

Abstract
Dipeptidyl peptidase 9 (DPP9) is a direct inhibitor of NLRP1, but how it impacts inflammasome regulation in vivo is not yet established. Here, we report three families with immune-associated defects, poor growth, pancytopenia and skin pigmentation abnormalities that segregate with biallelic DPP9 rare variants. Using patient-derived primary cells and biochemical assays, these variants were shown to behave as hypomorphic or knockout alleles that failed to repress NLRP1. Remarkably, the removal of a single copy of either Nlrp1a/b/c, Asc, Gsdmd, Il-1r, but not Il-18, was sufficient to rescue the lethality of Dpp9 mutant neonates in mice. Similarly, dpp9 deficiency was partially rescued by the inactivation of asc, an obligate downstream adapter of the NLRP1 inflammasome, in zebrafish. These experiments suggest that the deleterious consequences of DPP9 deficiency were mostly driven by the aberrant activation of the canonical NLRP1 inflammasome and IL-1β signaling. Collectively, our results delineate a Mendelian disorder of DPP9 deficiency driven by increased NLRP1 activity as demonstrated in patient cells and in two animal models of the disease.
One Sentence Summary:
Loss of DPP9 activity in humans, mice and zebrafish results in pathogenic NLRP1 overactivation.
INTRODUCTION
The human NLRP1 inflammasome is a multi-protein complex that is triggered by viral infections(1, 2) or danger-associated signals(3), resulting in the release of proinflammatory cytokines, including IL-1 family members, IL-1β and IL-18(4). Dysregulation of the human NLRP1 inflammasome is associated with a range of inflammatory Mendelian diseases such as Multiple Self-healing Palmoplantar Carcinoma (MSPC, MIM615225)(5), Juvenile Recurrent Respiratory Papillomatosis (JRRP, MIM618803)(6), and Autoinflammation with Arthritis and Dyskeratosis (AIADK, MIM617388)(7). In the absence of any cognate triggers, inflammasome sensors in the family of Nod-like receptors (NLRs) are maintained in an inactive state by a number of regulatory proteins. For example, NLRP1 is bound to, and repressed by, the enzymatic activity and scaffolding function of dipeptidyl peptidase 9 (DPP9) and DPP8 (7, 8). Recent CryoEM studies revealed that DPP9 restrains NLRP1 activation by directly binding to the NLRP1-FIIND domain (9, 10).Asingle monomer of a DPP9 dimer is bound to two molecules of NLRP1 of which one exists in the pre-auto-proteolysis conformation, whilst the other is the cleaved UPA-CARD fragment. Thus, DPP9, with the help of a full length uncleaved NLRP1 molecule, sequesters the bio-active C-terminal fragment of NLRP1, preventing inflammasome formation (9, 10). The role of DPP9 as a negative regulator of human or mouse NLRP1 has been established through pharmacological inhibition of DPP9 and inactivation of DPP9 in cell lines in vitro, but its function has not been documented in vivo.
Here we have identified four children displaying immune-associated defects and skin pigmentation abnormalities that harbor loss-of-function mutations in DPP9. Patient keratinocytes displayed spontaneous activation of the NLRP1 inflammasome. We employed a mouse model harboring a mutation in DPP9 that renders the protein catalytically inactive (Dpp9S759A/S759A) to model this condition. Dpp9S759A/S759A mice died within one day of birth through an unknown mechanism. We found that crossing Dpp9S759A/S759A mice to NLRP1 knockout mice (Nlrp1−/−) rescued neonate lethality with incomplete penetrance. The amelioration of phenotype in some mice suggests that a physiologic role of DPP9 is to prevent NLRP1 inflammasome assembly. Additionally, we have shown that the Dpp9S759A/S759A neonatal lethality could also be rescued in some cases by deletion of downstream components of the NLRP1 inflammasome. Similarly, we report that the early lethality of dpp9 knockout zebrafish could also be partly rescued by the concomitant inactivation of asc, an obligate downstream adapter of the NLRP1 inflammasome. Overall, we have shown the importance of DPP9 restraint of the NLRP1 inflammasome activation in vivo, with DPP9 deficiency resulting in NLRP1-driven inflammatory disease in both humans and two animal models.
RESULTS
Recessive loss-of-function DPP9 germline variants found in three families
As part of a cohort of patients with inborn errors of immunity, we studied a child (F2: II.4) with signs of susceptibility to infections consisting of recurrent facial HSV lesions and airway inflammation, including chronic intermittent asthma and primary pulmonary tuberculosis. Exome sequencing disclosed a germline homozygous stop-gained mutation (c.2551C>T; p.Q851*) in DPP9 (MIM608258) (Fig. 1A, B and E) which was heterozygous in the parents. A second proband, a 6-year-old child (F1: II.2) born to unrelated Ashkenazi parents, was identified and shown to carry compound heterozygous DPP9 variants with a paternal missense mutation (c.449G>A, p.G167S) and a maternal early truncating mutation (c.641C>G; p.S214*) (Fig. 1A–C). Through GeneMatcher (11), a third consanguineous Bedouin family from Israel with two similarly-affected cousins (F3: II:11 and III:1) was ascertained. Each propositus was shown to segregate a homozygous germline stop-gained variant (c.331C>T; p.R111*) while available parents and siblings were either heterozygous or non-carrier consistent with Mendelian recessive inheritance. All four affected children shared a triad of symptoms which we termed Hatipoğlu syndrome, the clinical presentation of which is summarized in Table 1. The symptoms associated with this monogenic disease partially overlapped with those of AIADK caused by an NLRP1 gain-of-function mutation (7). These included failure to thrive (consisting of poor weight gain, growing below the 3rd percentile), skin manifestations, pancytopenia and susceptibility to infections, which in the case of F1: II.2, F3: II:11 and III:1 required hematopoietic stem cell transplant. The stop-gain mutations found in these 3 probands from Families 2 and 3 were expected to completely abolish the enzymatic activity of DPP9, since its catalytic domain is situated at the C-terminus. The missense mutation p.G167S found in proband F1: II.2 affected a residue in a flexible loop that was located at the periphery of the DPP9 substrate binding site (Fig. 1D). Thus, the DPP9G167S mutation is predicted to diminish the substrate binding and/or catalytic efficiency. No homozygous damaging variants have been reported for DPP9 which is significantly averse to loss-of-function mutations according to genomAD (12).
Fig. 1: Three families segregating recessive loss-of-function DPP9 germline variants.
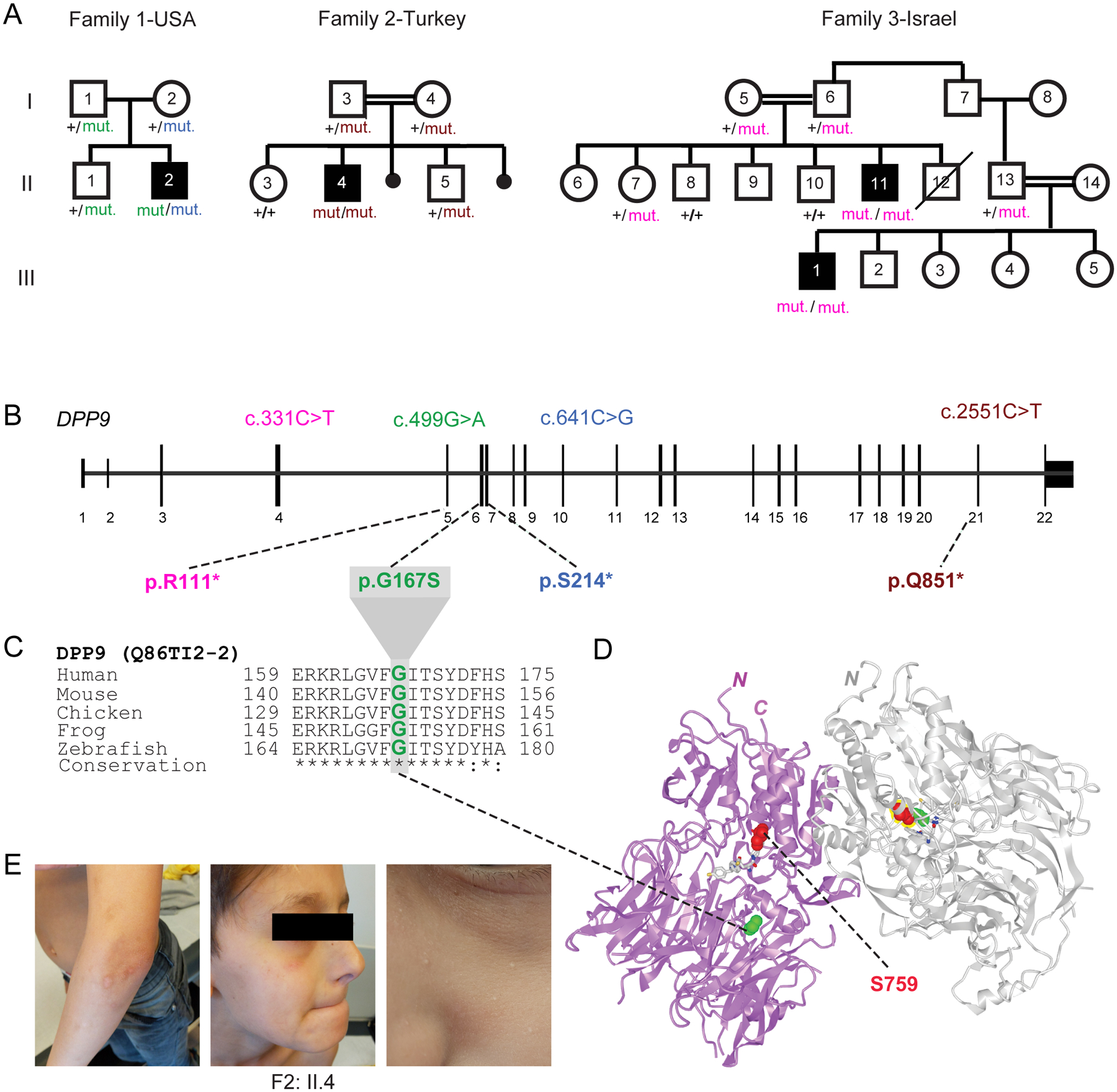
(A) Pedigrees of three families harboring germline DPP9 mutations. Proband from Family 1 (II.2, USA) is compound heterozygous, with paternal p.G167S and maternal p.S214* germline mutations. Proband from Family 2 (II.4, Turkey) carries a homozygous recessive p.Q851* mutation. Affected cousins from Family 3 (II.11 and III:1, Israel) carry a homozygous recessive p.R111* variant.
(B) Exon-intron genomic organization of DPP9 with positions of identified germline mutations.
(C) Phylogenetic conservation of the Gly167 residue, which is mutated in Family 1, across vertebrate species.
(D) Protein structure of a DPP9 homodimer (PDB ID: 6EOR) depicting location of p.G167 on the periphery of the substrate binding site and p.S759 in the catalytic residue.
(E) Photographs of proband from Family 2 (II.4) depicting recurrent HSV facial lesions, milia, and skin pigmentation anomalies on his arms.
Table 1:
Clinical summary of Hatipoğlu syndrome caused by biallelic DPP9 Loss-of-Function mutations
| Clinical synopsis and genetics | HPO Terms | Family 1 | Family 2 | Family 3 | Present in all cases | ||
|---|---|---|---|---|---|---|---|
| Country of origin | USA (Ashkenazi) | Turkey | Israel (Bedouin) | ||||
| Propositus number (refer to pedigrees) | II:2 | II:4 | II:11 | III:1 | |||
| Sex, (year of birth) | Male (2017) | Male (2008) | Male (2011) | Male (2012) | |||
| IUGR, (birth weight in kg) | HP:0001511 | + (2.3) | − (3.6) | − (3.2) | + (2.1) | ||
| Gene | DPP9 | DPP9 | DPP9 | NLRP1 | |||
| Inheritance | HP:0000007 | cpd htz | hmz | hmz | hmz | ✔ | htz |
| cDNA change (NM_139159) | maternal:c.641C>G | c.2551C>T | c.331C>T | ||||
| paternal:c.499G>A | |||||||
| Expected protein change (Q86TI2–2) | maternal: p.S214* | p.Q851* | p.R111* | p.P1214R | |||
| paternal: p.G167S | |||||||
| Observed protein change (Q86TI2–2) | maternal: p.0 | p.0 | n.d. | ||||
| paternal: p.G167S | |||||||
| Immune defects | |||||||
| Infantile eczema | HP:0000964 | + (after BM transplantation) | + | − | − | ||
| Recurrent fevers | HP:0032323 | + | bimonthly | + | + | ✔ | ✔ |
| Asthma | HP:0002099 | − | + | − | + | ||
| Repeated infections | HP:0002719 | + | + | + | + | ✔ | ✔ |
| Herpes infections | HP:0005353 | + | + (facial) | + | + | ✔ | |
| Bronchitis | HP:0012387 | + | + | − | + | ||
| Prone to acute otitis media | HP:0000403 | + | + | + | − | ||
| Pancytopenia | HP:0001876 | + | + | + | + | ✔ | |
| Anemia | HP:0001903 | + | + | + | + | ✔ | ✔ |
| Selective immunodeficiency | HP:0002721 | + | + | n.d. | n.d. | ||
| Severe allergies | HP:0012393 | + (sesame) | + | − | − | ||
| Behavioural/Neurological traits | |||||||
| Learning Disability | HP:0001328 | + | + | − | + | ||
| Speech delay | HP:0000750 | + | + | − | + | ||
| Slurred speech | HP:0001350 | + | + | − | − | ||
| Autism spectrum disorder | HP:0000729 | + | − | − | + | ||
| Skin findings and its appendages | |||||||
| Thick skin | HP:0001072 | + | − | − | − | ||
| Anhidrosis | HP:0000970 | − | + | − | − | ||
| Dry skin | HP:0000958 | − | + | − | − | ||
| Atopic dermatitis | HP:0001047 | + | + | − | − | ||
| Petechiae | HP:0000967 | + | + | + | + | ✔ | |
| Fair hair | HP:0002286 | + | + | − | − | ||
| Premature hair graying | HP:0002216 | − | + | − | − | ||
| Hypo- and hyper-pigmented macules | HP:0007441 | + | + | + | − | ||
| Delayed wound healing | HP:0001058 | − | + | − | − | ||
| Craniofacial dysmorphisms | |||||||
| Downslanted palpebral fissures | HP:0000494 | + | + | − | − | ||
| Broad forehead | HP:0002373 | + | − | − | − | ||
| Bone / skeletal abnormalities | |||||||
| Hemivertebrae | HP:0002937 | n.d. | n.d. | + | + | ||
| Other clinical manifestations | |||||||
| Proportionate short stature | HP:0003508 | + | + | + (−2.1 SD) | + (−1.8 SD) | ✔ | ✔ |
| Failure to thrive | HP:0001508 | + | + | + | + | ✔ | ✔ |
| Febrile seizures | HP:0002373 | + | − | − | − | ||
| Cryptorchidism / hypospadias | HP:0000028 | + | − | − | − | ||
| Inguinal hernia | HP:0000776 | + | − | − | + | ||
| Poor feeding | HP:0011968 | + | + | − | − | ||
n.d., not determined; +, affirmative; −, negative; BM, bone marrow; cpd, compound: hmz, homozygous; htz, heterozygous
Patients’ cells lack DPP9 or produce a strong enzymatic hypomorph
To investigate the impact of the DPP9 variants on mRNA expression and enzymatic function, primary skin cells were isolated from affected patients of families 1 and 2 (Fig. 2A). When compared with control primary fibroblasts, and those derived from the heterozygous parents of family 2 (F2: I.3 and I.4), a decrease in DPP9 protein was found in the proband’s (F1: II.2) fibroblasts. This was consistent with one allele not encoding the full-length protein due to an early stop mutation p.S214* (Fig. 2A, lanes 1 and 2). Notably, we did not observe any endogenous DPP9 protein in the proband of family 2 (F2: II.4), indicating that the stop-gain mutant (p.Q851*) was degraded via the nonsense-mediated decay (NMD) pathway (Fig. 2A, lanes 1 and 5). Thus, the p.Q851* DPP9 variant yielded a loss-of-expression, resulting in a protein-null allele. While we do not have access to patient cells from Family 3, we anticipate that the p.R111* allele, may also result in a whole body DPP9 knockout due to NMD of the mutant transcript. We next sought to assess the enzymatic activity of the DPP9G167S variant towards a model substrate Gly-Pro-AMC (Fig. 2B and Fig. S1A). The p.G167S mutation was found to reduce the enzymatic activity to ~15% of that of wild-type DPP9.
Fig. 2: Patients’ cells lack DPP9 or produce a strong enzymatic hypomorph.
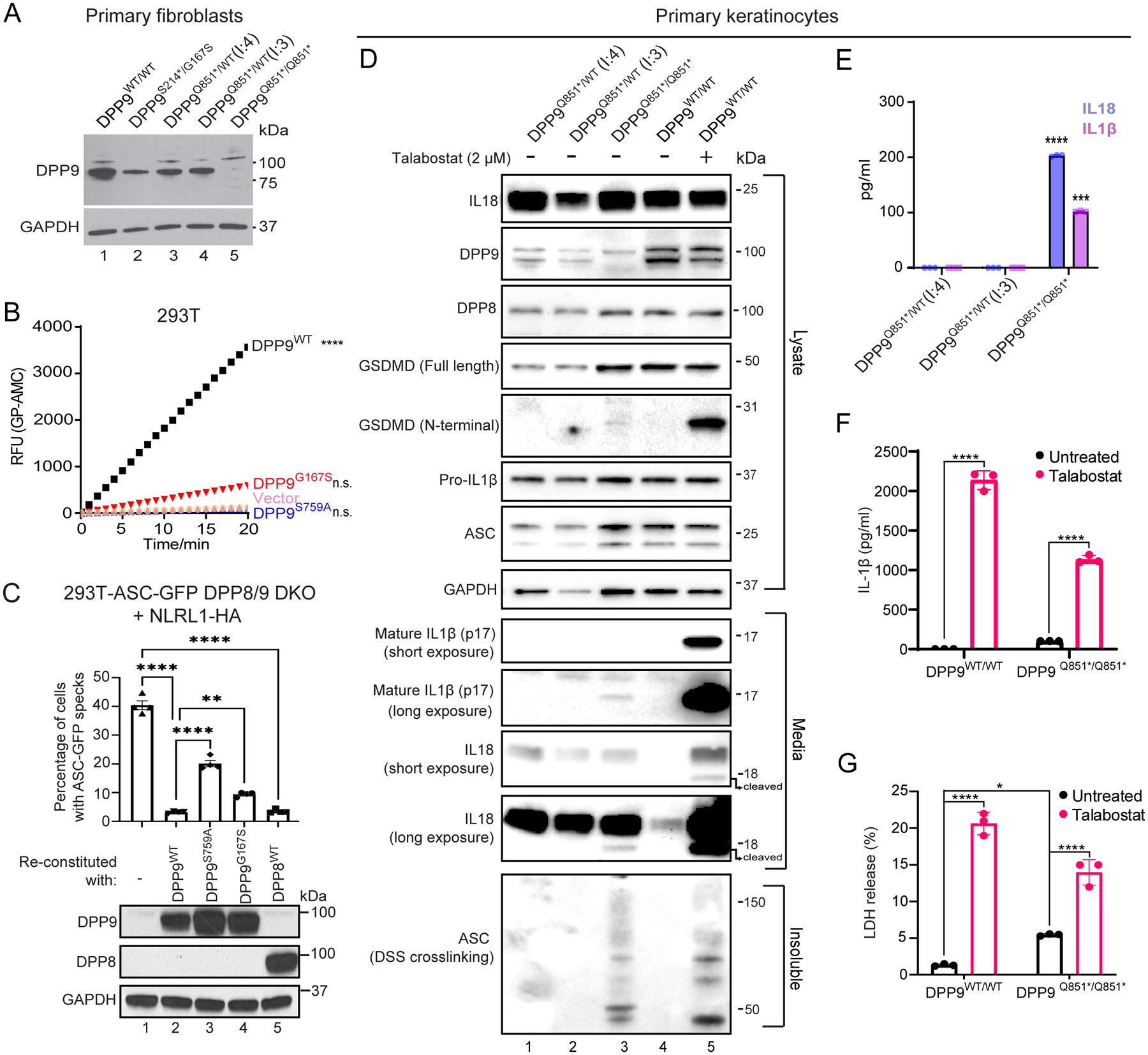
(A) Western blot analysis of DPP9 in primary dermal fibroblasts of an unrelated control (WT/WT) (lane 1), affected individual from family 1 (proband (II:2) S214*/G167S) (lane 2), two unaffected individuals from family 2 ((I:4) and (I:3) Q851*/WT) (lanes 3 and 4 respectively) and affected individual from family 2 (proband (II:4) Q851*/Q851*) (lane 5).
(B) 293T cells were transfected with either vector, wild-type DPP9, DPP9 S759A or DPP9 G167S and lysed in PBS 1% Tween 20, 48 h after transfection. 0.3 μg of total lysate was then incubated with Gly-Pro-AMC fluorescence substrate. AMC fluorescence was measured for 20 mins at 25 °C in a 50 μl reaction every minute on a spectrometer. One-way ANOVA. ****P<0.0001 calculated by comparing the vector control to wild-type DPP9, DPP9S759A or DPP9G167S (n = 3 independent replicates).
(C). Top, quantification of ASC-GFP speck formation in DPP8/9 DKO 293T-ASC-GFP cells reconstituted with WT DPP9, DPP9 S759A, DPP9 G167S or WT DPP8, and transfected with NLRP1. The percentage of cells with ASC-GFP specks were counted in three different fields after fixation. More than 100 cells were scored per condition. Data are mean ± SEM, n = 4 independent replicates. **p < 0.01; ****p < 0.0001. Bottom, western blot analysis validating DPP9 and DPP8 expression.
(D) Western blot analysis of inflammasome components in primary keratinocytes of two unaffected individuals from family 2 ((I:4) and (I:3) Q851*/WT) and the affected individual from family 2 (proband (II:4) Q851*/Q851*). Additionally an unrelated control (+/+), treated with or without the potent DPP8/9 inhibitor Talabostat (2 μM) was included as a positive control for NLRP1 inflammasome activation.
(E) IL1β and IL18 ELISA of the culture supernatant from primary keratinocytes from the two unaffected individuals from family 2 ((I:4) and (I:3) Q851*/WT) and affected individual from family 2 (proband (II:4) Q851*/Q851*). ****p < 0.001, ***p < 0.01 (one-way ANOVA). (n=3 replicates).
(F), (G) Levels of IL-1β (F) and lactate dehydrogenase (LDH; G) released from DPP9WT/WT control and DPP9Q851*/Q851* patient-derived primary keratinocytes in absence or presence of Talabostat. Data are mean ± SEM, n = 3 independent replicates. *p < 0.05; ****p < 0.0001.
Patients’ cells with DPP9 deficiency display NLRP1 inflammasome activation
Given that DPP9 is a specific interacting partner and inhibitor of human and mouse NLRP1(7, 8), we measured the ability of DPP9WT, catalytically-dead DPP9S759A or the patient DPP9G167S mutant to repress human NLRP1 when re-expressed in DPP8/DPP9 double KO 293T-ASC-GFP reporter cells (Fig. 2C, lanes 1 – 4, Fig. S1B and C). Exogenous DPP9WT repressed NLRP1-dependent ASC-GFP speck formation. In contrast, both DPP9S759A and DPP9G167S was less effective in inhibiting ASC speck formation, despite both variants retaining the ability to bind NLRP1 (Fig. S1D). This agrees with previous findings that both the catalytic activity of DPP9 as well as its scaffolding function, within the NLRP1:DPP9 complex, is important for inflammasome activation (10, 13). Taken together, these experiments suggest that these DPP9 patient variants are less able to maintain NLRP1 in the inactive state.
We and others have demonstrated that NLRP1 is the most prominent inflammasome sensor in human keratinocytes and that these cells readily undergo pyroptotic cell death in response to NLRP1 activation (5, 14). Using primary keratinocytes isolated from family 2 and the Luminex multiplex assay, we found that the cytokine and chemokine profile of the proband differed from those of his heterozygous parents (Fig. S1E). We demonstrated that the proband keratinocytes, as do keratinocytes from patients with gain-of-function NLRP1 mutations (5), spontaneously secreted active IL-1β and IL-18 into the media without any exogenous stimulus. Despite measurable IL-1 release visualized by western blot and ELISA (Fig. 2D and E respectively), the patient’s keratinocytes were viable in culture and could be serially cultured (Fig. S1F) for at least 4 passages. This cytokine release was also accompanied by endogenous ASC oligomerization, as demonstrated using a DSS crosslinking assay, and low levels of GSDMD cleavage (Fig. 2D). Of note, the amount of IL-1 and intact lactate dehydrogenase (LDH) released from patient keratinocytes were not as profound as that from WT or patient keratinocytes treated with Val-boro-Pro (Talabostat), a small molecule inhibitor of both DPP9 and its homologue, DPP8 (Fig. 2D, lanes 1 – 3 and 5, Fig. 2F and G). This could be accounted for by intact DPP8 expression in patient keratinocytes playing a partially redundant role with DPP9 to inhibit NLRP1 (Fig. 2D). Indeed, genetic loss of DPP9 alone in immortalized N/TERT keratinocytes was not sufficient to elicit IL-1β release at comparable levels to Val-boro-Pro treatment (Fig. S1G), while re-constitution of DDP8/DPP9-dKO 293T-ASC-GFP reporter cells with DPP8 alone was sufficient to repress NLRP1-induced ASC-GFP specks (Fig. 2C, lanes 1 and 5). These data indicate that loss of DPP9 function in patient-derived cells could indeed be partially compensated by DPP8, accounting for their modest yet significant IL-1β release and cell death.
NLRP1 deficiency rescues lethality of Dpp9S759A/S759A mice
To dissect the pathogenicity of the loss of DPP9 in vivo, we took advantage of the DPP9 mutant mouse line which carries the catalytic-dead DPP9 p.S759A mutation. Homozygous Dpp9S759A/S759A knock-in mice die within 24 hours of birth(15, 16). Based on our results obtained in patient-derived primary cells, we examined whether this lethality might be associated with overactivation of the NLRP1 inflammasome. Indeed, homozygous loss of all Nlrp1 alleles (Nlrp1a/b/c, herein referred to as Nlrp1) protected some Dpp9S759A/S759A mice from neonatal lethality. Furthermore, deletion of even a single Nlrp1 allele was also sufficient to generate viable animals which proved to be fertile (Fig. 3A and Fig. S2). Dpp9S759A/S759ANlrp1−/− mice were runted (small and lean) however without any overt signs of disease compared to Dpp9S759A/+Nlrp1−/− siblings (Fig. 3A and B). Short stature of Dpp9S759A/S759ANlrp1−/− was associated with decreased hip width but relatively conserved limb width (Fig. 3C and D).
Fig. 3: Deficiency of NLRP1 and downstream signaling molecules rescues neonatal lethality of Dpp9S759A/S759A mice.
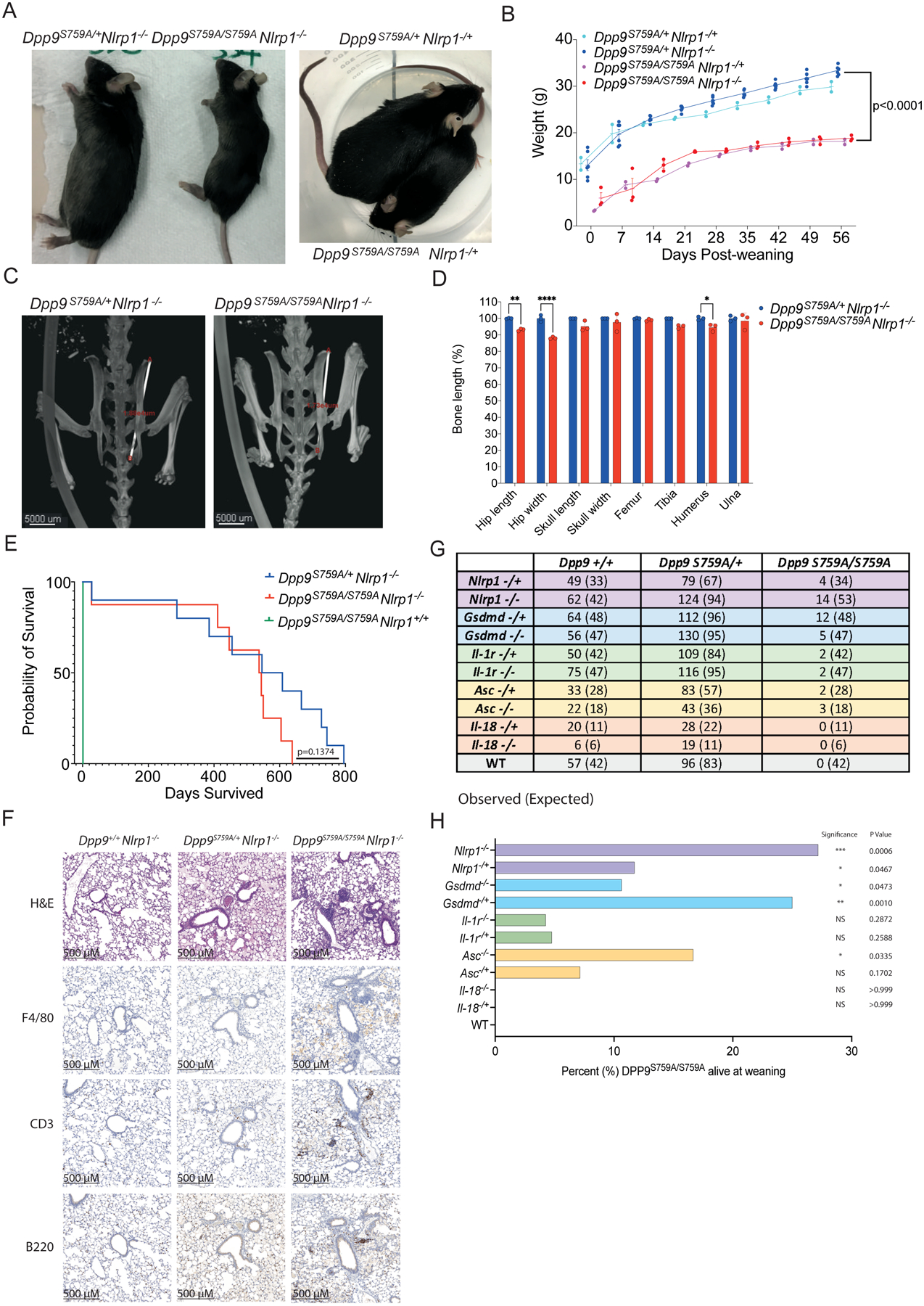
(A) Viable, 6-month-old male Dpp9S759A/S759A Nlrp1−/− and Dpp9S759A/S759A Nlrp1−/+ mice are runted compared to respective Dpp9S759A/+ Nlrp1−/− or Dpp9S759A/+ Nlrp1−/+ littermates.
(B) Male Dpp9S759A/S759A Nlrp1−/− and Dpp9S759A/+ Nlrp1−/− mice were weighed weekly for 8 weeks post weaning. n=2–6 mice per genotype, error bars represent mean ±SD. Statistical significance was calculated based on the Dpp9S759A/S759A Nlrp1−/− and Dpp9S759A/+Nlrp1−/− mice using a paired t-test.
(C) Example images of skeletal measurements and (D) quantification of 6-month-old Dpp9S759A/S759A Nlrp1−/− and Dpp9S759A/+ Nlrp1−/− mice imaged via MicroCT scanning. Measurements are normalised to the average Dpp9S759A/+ Nlrp1−/− values of mice of the same sex. n=3 mice per genotype, both male and female. Error bars represent mean ±SEM. *p<0.05, **p<0.005, ****p<0.0001 (2way ANOVA)
(E) Kaplan Meier survival curve of Dpp9S759A/S759A Nlrp1−/− and Dpp9S759A/+Nlrp1−/− mice, starting at weaning age. n=8–13 per genotype, both male and female. Statistical significance was determined by a Mantel-Cox test.
(F) Representative section of lung tissue from 6-month-old Dpp9S759A/S759A Nlrp1−/− and Dpp9S759A/+ Nlrp1−/− mice stained with Hematoxylin and eosin or immunohistochemistry of F4/80, B220 or CD3 antibodies. n=3 mice per genotype, both male and female.
(G) The number of viable mice at weaning age (~3 weeks old) of various genotypes were genotyped and tallied, this is the ‘observed’ number depicted on the left of each cell. Mendelian ratios were used to calculate ‘Expected’ number of mice per genotype, written in brackets on the right of each cell.
(H) Graphical representation of the percentage of Dpp9S759A/S759A mice surviving to weaning for each genetic cross. Statistical analysis was performed using Fisher’s exact test.
At birth (day 0), Dpp9S759A/S759A mice appeared normal and were suckling, as evidenced by a milk spot (Fig. S3A) and were present at normal Mendelian ratios (Fig. S3B). As both DPP9 deficient patients displayed pancytopenia, we used flow cytometry to characterize immune cell populations in the blood of Dpp9S759A/S759A mutant mice sufficient, heterozygous or deficient for NLRP1, along with relevant controls. Numbers of CD11b+ Ly6C+, CD11b+ Ly6G+, CD11b+ NK1.1+, CD11c+, CD3+ CD4+, CD3+ CD8+ and CD19+ cells in the bone marrow were comparable between all genotypes assessed (Fig. S3C and D, gating strategy Fig 4 3). This is consistent with viable fetal liver chimeras that have previously been generated from this line(17, 18). Similarly, numbers of the same immune cell populations collected from peripheral blood were also comparable between all genotypes accessed (Fig. S3E and F, gating strategy Fig S4). Thus, we do not observe significant haematological abnormalities similar to those observed in patients with germline loss of DPP9 activity.
Fig. 4: Deficiency of asc extends lifespan and abrogates pro-inflammatory il-1b upregulation in dpp9−/− zebrafish.
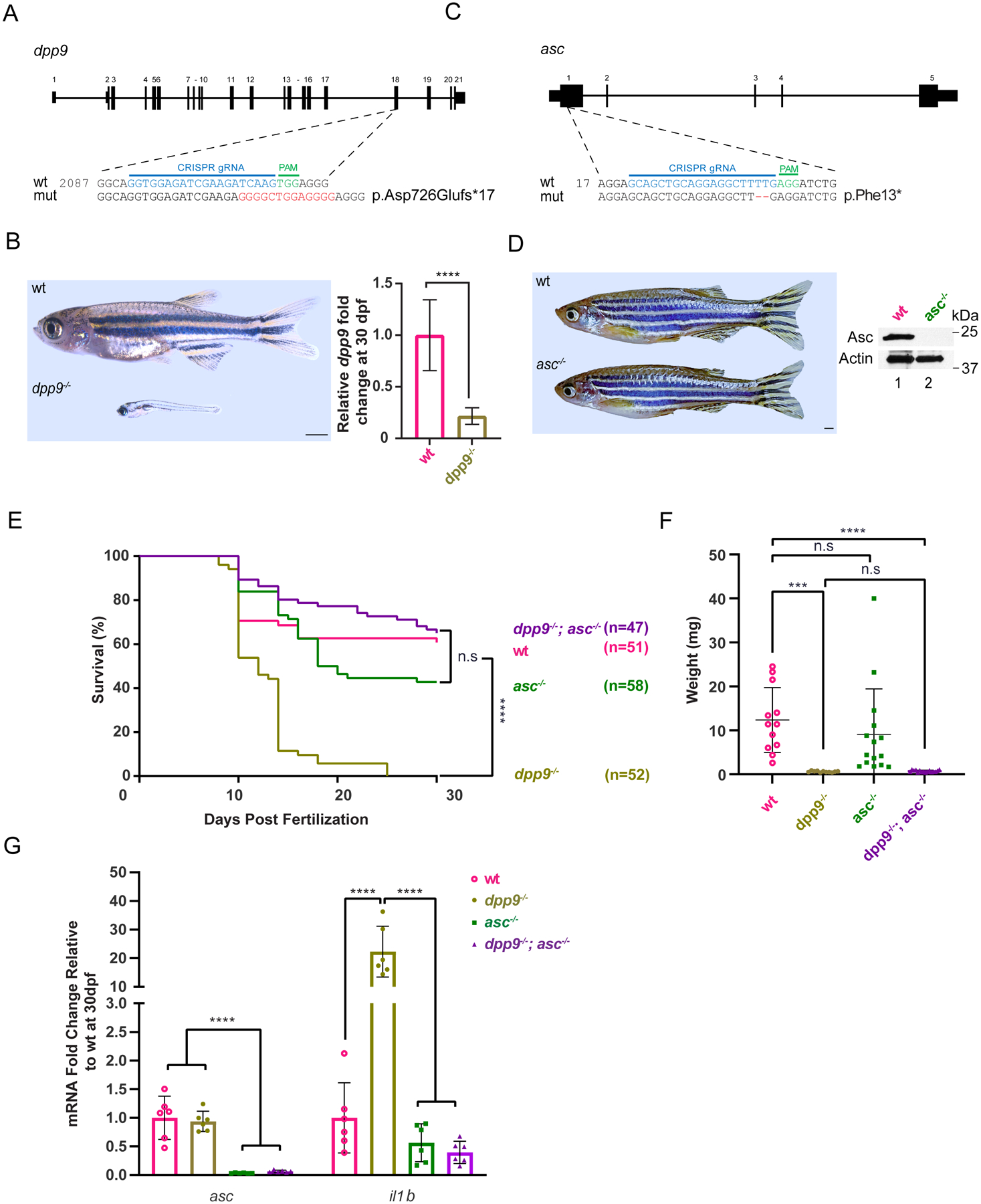
(A) Generation of dpp9 knockout zebrafish using CRISPR/Cas9-genome editing that resulted in an out-of-frame allele with premature stop codon. (B) Left, dpp9−/− zebrafish at 1 month of age exhibit a dramatic reduction in size (quantification shown in F). Right, validation of dpp9-KO by RT-qPCR. Data are mean ± SEM (n = 6). ****p<0.001 (one-way ANOVA).
(C) Generation of asc knockout zebrafish using CRISPR/Cas9 genome editing that resulted in a 2-bp deletion in exon 1 followed by a premature stop codon.
(D) Left, asc−/− zebrafish appeared indistinguishable from wt. Right, validation of complete asc-KO by western blotting.
(E) Kaplan-Meier survival curves of wt, dpp9−/−, asc−/− and double dpp9−/−; asc−/− zebrafish. Dpp9−/− zebrafish demonstrated a significant survival disadvantage that is rescued by inactivating asc. ****p<0.0001 (Logrank Mantel-Cox test).
(F) The weight of double dpp9−/−; asc−/− zebrafish is not rescued compared to that of dpp9−/− zebrafish. Data are mean ± SEM. ****p<0.001; ***p<0.01; n.s., non-significant (one-way ANOVA).
(G) In double dpp9−/−; asc−/− zebrafish, il1b expression is rescued to that of wt levels as demonstrated by RT-qPCR analysis. Data are mean ± SEM (n = 6). ****p<0.001 (one-way ANOVA).
In order to determine NLRP1-independent effects of DPP9 loss of function we further interrogated the Dpp9S759A/S759A Nlrp1−/− line. Aside from being runted, Dpp9S759A/S759A Nlrp1−/− mice had a trending but not significant (p=0.1374) decrease in life expectancy (Fig. 3E) compared to control lines. Dpp9S759A/S759A Nlrp1−/− mice also displayed immune infiltrate in their lungs, particularly in areas surrounding the bronchioles. The immune cell infiltrate contained foamy macrophages (F4/80+), B cells (B220+) and T cells (CD3+) at 6 months of age and in aged mice, (Fig. 3F, Fig. S5A). Histological analysis of other organs and automated blood cell analysis showed no overt differences between genotypes (Fig. S5-G). Overall, these results suggest inflammasome-independent roles of DPP9 relating to stature and lung inflammation, which is consistent with the broad range of processes regulated by DPP9. Decreased stature and lung inflammation were also recorded in DPP9-deficient patients (Table 1). Notably, two GWAS studies have implicated germline DPP9 variants in the development of pulmonary fibrosis(19, 20) and severe COVID-19 pathology(21). Therefore, lethality caused by loss of DPP9 function in mice can be partially rescued by the deletion of NLRP1, however independent pathology including runting and lung inflammation remain.
ASC, GSDMD, IL-1R, but not IL-18, -deficiency also rescues lethality of Dpp9S759A/S759A mice
To further dissect the contribution of downstream components of the NLRP1 signaling axis, we next crossed Dpp9S759A/S759A mice with strains deficient for effectors of the NLRP1 inflammasome. Both heterozygous and homozygous loss of Asc, Gsdmd and Il-1r could rescue Dpp9S759A/S759A neonatal lethality with incomplete penetrance (Fig. 3G and H). Interestingly, the biallelic loss of Il-18 did not rescue DPP9 loss of function (Fig. 3G and H) suggesting that Il-1 may play a more important role in disease. Rescued mice exhibited a similar phenotype to Dpp9S759A/S759A Nlrp1−/− mice, appearing healthy yet runted. For example, Dpp9S759A/S759A Gsdmd−/− mice did not gain weight at the same rate as controls, and developed the same immune cell infiltrate in their lungs as Dpp9S759A/S759A Nlrp1−/− mice at 6 months of age, despite normal blood cell counts (Fig. S6). Notably, the number of rescued Dpp9S759A/S759A mice that survived to weaning age fell significantly below Mendelian ratios (Fig. 3G). This was also true at 2 days of age, suggesting that a significant number of mice suffer from NLRP1 inflammasome independent disease causing events, which in some circumstances lead to death (Fig. S3G). Overall, the perinatal lethality of DPP9 can be rescued in a subset of mice, by the concomitant deletion of inflammasome components and IL-1 signaling, but not IL-18.
Asc inactivation also partially rescues lethality of Dpp9 knockout zebrafish
The rapid evolution of the NLRP1 gene across vertebrate species suggests selective pressure and adaptation in different phyla. For example, felines have lost NLRP1 homologues, while ancestral vertebrates, including zebrafish, express nlrp1, which plays a similar role as mammalian NLRP1 in canonical inflammasome activation characterized by recruitment of ASC, activation of proinflammatory caspases and IL-1β maturation (23, 24). In contrast to humans (expressing DPP8 and CARD8) and mice (expressing DPP8 but not CARD8), zebrafish does not possess DPP8 nor CARD8, making it a simpler vertebrate model to address the role of DPP9 in NLRP1 inflammasome regulation.
To that end, we generated dpp9--KO and asc-KO zebrafish using CRISPR/Cas9 (Fig. 4A and C). Similar to Dpp9S759A/S759A mice, homozygous dpp9-KO zebrafish also displayed a drastic reduction in body size and a significantly shorter lifespan that did not last more than 35 days (Fig. 4B and E). In contrast, asc−/− zebrafish were viable till adulthood and were indistinguishable from WT in appearance (Fig. 4D). To examine whether dpp9−/− lethality was associated with downstream NLRP1-inflammasome activation, we generated dpp9−/−; asc−/− double-KO zebrafish to prevent inflammasome assembly. Indeed, homozygous loss of asc sufficed to rescue Dpp9−/− lethality (Fig. 4E) but without ameliorating their diminished body size (Fig. 4F). Further, the absence of endogenous ASC restored the elevated IL-1β transcript level seen in dpp9--KO larvae (Fig. 4G). Of note, Dpp9S759A/S759ANlrp1−/− mice also exhibited a decreased stature despite being able to survive past birth. It is likely that DPP9 plays other roles independent of inflammasome regulation.
DISCUSSION
Here, we identified three unrelated families with Hatipoğlu syndrome, an autosomal recessive DPP9 deficiency, which is characterized by immune-associated defects and dermatological anomalies. This Mendelian disease bears some degree of resemblance with auto-immune disorders caused by NLRP1 activating mutations. In particular, the AIADK-causing NLRP1 p.P1214R mutation, which entirely abrogates DPP9 binding and inhibition of NLRP1, is expected to yield comparable phenotypes(7). Both diseases appear to manifest with recurrent episodes of fevers, auto-immune anemia and a general failure to thrive. The milder skin manifestations seen in DPP9 patients may be explained by the presence of active DPP8 which we have shown can compensate for DPP9 deficiency in human cells.
Using biochemical and cellular assays, we demonstrated that patient-derived DPP9 alleles had reduced catalytic activity and were defective in suppressing the NLRP1 inflammasome in vitro. These results suggest that the patients’ symptoms are at least in part due to the aberrant activation of the NLRP1 inflammasome. This notion was validated by in vivo experiments: the neonatal lethality of Dpp9 mutant mice was rescued in some cases by the deletion of Nlrp1 or downstream inflammasome signaling components Asc, Gsdmd and Il-1r, while early lethality of dpp9-KO zebrafish was also rescued by loss of Asc. However, the incomplete penetrance of this rescue makes it clear that there are NLRP1 independent roles for DPP9. Nevertheless, given that the deletion of a single allele of Il-1r was sufficient to yield an almost normal adult Dpp9S759A/S759A mouse, it raises the possibility that even partial inhibition of the NLRP1-inflammasome pathway, with IL-1Ra (anakinra) for example, would provide some therapeutic benefit for DPP9 deficient patients. In support of this, anti-IL-1β therapy was partially successful in a patient harboring a germline NLRP1 p.P1214R mutation which abrogated binding and therefore inhibition by DPP9(22).
It is notable that even at 2 days of age, there was incomplete penetrance for the rescue of Dpp9S759A/S759A neonatal lethality by deletion of NLRP1 inflammasome components. As DPP9 has been reported to be involved in multiple cellular processes, it is possible that an NLRP1 independent consequence of DPP9 loss-of-function is also contributing to neonatal lethality. As Dpp9S759A/S759A mice were born at Mendelian ratios and so only die after birth (15) we speculate that a process occurring at or soon after birth is triggering death in Dpp9S759A/S759A mice, where deficiency of Nlrp1, Asc, Gsdmd and IL-1r was sufficient for survival in some cases. This period of time is, of course, when neonates are first exposed to a wide variety of microbes integral in shaping their eventual microbiomes. This could be a critical event driving neonatal lethality in Dpp9S759A/S759A mice, perhaps associated with TLR priming of pro IL-1β expression which a constitutively active NLRP1 inflammasome would then process. However, it remains uncertain whether inflammasome activation is the main driver of death in Dpp9S759A/S759A mice. We do not know if Dpp9S759A/S759A driven activation of NLRP1 is sufficient to induce disease in the absence of other underlying issues related to the loss of DPP9 function. An alternative hypothesis is that an NLRP1 independent function of DPP9 is driving lethality and that reducing inflammasome signaling lowers the burden of disease enough that a subset of Dpp9S759A/S759A mice survive.
The neonatal lethality seen in Dpp9S759A/S759A mice was a more severe phenotype than that seen in mice with a gain-of-function mutation in Nlrp1a, which suffer from a systemic inflammatory disease that is lethal around 3–6 months of age (25). Furthermore, that disease did not require the inflammasome adaptor ASC, nor could be rescued by heterozygous deletion of Caspase-1 or IL-1R, unlike the deficiency of DPP9. However, disease in the Nlrp1aQ593P/Q593P mice was found to be driven by IL-1, but not IL-18, mirroring what we see in Dpp9S759A/S759A mice. Therefore, the underlying inflammatory pathway downstream of Nlrp1 is broadly similar in both cases, despite a wide divergence in phenotype, which could instead reflect cell and tissue expression profiles of Nlrp1a, Nlrp1b and Dpp9.
Human DPP9 has many cellular substrates and also represses an orthologous inflammasome sensor, CARD8, which is absent in both rodents and zebrafish(13, 26–29). These findings could account for the broad spectrum of symptoms in patients with DPP9 mutations, some of which might not be modeled in mice nor in zebrafish. Consequently, we cannot rule out the involvement of CARD8 in the pathogenesis of the DPP9-deficient patient disease. However, since NLRP1, but not CARD8, is responsible for pyroptosis induction upon Talabostat treatment in human keratinocytes, we speculate that at least in the skin of DPP9-deficient patients, NLRP1 plays a prominent role in DPP9-driven disease. A limitation of our study is that we do not have access to other patient-derived cell types, such as T cells, in which CARD8, but not NLRP1, is specifically activated by Talabostat (28). Overall, our results delineate a heretofore unknown Mendelian disease caused by germline DPP9 deficiency with pathology driven in part by aberrant activation of the NLRP1 inflammasome.
MATERIALS AND METHODS
Study Design
The aim of this study was to explore the in vivo consequences of DPP9 regulation of the NLRP1 inflammasome. Four patients displaying skin pigment abnormalities, poor growth, immune-associated defects and pancytopenia were identified to harbor biallelic DPP9 variants. We used patient-derived primary keratinocytes as well as biochemical assays to show the patient mutations result in a loss-of-function. Additionally, we modeled DPP9 loss in a mouse and zebrafish model to study the underlying cause of disease.
Human study participants
The study included 4 affected children from 3 distinct families. All are affected with recessive germline DPP9 mutations. Families ethnologically originated from the USA (Family 1), Turkey (Family 2) and Israel (Family 3) (Fig. 1A and Table 1). The study protocol was approved by A*STAR IRB (2019-087) and genetic analyses were performed in accordance with bioethics rules of national laws of The Rockefeller University Hospital, New York, USA; Stanford University School of Medicine, Stanford, California, USA; Necker-Enfants Malades Hospital, Paris, France, the Rabin Medical School, Petah Tikva, Israel, and Boston Children’s Hospital, Boston, MA. Informed consent was obtained for all patient and healthy control volunteers reported in the study.
DNA extraction, quantification, and quality control
Genomic DNA of the affected individuals and other available family members was extracted from either whole blood using a variety of extraction protocols. DNA concentration and quality were assessed using NanoDrop (Thermo Scientific) and Qubit (Life technologies) fluorometers. A260/A280 ratios of 1.8 to 2.0 and A260/A230 ratios >1.5 were accepted. DNA fragmentation was assessed using agarose gel (0.8%) electrophoresis.
Exome sequencing
Exome sequencing was employed independently for the detection of variants in Families 1, 2 and 3.
Family 1 - Trio exome sequencing was performed using DNA extracted from peripheral blood samples at the Baylor Medical Genetics Laboratories according to published methods(30, 31). Briefly, capture was performed with VCRome 2.1 in-solution exome probes, as well as additional probes for over 3,600 Mendelian-disease-related genes. Library DNA was sequenced on an Illumina HiSeq for 100 bp paired-end reads. Data analysis was performed with Mercury 1.0. Genes possibly associated with the patient’s phenotype under a recessive mode of inheritance were reported in DPP9, MS4A2, MXRA5 and PAPLN.
Family 2 - Genomic DNA (gDNA) was prepared from blood samples from patient II:1 by the standard phenol-chloroform extraction method. WES was performed with the SureSelect Human All Exon 71 Mb kit (Agilent Technologies). Paired-end sequencing was performed on a HiSeq 2500 machine (Illumina) generating 100-base reads. We aligned the sequences with the GRCh37 reference build of the human genome with the Burrows-Wheeler aligner(32). Downstream processing and variant calling were performed with the Genome Analysis Toolkit(33), SAMtools(34). Substitution and InDel calls were made with the GATK Unified Genotyper. All variants were annotated with annotation software developed in-house(35–38). Blacklisting variants common in private cohorts but not in public databases optimizes human exome analysis(38). Human Gene Mutation Database for any given gene(39). Based on the high consanguinity in this patient (1.66%), exome sequencing was analysed assuming autosomal recessive inheritance by keeping all homozygous non-synonymous coding variants and essential splice site variants with a MAF <10−2 in different ethnic subpopulations.
Family 3 - Whole exome sequencing of DNA from blood and data processing were performed by the Genomics Platform at the Broad Institute of MIT and Harvard with Twist exome capture (~38 Mb target) and sequenced (150 bp paired reads) to cover >85% of targets at 20x and a mean target coverage of >60x. Exome sequencing data was processed through a pipeline based on Picard and mapping done using the BWA aligner to the human genome build 38. Variants were called using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.5. Default filters were applied to SNV and indel calls using the GATK Variant Quality Score Recalibration (VQSR) approach. Annotation was performed using Variant Effect Predictor (VEP). Lastly, the variant call set was uploaded to seqr for collaborative analysis with the Broad Institute Center for Mendelian Genomics.
Isolation of human fibroblasts and keratinocytes
Primary human fibroblasts and keratinocytes from Family 2 proband and two unaffected parental controls were isolated from fresh skin biopsies. Briefly, biopsies were incubated in dispase (2.4 U/ml) overnight at 4°C to enable peeling of the epidermis from the dermal compartment by gentle scraping with a sterile scalpel. Keratinocytes from the epidermis were then isolated using mechanical disruption by pulling epidermis apart with sterile forceps and resuspending it vigorously in keratinocyte culture medium (DMEM High glucose supplemented with L-glutamine, pen/strep, 22% F12, 10% FBS, transferrin [5 mg/ml], adenine [9.72 mg/ml], insulin [5 mg/ml], liothyronine [2 × 10–6 M], hydrocortisone [200 μg/ml] and epidermal growth factor [10 μg/ml] followed by filtering through a 100 μm filter to remove collagen fragments and centrifugation at 1000 × g for 5 min. Pelleted keratinocytes were then grown on a 3t3 feeder layer. Fibroblasts were isolated by finely chopping up the dermis and incubating it overnight in collagenase D. The collagenase was then removed the next day and fibroblasts were allowed to migrate out of the dermal fragments.
Cell culture
All 293T (ATCC) and derivatives were cultured in DMEM supplemented with 10% FBS and confirmed to be mycoplasma-free. Any control primary fibroblasts or keratinocytes used as WT controls were obtained from the Skin Research Institute of Singapore (SRIS) Skin Cell Bank with informed consent and prior IRB approval.
SDS PAGE western blot, ELISA and LDH release
Protein lysate was quantified using Bradford assay and 20 ug of protein loaded unless stated otherwise. Western blotting was carried out by incubating the primary antibodies overnight in 3% Milk in TBST. The following primary antibodies were used: DPP9 (1:5000, abcam, #ab42080), DPP8 (400ng/ml, abcam, #ab96470), IL-18 (300 ng/ml, abcam, #ab207323), IL-1β (1:1000, R&D, #AB-201-NA), cleaved IL-1β (Asp116) (D3A3Z) (100 ng/ml, CST, #83186), GSDMD (400 ng/ml, Novus, #NB2P-33422), ASC (200 ng/ml, Adipogen, #AL177), HA (100 ng/ml, CST, #2367) FLAG (100 ng/ml, Santa Cruz Biotechnology, #SC-5288), NLRP1 (200 ng/ml, R&D, #AF6788) and GAPDH (1:1000, Santa Cruz Biotechnology, #sc47724).
IL-1β ELISA (BD, #557953), IL-18 ELISA (MBL Bioscience, #7620) and LDH release assay (Promega CytoTox96, #G1780) were carried out according to manufacturer’s instructions.
ASC speck assay
Images of ASC-GFP specks were acquired in 3 random fields in 4’,6-diaminidino-2-phenylindole (DAPI, 358 nm/461) and GFP (469 nm/ 525 nm) channels using the EVOS microscope (FL Auto M5000, #AMF5000) according to the manufacturer’s protocol. Quantification method of ASC-GFP specks was previously described in detail(1).
Luminex multiplex microbead-based immunoassay
Conditioned supernatants were collected for Luminex analysis using the ProcartaPlex, Human Customized 65-plex Panel (Thermo Fisher Scientific,) to measure the following targets: APRIL; BAFF; BLC; CD30; CD40L; ENA-78; Eotaxin; Eotaxin-2; Eotaxin-3; FGF-2; Fractalkine; G-CSF; GM-CSF; Gro a; HGF; IFN-a; IFN-g; IL-10; IL-12p70; IL-13; IL-15; IL-16; IL-17 A; IL-18; IL-1 A; IL-1 b; IL-2; IL-20; IL-21; IL-22; IL-23; IL-27; IL-2 R; IL-3; IL-31; IL-4; IL-5; IL-6; IL-7; IL-8; IL-9; IP-10; I-TAC; LIF; MCP-1; MCP-2; MCP-3; M-CSF; MDC; MIF; MIG; MIP1a; MIP-1 b; MIP-3 A; MMP-1; NGF beta; SCF; SDF-1 A; TNF b; TNF-a; TNF-R2; TRAIL; TSLP; TWEAK; VEGF-A.
Harvested supernatants and standards were incubated with fluorescent-coded magnetic beads pre-coated with respective antibodies in a black 96-well clear-bottom plate overnight at 4°C. After incubation, plates were washed 5 times with wash buffer (PBS with 1% BSA (Capricorn Scientific) and 0.05% Tween-20 (Promega)). Sample-antibody-bead complexes were incubated with Biotinylated detection antibodies for 1 hour and washed 5 times with wash buffer. Subsequently, Streptavidin-PE was added and incubated for another 30 mins. Plates were washed 5 times again, before sample-antibody-bead complexes were re-suspended in sheath fluid for acquisition on the FLEXMAP® 3D (Luminex) using xPONENT® 4.0 (Luminex) software. Data analysis was done on Bio-Plex Manager™ 6.1.1 (Bio-Rad). Standard curves were generated with a 5-PL (5-parameter logistic) algorithm, reporting values for both mean fluorescence intensity (MFI) and concentration data. Hierarchical clustering and generation of heatmap was performed using pheatmap package on RStudio 1.3.1093.
Mice
Dpp9S759A/S759A mice were provided by Prof Mark Gorrell (Centenary Institute). Nlrp1−/− (25), Gsdmd−/− (40), Ilr-1−/− (41), Asc−/− (42) and Il-18−/− (43) were generated on, or backcrossed at least 10 generations onto the C57BL/6 background. All experiments use littermate controls, however multiple litters have been used in each experiment to generate sufficient numbers. To generate survival data, mice that were Dpp9S759A/+ on either a heterozygous or homozygous Nlrp1, Gsdmd, Ilr-1, Asc or Il-18 knockout background were set up in either single breeding pairs or trios (1 male and 2 females), offspring were genotyped at either weaning age (3 weeks old) or P2–3. Statistical analysis of survival data at weaning was performed using Fisher’s exact test. All animal experiments complied with the regulatory standards of, and were approved by, the Walter and Eliza Hall Institute Animal Ethics Committee.
Histology
Organs were collected in 10% neutral buffered formalin. Organ sections were prepared from paraffin blocks and stained with hematoxylin and eosin. Immunohistochemistry was performed using antibodies against F4/80 (in house), B220 (in house) or CD3 (Agilent #A045229). Immunohistochemistry of representative areas was quantified manually, based on size, shape and pigmentmentation.
Micro-CT
For Micro-CT scans mice were euthanised and scanned on a Bruker Skyskan 1276 Micro-CT. Images were analysed using Imaris software to measure bone lengths.
Hematology
Automated cell counts were performed on blood collected from the sub-mandibular vein into Microtainer tubes containing EDTA (Sarstedt), using an Advia2120i hematological analyser (Siemens, Munich, Germany).
FACs analysis of newborn pup blood and bone marrow
Pups were euthanised 3–6 hours after birth via decapitation and blood collected from the neck into Microtainer tubes containing EDTA (Sarstedt). Limbs and sternum were collected, bones were crushed and passed through a 100 uM filter to obtain a single cell suspension. 1×106 cells of the bone suspension and 15 uL of blood were plated per sample before red cell depletion using RCR Buffer (156 mM NH4Cl, 11.9 mM NaHCO3, 0.097 mM EDTA). Cells were washed in PBS and incubated with rat anti-mouse CD16/CD23 (1:200 BioLegend #101302) at 4°C for 10 minutes to block Fc receptors. Cells were washed and stained for 1 hour at 4°C with fluorescently tagged antibodies: MHCII-FITC (1:200 in house), CD11c-PE (1:200 BioLegend #117307), CD4-PE-Cy7 (1:200 BioLegend #100528), CD3-PE-CF594 (1:200 BD Bioscience #562286), Ly6G-APC (1:200 in house), CD19-AF700 (1:200 BioLegend #15528), NK1.1-BV450 (1:400 BioLegend #108731), Zombie aqua viability Dye (1:500 Biolegend #77143), CD11b-BV605 (1:400 Biolegend 101257), CD8-BV650 (1:400 BioLegend #100742), Ly6C-BV711 (1:400 Biolegend #128037), CD45-BV786 (1:200 BioLegend #103149). Cells were washed and fixed with BD Phosflow Lyse/Fix buffer (BD Bioscience) for 20 min at RT and then stored at 4°C. Flow cytometry was performed on the BD LSRFortessa X-20 (BD Bioscience) and data acquired using BD FACSDiva (BD Bioscience) software. Data was analysed using FloJo (Tree Star). All cell populations shown were first gated to exclude small cellular debris and doublets, then gated on live cells (zombie negative) and CD45 positive, before gating on markers as shown in figure, representative gating strategy is shown in Figure S3.
Zebrafish strains and maintenance
All zebrafish husbandry procedures were performed in compliance with the Singapore National Advisory Committee on Laboratory Animal Research Guidelines from the Institutional Animal Care and Use Committee (IACUC, IACUC number 201514). The dpp9−/− and asc−/− mutant lines used for this study were maintained in the AB background. All embryos were raised in egg water (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4).
CRISPR/Cas9-mediated Zebrafish Knockout
A custom gBLOCK (Integrated DNA Technologies) was designed incorporating a guide RNA-targeting sequence preceded by a T7 promoter sequence. Guide RNAs against exon 18 of dpp9 and exon 1 of asc were used with the following sequences: 5′-GGTGGAGATCGAAGATCAAG-3′ and 5′-GCAGCTGCAGGAGGCTTTTG-3′ respectively. Guide RNAs were synthesized using MEGA shortscript™ Kit (Applied Biosystems), according to the manufacturer’s protocol, and were purified using RNeasy Mini Kit (Qiagen, #74106). Cas9 mRNA was synthesized using mMESSAGE mMACHINE® SP6 Transcription Kit (Thermo Fisher Scientific, #AM1340). Two nL of a mixture containing 250 ng/μL gRNA and 0.1 mg/mL Cas9 protein was injected into the yolk of 1-cell AB zebrafish embryos.
Live genotyping of zebrafish larvae
Zebrafish larvae were genotyped at 3–4 days post fertilization as described previously (44). In brief, zebrafish larvae were incubated in DNA collection buffer (10mM Tris-HCl pH 8.0, 0.6mM Tricaine, 25μg/ml Proteinase K) at 37 °C, 200 rpm for 20 min to shed skin tissues. DNA collection buffer was then collected and mixed with lysis buffer (10 mM Tris-HCl pH 8.0, 50 mM KCl, 0.3% Tween 20, 0.3% NP40, 1 mM EDTA), and was lysed at 98°C for 5 min to obtain genomic DNA. Zebrafish larvae were then placed in recovery solution (5 mM NaCl, 6.8 mM KCl, 0.3 mM CaCl2, 0.5 mM MgSO4) for 2 days and were subsequently transferred to egg water according to their genotype.
RT-qPCR analysis of zebrafish larvae
Total RNA was extracted from single zebrafish larvae at 3 weeks post-fertilization using the RNeasy Micro Kit (Qiagen #74004) following the manufacturer’s protocol, including the optional DNase RNase-free treatment. Prior to RNA extraction, embryos were first disrupted using a syringe and a 23G needle, and then homogenized using QIAshredder (Qiagen #79654). 200 ng of the extracted RNA were converted to cDNA using the Iscript Reverse Transcription Supermix (Bio-Rad #1708840) according to the manufacturer’s instructions. RT-qPCR reactions were carried out using the Power SYBR Green PCR Master Mix (Applied Biosystems #4309155) and specific primers for the target genes (below) on the 7900HT Fast Real-Time PCR System (Applied Biosystems). RT-qPCR assays involved six embryos per genotype and three technical replicates per sample (N = 6, n = 3). gapdh was used as the housekeeping gene to normalize gene expression.
il-1β Fwd: ATGGCGAACGTCATCCAAGA
il-1β Rev: GAGACCCGCTGATCTCCTTG
asc Fwd: ATGGCGGAATCTTTCAAGGAGC
asc Rev: TGAACACGCCGACCATTAAATC
dpp9 Fwd: CATCGCAAGAGCTGGGTGGA
dpp9 Rev: GGTATGAAGAGCGCCGGTGG
gapdh Fwd: GTGGAGTCTACTGGTGTCTTC
gapdh Rev: GTGCAGGAGGCATTGCTTACA
Statistics
Statistical analyses were performed using GraphPad Prism 9.0 (Graphpad Software). Differences between two groups were evaluated using a paired t test (parametric) or an unpaired t test (parametric) depending on experimental design. Where differences between- multiple groups were evaluated by a One-way or two-way ANOVA. Differences in long-term survival between groups was evaluated by a Mantel-Cox test. Differences with a p value <0.05 were considered significant.
Supplementary Material
Acknowledgments:
We are grateful to all members of the Masters, Zhong and Reversade laboratories for support. We thank Prof. Anthony Oro (Stanford University) for generous help in fibroblast derivation from family 1. We thank E. Kravets for study coordination and M.W. Allain for data collection (Stanford University). We thank S. Russo and Edan Azzopardi for outstanding animal husbandry. We thank Dr. Anne O’Donnell-Luria and Dr. Stephanie DeTroia (Center for Mendelian Genomics at the Broad Institute of MIT and Harvard) for discussion of the genomics analysis of Family 3.
Funding:
S.L.M acknowledges funding from NHMRC Project Grants 2003159, 2003756, the Victorian Endowment for Science Knowledge and Innovation Fellowship, the HHMI-Wellcome International Research Scholarship and the Sylvia and Charles Viertel Foundation Fellowship. F.L.Z. acknowledges funding from the National Research Foundation (NRF, Singapore): Fellowship. K.L. acknowledges funding from National Medical Research Council (NMRC, Singapore) Open Fund-Young Individual Research Grant (OF-YIRG, MOH-000328-00). J-L.C acknowledges funding from the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) of the National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program (UL1TR001866), the French National Research Agency (ANR) under the “Investments for the Future” ANR program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the “PNEUMOPID” project (Grant ANR 14-CE15-0009-01), the French Foundation for Medical Research (FRM) (EQU201903007798), the Howard Hughes Medical Institute, the The Rockefeller University, St. Giles Foundation, the Institut National de la Santé et de la Recherche Médicale (INSERM) and Université de Paris. S.D acknowledges funding from the Australian National Health and Medical Research Council (NHMRC) grants GNT1143412 and GNT2003756. C-H.Y acknowledges funding from the Walter and Eliza Hall Institute of Medical Research (WEHI) Centenary Fellowship and Ormond College’s Thwaites Gutch Fellowship in Physiology. J.G. acknowledges funding from the National Institutes of Health, National Institute of (A.S. and M.D.F.); National Institutes of Health (Grant R01HG009141) DAAD Care-For-Rare Fellowship. B.R. acknowledges funding from National Research Foundation (NRF, Singapore): Investigatorship, the Branco Weiss Foundation (Switzerland) Fellowship, the European Molecular Biology Organization (EMBO) Young Investigatorship and Agency for Science and the Technology and Research (A*STAR, Singapore): Use-Inspired Basic Research (UIBR) Fund and A*STAR Investigatorship.
Footnotes
Competing interests:
S.L.M. is a Scientific Advisor for IFM Therapeutics and NRG Therapeutics.
Data and material availability
All data needed to evaluate the conclusions in the paper are present in the paper or the supplementary material.
References and Notes
- 1.Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, Lim CK, Lay K, Au BV, Lew TS, Chu JJH, Chow VTK, Wang DY, Zhong FL, Reversade B, Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science. 370 (2020), doi: 10.1126/science.aay2002. [DOI] [PubMed] [Google Scholar]
- 2.Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V, Human NLRP1 is a sensor for double-stranded RNA. Science (2020), doi: 10.1126/science.abd0811. [DOI] [PubMed] [Google Scholar]
- 3.Fenini G, Grossi S, Contassot E, Biedermann T, Reichmann E, French LE, Beer H-D, Genome Editing of Human Primary Keratinocytes by CRISPR/Cas9 Reveals an Essential Role of the NLRP1 Inflammasome in UVB Sensing. J. Invest. Dermatol 138, 2644–2652 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Broz P, Dixit VM, Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, Szeverényi I, Takeichi T, Balaji R, Lau A, Tye H, Roy K, Bonnard C, Ahl PJ, Jones LA, Baker PJ, Lacina L, Otsuka A, Fournie PR, Malecaze F, Lane EB, Akiyama M, Kabashima K, Connolly JE, Masters SL, Soler VJ, Omar SS, McGrath JA, Nedelcu R, Gribaa M, Denguezli M, Saad A, Hiller S, Reversade B, Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell. 167, 187–202.e17 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Drutman SB, Haerynck F, Zhong FL, Hum D, Hernandez NJ, Belkaya S, Rapaport F, de Jong SJ, Creytens D, Tavernier SJ, Bonte K, De Schepper S, van der Werff Ten Bosch J, Lorenzo-Diaz L, Wullaert A, Bossuyt X, Orth G, Bonagura VR, Béziat V, Abel L, Jouanguy E, Reversade B, Casanova J-L, Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc. Natl. Acad. Sci. U. S. A 116, 19055–19063 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong FL, Robinson K, Teo DET, Tan K-Y, Lim C, Harapas CR, Yu C-H, Xie WH, Sobota RM, Au VB, Hopkins R, D’Osualdo A, Reed JC, Connolly JE, Masters SL, Reversade B, Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem 293, 18864–18878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH, Sanford DG, Arciprete MO, Golub TR, Bachovchin WW, Bachovchin DA, DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol 13, 46–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, Paulo JA, Gygi SP, Bachovchin DA, Wu H, DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature. 592, 778–783 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang M, Zhang X, Toh GA, Gong Q, Wang J, Han Z, Wu B, Zhong F, Chai J, Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature. 592, 773–777 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobreira N, Schiettecatte F, Valle D, Hamosh A, GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat 36, 928–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG, The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griswold AR, Ball DP, Bhattacharjee A, Chui AJ, Rao SD, Taabazuing CY, Bachovchin DA, DPP9’s Enzymatic Activity and Not Its Binding to CARD8 Inhibits Inflammasome Activation. ACS Chemical Biology. 14 (2019), pp. 2424–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sand J, Haertel E, Biedermann T, Contassot E, Reichmann E, French LE, Werner S, Beer H-D, Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis. 9, 24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gall MG, Chen Y, Vieira de Ribeiro AJ, Zhang H, Bailey CG, Spielman DS, Yu DMT, Gorrell MD, Targeted inactivation of dipeptidyl peptidase 9 enzymatic activity causes mouse neonate lethality. PLoS One. 8, e78378 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M, Minoux M, Piaia A, Kueng B, Gapp B, Weber D, Haller C, Barbieri S, Namoto K, Lorenz T, Wirsching J, Bassilana F, Dietrich W, Rijli FM, Ksiazek I, DPP9 enzyme activity controls survival of mouse migratory tongue muscle progenitors and its absence leads to neonatal lethality due to suckling defect. Dev. Biol 431, 297–308 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Kim M, von Muenchow L, Le Meur T, Kueng B, Gapp B, Weber D, Dietrich W, Kovarik J, Rolink AG, Ksiazek I, DPP9 enzymatic activity in hematopoietic cells is dispensable for mouse hematopoiesis. Immunol. Lett 198, 60–65 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Gall MG, Zhang HE, Lee Q, Jolly CJ, McCaughan GW, Cook A, Roediger B, Gorrell MD, Immune regeneration in irradiated mice is not impaired by the absence of DPP9 enzymatic activity. Sci. Rep 9, 7292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, Collard HR, Wolters PJ, Bradford WZ, Kossen K, Seiwert SD, du Bois RM, Garcia CK, Devine MS, Gudmundsson G, Isaksson HJ, Kaminski N, Zhang Y, Gibson KF, Lancaster LH, Cogan JD, Mason WR, Maher TM, Molyneaux PL, Wells AU, Moffatt MF, Selman M, Pardo A, Kim DS, Crapo JD, Make BJ, Regan EA, Walek DS, Daniel JJ, Kamatani Y, Zelenika D, Smith K, McKean D, Pedersen BS, Talbert J, Kidd RN, Markin CR, Beckman KB, Lathrop M, Schwarz MI, Schwartz DA, Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet 45, 613–620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen RJ, Guillen-Guio B, Oldham JM, Ma S-F, Dressen A, Paynton ML, Kraven LM, ‘en Obeidat M, Li X, Ng M, Braybrooke R, Molina-Molina M, Hobbs BD, Putman RK, Sakornsakolpat P, Booth HL, Fahy WA, Hart SP, Hill MR, Hirani N, Hubbard RB, McAnulty RJ, Millar AB, Navaratnam V, Oballa E, Parfrey H, Saini G, Whyte MKB, Zhang Y, Kaminski N, Adegunsoye A, Strek ME, Neighbors M, Sheng XR, Gudmundsson G, Gudnason V, Hatabu H, Lederer DJ, Manichaikul A, Newell JD Jr, O’Connor GT, Ortega VE, Xu H, Fingerlin TE, Bossé Y, Hao K, Joubert P, Nickle DC, Sin DD, Timens W, Furniss D, Morris AP, Zondervan KT, Hall IP, Sayers I, Tobin MD, Maher TM, Cho MH, Hunninghake GM, Schwartz DA, Yaspan BL, Molyneaux PL, Flores C, Noth I, Jenkins RG, Wain LV, Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 201, 564–574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, Walker S, Parkinson N, Fourman MH, Russell CD, Furniss J, Richmond A, Gountouna E, Wrobel N, Harrison D, Wang B, Wu Y, Meynert A, Griffiths F, Oosthuyzen W, Kousathanas A, Moutsianas L, Yang Z, Zhai R, Zheng C, Grimes G, Beale R, Millar J, Shih B, Keating S, Zechner M, Haley C, Porteous DJ, Hayward C, Yang J, Knight J, Summers C, Shankar-Hari M, Klenerman P, Turtle L, Ho A, Moore SC, Hinds C, Horby P, Nichol A, Maslove D, Ling L, McAuley D, Montgomery H, Walsh T, Pereira A, Renieri A, GenOMICC Investigators, ISARICC Investigators, COVID-19 Human Genetics Initiative, 23andMe Investigators, BRACOVID Investigators, Gen-COVID Investigators, Shen X, Ponting CP, Fawkes A, Tenesa A, Caulfield M, Scott R, Rowan K, Murphy L, Openshaw PJM, Semple MG, Law A, Vitart V, Wilson JF, Baillie JK, Genetic mechanisms of critical illness in Covid-19. Nature (2020), doi: 10.1038/s41586-020-03065-y. [DOI] [Google Scholar]
- 22.Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, Frouin E, Seyger M, Girard M, Puechberty J, Costes V, Rodière M, Carbasse A, Jeziorski E, Portales P, Sarrabay G, Mondain M, Jorgensen C, Apparailly F, Hoppenreijs E, Touitou I, Geneviève D, A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis 76, 1191–1198 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Cui H, Zhang L, Key Components of Inflammasome and Pyroptosis Pathways Are Deficient in Canines and Felines, Possibly Affecting Their Response to SARS-CoV-2 Infection. Front. Immunol 11, 592622 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J-Y, Gao K, Shao T, Fan D-D, Hu C-B, Sun C-C, Dong W-R, Lin A-F, Xiang L-X, Shao J-Z, Characterization of an NLRP1 Inflammasome from Zebrafish Reveals a Unique Sequential Activation Mechanism Underlying Inflammatory Caspases in Ancient Vertebrates. J. Immunol 201, 1946–1966 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ, Cengia LH, Henley KJ, Collinge JE, Kastner DL, Feigenbaum L, Hilton DJ, Alexander WS, Kile BT, Croker BA, NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 37, 1009–1023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Justa-Schuch D, Silva-Garcia M, Pilla E, Engelke M, Kilisch M, Lenz C, Möller U, Nakamura F, Urlaub H, Geiss-Friedlander R, DPP9 is a novel component of the N-end rule pathway targeting the tyrosine kinase Syk. Elife. 5 (2016), doi: 10.7554/eLife.16370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griswold AR, Cifani P, Rao SD, Axelrod AJ, Miele MM, Hendrickson RC, Kentsis A, Bachovchin DA, A Chemical Strategy for Protease Substrate Profiling. Cell Chem Biol. 26, 901–907.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson DC, Okondo MC, Orth EL, Rao SD, Huang H-C, Ball DP, Bachovchin DA, DPP8/9 inhibitors activate the CARD8 inflammasome in resting lymphocytes. Cell Death Dis. 11, 628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A, Bachovchin DA, DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med 24, 1151–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM, Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 312, 1870–1879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM, Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med 369, 1502–1511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Durbin R, Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR, A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J, A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng PC, Henikoff S, Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maffucci P, Bigio B, Rapaport F, Cobat A, Borghesi A, Lopez M, Patin E, Bolze A, Shang L, Bendavid M, Scott EM, Stenson PD, Cunningham-Rundles C, Cooper DN, Gleeson JG, Fellay J, Quintana-Murci L, Casanova J-L, Abel L, Boisson B, Itan Y, Blacklisting variants common in private cohorts but not in public databases optimizes human exome analysis. Proc. Natl. Acad. Sci. U. S. A 116, 950–959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, Cooper DN, Quintana-Murci L, Zhang S-Y, Abel L, Casanova J-L, The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods 13, 109–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, Bartfai T, Solorzano C, Moldawer LL, Chizzonite R, McIntyre KW, Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J. Immunol 159, 2452–2461 (1997). [PubMed] [Google Scholar]
- 42.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM, Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 430, 213–218 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S, Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 8, 383–390 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Zhang Z, Zhao Q, Lou X, Rapid and Efficient Live Zebrafish Embryo Genotyping. Zebrafish. 17, 56–58 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the supplementary material.