, damaged DNA can be discerned by long range PCR123, the decline in transcription through large genes resulting in a shift towards mRNAs of small genes in the ageing transcriptome of post-mitotic tissues43, or detection of transcription-blocking lesions by strand-biased, PCR-based next generation sequencing of DNA protected by elongating RNA polymerases124. Specific types of DNA lesions that are amenable either to antibody binding or enzymatic modification have been mapped by high throughput sequencing. Third generation sequencing technologies are rapidly advancing to detect specific DNA modifications even in low amounts of DNA125. Also the formation of DNA repair complexes such as foci formation of γH2AX, 53BP1, Rad51 and other repair or signaling proteins at DSB sites and at sites of DNA-damage-induced replication stress are useful indicators. When erroneous repair or lesion bypass during replication results in mutations, sequencing methods can be applied to detect the altered DNA sequence in single cells. Somatic mutations increase linearly during ageing in multiple tissues and species including humans82. However, quantitative estimates of the total landscape of spontaneous DNA damage in humans or animals are lacking.
A third, more recent argument against DNA damage-centric ageing theories is the dearth of DNA repair genes emerging from genome-wide association studies (GWAS) of ageing-related diseases or extreme longevity. However, the utter complexity of the genetics of ageing and longevity makes it highly unlikely to find genetic association with common variants in generally underpowered studies. Extreme longevity is rare and individual age-related diseases often involve genes not necessarily related to systemic ageing, e.g., lipoprotein genes. Nevertheless, in a meta-analysis of over 400 GWAS of five major categories of age-related diseases genome maintenance pathways were found111 and genome maintenance was also the top pathway found associated with the age of natural menopause112. Age of natural menopause is strongly linked with a wide variety of ageing-pathologies, including cardio-vascular disease, type II diabetes and osteoporosis, and importantly with longevity113. These findings are consistent with the observation in both humans and mice that the vast majority of rare genetic progeroid syndromes where multiple, bona fide ageing-associated diseases develop early in life, is caused by mutations in DNA repair genes6 (Text box 1).
Hence, while not invalid, all three arguments against a major role of DNA damage in ageing are unconvincing in view of the sheer complexity of DNA repair processes and the abundant evidence that only DNA repair dysfunction, not defects in proteostasis, antioxidant defense, immune response or any other physiological defense system, is associated with systemic premature ageing. Based on all the evidence, DNA damage is by far the most likely molecular driver of ageing. DNA damage and the DDR lead to broad cellular and physiological end points that can explain the entire spectrum of ageing phenotypes, from atrophy to inflammation and cancer (Figure 2). This understanding is far from new: It is known since the 1940s that rodents exposed to radiation show multiple symptoms of premature ageing114 and the first proposals that DNA damage was the main driver of ageing stem from the 1960s115. More recently, the validity of these old observations was dramatically underscored by the notion that the long-term consequences of DNA-targeting chemo- and radiotherapies of cancer are accelerated, multi-organ ageing7.
The causal relationship between DNA damage and ageing may go back in evolution to the first replicators. When DNA became the genetic material, it was already far more stable than RNA, the presumed initial carrier of genetic information. The subsequent increased length of DNA templates put a premium on faithful replication and repair, which became prerequisites for rejuvenation amid the intrinsic instability of nucleic acids even during early evolution when life was not much more than compartmentalized DNA and well before the various homeostatic alterations of ageing discussed here had evolved. Hence, DNA damage as a primary cause of ageing has probably been with us since the origin of life.
Future prospects
Time-dependent accumulation of DNA damage of endogenous and exogenous origin and its consequences progressively hamper cellular functionality and increase susceptibility to develop the chronic ailments of ageing. Interventions that aim at alleviating the root cause of ageing-associated multimorbidity should therefore be targeted at restoring genome integrity by reducing DNA damage and augmenting DNA repair. Reducing exogenous DNA damage for example through UV protection and avoidance of tobacco smoking has already proven to lower ageing-associated disease risks. Dietary interventions might be able to reign in some endogenous DNA damage sources, but the majority of spontaneous lesions will inevitably occur. Augmenting DNA repair has remained a great challenge due to the intricate complexity of repair machineries. An exception are the highly lesion-specific photolyase repair enzymes, active in many species but not placental mammals. Ectopic expression of this enzyme is indeed sufficient to prevent UV-induced carcinogenesis in mice116. However, those one-enzyme reactions are incapable of repairing the myriad of different lesions that require more sophisticated repair systems. Master regulators of DNA repair affecting multiple DNA repair systems have thus far remained elusive but might await discovery. Genetic screens using model organisms might be very suitable for the pursuit of such mechanisms augmenting genome stability.
Since the initial proposals that DNA damage was the main cause and DNA repair the main determinant of ageing117,118, and the subsequent discovery that DNA repair defects can accelerate the development of a wide range of age-related pathologies119, great strides have been made in unraveling the mechanistic links between DNA damage and nearly every aspect of the ageing process. Venturing further into the mechanisms through which DNA damage affects each of the major processes that causally contribute to pathologies occurring at old age opens perspectives to tackle the ageing process at its causal roots and thus counteract all ageing-associated diseases simultaneously.
Acknowledgements:
Research was supported by the Deutsche Forschungsgemeinschaft (SCHU 2494/3-1, SCHU 2494/7-1, SCHU 2494/10-1, SCHU 2494/11-1, KFO 286, KFO 329, GRK 2407 to BS, and CECAD, SFB 829 to BS and JH), Deutsche Krebshilfe (70112899), H2020-MSCA-ITN-2018 (Healthage and ADDRESS ITNs) and John Templeton Foundation Grant (61734) to BS, NIH grants (PO1 AG017242 to JV and JH, and U19 AG056278, U01 ES029519, P01AG047200, U01HL145560, P30AG038072 to JV), European Research Council Advanced Grants DamAge and Dam2Age, ONCODE (Dutch Cancer Society), Memorabel and Chembridge (ZonMW) and BBoL (NWO-ENW) to JP and JH.
Footnotes
Competing interests:
Jan Vijg is co-founder of SingulOmics Corp., USA
References
- 1.Schumacher B The Mystery of Human Aging: Surprising Insights from a Science That’s Still Young. (Algora Publishing, 2017). [Google Scholar]
- 2.Charlesworth B Fisher, Medawar, Hamilton and the evolution of aging. Genetics 156, 927–931 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams GC Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution (N. Y). 11, 398–411 (1957). [Google Scholar]
- 4.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G The Hallmarks of Aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindahl T Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993). [DOI] [PubMed] [Google Scholar]
- 6.Niedernhofer LJ et al. Nuclear Genomic Instability and Aging. Annu Rev Biochem 87, 295–322 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Lubberts S, Meijer C, Demaria M & Gietema JA Early ageing after cytotoxic treatment for testicular cancer and cellular senescence: Time to act. Crit. Rev. Oncol. Hematol. 151, 102963 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Gonzalo S, Kreienkamp R & Askjaer P Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 33, 18–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vijg J Aging of the Genome. (Oxford University Press, 2007). doi: 10.1093/acprof:oso/9780198569237.001.0001 [DOI] [Google Scholar]
- 10.Lundblad V & Szostak JW A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 57, 633–643 (1989). [DOI] [PubMed] [Google Scholar]
- 11.de Lange T Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 52, 223–247 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Fumagalli M et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdallah P et al. A two-step model for senescence triggered by a single critically short telomere. Nat. Cell Biol. 11, 988–993 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shay JW Role of telomeres and telomerase in aging and cancer. Cancer Discov. 6, 584–593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martínez P & Blasco MA Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 216, 875–887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaskelioff M et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469, 102–106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demanelis K et al. Determinants of telomere length across human tissues. Science (80-. ). 369, eaaz6876 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robin JD et al. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 28, 2464–2476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauer MH & Gasser SM Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 31, 2204–2221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Sullivan RJ, Kubicek S, Schreiber SL & Karlseder J Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 17, 1218–1225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Z et al. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 28, 396–408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu AT et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany. NY). 11, 303–327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortusewicz O, Schermelleh L, Walter J, Cardoso MC & Leonhardt H Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. U. S. A. 102, 8905–8909 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang S, Meyer DHDH & Schumacher B H3K4me2 regulates the recovery of protein biosynthesis and homeostasis following DNA damage. Nat. Struct. Mol. Biol. (2020). doi: 10.1038/s41594-020-00513-1 Wang and colleagues revealed that after the repair of DNA lesions, specific epigenetic marks, H3K4me2, are deposited to restore protein homeostasis and thus antagonize DNA damage-driven aging.
- 25.Ito T, Teo YV, Evans SA, Neretti N & Sedivy JM Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 22, 3480–3492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sedelnikova OA et al. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Rodier F et al. DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 124, 68–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russo G et al. DNA damage and Repair Modify DNA methylation and Chromatin Domain of the Targeted Locus: Mechanism of allele methylation polymorphism. Sci. Rep. 6, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang EF et al. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 157, 882–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oberdoerffer P et al. SIRT1 Redistribution on Chromatin Promotes Genomic Stability but Alters Gene Expression during Aging. Cell 135, 907–918 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bahar R et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 1011–1014 (2006). Bahar et al. demonstrated that cell-to-cell variation in gene expression increases in aged single cardiomyocytes suggesting that genome damage could trigger stochastic variations in transcript levels thus compromising cellular function during ageing.
- 32.Takada K & Becker LE Cockayne’s syndrome: Report of two autopsy cases associated with neurofibrillary tangles. Clin. Neuropathol. 5, 64–68 (1986). [PubMed] [Google Scholar]
- 33.Lopes AFC et al. A C. elegans model for neurodegeneration in Cockayne syndrome. Nucleic Acids Res. 1–13 (2020). doi: 10.1093/nar/gkaa795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bucholtz N & Demuth I DNA-repair in mild cognitive impairment and Alzheimer’s disease. DNA Repair (Amst). 12, 811–816 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Weissman L et al. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 35, 5545–5555 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leandro GS, Lobo RR, Oliveira DVNP, Moriguti JC & Sakamoto-Hojo ET Lymphocytes of patients with Alzheimer’s disease display different DNA damage repair kinetics and expression profiles of DNA repair and stress response genes. Int. J. Mol. Sci. 14, 12380–12400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sepe S, Payan-Gomez C, Milanese C, Hoeijmakers JH & Mastroberardino PG Nucleotide excision repair in chronic neurodegenerative diseases. DNA Repair (Amst). 12, 568–577 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Obulesu M & Rao DM DNA damage and impairment of DNA repair in Alzheimer’s disease. Int. J. Neurosci. 120, 397–403 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Sepe S et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson’s Disease. Cell Rep. 15, 1866–1875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones L, Houlden H & Tabrizi SJ DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 16, 88 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Gao R et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 8, 1–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lodato MA et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, eaao4426 (2018). Using single cell sequencing, this study revealed increasing somatic mutations in neurons during human ageing indicating that even post-mitotic cell types acrue mutations that might trigger age-associated functional decline and degeneration.
- 43. Vermeij WP et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537, 427–431 (2016). Vermeij et al. showed that premature aging in DNA repair deficient mice could be greatly alleviated by dietary restriction suggesting that DR is capable of reducing DNA damage infliction and promoting genome stability.
- 44.Wei YN et al. Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging. Genome Biol. 16, 1–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelmer Sacramento E et al. Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol. Syst. Biol. 16, 1–22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edifizi D et al. Multilayered Reprogramming in Response to Persistent DNA Damage in C. elegans. Cell Rep. 20, 2026–2043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rodier F et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009). This study established that senescent cells release cytokines and thus elicit non cell autonomous effects such as inflammatory responses.
- 48.Williams ABAB et al. Restoration of Proteostasis in the Endoplasmic Reticulum Reverses an Inflammation-Like Response to Cytoplasmic DNA in Caenorhabditis elegans. Genetics 212, 1259–1278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harman D Aging: a theory based on free radical and radiation chemistry. J Gerontol 11, 298–300 (1956). [DOI] [PubMed] [Google Scholar]
- 50.Kauppila TES, Kauppila JHK & Larsson NG Mammalian Mitochondria and Aging: An Update. Cell Metab. 25, 57–71 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Trifunovic A et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Kujoth GC et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–4 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Bua E et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wanagat J, Cao Z, Pathare P & Aiken JM Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 15, 322–332 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Kraytsberg Y et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Taylor RW et al. Mitochondrial DNA mutations in human colonic crypt stem cells Find the latest version : Mitochondrial DNA mutations in human colonic crypt stem cells. 112, 1351–1360 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vermulst M et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 39, 540–543 (2007). [DOI] [PubMed] [Google Scholar]
- 58.O’Hara R et al. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 29, 1878–1888 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ameur A et al. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet. 7, e1002028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoch NC et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541, 87–91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hayflick L & Moorhead PS The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961). [DOI] [PubMed] [Google Scholar]
- 62.Bodnar AG et al. Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–52 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Demaria M et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krtolica A, Parrinello S, Lockett S, Desprez P-Y & Campisi J Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. 98, 12072–12077 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Baker DJ et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). Baker et al. revealed that the elimination of senescent cells could delay ageing and provided the conceptual framework for eliminating senescence cells for therapeutic interventions aimed at extending healthspan.
- 66.Childs BG et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (80-. ). 354, 472–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeon OH et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kotsantis P, Petermann E & Boulton SJ Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov. 8, 537–555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gorgoulis V et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Andriani GA et al. Whole chromosome instability induces senescence and promotes SASP. Sci. Rep. 6, 1–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wiley CD et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McNeely T, Leone M, Yanai H & Beerman I DNA damage in aging, the stem cell perspective. Hum. Genet. (2019). doi: 10.1007/s00439-019-02047-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsumura H et al. Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science (80-. ). 351, aad4395–aad4395 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Alyodawi K et al. Compression of morbidity in a progeroid mouse model through the attenuation of myostatin/activin signalling. J. Cachexia. Sarcopenia Muscle 10, 662–686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geiger H, de Haan G & Florian MC The ageing haematopoietic stem cell compartment. Nat Rev Immunol 13, 376–389 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Beerman I, Seita J, Inlay MA, Weissman IL & Rossi DJ Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15, 37–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Flach J et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2015). This study established that increasing DNA replication stress in ageing hematopoietic stem cells leads to their functional decline.
- 78.Mohrin M et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 7, 174–185 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rossi DJ et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Lee-Six H et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zink F et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang L & Vijg J Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annu. Rev. Genet. 52, 397–419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franco I et al. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat. Commun. 9, 800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ju Z et al. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat. Med. 13, 742–747 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Mostoslavsky R et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Liu L et al. Impaired Notch Signaling Leads to a Decrease in p53 Activity and Mitotic Catastrophe in Aged Muscle Stem Cells. Cell Stem Cell 23, 544–556.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ou H-LH-L, Kim CSCSCS, Uszkoreit S, Wickström SASA & Schumacher B Somatic Niche Cells Regulate the CEP-1/p53-Mediated DNA Damage Response in Primordial Germ Cells. Dev. Cell 50, 167–183.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Kenyon C, Chang J, Gensch E, Rudner A & Tabtiang R A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464 (1993). [DOI] [PubMed] [Google Scholar]
- 89.Saxton RA & Sabatini DM mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prasad R & Katiyar SK Crosstalk Among UV-Induced Inflammatory Mediators, DNA Damage and Epigenetic Regulators Facilitates Suppression of the Immune System. Photochem. Photobiol. 93, 930–936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ermolaeva MA et al. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 501, 416–420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schumacher B et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 4, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Niedernhofer LJ et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038–1043 (2006). This study linked unrepaired DNA damage to genetic regulators of longevity by showing that insulin-like signaling is attenuated in DNA repair deficient progeroid mice.
- 94.Garinis GA et al. Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat. Cell Biol. 11, 604–615 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mueller MM et al. DAF-16/FOXO and EGL-27/GATA promote developmental growth in response to persistent somatic DNA damage. Nat. Cell Biol. 16, 1168–1179 (2014). Using C. elegans, Mueller et al. demonstrated that the longevity regulator DAF-16 responds to DNA damage and elevates the organism’s tolerance to persistent DNA lesions.
- 96.McCay CM, Maynard LA, Sperling G & Barnes LL Retarded Growth, Life Span, Ultimate Body Size and Age Changes in the Albino Rat after Feeding Diets Restricted in Calories. J. Nutr. (1939). doi: 10.1093/jn/18.1.1 [DOI] [PubMed] [Google Scholar]
- 97.López-Otín C, Galluzzi L, Freije JMP, Madeo F & Kroemer G Metabolic Control of Longevity. Cell 166, 802–821 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Matsuoka S et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Dominick G, Bowman J, Li X, Miller RA & Garcia GG mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell 16, 52–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alves-Fernandes DK & Jasiulionis MG The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int. J. Mol. Sci. 20, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu CL et al. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene 32, 2682–2689 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma Y, Vassetzky Y & Dokudovskaya S mTORC1 pathway in DNA damage response. Biochim. Biophys. Acta - Mol. Cell Res. 1865, 1293–1311 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Adamowicz M, Vermezovic J & d’Adda di Fagagna F NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 16, 2068–2076 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tian X et al. SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 177, 622–638.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kanfi Y et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221 (2012). This study established that in male mice transgenic expression of Sirt6 leads to reduced IGF-1 signaling and lifespan extension.
- 106.Shaposhnikov M, Proshkina E, Shilova L, Zhavoronkov A & Moskalev A Lifespan and Stress Resistance in Drosophila with Overexpressed DNA Repair Genes. Sci. Rep. 5, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhang L et al. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. U. S. A. 116, 9014–9019 (2019). Using single cell sequencing, Zhang and colleagues show increased somatic mutations during normal human aging occurring in genes and regulatory regions indicating their role in the age-dependent functional decline.
- 108.Brazhnik K et al. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci. Adv. 6, eaax2659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Franco I et al. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol. 20, 1–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yizhak K et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science (80-. ). 364, eaaw0726 (2019). This study used RNA sequencing to reveal the clonal expansion of somatic mutations in different human tissues.
- 111.Johnson SC, Dong X, Vijg J & Suh Y Genetic evidence for common pathways in human age-related diseases. Aging Cell 14, 809–817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Day FR et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 47, 1294–1303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Laven JSE, Visser JA, Uitterlinden AG, Vermeij WP & Hoeijmakers JHJ Menopause: Genome stability as new paradigm. Maturitas 92, 15–23 (2016). [DOI] [PubMed] [Google Scholar]
- 114.Henshaw PS, Riley EF & Stapleton GE The Biologic Effects of Pile Radiations. Radiology 49, 349–360 (1947). [DOI] [PubMed] [Google Scholar]
- 115.Alexander P The role of DNA lesions in the processes leading to aging in mice. Symp. Soc. Exp. Biol. 21, 29–50 (1967). [PubMed] [Google Scholar]
- 116.Jans J et al. Powerful skin cancer protection by a CPD-photolyase transgene. Curr. Biol. 15, 105–115 (2005). [DOI] [PubMed] [Google Scholar]
- 117.Hart RW & Setlow RB Correlation between deoxyribonucleic acid excision repair and life span in a number of mammalian species. Proc. Natl. Acad. Sci. U. S. A. 71, 2169–2173 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gensler HL & Bernstein H DNA damage as the primary cause of aging. Q. Rev. Biol. 56, 279–303 (1981). [DOI] [PubMed] [Google Scholar]
- 119. de Boer J et al. Premature aging in mice deficient in DNA repair and transcription. Science 296, 1276–1279 (2002). de Boer and colleagues established that genetic defects in DNA repair accelerate the mammalian ageing process.
- 120.Barzilai A, Schumacher B & Shiloh Y Genome instability: Linking ageing and brain degeneration. Mechanisms of Ageing and Development 161, 4–18 (2017). [DOI] [PubMed] [Google Scholar]
- 121.Traube FR et al. Isotope-dilution mass spectrometry for exact quantification of noncanonical DNA nucleosides. Nat. Protoc. 14, 283–312 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Mori T et al. High levels of oxidatively generated DNA damage 8,5′-cyclo-2′-deoxyadenosine accumulate in the brain tissues of xeroderma pigmentosum group A gene-knockout mice. DNA Repair (Amst). 80, 52–58 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Van Houten B, Cheng S & Chen Y Measuring gene-specific nucleotide excision repair in human cells using quantitative amplification of long targets from nanogram quantities of DNA. Mutat Res 460, 81–94 (2000). [DOI] [PubMed] [Google Scholar]
- 124.Nakazawa Y et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 180, 1228–1244.e24 (2020). [DOI] [PubMed] [Google Scholar]
- 125.Mingard C, Wu J, McKeague M & Sturla SJ Next-generation DNA damage sequencing. Chem. Soc. Rev. (2020). doi: 10.1039/D0CS00647E [DOI] [PubMed] [Google Scholar]