

SUMMARY
The molecular diversity of glia in the human hippocampus and their temporal dynamics over the lifespan remain largely unknown. Here we performed single-nucleus RNA sequencing to generate a transcriptome atlas of the human hippocampus across the postnatal lifespan. Detailed analyses of astrocytes, oligodendrocyte lineages, and microglia identified subpopulations with distinct molecular signatures and revealed their association with specific physiological functions, age-dependent changes in abundance, and disease relevance. We further characterized spatiotemporal heterogeneity of GFAP-enriched astrocyte subpopulations in the hippocampal formation using immunohistology. Leveraging glia subpopulation classifications as a reference map, we revealed the diversity of glial differentiated from human pluripotent stem cells, and identified dysregulated genes and pathological processes in specific glia subpopulations in Alzheimer’s disease (AD). Together, our study significantly extends our understanding of human glia diversity, population dynamics across the postnatal lifespan, and dysregulation in AD, and provides a reference atlas for stem cell-based glia differentiation.
ETOC
Su et al. performed single-nucleus RNA-sequencing analysis to map diversity, molecular properties, disease relevance, and age-dependent dynamics of glia subpopulations in the human hippocampus across the postnatal lifespan. The resulting glia reference atlas serves to assess human stem cell-based glia differentiation and transcriptomic dysregulation in brain disorders, such as Alzheimer’s.
INTRODUCTION
Glial cells, including neuroectoderm-derived astrocytes and oligodendrocytes, and hematopoietic lineage-derived microglia, comprise at least half of the cells in the adult human brain, and they play important roles in the nervous system and brain disorders (Barres, 2008). Compared to rodents, much less is known about glia diversity in humans and key questions regarding their molecular properties, functions, disease relevance, and dynamic changes across the lifespan remain unclear. In the human brain, astrogenesis and oligodendrogenesis peak during childhood, oligodendrocyte maturation and myelination persists throughout life (Zhu et al., 2018), and properties of microglia are shaped dynamically by the local environment (Bennett and Bennett, 2020). Therefore, comprehensive analysis throughout the postnatal lifespan is required to obtain a holistic view of molecular features and diversity of glial cells in the human brain.
The hippocampus supports many higher-level functions, such as memory, mood regulation, and spatial navigation (Small et al., 2011), which requires glia modulation (Bergles et al., 2000; Lee et al., 2021; Paolicelli et al., 2011). The molecular characterization of hippocampal glia remains elusive in rodents and much is less known in humans (Eroglu and Barres, 2010). The hippocampus has also long been implicated in many neuropsychiatric disorders in a spatiotemporally dependent manner, including epilepsy (EPI) and autism spectrum disorder (ASD) during childhood and adolescence, schizophrenia (SCZ) during young adulthood, Alzheimer’s disease (AD) during aging, and major depressive disorder (MDD) across ages (Small et al., 2011). Emerging studies have linked disease vulnerability to neuronal dysregulation, but much less is known about specific glia (sub)populations (Eroglu and Barres, 2010).
Recent large-scale transcriptomic profiling of human post-mortem brain specimens by single-nucleus RNA-sequencing (snRNA-seq) has revealed remarkable molecular diversity, often using the cortex as a model system. However, most human snRNA-seq studies focused on prenatal development, neuronal diversity in adults, and dysregulation in brain disorders (Zeng, 2022). Of the few studies profiling the human hippocampus, most focused on prenatal neural development (Zhong et al., 2020), neuronal diversity (Ayhan et al., 2021; Davila-Velderrain et al., 2021; Franjic et al., 2022; Habib et al., 2017; Tran et al., 2021; Zhou et al., 2022), and vascular properties (Sun et al., 2022; Yang et al., 2022) in adults, whereas glia are largely under-analyzed. Notably, almost all these studies examined only one age cohort. A lack of systematic characterization of glia diversity comparing their molecular properties and cellular abundance across ages poses a major gap in knowledge linking brain cell types to functions and disease traits.
Human pluripotent stem cell (hPSC)-derived 2D neural cultures and 3D brain organoids allow modeling of human brain development and disorders (Qian et al., 2019). Many recent protocols aim to model later developmental stages or etiologies and pathophysiology of adult-onset neuropsychiatric disorders (Zhang et al., 2021). In contrast to neurons, glia subpopulations are less clearly defined due to a lack of a standardized reference map. Such knowledge gaps in this rapidly evolving field preclude the assessment of how hPSC-based glia differentiation corresponds to in vivo glia subtypes and periods of brain development, maturation, and aging.
AD is a progressive neurodegenerative disorder with poorly understood etiology due to its complex pathophysiology (Scheltens et al., 2021). While bulk-level genomic measurements are likely affected by the averaging of gene expression among cell types, snRNA-seq analyses deconvolute cell type-specific pathology in the human AD cortex, including excitatory neuron vulnerability, weakened vascular cells and immune responses, aberrant oligodendrocyte cell lineage and myelination machinery, dysregulation of GFAP+ astrocytes, and microglia disease susceptibility (Saura et al., 2022). However, transcriptomic dysregulation of glia subpopulations in the hippocampus, a major site of pathology (Zakzanis et al., 2003), is unknown.
Here we performed snRNA-seq analyses of the post-mortem human hippocampi from infant, child, adolescent, adult, and aging stages to reveal the complete transcriptional landscapes of glia diversity (Figure 1A; Table S1). We identified molecular signatures, quantified cellular abundance of glia subpopulations across the postnatal lifespan, and validated several key findings in independent sets of human hippocampal specimens across ages (Table S1) using immunohistology and in situ analyses. Furthermore, we provide two examples of how our comprehensive temporal transcriptomic atlas of glia subpopulations can be utilized as a reference map to enhance future studies, first for assessing hPSC glia differentiation in multiple published studies, and second for identifying hippocampal glia dysregulation in AD.
Figure 1. snRNA-seq profiling of the postnatal human hippocampus across ages.

(A) A schematic of experimental design. HIPP: hippocampus; QC: quality control; Adoles.: adolescent.
(B-C) Uniform Manifold Approximation and Projection (UMAP) of integrated data of cross-age analysis, colored by cell type (B). Cell clusters identified by known marker genes, depicted in violin plots in (C). OPC: oligodendrocyte precursor cells.
(D) Heatmap showing transcriptomic correspondence of major cell types between published datasets of various brain regions and ours using a random forest classifier (Shekhar et al., 2016). PFC: prefrontal cortex; ACC: anterior cingulate cortex.
RESULTS
snRNA-seq profiling of human hippocampus across postnatal lifespan
To survey the complete transcriptional landscapes of the postnatal human hippocampus across ages, we generated profiles of 224,464 nuclei, detecting 1,083 genes with 1,893 UMIs per nucleus on average, of post-mortem neurotypical hippocampus from 32 subjects across infant (0–1 years, 47,139 nuclei), child (2–6 years, 40,721 nuclei), adolescent (13–18 years, 37,361 nuclei), adult (27–71 years, 56,857 nuclei), and aging (85–95 years, 42,386 nuclei) stages (Figure 1A; Table S1). We integrated all datasets (Hao et al., 2021) and identified ten major cell clusters based on established markers, including glutamatergic neurons, oligodendrocytes, astrocytes, oligodendrocyte progenitor cells (OPCs), GABAergic neurons, and microglia, as well as less abundant (< 1%) cell populations, including endothelial cells, ependymal cells, choroid plexus cells, and Cajal-Retzius cells (Figures 1B, 1C and S1A-S1D; Table S2A). The overall glia-to-neuron ratio in the hippocampus, 51: 47 (Figure S1C), agrees with a 1:1 ratio measured by isotropic nucleus fractionator counting across human brain regions (von Bartheld et al., 2016).
To provide a high-level comparison of cell-type transcriptomic divergence across brain regions and ages, we matched several published snRNA-seq datasets of various human brain areas, which only focused on one or two age stages (Jakel et al., 2019; Lake et al., 2018; Schirmer et al., 2019; Velmeshev et al., 2019), to the corresponding stage(s) of our hippocampal dataset. Analysis using a random forest classifier (Shekhar et al., 2016), trained with our age-matched hippocampal dataset, shows high similarity among all glial cell types across brain regions, whereas the similarity among neurons is much lower (Figure 1D). Thus, our systematic analysis of human hippocampal glia across ages may suggest common glia features across human brain regions.
Astrocyte transcriptomic diversity in postnatal human hippocampus
Astrocytes can be further classified based on their gene expression, location, morphology, function, and disease contribution (Ben Haim and Rowitch, 2017). To gain insight into their molecular diversity in the human hippocampus and relative abundance across ages, we took the astrocyte cluster (27,525 cells) (Figure 2A) for further partitioning and identified nine subpopulations (AST0–8) by their differential expression of gene signatures and transcription factors (Figures 2B, 2C and S2A; Table S2B). Gene Ontology (GO) analysis of enriched genes in each subpopulation suggests different identities and physiological functions (Figure 2D; Table S3A). For example, AST0 is most prominently associated with angiogenesis. AST1 and AST6, enriched for GFAP, are linked to injury responses, regeneration, autophagy, and amyloid fibril formation, likely representing reactive astrocytes (Liddelow and Barres, 2017). AST1, but not AST6, exhibits TGFβ signaling-related genes, whereas AST6 and AST7 are involved in phagocytosis. SOX2- and EGFR-enriched AST2 is associated with gliogenesis, representing a putative progenitor cell population (Zhang et al., 2016). AST3 is related to programmed cell death. Most subpopulations, including AST0, AST2, and AST4–8, are associated with synaptic regulation (Figure 2D; Table S3A).
Figure 2. Transcriptomic diversity of human hippocampal astrocytes across the postnatal lifespan.

(A and B) UMAP of integrated data highlighting astrocytes (A), which were sub-clustered and visualized in UMAP colored by subpopulation and generic marker expression (B).
(C and D) Characteristics of astrocyte subpopulations. Heatmap (C) and bubble plot (D) showing representative enriched gene expression and Gene Ontology (GO) terms, respectively. p(FDR): p-value controlled for false-discovery rate.
(E) Dot plots showing the proportion of each subpopulation among all astrocytes across ages. Dots for individual specimens are fitted with linear regression fitting (lines) with 95% confidence interval (grey shades).
(F and G) Sample confocal images (F) and quantification (G) of SOX2+ cells among all S100B+ cells in the human hippocampus across ages. Asterisks and arrowheads indicate SOX2+S100B+ and SOX2-S100B+ cells, respectively. Insets boxed in orange and cyan colors show enlarged view of representative S100B+ cells that were SOX2+ and SOX2-, respectively. Scale bars, 10 μm (F). Dots represent value of quantification for individual subjects and box values represent median ± quantiles with whiskers for max and min (n = 4 subjects per stage) (G).
(H and I) UMAP projection of “astroglia” in two query datasets of hPSC-derived long-term brain organoid cultures (Qian et al., 2020; Szebenyi et al., 2021) to our in vivo astrocyte reference map (H). Colors represent the assigned subpopulations and intensity represents the prediction score for each query cell. Bar plots in (I) show the proportions of query cells mapped to our in vivo glia reference. Cells with prediction scores lower than 0.5 to any in vivo subpopulation were categorized as “unclassified”.
The abundance of most subpopulations among astrocytes remains constant across ages, indicating cluster stability, whereas the GFAP-enriched AST1 and SOX2-enriched AST2 populations display a trend toward increasing or decreasing with age, respectively (Figures 2E, S2B and S2C). Validation with immunohistology using S100B as an empirical, generic astrocyte marker (Figure 2B; Table S2A) showed that the percentage of SOX2+ cells among all S100B+ cells decreases during early postnatal periods to a sustained level throughout adulthood (Figures 2F and 2G). Furthermore, the temporal pattern of SOX2+S100B+ glia progenitors is similar among hippocampal subregions.
The proportion of GFAP-enriched AST1 and AST6 subpopulations among all astrocytes shows a trend towards increasing with age (Figure S2D). We systematically examined their spatiotemporal expression pattern using immunohistology. GFAP is highly expressed in astrocytes in the outer layer of the entorhinal cortex throughout life, in contrast to a much lower enrichment in the inner layer and a complete absence in the molecular layer of the dentate gyrus (Figure 3). Interestingly, the proportion of GFAP+ cells among all S100B+ cells increases with age in the CA1, CA3, hilus/CA4, granule cell layer of the dentate gyrus, and the inner layer of the entorhinal cortex (Figure 3). In addition, individual cells show a gradual increase of GFAP levels with age in the snRNA-seq dataset (Figure S2E), consistent with bulk tissue-level studies (Nichols et al., 1993).
Figure 3. Spatiotemporal patterns of GFAP+ astrocytes in the human hippocampal formation across the postnatal lifespan.
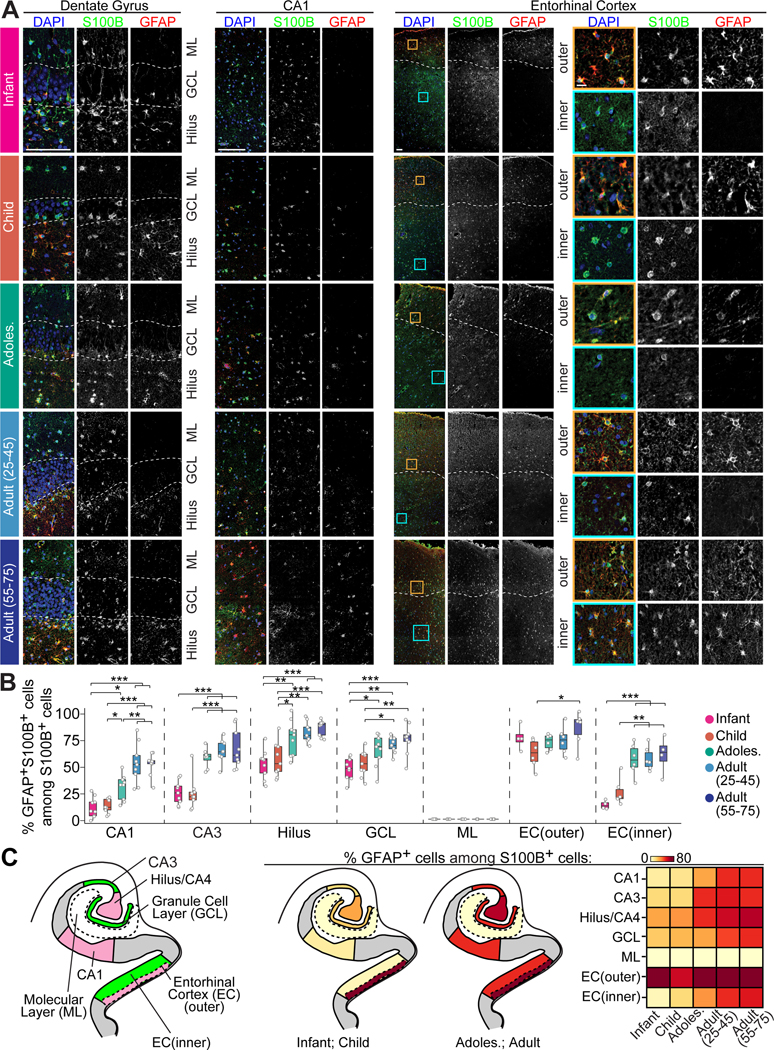
(A and B) Sample confocal images (A) and quantification (B) of GFAP expression patterns among S100B+ cells in hippocampal subregions across ages. Dashed lines in representative images of the dentate gyrus indicate the upper and lower borders of the granule cell layer (A). For representative images of the entorhinal cortex, dashed lines separate the outer and inner layers, and insets boxed in orange and cyan colors show an enlarged view of representative expression patterns of S100B and GFAP in the outer and inner layers, respectively (A). Scale bars, 100 μm for low-magnification images and 10 μm for insets (A). Individual dots represent the value of quantification for different sections (B). Box plots represent mean ± quantiles with whiskers for max and min (n = 3 specimens per age group; * p < 0.01, ** p < 0.001, *** p < 0.0001; Pairwise ANOVA with post-hoc Tukey HSD tests) (B).
(C) Schematic illustrations showing the human hippocampal formation colored by anatomical subregion (left panel) and summary of the proportion of GFAP+ cells among S100B+ cells in young and adult stages (middle two panels). Heatmap showing the percentage of GFAP+ cells among S100B+ cells in each subregion across ages (right panel).
See also Figure S2.
The temporal molecular landscapes for human astrocytes offer a transcriptomic reference map to benchmark glia differentiated from hPSCs to specific human developmental periods for modeling brain development or disorders. We picked two published scRNA-seq datasets of long-term sliced brain organoid cultures, where the “astroglia” clusters have a large number of cells that could be selected for re-annotation (Qian et al., 2020; Szebenyi et al., 2021). We quantitatively compared transcriptomic similarity of every query “astroglia” cell in organoids to each of the nine in vivo astrocyte subpopulations in our reference map by assigning prediction scores (Hao et al., 2021) (Figure 2H). Each query “astroglia” was projected to our reference UMAP with prediction scores for each subpopulation and was annotated based on the highest score if higher than 0.5 (Figures 2H and 2I). As a result, more than 70% (Qian et al., 2020) and 80% (Szebenyi et al., 2021) of query cells were similar to at least one subpopulation, indicating the capacity for long-term organoid cultures to model postnatal human astrocytes in vivo (Figure 2I). Among the mapped cells, 61.2% (Qian et al., 2020) and 82.4% (Szebenyi et al., 2021) matched GFAP+ astrocytes (AST1 and AST6), which is consistent with their respective immunohistology validation (Figure 2I). This may suggest a stress response in culture systems given that human GFAP+ astrocytes exhibit reactive astrocyte signatures. One system (Qian et al., 2020) appeared to exhibit higher diversity than the other (Szebenyi et al., 2021), where 8.2% show congruence to SOX2+ progenitors (AST2) and 1.6% are mapped to AST8, suggesting the emergence of synaptic modulation in long-term cultures (Figure 2I). In both cultures, no astrocytes were matched to AST0 or AST3–7, suggesting significantly reduced heterogeneity in brain organoids compared to the in vivo human brain (Figure 2I).
Together, these results reveal the molecular characteristics and cellular heterogeneity of astrocyte subpopulations in the human hippocampus and the spatiotemporal distribution of the GFAP-enriched astrocytes throughout the postnatal lifespan. Our reference map identifies astrocyte heterogeneity, although incomplete, in long-term cultured hPSC-derived brain organoids, including subpopulations present in adult and aging humans.
Oligodendrocyte lineage transcriptomic diversity in postnatal human hippocampus
Oligodendrocytes arising from OPCs ensheath axons and provide metabolic support to neurons (Nave and Werner, 2014), whereas their dysfunction leads to pathogenesis in aging and neurological disorders (Franklin and Ffrench-Constant, 2017), such as AD and multiple sclerosis (MS). Heterogeneity of the human oligodendrocyte lineage complicates disease prognosis and therapy development (Jakel et al., 2019). To examine their diversity and abundance in the human hippocampus across ages, we selected the OPC and oligodendrocyte populations (61,867 cells) for further partitioning (Figure 4A). We identified two OPC subpopulations (OPC1 and OPC2), an immature oligodendrocyte subpopulation (immatureOL), and three mature oligodendrocyte subpopulations (Oligo1, Oligo2 and Oligo3) (Figure 4B). OPC1, OPC2, and immatureOL share many progenitor genes (e.g., NG2/CSPG4, PCDH15, SOX6) related to cell growth and synapse organization (Figures 4C, 4D and S3A; Tables S2C and S3B). In addition, an immune response regulator, class II major histocompatibility complex (MHC-II)-associated gene, CD74, was highly enriched in OPC1, suggesting the non-progenitor roles of OPCs under physiological conditions (Figure 4C). Cytokine-conditioned OPCs have been shown to express MHC-I and MHC-II and play immunomodulatory roles under pathological conditions, such as MS, which further supports the functional roles of OPCs beyond serving as precursors for new oligodendrocytes (Falcao et al., 2018; Kirby et al., 2019). OPC2 is uniquely associated with Wnt signaling (Figure 4D), which may underlie Wnt-dependent crosstalk with endothelial tip cells in regulating white matter angiogenesis (Chavali et al., 2020). ImmatureOL, Oligo1, Oligo2, and Oligo3 are strongly associated with myelination processes (Figures 4B–4D and S3A; Tables S2C and S3B). Interestingly, BCAS1-enriched immatureOL shows enrichment of both myelination- and cell differentiation-related genes (Figures 4C and 4D). In addition, the Oligo1 population that is related to oligodendrocyte differentiation (Figures 4C and 4D) shares gene signatures (e.g., OPALIN) with the “intermediate oligodendrocytes” in human white matter that are susceptible to MS (Jakel et al., 2019). In contrast, genes enriched in Oligo2 and Oligo3 are associated with cell junction assembly, similar to the “end-state oligodendrocytes” in human white matter (Jakel et al., 2019).
Figure 4. Transcriptomic diversity of human hippocampal oligodendrocyte lineage cells across the postnatal lifespan.

(A and B) UMAP of integrated data highlighting oligodendrocyte lineage cells (A), which were sub-clustered and visualized in UMAP colored by subpopulation and generic marker expression (B).
(C-E) Characteristics of oligodendrocyte lineage subpopulations and their abundance across ages. Heatmap (C) and dot plots (D and E) similar as in Figures 2C-E. reg.: regulation.
(F and G) Sample confocal images (F) and quantification (G) of SOX6+ among all OLIG2+ oligodendrocyte lineage cells in the human hippocampus across ages. Asterisks and arrowheads indicate SOX6+ and SOX6- cells among OLIG2+ cells, respectively. Insets boxed in orange and cyan colors show enlarged view of representative OLIG2+ cells that were SOX6+ and SOX6-, respectively. Scale bars, 10 μm (F). Box plot similar as in Figure 2G (n = 4 subjects per stage) (G).
(H and I) Assessing hPSC-derived oligodendrocyte lineage cells in two query datasets (Chamling et al., 2021; Marton et al., 2019) with our in vivo reference map (H). UMAPs (H) and bar plots (I) similar as in Figures 2H and 2I.
We further assessed the abundance of each subpopulation among all oligodendrocyte lineages across ages and found that progenitors (OPC1, OPC2) and immatureOL, all enriched for SOX6, showed trends of declining with age, suggesting decreasing oligodendrogenesis (Figure 4E). We validated the trend of decreasing SOX6-enriched cells among OLIG2+ oligodendrocyte lineage cells across ages using immunohistology and found that it was similar among hippocampal subregions (Figures 4F and 4G; Table S1). In contrast, proportions of all mature oligodendrocytes trend towards a gradual increase with age (Figures 4E, S3B and S3C).
We leveraged the six in vivo subpopulations by benchmarking query hPSC-derived oligodendrocyte spheroids generated using different protocols (Chamling et al., 2021; Marton et al., 2019) to our glia reference map (Figure 4H). More than 80% of the query cells in both datasets matched at least one of the in vivo subpopulations with a prediction score of 0.5 or above (Figure 4I). Quantification among the classified cells shows that the two culture protocols were both able to yield mature oligodendrocytes despite very different cell compositions in terms of maturation level. The proportions of progenitors or immature oligodendrocytes to the more mature ones are around 3:1 (Marton et al., 2019) and 1:3 (Chamling et al., 2021), respectively (Figure 4I). Interestingly, only OPC1, but not OPC2, was observed in both cultures, suggesting the current protocols yield a subpopulation of OPCs with specific requirements for cell signaling (e.g., absence of Wnt signaling) or potentially niche cell types (e.g., absence of endothelial cells). Mature oligodendrocytes were transcriptomically similar to Oligo1 or Oligo3, but not Oligo2, suggesting the capacity for myelination in organoid systems but also room for improvement.
Together, these results reveal the molecular heterogeneity of human oligodendrocyte lineage subpopulations and changes in abundance in the hippocampus across the lifespan and substantial, but incomplete, diversity of oligodendrocyte lineage subpopulations in the current hPSC-derived brain organoid models.
Microglia transcriptomic diversity in postnatal human hippocampus
Microglia, brain resident macrophages, survey the local environment for phagocytosis and neuronal remodeling (Prinz et al., 2019). To characterize their molecular diversity and cellular abundance across ages, we selected the microglia cluster for further partitioning upon precluding specimens with less than 200 microglia (4,197 cells) and identified five subpopulations, each displaying distinct marker gene and transcription factor enrichment (Figures 5A–5C and S4A; Table S2D). For example, MG0 preferentially expresses CD83, EGR3, and CCL2, and is related to immune surveillance (Figure 5C and 5D; Table S2D and S3C), which was recognized in the cortex as a human-specific homeostatic subtype with a slightly activated state (Masuda et al., 2019; Olah et al., 2020). SPP1- and TREM2-enriched MG1 is related to autophagy and neuroinflammatory response, previously reported to be disease-associated and to play a role in de-/re-myelination in a mouse MS model (Keren-Shaul et al., 2017; Masuda et al., 2019). Both MG3 and MG4 are associated with axon and synaptic modulation, whereas MG3, but not MG4, is associated with myelination and glia differentiation (Figure 5D; Table S3C).
Figure 5. Transcriptomic diversity of human hippocampal microglia across the postnatal lifespan.

(A and B) UMAP of integrated data highlighting microglia (A), which were sub-clustered and visualized in UMAP colored by subpopulation and generic marker expression (B).
(C-E) Characteristics of microglia subpopulations and their abundance across ages. Heatmap (C) and dot plots (D and E) similar as in Figures 2C-E.
(F and G) Sample confocal images (F) and quantification (G) of CD83+ cells among all IBA1+ microglia in the human hippocampus across ages. Asterisks and arrowheads indicate CD83+ and CD83- cells among IBA1+ cells, respectively. Insets boxed in orange and cyan colors show enlarged view of representative IBA1+ cells that were CD83+ and CD83-, respectively. Scale bars, 10 μm (F). Box plot similar as in Figure 2G (n = 4 subjects per stage) (G).
(H and I) Assessing hPSC-derived microglia in three query datasets (Popova et al., 2021; Svoboda et al., 2019) with our in vivo reference map (H). UMAPs (H) and bar plots (I) similar as in Figures 2H and 2I.
We next compared the abundance of microglia subpopulations across ages (Figure 5E). Interestingly, CD83-, EGR3- and CCL2-enriched MG0 appears to be adult-specific (Figures 5C, 5E, S4B, and S4C). Immunohistology confirmed the absence of CD83 in IBA1+ microglia in the infant and child specimens despite its sparse expression in the adult ones (Figures 5F and 5G). The discrepancy in cellular abundance between snRNA-seq quantifications and immunostaining results is likely attributable to the reported post-transcriptional regulation of CD83 (Ehlers et al., 2013). In addition, in-situ hybridization analyses of EGR3 and CCL2 confirmed their expression in IBA1+ microglia in the adult, but not child, human hippocampus (Figure S4D). MG1 shows a trend toward decreasing with age, whereas MG2, MG3 and MG4 remain largely constant (Figure 5E).
We utilized our five in vivo subpopulation classifications as a reference to map three query datasets of hPSC-derived microglia, including commercially available cultured cells in vitro (iCell Microglia) (Popova et al., 2021) and “iMG” cultures in vitro and upon xenograft into mice (Svoboda et al., 2019) (Figure 5H). The vast majority of query microglia in vitro could be mapped to in vivo subpopulations with a prediction score over 0.5, suggesting that they maintained core microglia signatures under various culture protocols (Figures 5H and 5I). Almost all query microglia were transcriptomically similar to MG1, an immune- and inflammation-related subpopulation with elevated stress response, with very few matched MG2 (< 1%), and none to MG3 and MG4, indicating a lack of heterogeneity in culture (Figure 5I). Interestingly, despite a low proportion in culture, MG0, an immune-related, largely adult-specific subpopulation, appeared upon mouse xenograft at the expense of MG1 (Svoboda et al., 2019) (Figure 5I), indicating a strong environmental influence on cell identities.
Together, these results reveal the molecular and functional heterogeneity of human microglia and further identify a microglia subpopulation present largely only in the post-adolescent human hippocampus. Our analysis also suggests a lack of subpopulation heterogeneity for hPSC-derived microglia in culture and their dynamic properties upon xenograft into animals.
Cell type- and subpopulation-specific expression patterns of brain disorder risk genes
Glia play a crucial role in disease mitigation, whereas glia dysfunction contributes to brain disorders (Barres, 2008). We next examined risk gene enrichment of ASD, AD, SCZ, EPI, bipolar disorder, anxiety disorder, and MDD curated from genome-wide association studies (GWAS) in major hippocampal cell types, by calculating an enrichment score using MAGMA (de Leeuw et al., 2015) (Figure S5A) and aggregated expression (Figure S5B; Table S4) for each across ages. Neurons in general expressed more disease-associated risk genes than glia. Among all glia, risk genes were more enriched in astrocytes and OPCs than in mature oligodendrocytes and microglia.
We next measured each glia subpopulation for their relative risk gene enrichment and expression patterns (Figures S5C and S5D; Table S4). AST5, AST6, AST8, OPC1, OPC2, immatureOL, MG3, and MG4 expressed more disease-associated risk genes in their respective glia type, most of which do not have established disease associations, except for AST6 with neuroinflammation (Liddelow and Barres, 2017). Notably, almost all these subpopulations have been linked to neuronal synaptic regulation in our functional GO analyses (Figures 2D, 3D and 4D; Tables S3A-S3C).
Overall, our cell atlas of the neurotypical hippocampus enables us to uncover neuropsychiatric disease risk gene enrichment in neurons and selective glia subpopulations that are specialized in neuronal and synaptic modulation.
Transcriptomic dysregulation in glia subpopulations in Alzheimer’s disease
As another example to implement our atlas of glia subpopulations, we directly examined molecular pathology in post-mortem AD brains. We performed snRNA-seq analysis of the hippocampi of 8 AD patients (Braak stage III to VI) and 8 matched controls integrated with the 7 control specimens from the aging group above (Figures 6A and S6A). We detected 1,037 genes with 1,878 UMIs per nucleus on average. We re-identified all glia types and subpopulations in both AD and controls, whereas their abundance was not significantly altered (Figures S6B and S6C).
Figure 6. Subpopulation-specific transcriptomic dysregulation in glial cells in AD.

(A) UMAP of snRNA-seq of AD human hippocampus and matched controls, colored by major cell type.
(B) Heatmap showing the number of dysregulated genes in AD in each major cell type.
(C) Venn diagram showing comparison of dysregulated genes in different glial cells in AD among brain regions, including hippocampus (current study), PFC (combining all dysregulated genes in (Lau et al., 2020; Mathys et al., 2019; Sadick et al., 2022; Zhou et al., 2020); See Figure S6F), and entorhinal cortex (Grubman et al., 2019).
(D-F) Selective disruption in glia subpopulations in AD, showing the number of dysregulated genes (D), GO terms of biological processes (E), and exemplary gene expression (F).
See also Figures S5 and S6, Tables S4, S5 and S6.
Next, we analyzed differentially expressed genes (DEGs) between AD and controls for each major cell type (Figure 6B; Table S5A). Neurons, especially excitatory neurons, show a higher number of DEGs than other cell types (Figure 6B), which is similar to the findings from the AD cortex (Mathys et al., 2019) and likely due to the dominant proportion of neurons resulting in a greater statistical power to detect their DEGs. Among all glia, astrocytes and oligodendrocytes have more DEGs than microglia or OPCs (Figure 6B). While over 70% DEGs affect only one major cell type, there was a small group of genes affected in multiple (≥ 4) cell types, which are associated with cell adhesion, synaptic organization, endocytosis, and apoptosis (Figures S6D and S6E). We cross-compared genes disrupted by AD in our hippocampal dataset and several published snRNA-seq studies of the prefrontal cortex (PFC) and entorhinal cortex (Grubman et al., 2019; Lau et al., 2020; Mathys et al., 2019; Sadick et al., 2022; Zhou et al., 2020), where various gene comparison methods and thresholds were applied (Table S6A). Few DEGs are shared across studies of different brain regions, suggesting region-specific transcriptomic dysregulation in glial cells in AD (Figure 6C, Table S6A-S6D), although results from different previous PFC studies are variable (Figure S6F), indicating a need for additional confirmation.
We then assessed how AD alters gene expression in glia subpopulations. We found that only selected subpopulations exhibit DEGs in AD (Figure 6D). Only AST1 and AST3 among astrocytes show substantial DEGs in AD, which are the two subpopulations associated with disease response processes (Figures 2D and 6D). The GFAP-enriched AST1, which likely corresponds to reactive astrocytes known to be dysregulated in the AD cortex (Saura et al., 2022), is associated with autophagy and injury responses, whereas AST3 is related to programmed cell death (Figure 2D). Cell adhesion is downregulated in both AST1 and AST3 in AD, whereas VEGFR signaling and transcription regulation are upregulated in AST1 and AST3, respectively (Figure 6E). Oligodendrocyte lineages have been implicated in neurodegenerative diseases (Franklin and Ffrench-Constant, 2017). We found OPC1, Oligo1, Oligo2, and Oligo3 exhibit DEGs in AD (Figure 6D). OPALIN-enriched Oligo1 (Jakel et al., 2019) has the most DEGs. Interestingly, though dysregulated genes vary across brain regions (Figures 6C and S6F), many pathological processes are shared between the AD hippocampus and cortex (Mathys et al., 2019), including upregulation in response to unfolded protein (AST1), cell death (Oligo1), tau-protein kinase activities, response to heat (Oligo1 Oligo2), and ion transport (OPC1) (Figures 6E and 6F), indicating a common cross-region molecular pathology in AD. Many AD-perturbed subpopulations, including AST3, OPC1, Oligo1, and Oligo2, show synapse-related downregulation (transmission, ion transport, organization, myelination), while Oligo2 exhibits an increased expression of aging-relevant genes (Figures 6E and 6F). Finally, gene dysregulation in AD microglia occurred mostly in MG4 (Figures 6D and 6E), a subpopulation enriched for disease risk genes (Figures S5C and S5D) and related to cell adhesion and signaling transduction, suggesting a potential impairment in their surveillance function.
Overall, our analysis highlights the advantage of investigating AD pathology at single-cell resolution aided with new knowledge on glia diversity to identify differential gene dysregulation of specific glia subpopulations that may have higher disease vulnerability (Figures 6D-6F; Table S5B).
DISCUSSION
We present a comprehensive single-nucleus transcriptome atlas of a specific human brain region across the postnatal lifespan from infant, child, adolescent, adult, to aging stages. Using 224,464 high-quality nuclei from 32 postmortem specimens of neurotypical human hippocampus, detailed analyses revealed glia subpopulations, molecular characteristics, enrichment of pathways related to cell functions, disease relevance, and age-dependent changes in their abundance. Using this resource, we assessed subpopulation composition of various glia types arising from hPSC differentiation in 2D and 3D in vitro culture systems and upon xenograft into mice. We further revealed the impact of AD on specific glia subpopulations by analyzing 82,279 nuclei from post-mortem hippocampi of AD patients and matched controls. Together, our study provides a rich resource of a single-nucleus transcriptome atlas of glia diversity in the human hippocampus across the postnatal lifespan and in AD, as well as a glia reference map for annotating and assessing human stem cell glia differentiation and various brain pathologies. These data are freely available at the GEO database and can be explored using the UCSC Cell browser (https://hippo-lifespan.cells.ucsc.edu).
Molecular diversity and dynamics of glia subpopulations in human hippocampus across postnatal lifespan
A recent flurry of discoveries on human brain cell diversity has emerged from snRNA-seq studies, although nearly all provide snapshots of one age stage (Rajewsky et al., 2020), posing challenges to analyze cell dynamics across the lifespan. Such analysis is particularly critical for glia, many of which undergo prolonged maturation postnatally, and actively respond to environmental cues or postnatal brain disorders. Here we present a unique resource of the molecular landscapes of glia in the human hippocampus across the postnatal lifespan, with subpopulation characteristics and age-dependent alterations (Figure 1A). Interestingly, almost all glia subpopulations, except for MG0, are already present shortly after birth, but only a few exhibit age-dependent trends in their cellular abundance. We compared, at the global transcriptome level, several published datasets of the human cortex and other brain regions to our age-matched hippocampal dataset and found that regardless of age, glia display much less inter-regional divergence than neurons (Figure 1D), suggesting that our findings may represent general principles beyond the hippocampal region.
Glia modulate synaptic formation and transmission, as well as vasculature, inflammatory or injury response, phagocytosis, immune surveillance, and many other critical functions. Brain disorders, such as AD and MS, can alter their properties, leading to injury responses or reactivity in astrocytes, myelination defects, or expression of disease-related signatures in microglia. Their diverse roles suggest heterogeneity regarding cell subtype identities or molecular states. However, molecular identities of glia associated with various functions are largely unclear. Our systematic analyses across ages allow us to unbiasedly characterize human glia diversity based on their enriched genes, transcription factors and predicted functional characteristics. We identified several glia subpopulations in the human hippocampus that have been implicated in brain development or various disorders in other brain regions, such as the cortex or white matter. Our dataset spanning the postnatal lifespan allows us to further estimate age-dependent changes in cellular abundance. For example, the disease-associated OPALIN-enriched Oligo1 (Jakel et al., 2019) show a trend of steadily increasing with age (Figure 4E). The largely adult-specific CD83-enriched MG0 was suggested to contribute to homeostasis in the cortex (Olah et al., 2020) (Figure 5E). In contrast, the SOX2- (AST2) and SOX6- (OPC1, OPC2, and immatureOL) enriched progenitors of astrocytes and oligodendrocytes, respectively (Jakel et al., 2019; Zhang et al., 2016), both show a trend of decreasing with age (Figure 2E). The SPP1, TREM2-enriched MG1, which has been implicated in multiple degenerative diseases in the cortex (Masuda et al., 2020), also show a trend of decreasing with age (Figure 5E). We validated the age-dependent expression patterns in glial subpopulations enriched for SOX2, SOX6, CD83, EGR3, and CCL2 using immunohistology and in situ analyses (Figures 2F, 2G, 4F, 4G, 5F, 5G, and S4D).
As a part of the validation, we examined in detail the spatiotemporal divergence of GFAP+ astrocytes (AST1 and AST6) in the human hippocampus and found a continuous subregion-specific increase across ages in their abundance (Figure 3), which were identified as injury-responding, reactive astrocytes during neuroinflammation, normal aging, AD, MS, and other neurological disorders (Liddelow and Barres, 2017). This analysis presents an example of how our dataset can be used as a spatiotemporal reference map combined with other data modalities, such as immunohistology and in situ analyses, to reveal new biological insights.
A reference map assessing glia differentiation from human stem cells
hPSC-derived systems have emerged as a powerful platform to model human brain development and disorders. One of the current endeavors focuses on recapitulating late-stage brain development or adult-onset diseases, both consisting of more complex cell type composition, especially for glia. Our standardized classifications of glia subpopulations and systematic investigation of age-related changes provide a complete human glia transcriptomic reference map to benchmark hPSC-derived glia differentiation to specific human developmental, adult and aging periods. We quantitatively mapped several datasets containing glia derived from hPSCs using different protocols to our reference atlas and found that a significant portion are mapped to mature glia subpopulations in long-term cultures or after xenograft into mice, including GFAP+ mature astrocytes, mature oligodendrocytes, and adult-specific microglia (Figures 2I, 4I and 5I). These intriguing results demonstrate the potential for hPSC-based systems to model some aspects of mature human brains under physiological or pathological conditions. On the other hand, current protocols for hPSC-differentiation models require further optimization as many in vivo subpopulations are not detected in culture systems, including those with important functions (synaptic modulation, e.g., AST4, AST5, AST7, OPC2, MG4; myelination, e.g., Oligo2, MG3) or enriched for disease risk genes (e.g., AST5, OPC2, MG3, MG4) (Figures 2H, 2I, 4H, 4I, 5H, 5I and S5D). Furthermore, subpopulations that express stress signatures are over-represented in cultured astrocytes and microglia (e.g., AST1, AST6, MG1) (Figures 2I and 5I), suggesting imperfect culture conditions (Bhaduri et al., 2020). Overall, our reference map provides a resource to benchmark and improve differentiation of human stem cells into diverse glia subpopulations for better modeling of glia biology and associated disorders throughout human life.
Implication of specific glia subpopulations in neuropsychiatric disorders
GWAS of neuropsychiatric disorders identified many genetic variants that confer disease susceptibility and contribute to pathogenesis. We probed a curated database of brain disorder risk genes (Yu et al., 2010) in our transcriptome atlas across ages to assess distinct risk gene expression in specific cell types, in particular glia subpopulations (Figures S5B and S5D). Strong enrichment of risk genes was found in neurons and glia subpopulations performing neuronal and synaptic modulation functions, suggesting a molecular basis for the increased susceptibility of selected cell types to brain disorders based on physiological functions (Figure S5). These findings can inform future analyses of genetic variants for the development of disease mitigation strategies.
We also directly assessed the disease-associated changes in AD hippocampi and found a diverse impact on major cell types and glia subpopulations (Figures 6B and 6D). Only selected glia subpopulations exhibit transcriptomic dysregulation, with very different genes affected (Figures 6C-6F and S6F). However, many disrupted pathways and biological processes associated with these diverse genes are shared in the AD cortex or other degenerative disorders (such as a shared stress response), suggesting a convergence of pathological features among different brain regions in various disease contexts (Figures 6C and S6F). For example, OPALIN-enriched Oligo1 in our AD hippocampus has been implicated in the AD cortex (Lau et al., 2020) and the MS white matter (Jakel et al., 2019). Downregulation of myelination and upregulation of response to heat and cell death processes have been reported in the late-stage AD cortex (Saura et al., 2022) and in MS (Jakel et al., 2019). GFAP+ astrocytes (AST1 and AST6) are also dysregulated in the human AD cortex (Grubman et al., 2019; Lau et al., 2020; Leng et al., 2021; Morabito et al., 2021; Sadick et al., 2022), the cortex and hippocampus of an AD mouse model (Habib et al., 2020), and MS (Jakel et al., 2019) (Figures 6D and 6E). Notably, several AD-affected glia subpopulations (AST3, OPC1, Oligo1, Oligo2, and MG4) display impaired synaptic modulation, a process where genes are susceptible to disease disruption, as was predicted by our risk gene analysis (Figure S5). As a rapidly evolving field, hPSC-derived 2D cultures and 3D brain organoids have been used to model AD or other neuropsychiatric disorders, where our in vivo map for diseased glia could further be applied to assess molecular dysregulation of glia subpopulations in in vitro or ex vivo cultures. The two applications that we demonstrated in this study may converge in the near future to fuel our understanding of neuropsychiatric disorders using hPSC-based modeling.
A combination of our longitudinal analysis across ages and in AD provides a holistic picture of glial cells under physiological and pathological conditions. The glia subpopulations we classified across ages using distinct gene signatures can be used as a high-confidence reference to map their alterations in diseases. This contextualization allows us to investigate the impact of AD on each glia subpopulation with known distinct functions, in contrast to assessing the gene or pathway changes in each major glia type as a whole population in the AD cortex (Saura et al., 2022). Indeed, we found that only specific glia subpopulations are affected in AD and genes dysregulated in each subpopulations have very limited overlap (Figure S6G). These findings would be otherwise masked without subpopulation resolution, emphasizing the value of our comprehensive atlas across ages in understanding cellular mechanisms underlying brain homeostasis and disorders.
Limitations of the study
First, we examined a total of 48 hippocampal specimens across ages, a limited sample size for analyzing sex differences or providing statistical significance to the observed trends in age-dependent changes in cell abundance. We may also have missed some rare subpopulations due to limited cells and specimens sequenced and the sequencing depth. Future larger-scale sequencing efforts, expanded cohorts, or dataset integration and cross comparison may validate and enhance the resolution of our current study. Second, despite unbiased cell counting and whole-transcriptome characterization of glia subpopulations, our snRNA-seq analysis does not reveal their spatial distribution, which can be improved by the emerging spatially resolved scRNA-seq methods. Our analysis of GFAP+ astrocytes in the human hippocampus provided an example of integrative analysis combining other data modalities, such as immunohistology and in situ analyses, to reveal spatial and temporal cell dynamics. Third, as a common issue in snRNA-seq analyses, it remains challenging to determine whether cell subpopulations assigned by unsupervised clustering represent unique cell subtypes or distinct states of the same type, although almost all glia subpopulations are present in almost all human subjects, indicating cluster stability. Fourth, current standardized protocols to retrieve frozen specimens from biobanks and prepare for snRNA-seq may lead to aberrant gene expression particularly in microglia (Marsh et al., 2022) and a preferential enrichment of neuronal transcripts over glial ones, respectively (Lake et al., 2018). Though we have independently validated several key findings by immunohistology and in-situ analyses, more should be done in the future to rule out potential bias.
Overall, our study generated a rich resource of single-nuclei transcriptome atlas of glia subpopulations in the human hippocampus across the postnatal lifespan and in AD that not only provides a holistic view of glial cell molecular diversity, but can be used as a reference map for glia subpopulation identification across brain regions, ages, species, and disease conditions, and to benchmark hPSC differentiation into various glial cells in 2D cultures, 3D brain organoids and upon xenograft into animals.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Guo-li Ming (gming@pennmedicine.upenn.edu).
Materials availability
There are no restrictions on any materials presented in this paper.
Data and code availability
All snRNA-seq data are available at the GEO database (accession numbers GSE185553, GSE198323 and GSE199243) and UCSC Cell Brower (https://hippo-lifespan.cells.ucsc.edu). Information on the de-identified human specimens used in this study and their sequencing characteristics are described in Table S1. Scripts used in this study are available at https://github.com/ysu2015/HumanHippocampus_scRNAseq, maintained by Dr. Yijing Su (yijingsu@pennmedicine.upenn.edu). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tissue specimens
De-identified human hippocampal tissue specimens were collected and processed under protocols approved by the Institutional Review Boards of the University of Pennsylvania and the Children’s Hospital of Philadelphia. A total of 75 human hippocampal specimens between the ages of 0.1 to 95 years old were used in this study, including 40 post-mortem specimens from subjects free from neurological disorders and 8 post-mortem specimens from AD patients (Braak stage ranging from III to VI) for snRNA-seq, and 25 post-mortem specimens from subjects free from neurological disorders and 2 surgical specimens from epilepsy patients for immunohistological and in situ analyses (Table S1). Samples were from tissue banks at the Children’s Hospital of Philadelphia, the Johns Hopkins University Pathology Archive, the Lieber Institute for Brain Development, the NIH NeuroBioBank at the University of Pittsburgh Brain Tissue Donation Program, the University of Maryland Brain and Tissue Bank, the University of Miami Brain Endowment Bank, the Harvard Brain Tissue Resource Center, the Human Brain and Spinal Fluid Resource Center at the VA West Los Angeles Healthcare Center, and the Mount Sinai School of Medicine (Table S1). Informed consent for each specimen was obtained by its corresponding institution prior to tissue collection.
METHODS DETAILS
Single nucleus isolation
Single-nucleus RNA sequencing was performed using the SPLiT-seq platform with modifications (Qian et al., 2020; Rosenberg et al., 2018). Nuclei isolation from frozen hippocampal tissue was performed as previously described with minor modifications (Su et al., 2017). Briefly, tissue was minced with a razor blade and homogenized for 5 to 10 strokes using a tissue grinder (Fisher Scientific, 8853000002) in a 1 mL of cold homogenization buffer (1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, EDTA-free protease inhibitor, 0.3% IGEPAL-630, 0.25 M sucrose, 25 mM MgCl2, 20 mM Tricine-KOH). Homogenates were filtered through a 40-μm strainer and mixed with 200 μL of Myelin Removal Beads II (Miltenyi Biotec, 130–096-733) for a 15-minute incubation on ice. The mixture was transferred on top of a sucrose cushion buffer (0.5 mM MgCl2, 0.5 mM DTT, EDTA-free protease inhibitor, 0.88 M sucrose) at a 1:1 ratio (vol / vol) and centrifuged at 2,800 g for 10 minutes in a swinging bucket centrifuge at 4 °C. Nuclei were collected as pellets and resuspended with Phosphate-Buffered Saline (PBS, Corning, 21–040-CV) containing 0.01% Bovine serum albumin (BSA, Sigma-Aldrich, B6917). Nuclei were spun down for 3 minutes at 500 g at 4 °C, resuspended in 1 mL of cold PBS-RI (1x PBS, 0.05U/μl RNase Inhibitor) and passed through a 40-μm strainer. 3 mL of cold 1.33% formaldehyde solution was added to the nuclei suspension for fixation on ice for 10 minutes. Next, nuclei were permeabilized with 160 μL of 5% Triton X-100 for 3 minutes and centrifuged at 500 g for 3 minutes at 4 °C. Nuclei were resuspended in 500 μL cold PBS-RI before 500 μL of cold 100 mM Tris-HCl (pH 8.0) was added. Then, nuclei were spun down at 500 g for 3 minutes at 4 °C and resuspended in 300 μL of cold 0.5 X PBS-RI. Finally, nuclei were passed through a 40-μm strainer again, counted with a hemocytometer and diluted to 1,000,000 nuclei/mL with cold 0.5 X PBS-RI.
Sequencing library preparation
Library preparation was performed as previously described (Qian et al., 2020; Rosenberg et al., 2018). Briefly, mRNA from single nuclei were tagged in three rounds with barcoded primers (Integrated DNA Technologies), with in-cell ligations using T4 DNA ligase (New England Biolabs, M0202S). After adding barcodes, nuclei were washed with 4 mL of wash buffer (4 mL of 1X PBS, 40 μL of 10% Triton X-100 and 10 μL of SUPERase In RNase Inhibitor), spun down at 1000 g for 5 minutes at 4 °C and resuspended with 50 μL PBS-RI. Nuclei were counted, diluted and aliquoted into 10,000 nuclei per sublibrary in 50 μL PBS-RI. Lysate of each sublibrary was prepared by adding 50 μL of 2X lysis buffer (20 mM Tris (pH 8.0), 400 mM NaCl, 100 mM EDTA (pH 8.0), 4.4% SDS and 10 μL proteinase K solution) and incubating at 55 °C for 2 hours to reverse formaldehyde crosslinks. Ligation products in each lysate were purified with Dynabeads MyOne Streptavidin C1 beads, resuspended with a solution containing 44 μL of 5X Maxima RT buffer, 44 μL of 20% Ficoll PM-400 solution, 22 μL of 10 mM dNTPs, 5.5 μL of RNase Inhibitor, 11 μL of Maxima H Minus Reverse Transcriptase, and 5.5 μL of 100 μM of a template switch primer (BC_0127) (Rosenberg et al., 2018) and incubated at room temperature for 30 minutes and then 42 °C for 90 minutes for template switching. Then, beads were washed and resuspended with a solution containing 110 μL of 2X Kapa HiFi HotStart Master Mix, 8.8 μL of 10 μM stocks of primers BC_0062 and BC_0108 (Rosenberg et al., 2018), and 92.4 μL of water for PCR thermocycling with following parameters: 95 °C for 3 minutes, then five cycles at 98 °C for 20 seconds, 65 °C for 45 seconds, 72 °C for 3 minutes. Next, EvaGreen dye was added into PCR solution after beads were removed for additional qPCR thermocycling with the following parameters: 95 °C for 3 minutes, cycling at 98 °C for 20 seconds, 65 °C for 20 seconds, and then 72 °C for 3 minutes. The qPCR cycle was determined by the qPCR signal. Once the qPCR signal began to plateau, cycling was stopped and followed by a 5-minute incubation at 72 °C. In our experience, an additional 6–7 cycles were needed for sublibraries that contained 10,000 cells. PCR products were purified using a 0.8X ratio of SPRI Beads and cDNA concentration was measured by Qubit.
Tagmentation was performed with Nextera XT Library Prep Kit on 600 pg of purified cDNA following the manufacturer’s protocol. The tagmented cDNA libraries were further amplified with 12 enrichment PCR cycles using the indexed primers (P5 primer: BC_0118, one of indexed P7 primer: BC_0076-BC_0083) (Rosenberg et al., 2018). PCR products were purified with a 0.7X ratio of SPRI beads to generate an Illumina-compatible sequencing library.
Sequencing, reads alignment, and preprocessing
After quality control analysis by a Qubit Fluorometer (ThermoFisher Scientific, Q33238) and a Bioanalyzer (Agilent), libraries were sequenced on an Illumina NextSeq 550 instrument using Illumina 150-cycle High Output Kit v2.5 (20024907) with a modified 150bp paired-end protocol where R1 = 66bps and R2 = 94bps to maximize mapping. The raw data was converted to “.fastq” files using bcl2fastq (v2.17.1) software. Based on the design, individual cell barcodes and Unique Molecular Identifiers (UMIs) were embedded on Read2 in the positions of (1–10 UMIs; 11–18 Round3; 49–56 round2; 87–94 round1). Paired-end sequencing reads were pre-processed using Drop-seq-1.13 (Macosko et al., 2015) with some modifications. Briefly, each mRNA read was tagged with a cell barcode and a UMI, trimmed off sequencing adaptors and poly-A sequence, and aligned to the human reference genome assembly (hg38, Gencode release V28). Both exonic and intronic reads mapped to the predicted strands of annotated genes were retrieved for the cell type classification (Hu et al., 2017). Uniquely mapped reads were grouped by cell barcodes. To digitally count gene transcripts, a list of UMIs in each gene, within each nucleus, was assembled, and UMIs within ED = 1 were merged. The total number of unique UMI sequences was counted and reported as the number of transcripts of that gene for a given nucleus. Raw digital expression matrices were generated for each sequencing run. We observed a range of UMIs in different samples due to variability of sequencing depth (Table S1).
Quality control, cell clustering and dataset integration
Raw expression matrices of each individual specimen were loaded as Seurat objects (v 4.0.0) (Hao et al., 2021) in R (v4.0.4) using the function “CreateSeuratObject”. For each object, genes expressed in < 10 nuclei were discarded; nuclei with > 5% UMIs mapped to mitochondrial genes were discarded. Doublets were identified and removed using scDbiFinder (v1.4.0) (Germain et al., 2021). For analyzing glial cells across ages, all 32 objects from specimens free from neurological disorders were merged, nuclei with genes < 200 or > 8,000 were discarded and integrated using the “reciprocal PCA” method (RPCA) (Hao et al., 2021). For comparing AD and control specimens, objects from 8 AD specimens and all 15 control specimens older than 73 years old were merged, nuclei with genes < 400 or > 8,000 were discarded and integrated using canonical correlation analysis (CCA) (Hafemeister and Satija, 2019; Stuart et al., 2019). Integrated datasets for both analyses were respectively scaled and regressed out effects of library size (total UMI counts) and percentage of mitochondrial genes to mitigate specimen and preparation heterogeneity prior to further analyses.
Cell clustering, visualization, and marker gene identification
Principal component analysis (PCA) was used to reduce dimensionality of the dataset using the ‘RunPCA’ function in Seurat. Two methods were used to determine the optimal number of principal components (PCs) used for clustering analysis and visualization: (1) The cumulative standard deviations of each PC were plotted using the function “PCElbowPlot” in Seurat to identify the ‘knee’ point at a PC number after which successive PCs explain diminishing degrees of variance; (2) The significance of each gene’s association with each PC was assessed by the function “ScoreJackStraw” in Seurat. For analyzing subpopulations of each major glial cell type, UMI count matrices of each subset were loaded as a new Seurat object for further partitioning using the same methods and criteria described above. Specimens or batches with the total number of microglia less than 200 were excluded from the microglia subpopulation analysis. This is because the integration pipeline (Hao et al., 2021) we used requires a minimal cell number for each dataset to ensure a statistically confident data integration, and will produce errors during the “IntegrateData” step if this requirement is not met. Marker genes for each major cell type and for each glia subpopulation were identified with a Wilcoxon rank sum test implemented in the ‘FindAllMarkers’ function with the following criteria: false-discovery rate (FDR)-adjusted p-value < 0.05, log-fold change ≥ 0.25 (selecting positive markers only), and were detected in ≥ 10% of the cells within their respective clusters (Table S2). Differentially expressed genes of major cell types and glia subpopulations between AD and controls were identified using the “FindMarkers” function in Seurat using the following criteria: FDR-adjusted p-value < 0.05, log-fold change ≥ 0.25 or ≤ −0.25, and were detected in ≥ 10% of the cells within their respective clusters (Table S5).
Benchmarking cell types and subpopulations and comparing transcriptomic similarities to published datasets
Published single-cell transcriptomic datasets were obtained from UCSC Cell Brower (https://cells.ucsc.edu/) and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.go/geo/). Only specimens free from neurological diseases were used. Datasets were individually prepared using the same parameters described above. Clusters of the major cell types were identified using common marker genes (Table S2).
To compare cell-type transcriptomic similarity across brain regions (Figure 1D), published datasets from different brain regions, including the prefrontal cortex (PFC), visual cortex, cerebellum, anterior cingulate cortex (ACC), motor and premotor cortex, and white matter (Jakel et al., 2019; Lake et al., 2018; Schirmer et al., 2019; Velmeshev et al., 2019) were individually prepared and six major cell types were identified in each. Each dataset was matched to a subset of our hippocampal dataset that spans across the entire lifespan based on its corresponding age for comparison using a random forest classifier as previously described (Breiman, 2001; Shekhar et al., 2016) (Figure 1D).
To characterize glia subpopulations from human pluripotent stem cell-based 2D or 3D differentiation models using our glia subpopulations as a reference, published datasets, containing a sufficient number of cell populations of interest, from human pluripotent stem cell-derived astrocytes (Qian et al., 2020; Szebenyi et al., 2021), oligodendrocyte lineage cells in 3D brain organoids (Chamling et al., 2021; Marton et al., 2019), and microglia in 2D culture and upon xenograft into mice (Popova et al., 2021; Svoboda et al., 2019) were prepared separately to identify major cell types. The corresponding glia cluster(s) were subset out for further analyses. For each glia type, cluster(s) from the query datasets were projected to our glia reference map using a Seurat CCA integration-based method with default settings (https://satijalab.org/seurat/articles/integration_mapping.html) (Hao et al., 2021). Each query cell was projected back to the previously computed UMAP visualization plots of its respective glia type (Figures 2H, 4H and 5H). We applied an empirical prediction score cut-off of 0.5 to exclude query cells of low similarity to any of the in vivo subpopulations, labeling as “unclassified” cells. For each glia type, we then used the highest prediction scores to annotate glial cells to specific subpopulations and quantified their proportion among all glial cells (Figures 2I, 4I and 5I).
Gene ontology, disease risk gene, and transcription factor enrichment analyses
To map enrichment patterns of gene ontology (GO) terms of biological processes onto different subpopulations for each glial type, we applied the “compareCluster” function (“clusterProfiler” R package) (Wu et al., 2021) with default parameters (Figures 2D, 4D and 5D). Enriched marker genes for each glia subpopulation were used as input. A p-value, controlled for FDR, less than 0.05 is considered significantly enriched. A full list of GO terms was summarized in Table S3. To identify the biological processes significantly dysregulated in AD, we input the differential expression genes between AD and control specimens for each glia subpopulation to DAVID Knowledge Base (v2021q4, https://david-d.ncifcrf.gov). A p-value of less than 0.05 is considered significantly enriched. A full list of GO terms was summarized in Table S5.
We analyzed the enrichment of significantly regulated genes in each category with disease annotations curated from GWAS studies (Giannakopoulou et al., 2021; Grove et al., 2019; International League Against Epilepsy Consortium on Complex, 2018; Mullins et al., 2021; Otowa et al., 2016; Trubetskoy et al., 2022; Wightman et al., 2022) by calculating effect sizes (BETA) and the enrichment p-values using MAGMA (de Leeuw et al., 2015) (Figures S5A and S5C). The aggregated expression of curated disease associated genes from Phenopedia (Yu et al., 2010) (accessed on March 25, 2021) in each major cell type and subpopulation is shown in Figures S5B and S5D and summarized in Table S4.
A list of human transcription factors (TFs) was downloaded from AnimalTFDB3.0 (Hu et al., 2019) for performing TF expression analysis. For a given glia type, selected TFs among marker genes of each subpopulation (Table S2) were plotted as a heatmap for their average expression (Figures S2A, S3A and S4A).
Immunostaining and confocal microscopy
Immunohistology on brain tissue sections was performed as previously described (Zhou et al., 2018). For formalin-fixed, paraffin-embedded (FFPE) tissue sections, prior to further processing, they were deparaffinized in 4 times xylene (Fisher Scientific, X5–1), 4 times 100% ethanol, and 4 times 95% ethanol, each for 5 minutes. For paraformaldehyde (PFA)-fixed sections, brain tissue blocks were fixed with 4% PFA at 4 °C for 24–48 hours, and cryoprotected with 30% sucrose (wt / vol). 40-μm-thick sections were cut on a frozen sliding microtome (Leica, SM2010R). The sections then underwent antigen retrieval prior to antibody application by being incubated in 1X target retrieval solution (DAKO) at 95 °C for 12.5 minutes, followed by a 15-minute cooling to room temperature. Antibodies were diluted in Tris buffered saline (TBS) with 0.1% Triton X-100, 5% (vol / vol) donkey serum (Millipore, S30), and sodium azide (Sigma, S2002, 1:100). Sections were incubated with primary antibodies at 4 °C for two nights. The following primary antibodies were applied: Cd83 (mouse, Bio-rad, MCA1582, 1:50), Cd83 (mouse, BioLegend, 305302, 1:50), Gfap (sheep, R&D Systems, AF2594, 1:1000), Iba1 (rabbit, WAKO, 019–19741, 1:500), Olig2 (goat, R&D Systems, AF2418, 1:500), S100b (rabbit, Sigma, s2644, 1:500), Sox2 (goat, Santa Cruz, sc-17320, 1:200), Sox2 (goat, R&D Systems, AF2018, 1:200), Sox2 (mouse, Abcam, ab79351, 1:250), and Sox6 (rabbit, Millipore, AB5805, 1:250). The Cy2- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch; 1:300) to the appropriate species and DAPI (Thermo Fisher Scientific, D1306) were incubated at room temperature for 2 hours. After washing with TBS, sections were incubated with 1X TrueBlack (Biotium, 23007; diluted 1:20 in 70% ethanol) for 1 minute to block the auto-fluorescent lipofuscin and blood components. After washing with PBS, stained sections were mounted and imaged as Z-stacks on a Zeiss LSM 800 confocal microscope (Carl Zeiss) using a 20X or 40X objective with Zen 2 software (Carl Zeiss).
In-situ hybridization with immunostaining
PFA-fixed and cryopreserved 40 μm-thick hippocampal tissue sections were mounted on Superfrost™ Plus slides and dried at 60 °C for 10 minutes followed by dehydration with ethanol, blocking of endogenous peroxidases with hydrogen peroxide, antigen retrieval, protease treatment, and in-situ hybridization using the RNAscope™ Multiplex Fluorescent Reagent kit v2 according to manufacturer specifications (Advanced Cell Diagnostics (ACD), 323100). Tissue was probed with either EGR3 (ACD, 470161) or CCL2 (ACD, 423811) probes and developed with TSA plus Cyanine 3 (Akoya Biosciences, NEL744001KT, 1:100). Following in-situ hybridization, sections were washed in phosphate-buffer saline (PBS) with 0.1% TritonX-100 (PBST), and subsequently blocked with 10% donkey serum-PBST for 1 hour at room temperature. Slides were then incubated with anti-Iba1 antibodies (rabbit, WAKO, 019–19741, 1:500) in 1% donkey serum PBST overnight at 4 °C, washed and then incubated with fluorescently conjugated secondary (donkey anti-rabbit AlexaFluor 647, Thermofisher, A-31573, 1:500) for 2 hours at room temperature. Slides were incubated with 1x TrueBlack for 30 seconds to block autofluorescence, washed with PBS and coverslipped with DAPI. Sections were imaged by confocal as described above.
Image processing and data analyses
All confocal images were blindly acquired among different specimens under the same laser power and gain, analyzed using Imaris 9.0 software (BitPlane) as previously described (Sun et al., 2015a; Sun et al., 2015b). The Spots module in Imaris was used to digitize cell-nucleus locations in 3D space and to code cell type classifications according to distinct morphological and molecular markers. A minimum of three randomly chosen areas of equal dimensions in each section were quantitated. To minimize bias caused by sparse sampling of lowly abundant cells (e.g., CD83+ microglia), quantifications of all areas across three sections per patient were averaged and considered as one data point. We counted at least 100 cells to confidently quantify the ratio. No statistical methods were used to predetermine sample size. To quantify the spatio-temporal pattern of GFAP expression among S100B+ cells (Figure 3), subregions of the hippocampal formation were identified based on its distinct anatomical structure. Three randomly chosen areas of equal dimensions within each sub-region were quantitated and the sum of quantifications of these areas per section was considered as one data point.
QUANTIFICATION AND STATISTICAL ANALYSIS
The studies were blinded during data collection and quantification. Data in figure panels reflect several independent experiments performed on different days. No data were excluded. All data are shown as median ± quantiles. All statistical analyses are indicated in the text or figure legends and performed with the R language for statistical computing (https://www.r-project.org/).
Supplementary Material
Table S1. Human brain specimens used in the current study (A) and summary of sequencing characteristics of the snRNA-seq datasets (B), related to Figure 1.
This Table includes lists of significantly enriched genes for all major cell types (A), astrocyte subpopulations (B), oligodendrocyte lineage subpopulations (C), and microglia subpopulations (D).
This Table includes Gene Ontology terms related to enriched genes of astrocyte subpopulations (A), oligodendrocyte lineage subpopulations (B), and microglia subpopulations (C).
Table S5. Lists of genes dysregulated in AD in major cell types (A) and specific glia subpopulations (B) and their Gene Ontology terms (C), related to Figure 6.
Table S6. Summary of statistical methods used in different studies (A) and lists of genes dysregulated in AD shared among human hippocampus, prefrontal cortex and entorhinal cortex in astrocytes (B), oligodendrocytes (C), and microglia (D), related to Figure 6.
Table S4. Glia subpopulation-specific expression patterns across ages of risk genes in brain disorders, related to Figures 6 and S5.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD83 | Bio-rad | Cat# MCA1582; RRID: AB_321773 |
| Mouse anti-CD83 | BioLegend | Cat# 305302; RRID: AB_314510 |
| Sheep anti-GFAP | R&D Systems | Cat# AF2594; RRID: AB_2109656 |
| Rabbit anti-IBA1 | WAKO | Cat# 019-19741; RRID: AB_839504 |
| Goat anti-OLIG2 | R&D Systems | Cat# AF2418; RRID: AB_2157554 |
| Rabbit anti-S100B | Sigma | Cat# S2644; RRID: AB_477501 |
| Goat anti-SOX2 | Santa Cruz | Cat# sc-17320; RRID: AB_2286684 |
| Goat anti-SOX2 | R&D Systems | Cat# AF2018; RRID: AB_355110 |
| Mouse anti-SOX2 | Abcam | Cat# ab79351; RRID: AB_10710406 |
| Rabbit anti-SOX6 | Millipore | Cat# AB5805; RRID: AB_2302618 |
| Donkey anti-goat secondary antibody, Cyanine 2 | Jackson ImmunoResearch | Cat# 705-225-147; RRID: AB_2307341 |
| Donkey anti-mouse secondary antibody, Cyanine 2 | Jackson ImmunoResearch | Cat# 715-225-151; RRID: AB_2340827 |
| Donkey anti-rabbit secondary antibody, Cyanine 2 | Jackson ImmunoResearch | Cat# 711-225-152; RRID: AB_2340612 |
| Donkey anti-sheep secondary antibody, Cyanine 2 | Jackson ImmunoResearch | 713-225-147; RRID: AB_2340735 |
| Donkey anti-goat secondary antibody, Cyanine 3 | Jackson ImmunoResearch | Cat# 705-165-147; RRID: AB_2307351 |
| Donkey anti-mouse secondary antibody, Cyanine 3 | Jackson ImmunoResearch | Cat# 715-165-151; RRID: AB_2315777 |
| Donkey anti-rabbit secondary antibody, Cyanine 3 | Jackson ImmunoResearch | Cat# 711-165-152; RRID: AB_2307443 |
| Biological samples | ||
| Human post-mortem hippocampal specimens | Children’s Hospital of Philadelphia; Johns Hopkins University Pathology Archive; Lieber Institute for Brain Development; Multiple repositories from the NIH NeuroBioBank | Full list in Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| Bovine serum albumin (BSA) | Life Technologies | Cat# 37525 |
| DAKO target retrieval solution (10X) | DAKO | Cat# S1699 |
| DAPI | Thermo Fisher Scientific | Cat# D1306; RRID: AB_2629482 |
| DL-Dithiothreitol (DTT) | Sigma | Cat# D0632 |
| dNTP | Thermo Fisher | Cat# R0192 |
| Donkey serum | Millipore | Cat# S30 |
| EDTA | cellgro | Cat# 46034Cl |
| EDTA-free protease inhibitor | Roche | Cat# 11836170001 |
| EvaGreen dye, 20X in Water | Biotium | Cat# 31000 |
| Ficoll solution, type 400 | Sigma | Cat# F5415-25ML |
| IGEPAL-630 | Sigma-Aldrich | Cat# I8896-50ML |
| Maxima H Minus Reverse Transcriptase | Fisher Scientific | Cat# EP0753 |
| MgCl2 | Thermo Fisher Scientific | Cat# AM9530G |
| Myelin Removal Beads II | Miltenyi Biotec | Cat# 130-096-733 |
| NaCl | Ambion | Cat# AM9760G |
| NEBuffer 3.1 | New England Biolabs | Cat# B7203s |
| Paraformaldehyde (PFA), 16%, methanol-free | Fisher Scientific | Cat# 50-980-487 |
| Phosphate-Buffered Saline (PBS) | Corning | Cat# 21-040 |
| PMSF protease inhibitor | Thermo Fisher | Cat# 36978 |
| Proteinase K Solution (20 mg/mL) | Thermo Fisher | Cat# AM2546 |
| RNase Inhibitor | Enzymatics | Cat# Y924L |
| SDS | Thermo Fisher | Cat# 15553027 |
| Sodium azide | Sigma-Aldrich | Cat# S2002 |
| Spermidine | Sigma-Aldrich | Cat# S0266-1G |
| Spermine | Sigma-Aldrich | Cat# S4264-1G |
| SPRIselect Beads | BECKMAN COULTER | Cat# B23318 |
| Sucrose | Sigma-Aldrich | Cat# S5016-500G |
| Superase-In RNase Inhibitor | Thermo Fisher | Cat# AM2694 |
| T4 DNA Ligase | New England Biolabs | Cat# M0202S |
| Tricine-KOH | Sigma-Aldrich | Cat# T5816-100G |
| Tris-HCl, pH8.0 | Ambion | Cat# AM9855G |
| Triton X-100 | Sigma-Aldrich | Cat# T9284 |
| Tween-20 | Thermo Fisher | Cat# 85113 |
| Xylene | Fisher Scientific | Cat# X5-1 |
| Critical Commercial Assays | ||
| Agilent High Sensitivity DNA Kit | Aligent | Cat# 5067-4626 |
| Dynabeads™ MyOne™ Streptavidin C1 Kit | Thermo Fisher | Cat# 65001 |
| Fuchs-Rosenthal hemocytometer | Incyto | Cat# DHCF015 |
| Kapa HiFi HotStart Master Mix | KAPA Biosystems | Cat# KK2601 |
| NextSeq 500/550 High Output v2.5 kit (150 cycles) | Illumina | Cat# 20024907 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131-1024 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Cat# Q32854 |
| TrueBlack Lipofuscin Autofluorescence Quencher | Biotium | Cat# 23007 |
| Oligonucleotides | ||
| Split barcode primer: Round 1/2/3 | IDT | (Rosenberg et al., 2018) |
| Deposited data | ||
| Human hippocampus snRNA-seq data | This paper | GSE185553, GSE198323 and GSE199243 |
| Human anterior cingulate cortex (ACC) and prefrontal cortex (PFC) snRNA-seq data (controls) | (Velmeshev et al., 2019) | PRJNA434002 |
| Human cerebellum, frontal cortex and visual cortex snRNA-seq data | (Lake et al., 2018) | GSE97930 |
| Human motor cortex snRNA-seq data (controls) | (Schirmer et al., 2019) | PRJNA544731 |
| Human white matter snRNA-seq data (controls) | (Jakel et al., 2019) | GSE118257 |
| Sliced human cortical organoid snRNA-seq data | (Qian et al., 2020) | GSE137941 |
| Sliced human brain organoid scRNA-seq data | (Szebenyi et al., 2021) | GSE180122 |
| iPSC-derived microglia cells scRNA-seq data | (Popova et al., 2021) | GSE180945 |
| iPSC-derived microglia cells scRNA-seq data | (Svoboda et al., 2019) | GSE139193 |
| iPSC-derived oligodendrocyte scRNA-seq data | (Chamling et al., 2021) | GSE146373 |
| iPSC-derived oligodendrocyte scRNA-seq data | (Marton et al., 2019) | GSE115011 |
| Software and algorithms | ||
| Adobe Illustrator CS6 | Adobe | https://www.adobe.com/products/illustrator.html; RRID: SCR_010279 |
| Adobe Photoshop CS6 | Adobe | https://www.adobe.com/products/photoshop.html; RRID:SCR_014199 |
| Bamtools (v2.3.0) | NA | https://bioinformatics.readthedocs.io/en/latest/bamtools/; RRID: SCR_015987 |
| bcl2fastq (v2.17.1) | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html; RRID: SCR_015058 |
| clusterProfiler (v4) | (Wu et al., 2021) | https://github.com/YuLab-SMU/clusterProfiler/; RRID: SCR_016884 |
| DAVID Knowledge Base (v2021q4) | https://david-d.ncifcrf.gov; RRID:SCR_001881 | |
| Drop-seq tools (v1.13) | (Macosko et al., 2015) | http://mccarrolllab.org/dropseq/; RRID: SCR_018142 |
| GeneOverlap | (Shen, 2021) | https://bioconductor.org/packages/release/bioc/html/GeneOverlap.html; RRID:SCR_018419 |
| Imaris 9.0 | Bit Plane | http://www.bitplane.com/imaris/imaris/; RRID: SCR_007370 |
| JAVA (v 1.8.0) | JAVA | https://www.java.com/en/download/ |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/p/excel/cfq7ttc0k7dx?activetab=pivot%3aoverviewtab; RRID: SCR_016137 |
| PicardTools (v2.13.2) | Broad Institute | http://broadinstitute.github.io/picard/; RRID: SCR_006525 |
| R (v4.0.4) | R | https://cran.r-project.org/; RRID: SCR_001905 |
| R Studio | RStudio | https://rstudio.com/; RRID: SCR_000432 |
| randomForest (v4.6.14) | (Breiman, 2001) | https://cran.r-project.org/web/packages/randomForest/index.html; RRID: SCR_015718 |
| Samtools (v1.1) | NA | http://www.htslib.org/; RRID:SCR_005227 |
| scDbiFinder (v1.4.0) | (Germain et al., 2021) | https://bioconductor.org/packages/release/bioc/html/scDblFinder.html |
| sctransform (v0.3) | (Hafemeister and Satija, 2019) | https://github.com/ChristophH/sctransform |
| Seurat (v 4.0.0) | (Hao et al., 2021) | https://satijalab.org/seurat/; RRID: SCR_007322 |
| STAR (v2.5.2a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR; RRID: SCR_015899 |
| UMAP | R package | https://umap-learn.readthedocs.io/en/latest/; RRID:SCR_018217 |
| Zen 2 | Carl Zeiss | https://www.zeiss.com; RRID: SCR_013672 |
| Other | ||
| 40-μm cell strainer | Fisher Scientific | 22-363-547 |
| Bioanalyzer 2100 | Aligent | G2939BA |
| Confocal microscope | Carl Zeiss | Zeiss LSM 800 |
| Dounce homogenizer | Fisher Scientific | 8853000002 |
| Frozen sliding microtome | Leica | Cat# SM2010R |
| Illumina NextSeq 550 sequencer | Illumina | |
| Qubit Fluorometer | Thermo Fisher Scientific | Q33238 |
| T100 Thermal Cycler | Bio-rad | 1861096EDU |
HIGHLIGHTS.
A single-nucleus transcriptome atlas of human glia diversity across postnatal life
A transcriptome reference map assessing glia differentiation of human stem cells
Spatiotemporal heterogeneity of GFAP-enriched astrocytes within human hippocampus
Transcriptomic dysregulation in hippocampal glia subpopulations in Alzheimer’s
ACKNOWLEDGMENTS
We thank all brain donors, patients, and their families for the tissue specimens used in this study and bio-tissue banks for coordination. We thank J.R. Glausier, A.N. LeFevre, J. Cottrell, W.K. Scott, and O. Spicer for help in obtaining tissue samples. We thank members of Ming and Song laboratories for discussion, K.M. Christian for comments, B. Temsamrit, E. LaNoce, and A. Garcia for technical support, and A. Angelucci and J. Schnoll for lab coordination. Some schematic illustrations were modified using images from biorender.com. This work was supported by grants from the National Institutes of Health (R35NS097370 and RF1MH123979 to G-l.M., R35NS116843, U19MH106434, R01AG061852, R01AG057497 and RF1AG079557 to H.S., R01NS127913 and R21MH122239 to Y.S.), The Lieber Institute for Brain Development (to J.E.K., T.M.H., and D.R.W.), Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G.-l.M.). M.L.B was supported by R25NS065745. M.C.-C. was supported by a fellowship from the “La Caixa” Foundation.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographic location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Footnotes
DECLARATION OF INTERESTS
G.-l.M. is a member of the Advisory Board for Cell Stem Cell. The authors declare no other competing interests.
REFERENCES
- Ayhan F, Kulkarni A, Berto S, Sivaprakasam K, Douglas C, Lega BC, and Konopka G. (2021). Resolving cellular and molecular diversity along the hippocampal anterior-to-posterior axis in humans. Neuron 109, 2091–2105 e2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Ben Haim L, and Rowitch DH (2017). Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18, 31–41. [DOI] [PubMed] [Google Scholar]
- Bennett ML, and Bennett FC (2020). The influence of environment and origin on brain resident macrophages and implications for therapy. Nat Neurosci 23, 157–166. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, and Jahr CE (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191. [DOI] [PubMed] [Google Scholar]
- Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, et al. (2020). Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (2001). Random Forests. Machine Learning 45, 5–32. [Google Scholar]
- Chamling X, Kallman A, Fang W, Berlinicke CA, Mertz JL, Devkota P, Pantoja IEM, Smith MD, Ji Z, Chang C, et al. (2021). Single-cell transcriptomic reveals molecular diversity and developmental heterogeneity of human stem cell-derived oligodendrocyte lineage cells. Nature communications 12, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavali M, Ulloa-Navas MJ, Perez-Borreda P, Garcia-Verdugo JM, McQuillen PS, Huang EJ, and Rowitch DH (2020). Wnt-Dependent Oligodendroglial-Endothelial Interactions Regulate White Matter Vascularization and Attenuate Injury. Neuron 108, 1130–1145 e1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila-Velderrain J, Mathys H, Mohammadi S, Ruzicka B, Jiang X, Ng A, Bennett DA, Tsai L-H, and Kellis M. (2021). Single-cell anatomical analysis of human hippocampus and entorhinal cortex uncovers early-stage molecular pathology in Alzheimer’s disease. 2021.2007.2001.450715.
- de Leeuw CA, Mooij JM, Heskes T, and Posthuma D. (2015). MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers C, Schirmer S, Kehlenbach RH, Hauber J, and Chemnitz J. (2013). Post-transcriptional regulation of CD83 expression by AUF1 proteins. Nucleic Acids Res 41, 206–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, and Barres BA (2010). Regulation of synaptic connectivity by glia. Nature 468, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcao AM, van Bruggen D, Marques S, Meijer M, Jakel S, Agirre E, Floriddia E.M. Samudyata, Vanichkina DP, Ffrench-Constant C, et al. (2018). Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med 24, 1837–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franjic D, Skarica M, Ma S, Arellano JI, Tebbenkamp ATN, Choi J, Xu C, Li Q, Morozov YM, Andrijevic D, et al. (2022). Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron 110, 452–469 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM, and Ffrench-Constant C. (2017). Regenerating CNS myelin - from mechanisms to experimental medicines. Nature reviews Neuroscience 18, 753–769. [DOI] [PubMed] [Google Scholar]
- Germain P-L, Lun A, Macnair W, and Robinson M. (2021). Doublet identification in single-cell sequencing data using scDblFinder (F1000Res). [DOI] [PMC free article] [PubMed]
- Giannakopoulou O, Lin K, Meng X, Su MH, Kuo PH, Peterson RE, Awasthi S, Moscati A, Coleman JRI, Bass N, et al. (2021). The Genetic Architecture of Depression in Individuals of East Asian Ancestry: A Genome-Wide Association Study. JAMA Psychiatry 78, 1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, et al. (2020). Disease-associated astrocytes in Alzheimer’s disease and aging. Nature neuroscience 23, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R. (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, and Guo AY (2019). AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res 47, D33–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Fabyanic E, Kwon DY, Tang S, Zhou Z, and Wu H. (2017). Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol Cell 68, 1006–1015 e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International League Against Epilepsy Consortium on Complex, E. (2018). Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat Commun 9, 5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel S, Agirre E, Mendanha Falcao A, van Bruggen D, Lee KW, Knuesel I, Malhotra D, Ffrench-Constant C, Williams A, and Castelo-Branco G. (2019). Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, Strasburger H, Herbst L, Alexis M, Karnell J, et al. (2019). Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun 10, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SF, Cao H, Fu AKY, and Ip NY (2020). Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci U S A 117, 25800–25809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Kim JY, Noh S, Lee H, Lee SY, Mun JY, Park H, and Chung WS (2021). Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 590, 612–617. [DOI] [PubMed] [Google Scholar]
- Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nature neuroscience 24, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, and Barres BA (2017). Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SE, Walker AJ, Kamath T, Dissing-Olesen L, Hammond TR, de Soysa TY, Young AMH, Murphy S, Abdulraouf A, Nadaf N, et al. (2022). Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat Neurosci 25, 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton RM, Miura Y, Sloan SA, Li Q, Revah O, Levy RJ, Huguenard JR, and Pasca SP (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat Neurosci 22, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Scheiwe C. Sagar, Nessler S, Kunz P, van Loo G, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. [DOI] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, and Prinz M. (2020). Microglia Heterogeneity in the Single-Cell Era. Cell Rep 30, 1271–1281. [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, Silva J, Leavy K, Perez-Rosendahl M, and Swarup V. (2021). Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, Als TD, Bigdeli TB, Borte S, Bryois J, et al. (2021). Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet 53, 817–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, and Werner HB (2014). Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol 30, 503–533. [DOI] [PubMed] [Google Scholar]
- Nichols NR, Day JR, Laping NJ, Johnson SA, and Finch CE (1993). GFAP mRNA increases with age in rat and human brain. Neurobiol Aging 14, 421–429. [DOI] [PubMed] [Google Scholar]
- Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nature communications 11, 6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otowa T, Hek K, Lee M, Byrne EM, Mirza SS, Nivard MG, Bigdeli T, Aggen SH, Adkins D, Wolen A, et al. (2016). Meta-analysis of genome-wide association studies of anxiety disorders. Mol Psychiatry 21, 1485. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Popova G, Soliman SS, Kim CN, Keefe MG, Hennick KM, Jain S, Li T, Tejera D, Shin D, Chhun BB, et al. (2021). Human microglia states are conserved across experimental models and regulate neural stem cell responses in chimeric organoids. Cell Stem Cell 28, 2153–2166 e2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Jung S, and Priller J. (2019). Microglia Biology: One Century of Evolving Concepts. Cell 179, 292–311. [DOI] [PubMed] [Google Scholar]
- Qian X, Song H, and Ming GL (2019). Brain organoids: advances, applications and challenges. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, Su K, Li S, Lu L, Jacob F, et al. (2020). Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 26, 766–781 e769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N, Almouzni G, Gorski SA, Aerts S, Amit I, Bertero MG, Bock C, Bredenoord AL, Cavalli G, Chiocca S, et al. (2020). LifeTime and improving European healthcare through cell-based interceptive medicine. Nature 587, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, and Liddelow SA (2022). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 110, 1788–1805 e1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Deprada A, Capilla-Lopez MD, and Parra-Damas A. (2022). Revealing cell vulnerability in Alzheimer’s disease by single-cell transcriptomics. Seminars in cell & developmental biology. [DOI] [PubMed]
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, Cummings J, and van der Flier WM (2021). Alzheimer’s disease. Lancet 397, 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer L, Velmeshev D, Holmqvist S, Kaufmann M, Werneburg S, Jung D, Vistnes S, Stockley JH, Young A, Steindel M, et al. (2019). Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323 e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, and Barnes CA (2011). A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nature reviews Neuroscience 12, 585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H. (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GJ, Zhou Y, Ito S, Bonaguidi MA, Stein-O’Brien G, Kawasaki NK, Modak N, Zhu Y, Ming GL, and Song H. (2015a). Latent tri-lineage potential of adult hippocampal neural stem cells revealed by Nf1 inactivation. Nat Neurosci 18, 1722–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GJ, Zhou Y, Stadel RP, Moss J, Yong JH, Ito S, Kawasaki NK, Phan AT, Oh JH, Modak N, et al. (2015b). Tangential migration of neuronal precursors of glutamatergic neurons in the adult mammalian brain. Proc Natl Acad Sci U S A 112, 9484–9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Akay LA, Murdock MH, Park Y, Bubnys A, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, et al. (2022). Single-cell multi-region dissection of brain vasculature in Alzheimer’s Disease. 2022.2002.2009.479797.
- Svoboda DS, Barrasa MI, Shu J, Rietjens R, Zhang S, Mitalipova M, Berube P, Fu D, Shultz LD, Bell GW, et al. (2019). Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proceedings of the National Academy of Sciences of the United States of America 116, 25293–25303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi K, Wenger LMD, Sun Y, Dunn AWE, Limegrover CA, Gibbons GM, Conci E, Paulsen O, Mierau SB, Balmus G, et al. (2021). Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nature neuroscience 24, 1542–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MN, Maynard KR, Spangler A, Huuki LA, Montgomery KD, Sadashivaiah V, Tippani M, Barry BK, Hancock DB, Hicks SC, et al. (2021). Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 109, 3088–3103 e3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trubetskoy V, Pardinas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, Bryois J, Chen CY, Dennison CA, Hall LS, et al. (2022). Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Bahney J, and Herculano-Houzel S. (2016). The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol 524, 3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, Rongve A, Borte S, Winsvold BS, Drange OK, et al. (2022). A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease (Sep, 10.1038/s41588-021-00921-z, 2021). Nat Genet 54, 1062–1062. [DOI] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y) 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, et al. (2022). A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Clyne M, Khoury MJ, and Gwinn M. (2010). Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 26, 145–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakzanis KK, Graham SJ, and Campbell Z. (2003). A meta-analysis of structural and functional brain imaging in dementia of the Alzheimer’s type: a neuroimaging profile. Neuropsychol Rev 13, 1–18. [DOI] [PubMed] [Google Scholar]
- Zeng H. (2022). What is a cell type and how to define it? Cell 185, 2739–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Song H, and Ming GL (2021). Modeling neurological disorders using brain organoids. Seminars in cell & developmental biology 111, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Ding W, Sun L, Lu Y, Dong H, Fan X, Liu Z, Chen R, Zhang S, Ma Q, et al. (2020). Decoding the development of the human hippocampus. Nature 577, 531–536. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Bond AM, Shade JE, Zhu Y, Davis CO, Wang X, Su Y, Yoon KJ, Phan AT, Chen WJ, et al. (2018). Autocrine Mfge8 Signaling Prevents Developmental Exhaustion of the Adult Neural Stem Cell Pool. Cell Stem Cell 23, 444–452 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Su Y, Li S, Kennedy BC, Zhang DY, Bond AM, Sun Y, Jacob F, Lu L, Hu P, et al. (2022). Molecular landscapes of human hippocampal immature neurons across lifespan. Nature 607, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Sousa AMM, Gao T, Skarica M, Li M, Santpere G, Esteller-Cucala P, Juan D, Ferrandez-Peral L, Gulden FO, et al. (2018). Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Human brain specimens used in the current study (A) and summary of sequencing characteristics of the snRNA-seq datasets (B), related to Figure 1.
This Table includes lists of significantly enriched genes for all major cell types (A), astrocyte subpopulations (B), oligodendrocyte lineage subpopulations (C), and microglia subpopulations (D).
This Table includes Gene Ontology terms related to enriched genes of astrocyte subpopulations (A), oligodendrocyte lineage subpopulations (B), and microglia subpopulations (C).
Table S5. Lists of genes dysregulated in AD in major cell types (A) and specific glia subpopulations (B) and their Gene Ontology terms (C), related to Figure 6.
Table S6. Summary of statistical methods used in different studies (A) and lists of genes dysregulated in AD shared among human hippocampus, prefrontal cortex and entorhinal cortex in astrocytes (B), oligodendrocytes (C), and microglia (D), related to Figure 6.
Table S4. Glia subpopulation-specific expression patterns across ages of risk genes in brain disorders, related to Figures 6 and S5.
Data Availability Statement
All snRNA-seq data are available at the GEO database (accession numbers GSE185553, GSE198323 and GSE199243) and UCSC Cell Brower (https://hippo-lifespan.cells.ucsc.edu). Information on the de-identified human specimens used in this study and their sequencing characteristics are described in Table S1. Scripts used in this study are available at https://github.com/ysu2015/HumanHippocampus_scRNAseq, maintained by Dr. Yijing Su (yijingsu@pennmedicine.upenn.edu). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.