

Abstract
Particle-based systems provide a capability for the delivery of imaging and/or therapeutic payloads. We have engineered constructs derived from erythrocytes, and doped with the FDA-approved near infrared dye, indocyanine green (ICG). We refer to these optical particles as NIR erythrocyte-mimicking transducers (NETs). A particular feature of NETs is that their diameters can be tuned from micron- to nano-scale. Herein, we investigated the effects of micron- (≈2.6 μm diameter), and nano- (≈145 nm diameter) sized NETs on their biodistribution, and evaluated their acute toxicity in healthy Swiss Webster mice. Following tail vein injection of free ICG and NETs, animals were euthanized at various time points up to 48 hours. Fluorescence analysis of blood showed that nearly 11% of the injected amount of nano-sized NETs (nNETs) remained in blood at 48 hours post-injection as compared to ≈5% for micron-sized NETs (μNETs). Similarly, at this time point, higher levels of nNETs were present in various organs including the lungs, liver, and spleen. Histological analyses of various organs, extracted at 24 hours post-injection of NETs, did not show pathological alterations. Serum biochemistry profiles, in general, did not show elevated levels of the various analyzed biomarkers associated with liver and kidney functions. Values of various hematological profiles remained within the normal ranges following the administration of μNETs and nNETs. Results of this study suggest that erythrocyte-derived particles can potentially provide a non-toxic platform for delivery of ICG.
Introduction
Particle-based systems play a prominent role in the field of clinical medicine as they provide a capability to deliver imaging or therapeutic agents to specific sites within the body.1,2 In relation to particles that deliver optical agents for imaging or phototherapeutic applications, the use of materials that are photo-activated by near infrared (NIR) excitation wavelengths (≈700–1300 nm) is especially advantageous since in this optically transparent window there is minimal light absorption by water and proteins, and diminished scattering by biological components, leading to increased depth of penetration on the order of a few centimeters.3 Furthermore, given that there is negligible tissue autofluorescence over this spectral window, the use of exogenous fluorescent materials can enhance the image contrast.
One particular NIR exogenous dye is indocyanine green (ICG). It is a tricarbocyanine molecule with maximum spectral peak in the range of ≈780–810 nm, depending on the solvent and concentration.4,5 To date, ICG remains as the only NIR-activated agent approved by the United States Food and Drug Administration for specific applications such as ophthalmic angiography, and assessment of liver and cardiovascular functions.6 Additionally, considerable efforts have been devoted to utilize ICG as a photothermal therapy reagent and photosensitizer for photodynamic therapy.7–10
Despite its effective usage in clinical medicine, the major limitations of ICG are its short half-life within plasma (2–4 minutes), with nearly exclusive uptake by hepatocytes and elimination from the body through the hepatobiliary mechanism.11,12 Encapsulation of ICG has been used as a strategy to reduce its non-specific interactions with plasma biomolecules, and increase its circulation time.13 Previously, we reported the encapsulation of ICG into a synthetic polymer, poly (allylamine) hydrochloride cross-linked to sodium phosphate.14 When coated with 5 kDa poly (ethylene) glycol, these ICG-containing nanoparticles with peak diameter of ≈80 nm remained detectable within the circulation at 90 minutes post intravenous injection in mice.
Apart from synthetic polymers,14,15 and liposomes16 attention has been given to the use of mammalian cells as carriers for delivery of imaging/contrast reagents or therapeutic payloads.16,17 We previously provided the first report on the engineering of erythrocyte-derived nano-sized vesicles loaded with ICG, and their utility for fluorescence imaging and photo-destruction of human mammalian cells.18 We refer to these constructs as NETs (NIR erythrocyte-derived transducers) as they can convert the absorbed NIR light to emit fluorescence, generate heat, or induce photochemistry.19
Recently, other investigators have encapsulated ICG or other agents such as gold nanoparticle and doxorubicin into erythrocytes-derived nano-constructs.20–24 For example, nanoconstructs (≈80 nm diameter) composed of poly(lactic-co-glycolic acid) (PLGA) core fused with erythrocyte-derived membranes were retained in mice blood at two days post tail vein injection.25 Piao et al. have reported that gold nanocages cloaked with erythrocyte membranes (≈90 nm diameter) were present in mice blood 24 hours after tail vein injection.23 Godfrin et al. have demonstrated that erythrocytes encapsulating L-asparaginase for treatment of acute lymphocytic leukemia were still detectable at ~28 days after delivery.26 Hence, constructs derived from erythrocytes may offer a promising approach for increased circulation time within the vasculature.
A particular feature of NETs is that their diameters can be tuned from nano- to micron-scale levels based on appropriate mechanical manipulation methods.27 The nano-sized NETs (nNETs) have relevance to optical imaging and phototherapy of tumors since particles with diameters < ≈200 nm are effective for extravasation into tumors by the enhanced permeability and retention effect resulting from leaky tumor vasculature and impaired lymphatic drainage.28,29 Micron-sized NETs (μNETs) have relevance to phototherapy of vasculature. An example of abnormal vasculature is associated with cutaneous capillary malformations such as port wine stain lesions where NETs can potentially serve as the target of laser irradiation to induce photo-destruction of the abnormal vasculature plexus.30 To the best of our knowledge, there have not been prior studies to investigate the effects of the diameter of erythrocyte-derived constructs such as NETs on their resulting circulation kinetics. Additionally, we investigate the effects of NETs diameter on toxicology profiles of these particles in healthy mice. Results of this study provide important information not only towards identifying the window of time over which NETs-based optical imaging or phototherapy can be performed, but may also be useful for investigators interested in various optical sensing, imaging, or photo-therapeutic applications of erythrocyte-derived platforms.
Experimental
Fabrication of μNETs and nNETs
A schematic of the NETs fabrication process is presented in Fig. 1. Erythrocytes were separated from bovine whole blood (Rockland Immunochemicals, Inc., Limerick, PA, USA) by centrifugation process. Approximately, 1 ml of bovine whole blood was taken in an Eppendorf, and centrifuged for 10 minutes (1600g at 4 °C). The supernatant containing the plasma and buffy coat were discarded, and the resulting packed erythrocytes were washed twice with 310 mOsm, phosphate buffer solution (PBS) (referred to as the 1× solution) (Fisher Scientific, Hampton, NH, USA) at pH ~ 8.0. The erythrocytes were then subjected to sequential hypotonic treatment with 0.5 × (155 mOsm, pH ~ 8.0) and 0.25 ~ (80 mOsm, pH ~ 8.0) PBS, respectively. The centrifugation process (20 000g, 15 minutes, 4 °C) was repeated until all the hemoglobin was depleted, resulting in an opaque white pellet. The obtained pellet containing micron-sized erythrocyte ghosts (μEGs) were resuspended in 1 ml of 1× PBS. To obtain nano-sized erythrocyte ghosts (nEGs), μEGs were extruded 20 times through 400 nm polycarbonate porous membranes, followed by 20 more extrusions through 100 nm polycarbonate porous membranes using an extruder (Avanti mini extruder, Avanti Polar Lipids, Alabaster, AL, USA).
Fig. 1.
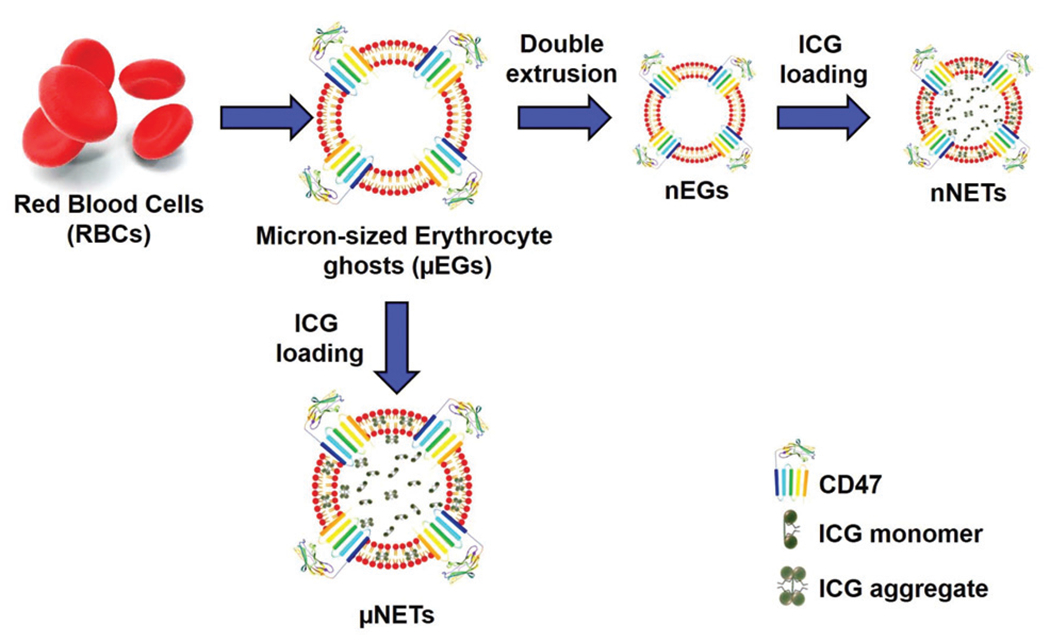
Schematic representation for fabrication of μNETs and nNETs.
To load ICG (MP Biomedicals, Santa Ana, CA, USA) into micron/nano-sized EGs, 350 μl of μEGs or nEGs suspended in 1× PBS were incubated with 23 μl of ICG stock solution (645 μM), 350 μl of Sorenson’s buffer (Na2HPO4/NaH2PO4, 140 mOsm, pH ~ 8.0) and 280 μl nanopure water for 5 minutes at 4 °C in the dark. Resulting concentration of ICG in this loading buffer was 15 μM. Suspension was then centrifuged and washed twice with 1× PBS at 20 000g for 20 minutes at 4 °C, or 100 000g for 60 minutes to form μNETs and nNETs, respectively. The resulting μNETs and nNETs formulations were then re-suspended in 1 ml of 1× cold PBS. We note that the use of 15 μM ICG in the loading buffer can result in production of μNETs and nNETs with near maximum values of spectrally-integrated fluorescence emission.27
Characterizations
Absorption spectra of free 15 μM ICG, μNETs and nNETs suspended in 1× PBS were obtained using a UV-visible spectrophotometer (Jasco-V670 UV-vis spectrophotometer, JASCO, Easton, MD, USA) with optical path length of 1 cm. Fluorescence emission spectra of NETs in response to 720 ± 2.5 nm excitation light, spectrally filtered from a 450 W xenon lamp, were recorded in the range of 735–900 nm using a fluorometer (Fluorolog-3 spectrofluorometer, Horiba Jobin Yvon, Edison, NJ, USA). We normalized the fluorescence emission spectra as:
| (1) |
where A and F are the respective wavelength (λ)-dependent absorbance and intensity of the emitted fluorescence light, and λex is the excitation wavelength. The hydrodynamic diameters and zeta potentials of μNETs and nNETs suspended in 1× PBS were measured by dynamic light scattering (DLS) (Zetasizer Nano ZS90, Malvern Instruments Ltd, Westborough, MA, USA). We fitted lognormal functions to the DLS-based estimates of NETs’ hydrodynamic diameters.
We imaged the μNETs by confocal laser scanning microscopy (CLSM). Sample solution (~10 μl) was added to poly-l-lysine coated glass slide, followed by placing a coverslip on the top of the glass slide, and then imaging it with a confocal microscope (ZEISS 510, Carl Zeiss AG, Oberkochen, Germany). We imaged the nNETs by scanning electron microscopy (SEM). nNETs were fixed with 2.5% glutaraldehyde (Sigma Aldrich, St Louis, MO, USA) overnight. Subsequently, 10 μl of the nNETs solution was added to poly-l-lysine coated slide and dried (Critical-point-dryer, Balzers CPD0202) for 30 minutes followed by sputter coating with platinum for 20 s, and then imaging with SEM (FEI NNS450).
Assessment of ICG leakage from μNETs and nNETs under physiological temperature
Approximately, 2 ml of μNETs and nNETs suspensions were transferred into various Eppendorf tubes, and incubated at 37 °C in the dark. After specific incubation times (0, 2, 4, 6, 24 and 48 hours), the NETs suspensions were centrifuged, and the pellet was then resuspended in 1 ml of fresh 1× PBS. Subsequently, the absorption spectra of re-suspended pellet and supernatant were recorded and compared to those for day 0 to assess the ICG leakage from μNETs or nNETs. The percentage of ICG leakage (υ) in μNETs and nNETs as a function of time were calculated as:
| (2) |
where Apellet and Asupernatant are the absorbance values of the pellet and supernatant recorded at 804 nm for each specific time point. Results of these experiments are presented as ESI (Fig. S1†).
Biodistribution experimental design
Female Swiss Webster mice (≈20–25 g; ≈8–10 weeks old) were procured from Taconic Biosciences (Rensselaer, NY, USA). All animal maintenance and procedures were performed in accordance with the Public Health Service Policy, U.S. Department of Agriculture (USDA), and American Veterinary Medical Association (AVMA). Animal studies were approved by the University of California, Riverside Institutional Animal Care and Use Committee (protocol A-20170038). Animals were anesthetized by inhalation of 2% isoflurane in oxygen. Free ICG, μNETs or nNETs were administered intravenously via tail-vein injection while the animal was anesthetized. The injection volume for all samples was ≈100 μl. Injected dosages of ICG in our experiments were estimated as ≈58, 26.16, 14.5 μg kg−1 weight of the mouse for free ICG, μNETs, and nNETs respectively. For example, the injection dosage of free ICG used was estimated as follows: based on the molecular weight of ICG (775 Da), injection concentration of 15 μM free ICG corresponds to ≈11.6 μg ml−1. Given the injection volume of ≈0.1 ml into each mouse, and an average mouse weight of ≈20 g, dosage of free ICG administered into an animal was ≈0.058 μg g−1 (11.6 μg ml−1 × 0.1 ml)/20 g, or ≈58 μg kg−1. Taking into account the respective loading efficiency of 15 μM ICG into μNETs and nNETs as ≈45% and 25%,27 the administered dosage of ICG in μNETs and nNETs formulations were estimated as ≈26.16 μg kg−1 and 14.5 μg kg−1, respectively. These injected dosages of ICG were much lower than the lethal dosage in 50% of animals (LD50) of ~62 mg kg−1 in mice.31 We also note that loading efficiency of ICG is not the only metric that constitutes the ideal preparation of NETs. Other important metrics are the relative fluorescence quantum yield of NETs and their total fluorescence emission over a spectral band of interest. As we have reported previously, concentration of ICG in the loading buffer, ICG loading efficiency, and NETs’ fluorescence quantum yield and total emission are interrelated parameters.27 Increased levels of the indicated optical metrics can be associated with lower values of ICG loading efficiency.27
Mice were euthanized with compressed CO2 gas at various times (5 minutes, 45 minutes, 2, 6, 24, and 48 hours) following injection with free ICG, μNETs or nNETs. Three mice were used for 5 minutes, 45 minutes and 2 hours time points, whereas, five mice were used for 6, 24 and 48 hours time points for each of the imaging agents, giving a total of 72 animals for the biodistribution studies.
Fluorescence imaging and analysis of extracted organs
Following euthanasia, liver, spleen, kidney (single), stomach, intestine, heart and lungs (two) were extracted and imaged fluorescently in a luminescence dark box. Two light emitting diodes (LEDs) delivering excitation light in the range of 700 ± 30 nm were used for illumination. Fluorescence emission from the organs was captured using a charge-coupled device (CCD) camera (Pixis 1024B, Roper Scientific, Trenton, NJ, USA) equipped with a long pass filter transmitting wavelengths greater than 810 nm. Camera exposure time was set to 90 s. Acquired fluorescent images were analyzed using ImageJ software. Regions of interests (ROIs) were selected for each organ. The mean intensity (Ī) values per gram of each organ acquired from a given ROI at different post-injection times were calculated as:
| (3) |
where p is the total number of pixels in the ROI, m is the mass of a given organ, n is the numbers of a given organ, and Ij is the pixel intensity at the jth pixel of a given image.
Biodistribution analysis
Extracted organs were grinded using Omni Tissue Homogenizer (Omni International, Inc., Kennesaw, GA, USA), and then incubated in 4 ml of sodium dodecyl sulfate (SDS) (Sigma Aldrich, St Louis, MO, USA) solution (5% w/v in water) for one hour to lyse the cells causing the release of ICG. We also collected 300 μl of blood from the heart by cardiac puncture. The blood sample was mixed with 1 ml of SDS solution and incubated for 30 minutes. This approach would ensure that any ICG or NETs uptaken by blood cells would also be released. Lysed organs and blood samples were centrifuged in the SDS solution at 16 000g for one hour at 10 °C. Then the supernatants of the blood samples and homogenized organs were collected, and the corresponding fluorescence emission spectra in response to 720 ± 2.5 nm excitation wavelength were recorded using the fluorometer. ICG concentration in each organ was estimated by comparing the integrated fluorescence emission signal over the 735–900 nm spectral band with a calibration curve that related the integrated fluorescence emission over the same wavelength range to various concentrations of ICG in SDS solution. Specifically, we present the percentage of ICG recovered from each organ with respect to the initial dose (ID) injected per gram of organ (% ID g−1).
For blood samples, the calibration curve related the spectrally-integrated fluorescence emission over the same wavelength range to various concentrations of ICG and blood in SDS solution. The integrated fluorescence emission value of whole blood in SDS solution without free ICG or NETs was subtracted from the integrated fluorescence emission values obtained for blood samples containing free ICG or NETs. The fluorescence profiles of ICG extracted from SDS-treated organs closely resembled that of free ICG dissolved in SDS (data not shown). This result validated that the fluorescence of ICG extracted from the various organs was not degraded in the presence of 5% SDS, and that autofluorescence was not a contributor to fluorescence emission at 720 ± 2.5 nm excitation wavelength.
Hematological, enzymatic, and histological evaluations
We injected ≈100 μl of μNETs or nNETs solutions via the tail-vein into anesthetized mice. At 24 hours post-injection, mice were euthanized, and blood samples were collected for serum biochemistry and enzymatic evaluations. This included three assays of hepatic function (Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST) and Alkaline Phosphatase (ALP)), and two renal assays (Urea nitrogen and creatinine) performed by a blood biochemical autoanalyzer (Roche Integra 400 Plus, Roche, Basel, Switzerland). A complete blood count (CBC) was also performed, which included white blood cells (WBCs) count, red blood cells (RBCs) count, mean corpuscular volume (MCV), hemoglobin, hematocrit, and platelets performed by a blood count analyzer (Drew HemaVet 950 FS, Drew Scientific, Miami Lakes, FL, USA).
A complete necropsy was performed and major organs including heart, liver, spleen, lungs and kidneys were collected. Tissues were fixed in 10% neutral buffered formalin (Fisher Scientific, Waltham, MA, USA), processed routinely into paraffin blocks and sectioned at 4 μm. These sections were stained with hematoxylin and eosin (H&E), and examined histologically for toxicity changes including thrombosis, inflammation, and necrosis. Ten mice were used for each agent for CBC and serum biochemistry, giving a total of 30 animals. Out of these, five mice were used for each agent for histological evaluations.
Statistical analysis
Statistical analyses were conducted using the Graphpad Instat 3.0 software by the one-way ANOVA analysis. We defined statistically significant differences at *p < 0.05.
Results and discussion
Characterizations
Based on the analysis of CLSM images (Fig. 2A), average diameter of μNETs was ~3 μm. The SEM image of nNETs demonstrated the nano-sized dimensions of these NETs (Fig. 2B). The estimated mean peak values of hydrodynamic diameters for μNETs and nNETs, as estimated by the DLS method were 2.6 μm and 145 nm, respectively (Fig. 2C and D).
Fig. 2.
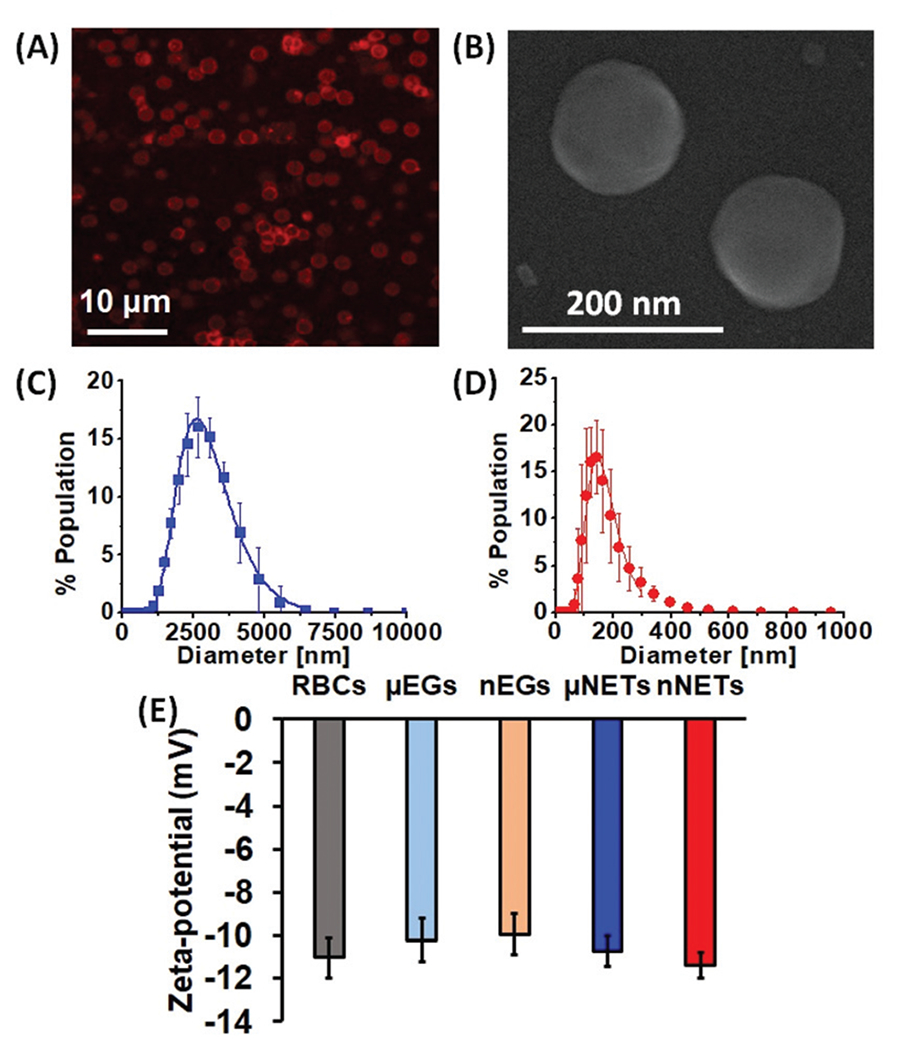
(A) Confocal laser scanning microscopy image of μNETs. A 633 nm laser was used for photo-excitation and a long-pass filter transmitting λ > 650 nm was used to collect the emitted NIR fluorescent light, falsely-colored in red. (B) Scanning electron microscopy (SEM) image for nNETs. Panels (C) and (D) represent the dynamic light scattering-based measurements of diameter distributions for μNETs and nNETs in 1× PBS, respectively. We present the mean of each measurement with errors bars representing standard deviations from the mean values. We fitted Lognormal functions to the measured diameter distributions. (E) Zeta-potentials for RBCs, μEGs, nEGs, μNETs and nNETs recorded in 1× PBS. Error bars represent the standard deviation for the three independent measurements.
The mean ± standard deviation (SD) zeta-potential values for RBCs, μEGs, nEGs, μNETs and nNETs formulations in 1× PBS (pH ~ 7.4) were −11.03 ± 1.91 mV, −10.2 ± 1.93 mV, −9.96 ± 1.74 mV, −10.7 ± 1.42 mV and −11.4 ± 1.13 mV, respectively (Fig. 2E). Statistical analysis showed that the mean values of these zeta-potentials were not significantly different from each other. The similar zeta-potential values of μEGs, nEGs, μNETs and nNETs suggest that the sulfonate portions of ICG molecules, which carry the negative charge of ICG, were not exposed to the extracellular environment, but rather localized within the membrane or within the interior core of NETs. The overall negative charge of erythrocyte-derived constructs can be attributed to the presence of the carboxylic groups of the sialic acid on erythrocytes membranes,32–34 suggesting that these groups were preserved on both EGs and NETs.
The peak of the absorption spectrum for 15 μM free ICG dissolved in nanopure water was at 780 nm, and is associated with the monomeric form of ICG (Fig. 3A).4 The absorption peak at 280 nm for μNETs and nNETs corresponds to the membrane proteins present on the surface of the NETs. The absorption spectra of μNETs and nNETs showed bathochromic (red) shifts in the monomeric peak of ICG from 780 nm to 804 nm, compared to free ICG.
Fig. 3.
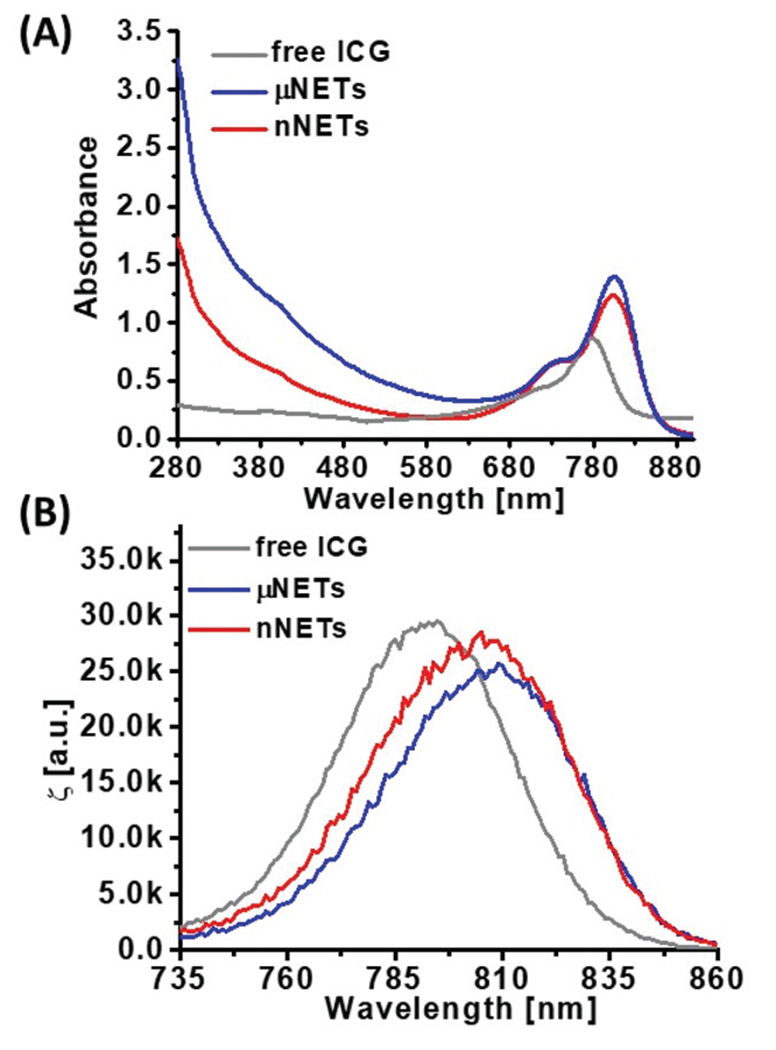
Representative (A) absorption and (B) normalized fluorescence emission spectra of 15 μM free ICG, μNETs, and nNETs. Both μNETs and nNETs were fabricated using 15 μM ICG in the loading buffer. Spectra for μNETs and nNETs were recorded in 1× PBS, and in nanopure water for free ICG. Prior to absorption and fluorescence spectral recordings, ICG and NETs solution were diluted by 50 and 10 times, respectively. Emission spectra were obtained in response to photo-excitation at 720 ± 2.5 nm.
We attribute this shift to encapsulation-induced changes in ICG including the formation of membrane protein-bound and phospholipid-bound forms with altered electronic states as compared to free form of ICG.18,27 Normalized fluorescence emission spectra of 15 μM free ICG, μNETs and nNETs in response to 720 nm excitation wavelength are presented in Fig. 3B. The respective spectral peak emission values for free ICG, μNETs and nNETs were at 793 nm, 809 nm, and 806 nm, corresponding to the monomeric forms of ICG.4,18
Absorption spectra of μNETs and nNETs and supernatant after centrifugation at physiological temperature in dark over a period of 48 hours are shown in Fig. S1A and B.† Using eqn (2), we estimate the percentage leakage of ICG from μNETs and nNETs as ≈9.1% and 5.6%, respectively, at physiological temperature over time period of 48 hours (Fig. S1C†).
Quantitative fluorescence imaging of extracted organs
Fluorescence emission from extracted liver was relatively weak at 6 hours post-injection of free ICG (Fig. 4A). This result suggests that by this time most of ICG was eliminated from the liver, its primary accumulation site. The weak fluorescence emission from the stomach could be due to the presence of chlorophyll (alfalfa) in the rodent diet.35 Decreased levels of free ICG emission from the liver at 24 and 48 hours combined with minimal signal from the intestine and other organs suggest that most of the free ICG was eliminated from the body through excretion from the liver into the intestine via bile.36
Fig. 4.
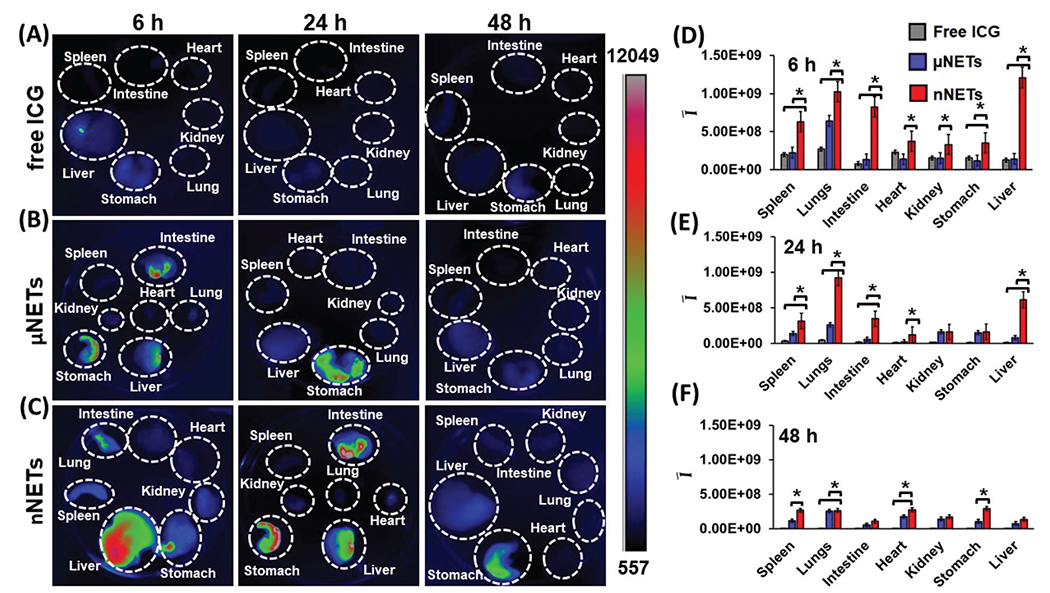
Fluorescence images of organs extracted from healthy mice at 6, 24 and 48 hours post tail-vein injection with (A) indocyanine green (ICG) (15 μM), (B) μNETs, and (C) nNETs. Scale bar on the right (in arbitrary units) corresponds to fluorescence emission intensity. (D–F) Mean ROI fluorescence intensities per gram of extracted organ (see eqn (3)) from healthy mice at (D) 6, (E) 24, and (F) 48 hours post tail-vein injection of ICG (15 μM), μNETs and nNETs. Error bars represent standard deviation and the single asterisks denote statistically significant differences (p < 0.05) between the indicated pairs.
μNETs and nNETs showed higher fluorescence signals from the extracted liver, intestine, and stomach at 6 hours post-injection (Fig. 4B and C), suggesting a lower clearance rate of these particles from systemic circulation. Fluorescence emissions associated with μNETs and nNETs were detectable in liver up to 48 hours post-injection, suggesting that NETs had prolonged the availability of ICG within the body.
The mean fluorescence intensity (Ī) values per gram of each extracted organ for free ICG, μNETs, and nNETs at 6, 24, and 48 hours post-injection time points are shown in Fig. 4D–F. When compared to free ICG and μNETs, nNETs showed statistically significant (p < 0.05) higher values of Ī in all extracted organs at 6 hours post-injection (Fig. 4D), indicating the higher accumulation of nNETs in these organs. At 24 hours post-injection, value of Ī associated with nNETs was significantly higher in spleen, lungs, intestine, heart and liver as compared to free ICG and μNETs (Fig. 4E).
At 48 hours post-injection, value of Ī associated with nNETs remained significantly higher in spleen, lungs, heart, and stomach than those for free ICG and μNETs (Fig. 4F). In contrast, there were no statistically significant differences between free ICG and μNETs at 48 hours in all organ samples.
Quantification of ICG content in blood
Fluorescence emission spectra of blood collected from healthy mice at 6 and 48 hours post-injection with μNETs and nNETs are presented in Fig. 5A. Fluorescence emission intensity of μNETs at 6 hours post-injection was higher than that at 48 hours post-injection over the spectral band 735–900 nm, indicating that the fraction of these particles in the bloodstream was reduced between 6–48 hours. Nano-encapsulation improved the longevity of ICG in bloodstream as compared to microencapsulation as evidenced by higher fluorescence emission intensity of nNETs at 48 hours post-injection when compared to that of μNETs at 48 hours post-injection (Fig. 5A). Fluorescence emission spectrum of nNETs at 48 hours post-injection was comparable to that of μNETs at 6 hours post-injection.
Fig. 5.
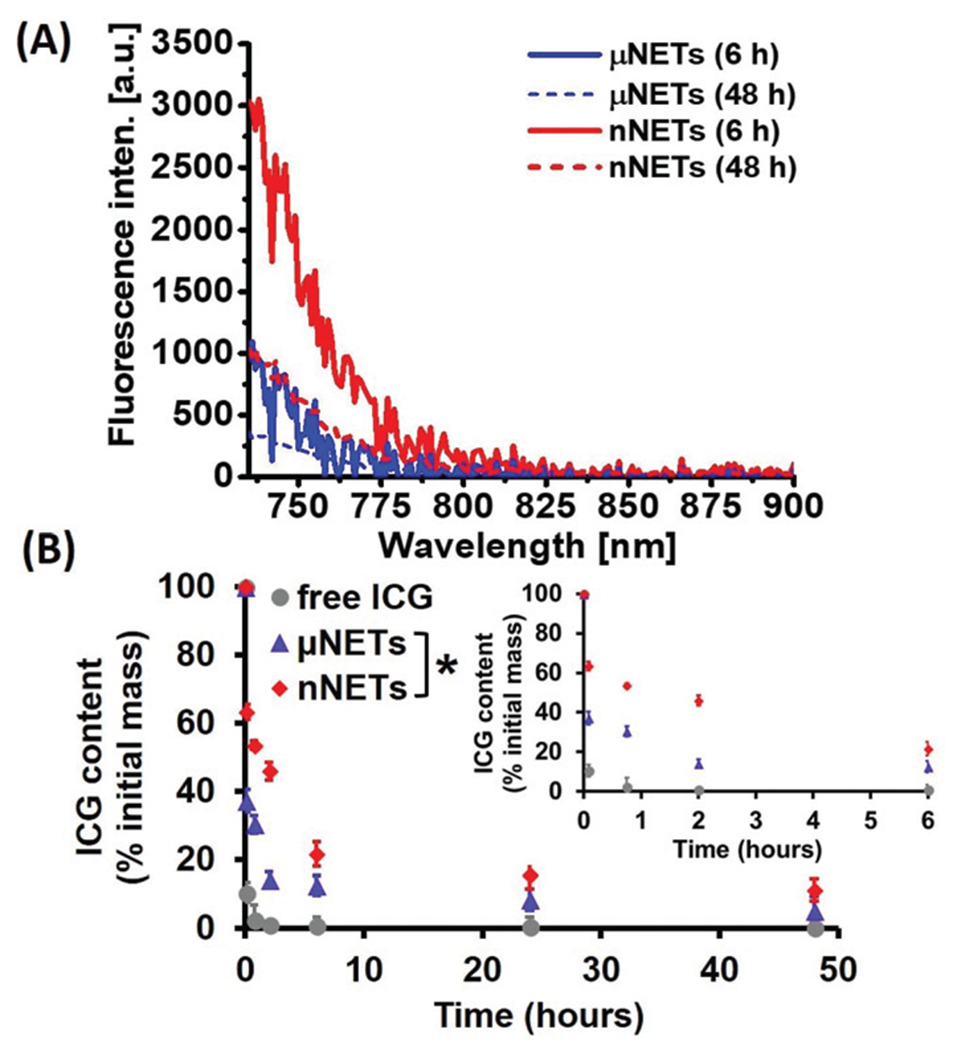
(A) Fluorescence emission spectra of blood collected from healthy mice at 6 and 48 hours post tail-vein injection with μNETs and nNETs (λex = 720 ± 2.5 nm) after baseline subtraction of blood emission spectra in SDS. (B) Estimated percentages of ICG recovered from mice blood after tail vein injection of ICG (circles  ), μNETs (triangles
), μNETs (triangles  ) and nNETs (diamonds
) and nNETs (diamonds  ) as a function of time. The inset represents the estimated percentages of ICG recovered from mice blood for the three agents at 0 min, 5 min, 45 min, 2 and 6 hours respectively. Error bars represent standard deviations, and the single asterisk denotes statistically significant differences (p < 0.05) between the indicated pairs for all the time points.
) as a function of time. The inset represents the estimated percentages of ICG recovered from mice blood for the three agents at 0 min, 5 min, 45 min, 2 and 6 hours respectively. Error bars represent standard deviations, and the single asterisk denotes statistically significant differences (p < 0.05) between the indicated pairs for all the time points.
We estimated the relative ICG content of blood at 5 min, 45 min, 2, 6, 24 and 48 h post-injection of free ICG, μNETs and nNETs formulations (Fig. 5B). The relative ICG content of blood at 6 hours post-injection for nNETs was ≈21.6% as compared ≈12.5% for μNETs. At 48 hours post-injection, the relative amounts of ICG for nNETs and μNETs were ≈11.3% and 5.3%, respectively. The lower circulation time of μNETs as compared to nNETs can result from the increased uptake of μNETs by the mononuclear phagocytic system (MPS) that include the spleen and liver macrophages.
Biodistribution profiles
We quantified the relative amounts of free ICG, μNETs and nNETs in various organs at 6, 24 and 48 hours post-injections after homogenizing the organs (Fig. 6). In all organs, there were significantly greater levels of nNETs than μNETs and free ICG at 48 hours post-injection (p < 0.05). For example, at 48 hours post-injection, ≈18.8%, 12.8%, and 8% ID g−1 of nNETs, μNETs, and free ICG were present in the heart, respectively (Fig. 6A).
Fig. 6.
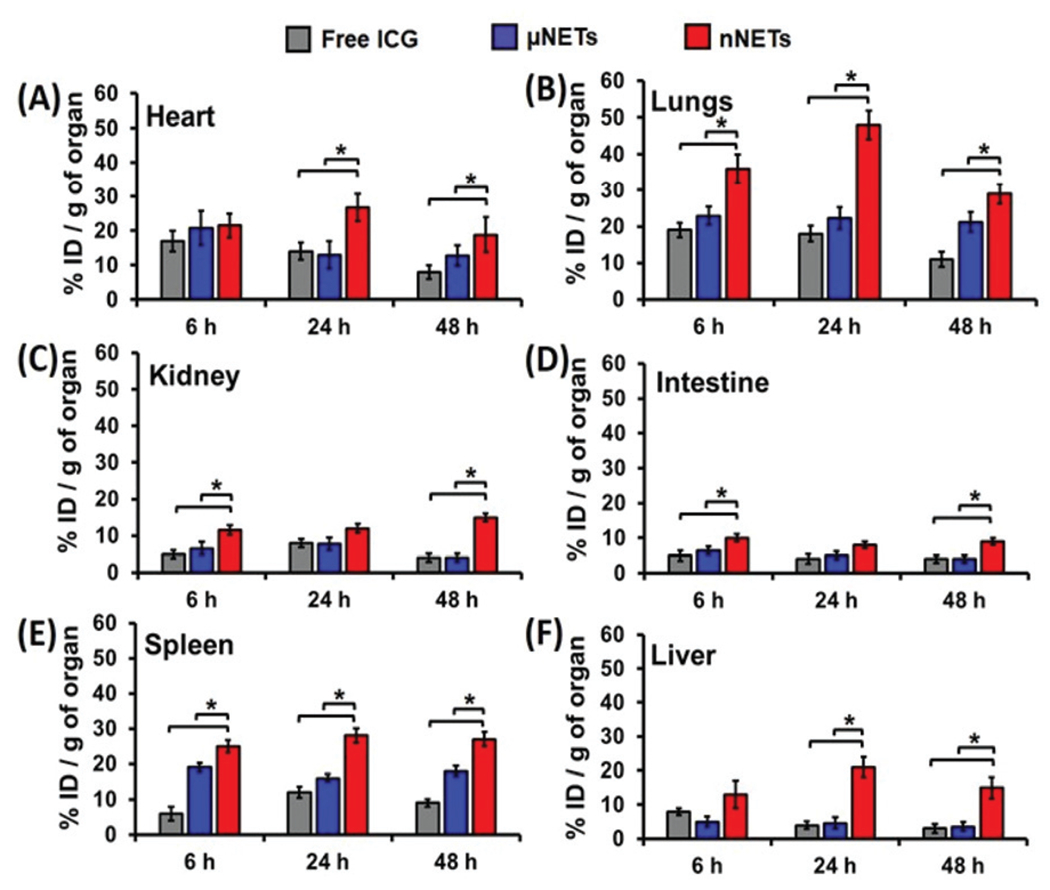
Estimated percentage of ICG recovered from organs of mice with respect to the initial dose injected per gram of organ at 6, 24 and 48 hours post tail-vein injection with indocyanine green (ICG) (15 μM), μNETs and nNETs for (A) heart, (B) lungs, (C) kidney, (D) intestine, (E) spleen, and (F) liver. Error bars represent standard deviations, and the single asterisks denote statistically significant differences (p < 0.05) between the indicated pairs.
Lungs were another organ site for significantly higher accumulation of nNETs (Fig. 6B). The higher accumulation level of the nNETs in the lungs can be attributed due to their effective entrapment in the pulmonary capillaries. It is believed that endocytosis process by endothelial cells of the lungs capillaries is more effective in internalization of nano-sized particles as compared with micron-sized particles.13,37 At 48 hours post-injection time point, as compared to μNETs and free ICG, statistically significantly higher levels of nNETs were also detected in kidney (≈15% ID g−1) and intestine (9% ID g−1) (Fig. 6C and D).
At 48 hours post-injection, there were ≈27% and 15% ID g−1 of nNETs in spleen and liver, respectively. In contrast, at this time point, there were only about ≈18% and 3.6% ID g−1 of μNETs, and ≈9% and 3% ID g−1 of free ICG in spleen and liver, respectively (Fig. 6E and F). A higher level of nNETs in spleen at 48 hours post-injection suggests that nNETs were still within the circulation and passing through the spleen.
In addition to filtering foreign body materials, spleen is also involved in removal of and damaged or aged erythrocytes from the blood.38 In the open blood circulation system of spleen, the arterial blood arrives into cords in the red pulp, which contain the splenic macrophages. Blood from the cords enters the venous sinuses of the red pulp through interendothelial slits with an average width of ≈650 nm.39,40 Due to their smaller diameters, nNETs (~145 nm mean peak diameter) can pass through these slits, and gain re-entry into the circulatory system. Some fraction of nNETs may also be uptaken by splenic macrophages of the red pulp located in the cords.
The circulation time of red blood cells (RBCs) depends on various biochemical and physical characteristics. For example, the presence of key membrane glycoproteins such as CD47, decay-accelerating factor (DAF), CD59, and CR1 on the RBC surface can impede phagocytosis and prolong the circulation time of RBCs.41–44 Our previous results indicate that CD47 remains on the surface of μEGs and nNETs, suggesting that CD47 can survive the mechanical extrusion of μEGs,45 and contribute to the extended circulation time of NETs by keeping them shielded from phagocytosis.
In addition to the presence of appropriate biochemical markers, healthy RBCs also have appropriate mechanical properties that allow them to deform and pass through the splenic slits. In contrast, senescent RBCs have reduced deformability.46,47 For example, nearly 35% increase in membrane surface viscosity is reported for aged RBCs as compared to young cells.48 A reduction in deformability of senescent RBCs impairs their capability to pass through interendothelial slits. Aged RBCs are eventually phagocytosed by the red pulp macrophages,39 and eliminated from circulation. Similarly, μNETs (~2.6 μm mean peak diameter) may have reduced deformability to allow them to squeeze through splenic slits. Nash and Meiselman have reported that fresh micron-sized EGs with average reduction of approximately 125 times in cellular hemoglobin content had nearly 68% increase in average elastic shear modulus as compared to young intact RBCs.48 With compromised mechanical integrity, specifically due to lowered deformability, μNETs are likely to remain within the cords and ultimately phagocytosed by the splenic macrophages. Combined with slowed blood flow rate in spleen,40 resident red pulp macrophages can efficiently recognize μNETs and remove them from circulation. Deformability of NETs can also be altered as a result of changes to specific membrane integral proteins.49 Such changes can subsequently affect the interactions of the membrane proteins with the membrane lipids, and with the cytoskeletal spectrin network.49,50
Liver is another site for the elimination of foreign agents. Endothelial cells in liver sinusoids form a fenestrated lining containing pores with diameter of ≈170 nm,51 which allow for passage of blood plasma to interact with hepatocytes.52 A fraction of nNETs with diameters less than the pores’ diameter are likely to leave the sinusoids and extravasate into the space of Disse where they can go undergo endocytosis by hepatocytes, and subsequently, secreted into the bile ducts, passaged into duodenum, and finally eliminated from the body. This mechanism of nNETs elimination is consistent with the hepatobiliary clearance of nanoparticles with diameters in the range of 150–200 nm,52 and resembles hepatocytes-mediated elimination of liposomes and lipid-coated PLGA nanoparticles with similar characteristics consisting of mean diameters in the range of ≈150–195 nm and average zeta potentials between ≈−19 to −10.6 mV.53–55 Nanoerythrosomes with diameters < 100 nm have also been reported to accumulate in hepatic parenchymal cells.56
In addition to hepatocytes, hepatic stellate cells located in the space of Disse between parenchymal and sinusoidal endothelial cells, can also uptake the nNETs extravasated out of the sinusoids. This mechanism of nanoparticles elimination has been reported for liposomes with average diameter of 92 nm and zeta potential of ≈−20 mV.57 The fraction of nNETs with diameters greater than the diameter of sinusoid pores, and μNETs can be uptaken by the Kupffer cells, macrophages adherent to the endothelial lining of the liver sinusoids.52 This phagocytic uptake is consistent with the reported accumulation of nanoerythrosomes in Kupffer cells.56
A particular hydrodynamic phenomenon resulting from the high (≈50%) occupancy of blood volume by RBCs is the margination of particles to the vessels wall.42 Under this phenomenon, RBCs avoid the micron-scale marginal layer of the blood that directly contacts the vessel wall and allow for other circulating particles to come in contact with the wall.58,59 It has been reported that 2 μm diameter hydrogel particles had enhanced margination when compared to their 500 nm diameters counterparts.60 In relation to NETs, the margination phenomenon implies that μNETs may be in closer approximately to Kupffer and endothelial cells that line the sinusoids, resulting in greater uptake of these particles as compared to nNETs. Ultimately, degradation of μNETs by the splenic and hepatic MPS can result in their reduced circulation time.
Pathology, serum biochemistry and hematology
H&E stained sections of heart, liver, spleen, lung and kidney after 24 hours post intravenous injection of μNETs and nNETs were similar to PBS-injected (control) mice (Fig. 7A), indicating that NETs did not induce pathological alterations in these organs. The levels of blood biomarker enzymes (ALP, ALT, and AST) associated with the liver, and urea nitrogen and creatinine, associated with kidney functions are presented in Fig. 7B and C, respectively. Given that less than 20% of the injected dosages of NETs per gram of liver were detected at 48 hours (Fig. 6F), there may not be concerns with chronic toxicity to liver parenchyma that may otherwise arise as a result of prolonged retention of particles in liver. In contrast, nearly 53% of the injected dose of 20 nm diameter zinc oxide,61 and 80% of 30 nm diameter superparamagnetic iron oxide nanoparticles62 were retained in liver for at least 21 and 84 days, respectively.
Fig. 7.
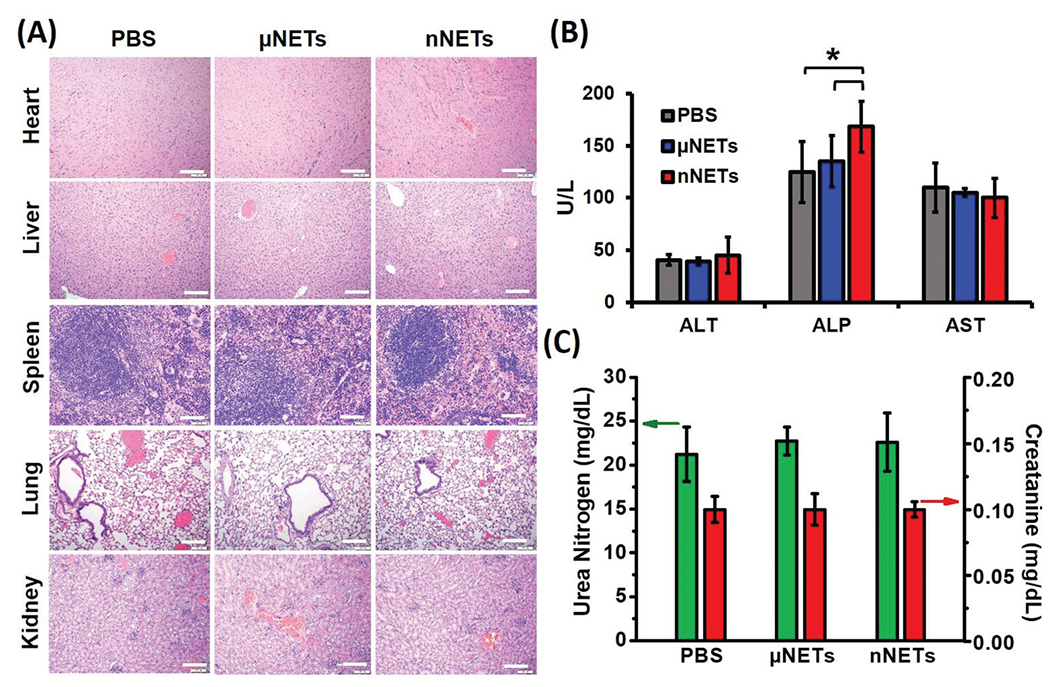
(A) Representative H&E stained images of major organs collected from healthy mice at 24 hours post tail-vein injection with PBS, μNETs, and nNETs derived from bovine blood. Scale bars indicate 100 μm. (B) Levels of Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST) and Alkaline Phosphatase (ALP), associated with hepatic function. (C) Levels of urea nitrogen and creatinine associated with renal function. Error bars represent standard deviations and the single asterisks denote statistically significant differences (p < 0.05) between the indicated pairs.
ALT and AST levels for μNETs and nNETs were similar to those for PBS-injected (control) mice (Fig. 7B). However, ALP levels were higher (p < 0.05) for nNETs when compared to those for PBS and μNETs. Levels of urea nitrogen and creatinine did not statistically differ as a function of the administered agent (Fig. 7C).
While the values for various hematological parameters can vary with the mouse strain, age, sex, and other factors, our measurements are within the normal ranges reported for these parameters,63,64 regardless of the administered agent (Table 1). Values of RBCs, MCV, hemoglobin and % hematocrit did not statistically differ as a function of the administered agent (Table 1). Despite our results that the levels of WBCs and platelets were significantly lower (p < 0.05) in response to administration of μNETs, as compared to those following the administration of PBS and nNETs, they were still within the normal ranges. Similarly, Marshall et al. have also reported a reduction in WBCs and platelets counts, but still within the normal range, upon injection of 20 mg kg−1 of free ICG into Sprague-Dawley rats (Table 1),65 a dosage substantially higher than our estimated dosages of 58 μg kg−1 for free ICG, and 26.16 and 14.5 μg kg−1 of ICG in μNETs and nNETs formulations, respectively.
Table 1.
Hematology profiles of mice at 24 hours post tail vein injection of PBS (control), μNETs, and nNETs derived from bovine blood. Each experiment was repeated five times. Values represent the mean ± standard deviations. WBCs: white blood cells; RBCs: red blood cells; MCV: mean corpuscular volume. K denotes 1000. Single asterisks denote statistically significant differences of p < 0.05 between the indicated pairs. Reported ranges from ref. 63 are based on values from various mouse strains, sex, and age. Reported ranges from ref. 64 are for 16 weeks female Swiss Webster mice from Charles River Laboratories, Inc. Reported values from ref. 65 are in response to 20 mg kg−1 of free ICG injection into Sprague-Dawley rats
| Parameter | Normal range | Free ICG | PBS | μNETs | nNETs | ||
|---|---|---|---|---|---|---|---|
| WBCs (K μL−1) | 3.0–14.263 | 5.0 ± 2.365 | 10.2 ± 1.4 | 6.0 ± 1.3 | 9.0 ± 1.6 | ||
|
|
|||||||
| * | * | ||||||
| RBCs (M μL−1 | 5.0–9.563 | 6.1 ± 1.165 | 8.9 ± 0.6 | 9.1 ± 1.0 | 9.0 ± 0.3 | ||
| MCV (fL) | 48.0–56.063 | 57.2 ± 1.965 | 48.6 ± 5.1 | 48.6 ± 3.9 | 50.4 ± 5.4 | ||
| Hemoglobin (g dL−1) | 10.9–16.363 | 12.8 ± 2.265 | 14 ± 0.7 | 12.8 ± 1.2 | 13.4 ± 0.6 | ||
| Hematocrit (%) | 41.5–48.264 | 34.9 ± 6.365 | 43 ± 3.5 | 42.4 ± 5.0 | 43.5 ± 3.1 | ||
| Platelets (K μL−1) | 930–162664 | 721.2 ± 462.765 | 1167 ± 155 | 1005 ± 145 | 1148 ± 193 | ||
|
|
|||||||
| * | * | ||||||
Activation of Kupffer cells through interaction with NETs in the liver can induce the secretion of various cytokines for recruitment of circulating neutrophils to engulf NETs and become activated apoptotic cells. Kupffer cells then recognize and phagocytose apoptotic neutrophils expressing phosphatidylserine through p-selectin-medicated hepatic sequestration.66 These endocytosed apoptotic cells are then degraded through phagosome maturation.67,68 This process results in elimination of neutrophils from circulation, corresponding to a decrease in WBCs count. Based on our results, this mechanism for elimination of neutrophils, which includes activation of Kupffer cells and interaction of neutrophils with NETs, appears to be more sensitive to μNETs as compared to nNETs.
Platelets interact with leukocytes in liver and spleen.69 They bind to the hepatic sinusoidal endothelium in an integrin-dependent manner, leading to platelet and endothelial activation and leukocyte recruitment.70 Activated platelets overexpress p-selectin glycoprotein ligand-1 (PSGL-1) can bind to leukocytes, neutrophils and monocytes resulting in platelet aggregation followed by the release of pro-inflammatory cytokines leading to clearance by the macrophages present in the complement system.69,71,72 Our findings appear to suggest that the mechanisms leading to platelets activation, aggregation, and their ultimate clearance may have been more prone to μNETs as compared to nNETs. It is also possible that platelets aggregation and clumping, which may have been more prevalent in response to presence of μNETs, could have led to a decrease in the automated platelets count, as suggested in literature.73 Finally, our findings are consistent with another study where noticeable signs of toxicity as determined by blood biochemical, hematological and histological assays were not observed in mice at 15 days post tail-vein injection of up-conversion nanoparticles coated with RBC membranes.22
Conclusions
We have engineered erythrocyte-derived optical particles doped with ICG. The diameter of these constructs, referred to as NETs, can be adjusted from micron to nano scale. Nano-sized NETs were retained at higher levels in blood and various organs at 48 hours post tail vein injection as compared to free ICG and micron-sized NETs. Histological analyses of various organs, extracted at 24 hours post intravenous injection of NETs, did not show any pathological alterations. Serum biochemistry, in general, did not show elevated levels of the various analyzed biomarkers associated with liver and kidney functions. Values of various hematological profiles remained within the normal ranges following the administration of μNETs and nNETs. Results of this study suggest that erythrocyte-derived particles can potentially provide a non-toxic platform for delivery of payloads, and when constructed at nano-size scale, can extend the circulation time of their cargo. As optical platforms, NETs offer a great potential for clinical translation for light-based theranostic applications.
Supplementary Material
Acknowledgements
This study was supported in parts by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (1R01-AR068067-01A1), and the National Science Foundation (CBET-1509218). The electron microscopy images were obtained at the Central Facility for Advanced Microscopy and Microanalysis (CFAMM) at University of California, Riverside.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01448e
Conflicts of interest
Authors R. V. and B. A. have a financial interest in Radoptics LLC, which is pursuing the commercial development of the particles reported in this study. This interest did not interfere with the scientific work, judgement, and the objectivity of these authors with regards to the experimental procedures, data analysis, interpretation of results, reporting, and any other aspect of the study.
References
- 1.Kelkar SS and Reineke TM, Bioconjugate Chem, 2011, 22, 1879–1903. [DOI] [PubMed] [Google Scholar]
- 2.Lim EK, Kim T, Paik S, Haam S, Huh YM and Lee K, Chem. Rev, 2015, 115, 327–394. [DOI] [PubMed] [Google Scholar]
- 3.Smith AM, Mancini MC and Nie SM, Nat. Nanotechnol, 2009, 4, 710–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Philip R, Penzkofer A, Bäumler W, Szeimies RM and Abels C, J. Photochem. Photobiol., A, 1996, 96, 137–148. [Google Scholar]
- 5.Haritoglou C, Gandorfer A, Schaumberger M, Tadayoni R, Gandorfer A and Kampik A, Invest. Ophthalmol. Visual Sci, 2003, 44, 2722–2729. [DOI] [PubMed] [Google Scholar]
- 6.Frangioni JV, Curr. Opin. Chem. Biol, 2003, 7, 626–634. [DOI] [PubMed] [Google Scholar]
- 7.Lim HJ and Oh CH, Photodiagn. Photodyn. Ther, 2011, 8, 337–342. [DOI] [PubMed] [Google Scholar]
- 8.Smretschnig E, Ansari-Shahrezaei S, Hagen S, Glittenberg C, Krebs I and Binder S, Retina-J. Ret. Vit. Dis, 2013, 33, 316–323. [DOI] [PubMed] [Google Scholar]
- 9.Tuchin VV, Genina EA, Bashkatov AN, Simonenko GV, Odoevskaya OD and Altshuler GB, Lasers Surg. Med, 2003, 33, 296–310. [DOI] [PubMed] [Google Scholar]
- 10.Klein A, Szeimies RM, Baumler W, Zeman F, Schreml S, Hohenleutner U, Landthaler M, Koller M and Babilas P, Br. J. Dermatol, 2012, 167, 333–342. [DOI] [PubMed] [Google Scholar]
- 11.Yoneya S, Saito T, Komatsu Y, Koyama I, Takahashi K and Duvoll-Young J, Invest. Ophthalmol. Visual Sci, 1998, 39, 1286–1290. [PubMed] [Google Scholar]
- 12.Ho C-M, Dhawan A, Hughes RD, Lehec SC, Puppi J, Philippeos C, Lee P-H and Mitry RR, Asian J. Surg, 2012, 35, 9–15. [DOI] [PubMed] [Google Scholar]
- 13.Yaseen MA, Yu J, Jung BS, Wong MS and Anvari B, Mol. Pharmaceutics, 2009, 6, 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bahmani B, Lytle CY, Walker AM, Gupta S, Vullev VI and Anvari B, Int. J. Nanomed, 2013, 8, 1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Daly SM, Gamal-Eldeen AM, Abo-Zeid MAM, Borai IH, Wafay HA and Abdel-Ghaffar ARB, Photodiagn. Photodyn. Ther, 2013, 10, 173–185. [DOI] [PubMed] [Google Scholar]
- 16.Lajunen T, Kontturi LS, Viitala L, Manna M, Cramariuc O, Rog T, Bunker A, Laaksonen T, Viitala T, Murtomaki L and Urtti A, Mol. Pharmaceutics, 2016, 13, 2095–2107. [DOI] [PubMed] [Google Scholar]
- 17.Couto D, Freitas M, Porto G, Lopez-Quintela MA, Rivas J, Freitas P, Carvalho F and Fernandes E, Arch. Toxicol, 2015, 89, 1759–1769. [DOI] [PubMed] [Google Scholar]
- 18.Bahmani B, Bacon D and Anvari B, Sci. Rep, 2013, 3, 2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burns JM, Vankayala R, Mac JT and Anvari B, ACS Appl. Mater. Interfaces, 2018, 10, 27621–27630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pei Q, Hu XL, Zheng XH, Liu S, Li YW, Jing XB and Xie ZG, ACS Nano, 2018, 12, 1630–1641. [DOI] [PubMed] [Google Scholar]
- 21.Wan GY, Chen BW, Li L, Wang D, Shi SR, Zhang T, Wang Y, Zhang LY and Wang YS, Biomaterials, 2018, 155, 25–40. [DOI] [PubMed] [Google Scholar]
- 22.Rao L, Meng QF, Bu LL, Cai B, Huang QQ, Sun ZJ, Zhang WF, Li A, Guo SS, Liu W, Wang TH and Zhao XZ, ACS Appl. Mater. Interfaces, 2017, 9, 2159–2168. [DOI] [PubMed] [Google Scholar]
- 23.Piao JG, Wang LM, Gao F, You YZ, Xiong YJ and Yang LH, ACS Nano, 2014, 8, 10414–10425. [DOI] [PubMed] [Google Scholar]
- 24.Peng JR, Yang Q, Li WT, Tan LW, Xiao Y, Chen LJ, Hao Y and Qian ZY, ACS Appl. Mater. Interfaces, 2017, 9, 44410–44422. [DOI] [PubMed] [Google Scholar]
- 25.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH and Zhang L, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 10980–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godfrin Y, Thomas X, Bertrand Y and Duget C, Blood, 2007, 110, 4325–4325. [Google Scholar]
- 27.Tang JC, Partono A and Anvari B, IEEE Trans. Biomed. Eng, 2018, DOI: 10.1109/TBME.2018.2866368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R and Langer R, Nat. Nanotechnol, 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- 29.Blanco E, Shen H and Ferrari M, Nat. Biotechnol, 2015, 33, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns JM, Jia W, Nelson JS, Majaron B and Anvari B, J. Biomed. Opt, 2018, 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebert B, Riefke B, Sukowski U and Kai LC, J. Biomed. Opt, 2011, 16, 066003. [DOI] [PubMed] [Google Scholar]
- 32.Kuo YC, Wu HC, Hoang D, Bentley WE, D’Souza WD and Raghavan SR, Langmuir, 2016, 32, 171–179. [DOI] [PubMed] [Google Scholar]
- 33.Eylar EH, Madoff MA, Brody OV and Oncley JL, J. Biol. Chem, 1962, 237, 1992–2000. [PubMed] [Google Scholar]
- 34.Durocher JR, Payne RC and Conrad ME, Blood, 1975, 45, 11–20. [PubMed] [Google Scholar]
- 35.Kwon S, Davies-Venn C and Sevick-Muraca EM, Neurogastroenterol. Motil, 2012, 24, 494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eguchi S, Takatsuki M, Yamanouchi K, Hidaka M, Soyama A, Tomonaga T, Tajima Y and Kanematsu T, Transplantation, 2009, 88, 747–748. [DOI] [PubMed] [Google Scholar]
- 37.Koren E and Torchilin VP, IUBMB Life, 2011, 63, 586–595. [DOI] [PubMed] [Google Scholar]
- 38.Moghimi SM, Adv. Drug Delivery Rev, 1995, 17, 103–115. [Google Scholar]
- 39.Mebius RE and Kraal G, Nat. Rev. Immunol, 2005, 5, 606–616. [DOI] [PubMed] [Google Scholar]
- 40.Pivkin IV, Peng ZL, Karniadakis GE, Buffet PA, Dao M and Suresh S, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 7804–7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muzykantov VR, Murciano JC, Taylor RP, Atochina EN and Herraez A, Anal. Biochem, 1996, 241, 109–119. [DOI] [PubMed] [Google Scholar]
- 42.Villa CH, Anselmo AC, Mitragotri S and Muzykantov V, Adv. Drug Delivery Rev, 2016, 106, 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagmann M, Science, 2000, 288, 1945–1946. [DOI] [PubMed] [Google Scholar]
- 44.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD and Lindberg FP, Science, 2000, 288, 2051–2054. [DOI] [PubMed] [Google Scholar]
- 45.Mac JT, Nunez V, Burns JM, Guerrero YA, Vullev VI and Anvari B, Biomed. Opt. Express, 2016, 7, 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohanty JG, Nagababu E and Rifkind JM, Front. Physiol, 2014, 5, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J, Lee H and Shin S, J. Cell. Biotechnol, 2015, 1, 63–79. [Google Scholar]
- 48.Nash GB and Meiselman HJ, Biophys. J, 1983, 43, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chien S, Annu. Rev. Physiol, 1987, 49, 177–192. [DOI] [PubMed] [Google Scholar]
- 50.Bratosin D, Mazurier J, Tissier JP, Estaquier J, Huart JJ, Ameisen JC, Aminoff D and Montreuil J, Biochimie, 1998, 80, 173–195. [DOI] [PubMed] [Google Scholar]
- 51.Wisse E, Braet F, Luo DZ, DeZanger R, Jans D, Crabbe E and Vermoesen A, Toxicol. Pathol, 1996, 24, 100–111. [DOI] [PubMed] [Google Scholar]
- 52.Zhang YN, Poon W, Tavares AJ, McGilvray ID and Chan WCW, J. Controlled Release, 2016, 240, 332–348. [DOI] [PubMed] [Google Scholar]
- 53.Yu FB, Jiang TY, Zhang JH, Cheng LH and Wang SL, Pharmazie, 2007, 62, 528–533. [PubMed] [Google Scholar]
- 54.Zhang XM, Zhang Q, Peng Q, Zhou J, Liao LF, Sun X, Zhang L and Gong T, Biomaterials, 2014, 35, 6130–6141. [DOI] [PubMed] [Google Scholar]
- 55.Gao DY, Lin TT, Sung YC, Liu YC, Chiang WH, Chang CC, Liu JY and Chen YC, Biomaterials, 2015, 67, 194–203. [DOI] [PubMed] [Google Scholar]
- 56.Desilets J, Lejeune A, Mercer J and Gicquaud C, Anticancer Res., 2001, 21, 1741–1747. [PubMed] [Google Scholar]
- 57.Patel G, Kher G and Misra A, J. Drug Targeting, 2012, 20, 155–165. [DOI] [PubMed] [Google Scholar]
- 58.Tan JF, Thomas A and Liu YL, Soft Matter, 2012, 8, 1934–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gentile F, Curcio A, Indolfi C, Ferrari M and Decuzzi P, J. Nanobiotechnol, 2008, 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fish MB, Fromen CA, Lopez-Cazares G, Golinski AW, Scott TF, Adili R, Holinstat M and Eniola-Adefeso O, Biomaterials, 2017, 124, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watson CY, Molina RM, Louzada A, Murdaugh KM, Donaghey TC and Brain JD, Int. J. Nanomed, 2015, 10, 4173–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bourrinet P, Bengele HH, Bonnemain B, Dencausse A, Idee JM, Jacobs PM and Lewis JM, Invest. Radiol, 2006, 41, 313–324. [DOI] [PubMed] [Google Scholar]
- 63.Suckow MA, Danneman P and Brayton C, The laboratory mouse, CRC Press, Boca Raton, Fla., 2001. [Google Scholar]
- 64.Serfilippi LM, Pallman DR and Russell B, Contemp. Top. Lab. Anim. Sci, 2003, 42, 46–52. [PubMed] [Google Scholar]
- 65.Marshall MV, Draney D, Sevick-Muraca EM and Olive DM, Mol. Imaging Biol, 2010, 12, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi J, Gilbert GE, Kokubo Y and Ohashi T, Blood, 2001, 98, 1226–1230. [DOI] [PubMed] [Google Scholar]
- 67.Hochreiter-Hufford A and Ravichandran KS, Cold Spring Harbor Perspect. Biol, 2013, 5, a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu N and Zhou Z, Int. Rev. Cell Mol. Biol, 2012, 293, 269–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Semple JW, Italiano JE Jr. and Freedman J, Nat. Rev. Immunol, 2011, 11, 264–274. [DOI] [PubMed] [Google Scholar]
- 70.Lalor PF, Herbert J, Bicknell R and Adams DH, Am. J. Physiol.: Gastrointest. Liver Physiol, 2013, 304, G469–G478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Radomski A, Jurasz P, Alonso-Escolano D, Drews M, Morandi M, Malinski T and Radomski MW, Br. J. Pharmacol, 2009, 146, 882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yun S-H, Sim E-H, Goh R-Y, Park J-I and Han J-Y, BioMed Res. Int, 2016, 2016, 9060143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Connell KE, Mikkola AM, Stepanek AM, Vernet A, Hall CD, Sun CC, Yildirim E, Staropoli JF, Lee JT and Brown DE, Comp. Med, 2015, 65, 96–113. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.