

Abstract
CD4+ T cell differentiation requires metabolic reprogramming to fulfil the bioenergetic demands of proliferation and effector function, and enforce specific transcriptional programmes1–3. Mitochondrial membrane dynamics sustains mitochondrial processes4, including respiration and tricarboxylic acid (TCA) cycle metabolism5, but whether mitochondrial membrane remodelling orchestrates CD4+ T cell differentiation remains unclear. Here we show that unlike other CD4+ T cell subsets, T helper 17 (TH17) cells have fused mitochondria with tight cristae. T cell-specific deletion of optic atrophy 1 (OPA1), which regulates inner mitochondrial membrane fusion and cristae morphology6, revealed that TH17 cells require OPA1 for its control of the TCA cycle, rather than respiration. OPA1 deletion amplifies glutamine oxidation, leading to impaired NADH/NAD+ balance and accumulation of TCA cycle metabolites and 2-hydroxyglutarate–a metabolite that influences the epigenetic landscape5,7. Our multi-omics approach revealed that the serine/threonine kinase liver-associated kinase B1 (LKB1) couples mitochondrial function to cytokine expression in TH17 cells by regulating TCA cycle metabolism and transcriptional remodelling. Mitochondrial membrane disruption activates LKB1, which restrains IL-17 expression. LKB1 deletion restores IL-17 expression in TH17 cells with disrupted mitochondrial membranes, rectifying aberrant TCA cycle glutamine flux, balancing NADH/NAD+ and preventing 2-hydroxyglutarate production from the promiscuous activity of the serine biosynthesis enzyme phosphoglycerate dehydrogenase (PHGDH). These findings identify OPA1 as a major determinant of TH17 cell function, and uncover LKB1 as a sensor linking mitochondrial cues to effector programmes in TH17 cells.
Mitochondrial function is linked to mitochondrial membrane morphology4. Remodelling of the inner mitochondrial membrane and outer mitochondrial membrane (OMM) is coordinated by several GTPases that control the opposing and dynamic processes of mitochondrial fusion and fission8 (division). Mitochondrial membrane rearrangements sustain mitochondrial function by altering cristae architecture, respiratory complex organization, mtDNA metabolism and organelle distribution8. Mitochondrial membrane remodelling is especially relevant during metabolic or environmental fluctuations9 and diverse signalling pathways regulate pro-fusion and pro-fission proteins to shape mitochondrial structure and support metabolic adaptations. Conversely, mitochondria and mitochondrial membrane remodelling participate in intracellular signalling events, modulating transcription and epigenetics during cell differentiation, immune responses, tumour angiogenesis and cell death4,10,11. Given the role of mitochondrial membrane dynamics and metabolism in cell function, we investigated how mitochondrial membrane morphology affects CD4+ T cell differentiation.
TH17 cells have fused mitochondria
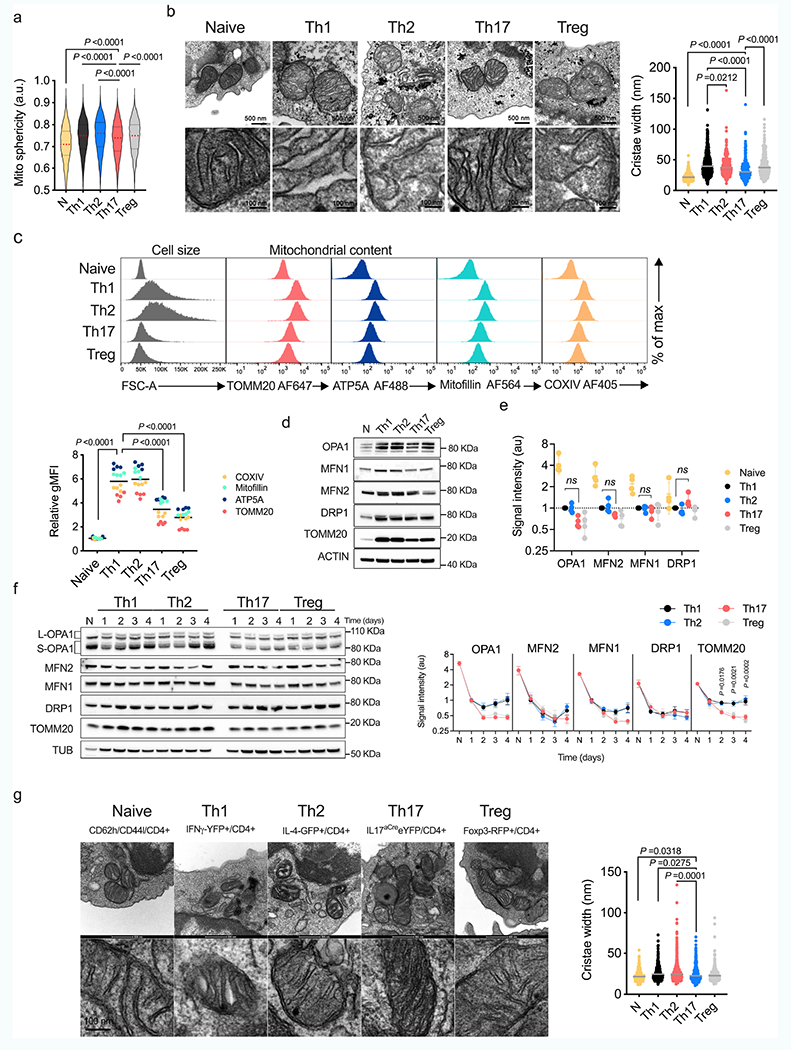
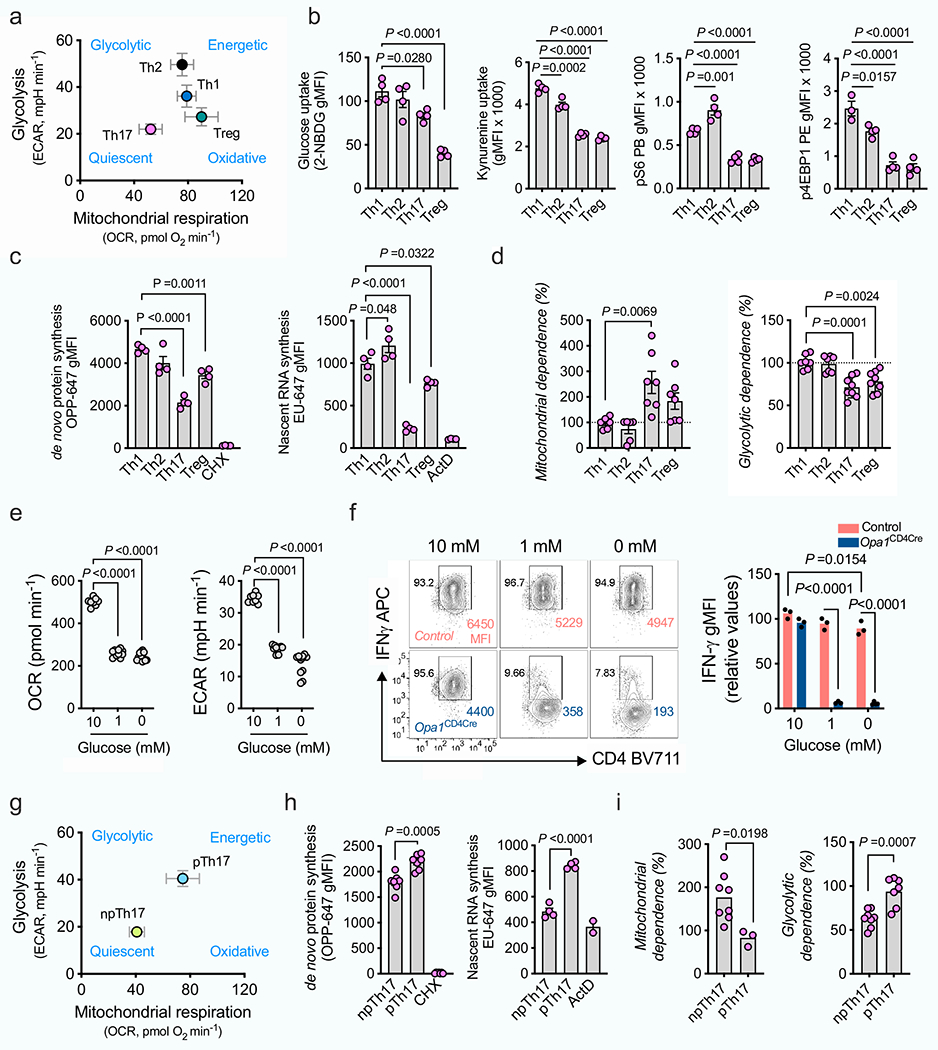
We polarized naive CD4+ T (TN) cells expressing a mitochondria-targeted fluorescent protein, into TH1, TH2, TH17 and T regulatory (Treg) cell subsets in vitro and assessed their mitochondrial morphology. TH17 cells contained elongated mitochondria with tight cristae (Fig. 1a and Extended Data Fig. 1a,b). Although in vitro-generated TH1 and TH2 cells had increased mitochondrial content and cell size compared with TH17 and Treg cells, all cells had similar amounts of proteins associated with mitochondrial membrane remodelling (Fig. 1b and Extended Data Fig. 1c–e). OPA1 regulates cristae morphology and inner mitochondrial membrane fusion6,12. OPA1 isoforms affect pro-fusion activity13,14, with long isoforms associated with tighter, fused mitochondrial membrane. Accumulation of long and short OPA1 isoforms (l-OPA1 and s-OPA1, respectively) varied across cell subsets (Fig. 1b)–l-OPA1 accumulated in TH17 cells, whereas s-OPA1 accumulated in TH1 and TH2 cells (Fig. 1c and Extended Data Fig. 1f).
Fig. 1 |. OPA1 is required for TH17 cell effector function.
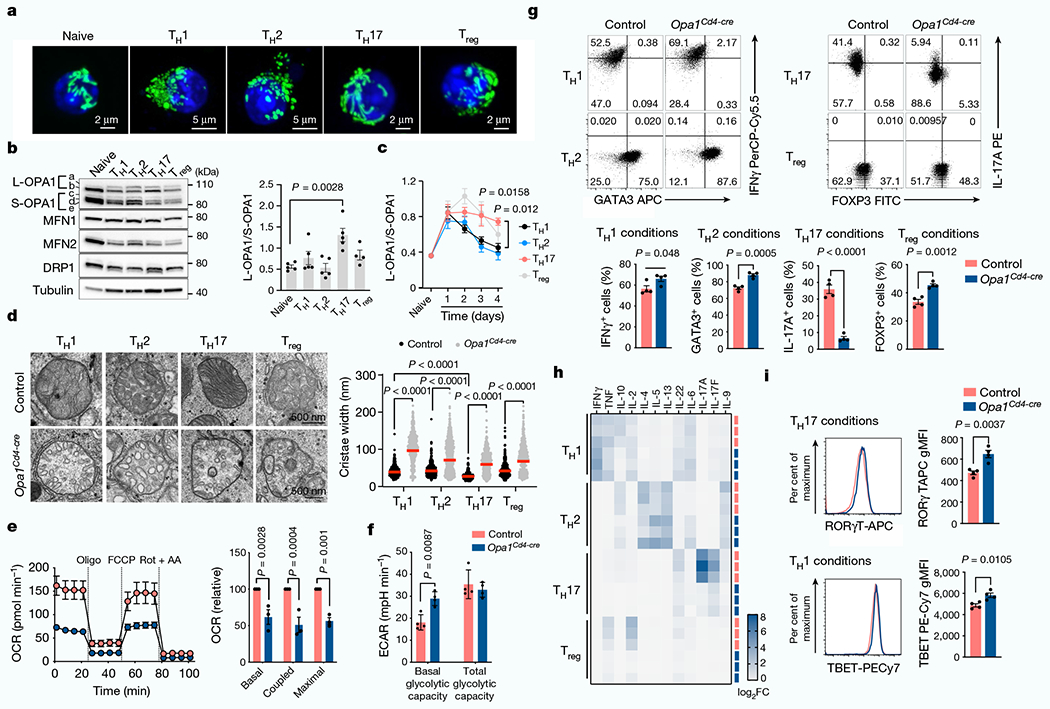
a, Representative live-cell spinning disk microscopy images showing mitochondria (GFP, green) and nuclei (Hoechst 42, blue) in CD4+ T cells from PhAM mice cultured in T helper (TH) and Treg cell conditions on day 4. Two separate experiments. b, Immunoblot of OPA1, MFN2, MFN1, DRP1 (left) and ratio of l- and s-OPA1 isoforms (right) in TN, TH and Treg T cells on day 4. Tubulin is used as a sample processing control. c, Kinetics of OPA1 isoforms during TH and Treg differentiation. In b,c, TH1, TH2 and TH17: n = 5 biological replicates, Treg: n = 4 biological replicates and 3 independent experiments. d, Electron microscopy (left) and width of mitochondrial cristae (right). Control TH1: n = 442, control TH2: n = 401, control TH17: n = 418, control Treg: n = 421, Opa1Cd4-cre TH1: n = 615, Opa1Cd4-cre TH2: n = 786, Opa1Cd4-cre TH17: n = 554 and Opa1Cd4-cre Treg: n = 433 cristae, and 3 biological replicates. Data are shown as individual measurements and geometric mean (red bar). e, Left, oxygen consumption rate (OCR) on addition of oligomycin (oligo), fluoro-carbonyl cyanide phenylhydrazone (FCCP) and rotenone plus antimycin A (Rot + AA) (n = 7 injections, 3 biological replicates). Right, baseline OCR, ATP-coupled respiration and maximal respiration (n = 3 independent experiments). f, Basal glycolytic rate and total glycolytic capacity (n = 4 biological replicates), measured by extracellular acidification rate (ECAR). g, Cells expressing IFNγ, 1L-17A, GATA3 and FOXP3 following restimulation (n = 4 biological replicates). The percentage of cells in each quadrant is indicated. h, Heat map of cytokine expression in the supernatant of indicated cells types, shown as log2 fold change (FC) (n = 3 biological replicates). i, TBET and ROR-γT expression (n = 4 biological replicates). gMFI, geometric mean fluorescence intensity. Representative plots and graphs summarize results of at least two independent experiments except where noted otherwise. Data in b,c,e-g,i are mean ± s.e.m. Two-sided Tukey’s test (b,d), Dunnett’s test (c), Šidak’s test (e,f) or unpaired two-tailed f-test (g,i). Exact P-values are indicated.
Using distinct fluorescent reporter mice, we next analysed mitochondria ultrastructure in vivo in CD4+ T cells after infection or experimental autoimmune encephalomyelitis (EAE), a TH17 cell-driven disease15. Interferon-γ (IFNγ)–YFP-expressing TH1 cells infected with Listeria monocytogenes exhibited looser cristae than CD4+ TN cells (Extended Data Fig. 1g). In mice co-expressing IL-4-GFP and FOXP3-RFP, cristae loosening was found in TH2 cells expressing IL-4-GFP, but not in those expressing FOXP3-RFP (that is, Treg cells) after infection with the helminth Heligmosomoides polygyrus (Extended Data Fig. 1g). Looser cristae in TH1 and TH2 cells contrasted with tighter cristae in IL-17A-cre-eYFP-expressing TH17 cells at the peak of EAE disease (Extended Data Fig. 1g), which exhibited a mitochondrial membrane configuration resembling CD4+ TN cells.
OPA1 is needed for IL-17 production
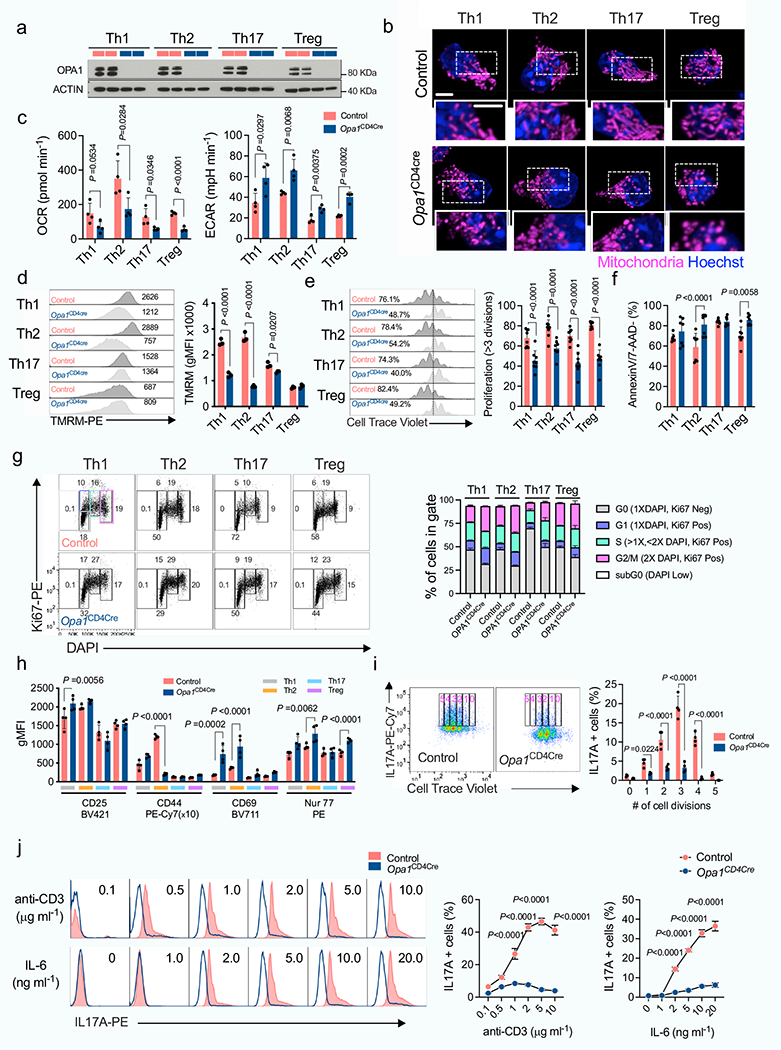
We next crossed Opa1-floxed mice with Cd4-cre mice (generating Opa1Cd4-cre mice) to delete OPA1 in T cells. In vitro-generated T cell subsets from these mice showed disrupted mitochondrial morphology and function upon OPA1 loss (Fig. 1d–f and Extended Data Fig. 2a–d). Whereas Opa1Cd4-cre TH17 cells expressed less interleukin-17A (IL-17A), Opa1Cd4-cre TH1 cells did not show decreased expression of IFNγ and tumour necrosis factor (TNF) and Opa1Cd4-cre TH2 cells did not show decreased expression of IL-4, IL-5 and IL-13 (Fig. 1g,h). Lineage-specifying transcription factor expression remained intact across subsets regardless of genotype (Fig. 1g,i).
OPA1 loss decreased cell proliferation, which was not associated with reduced viability, but rather defective cell cycle progression, as more cells expressed the proliferation marker Ki67 in the G2 and M phase (Extended Data Fig. 2e–g). Reduced IL-17A expression in Opa1Cd4-cre was independent of cell division, early activation marker expression, or T cell receptor (TCR) or IL-6 signal strength (Extended Data Fig. 2h–j). Thus, OPA1 sustains cytokine production in TH17 cells in vitro, but not early differentiation events associated with transcription factor expression.
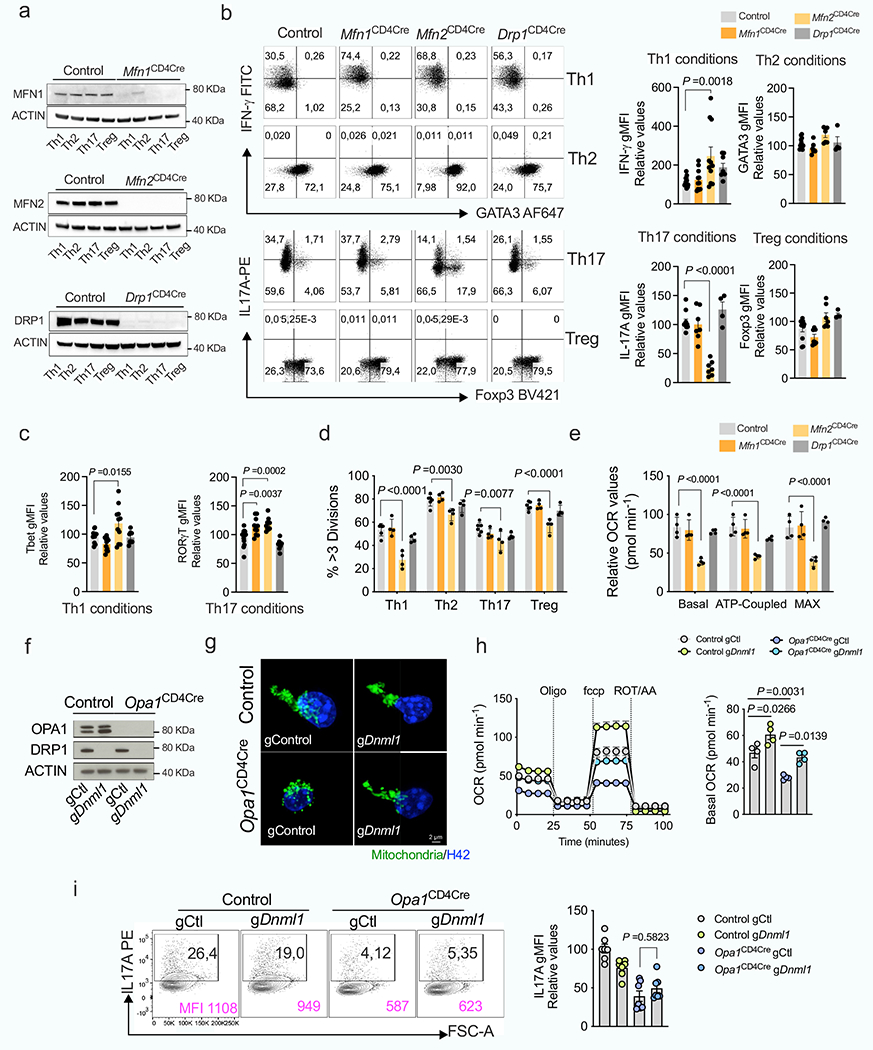
Loss of mitofusin 1 (MFN1) or dynamin-related protein 1 (DRP1)–which mediate OMM fusion and fission, respectively–did not affect T cell function (Extended Data Fig. 3a–e), whereas loss of MFN2–which controls OMM fusion, interorganelle tethering and calcium signalling–altered immunometabolic function across subsets (Extended Data Fig. 3a–e). Thus, OMM remodelling per se does not sustain TH17 effector function, and fusion-independent roles of MFN2 probably affect mitochondrial function to control TH17 effector function in a similar manner to OPA1. Notably, DRP1 deletion in Opa1Cd4-cre cells re-established OMM configuration and respiration but not IL-17A expression (Extended Data Fig. 3f–i), indicating that OPA1 supports TH17 effector function independently of its role in respiration.
OPA1 sustains TH17 cells in vivo
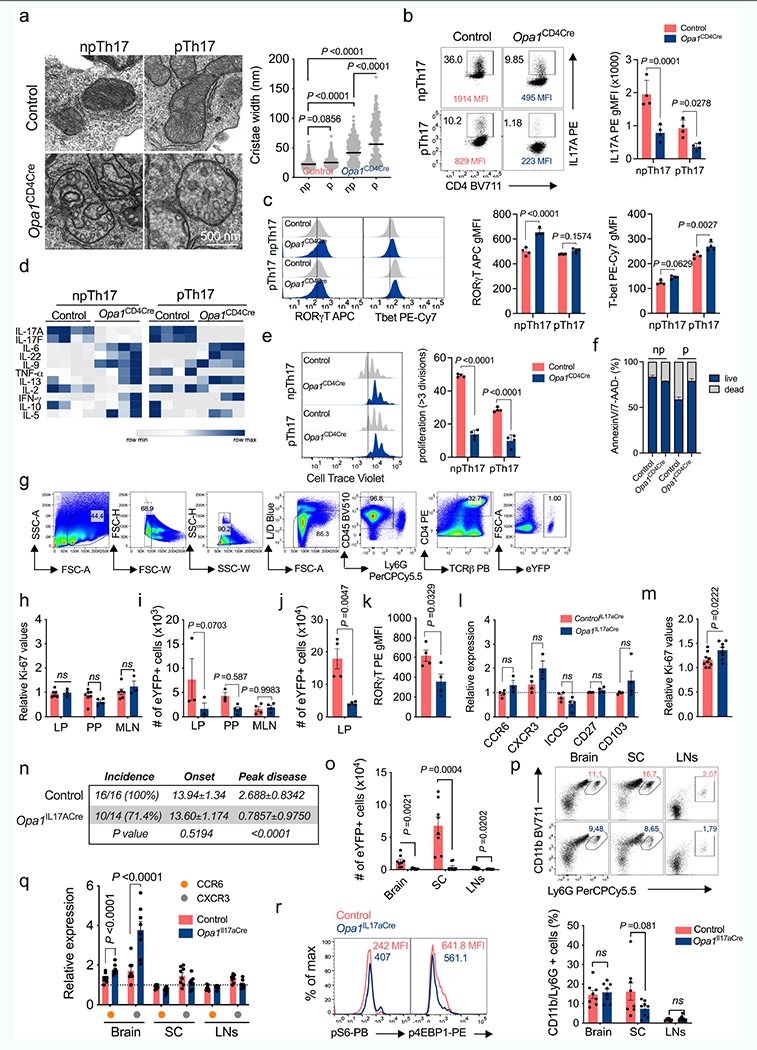
TH17 cells maintain gut homeostasis and protect against pathogens, as well as participate in inflammatory diseases15. These different functional states–non-pathogenic (npTH17) and pathogenic (pTH17)–exhibit distinct cytokine expression and metabolism16. OPA1 deletion disrupted cristae, cytokine expression, proliferation and cell cycle progression, but not viability or transcription factor expression in CD4+ TN cells cultured under npTH17 (with TGFβ and IL-6) or pTH17 (with IL-23, IL-6 and IL-1β) conditions (Extended Data Fig. 4a–f), illustrating that both cellular states rely on OPA1.
TH17 cells respond to segmented filamentous bacteria in the mesenteric lymph nodes (MLNs) and then migrate to the gut lamina propria for effector function17,18. Small intestine lamina propria cells isolated from reporter mice in which OPA1 is deleted in IL-17A-expressing cells (Il17a-cre × Rosa26R-eYFP19 × Opa1fl, (Opa1Il17a-cre)) showed similar expression of markers of tissue-resident TH17 cells (CD44, CD25, PD-1 and CCR6) and Ki67 to cells from control mice (Il17a-cre × Rosa26R-eYFP) (Fig. 2a,b and Extended Data Fig. 4g–h). Opa1Il17a-cre mice had a decreased frequency of TH17 cells in the lamina propria, but not in the Peyer’s patches or MLNs (Fig. 2c and Extended Data Fig. 4i), suggesting that OPA1 maintains small intestine lamina propria TH17 cells.
Fig. 2 |. OPA1 deficiency in IL-17A-expressing cells ameliorates TH17-mediated autoimmune pathogenesis.
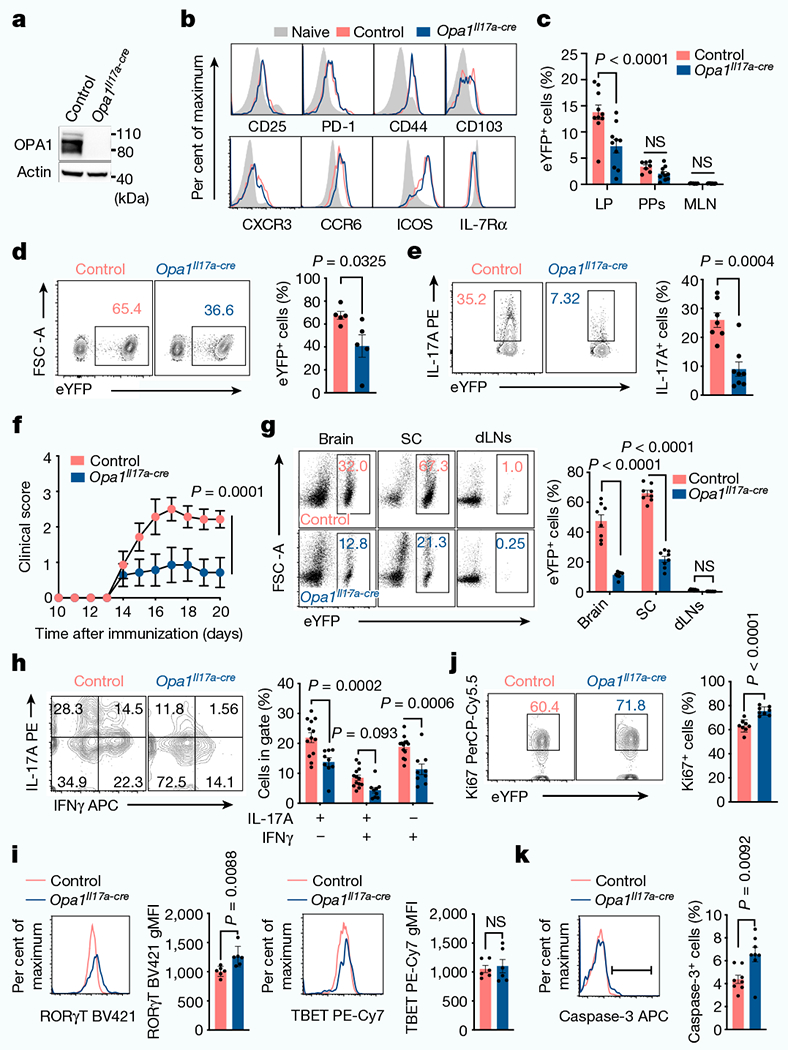
a, Immunoblot of CD4+TCRβ+ cells expressing enhanced yellow fluorescent protein (eYFP) (TH17 cells) isolated from control and Opa1Il17a-cre reporter mice. Actin is used as a loading control. b, Surface markers on CD4+TCRβ+eYFP+ cells and CD4+ TN cells (CD4+CD44lowCD62Lhigh) from small intestine lamina propria. c, Frequency of live CD4+TCRβ+eYFP+ cells in the small intestine lamina propria (LP) (control: n = 10, Opa1Il17a-cre: n = 10 mice), Peyer’s patches (PPs) (control: n = 7, Opa1Il17a-cre: n = 10 mice) and MLN (control: n = 8, Opa1Il17a-cre: n = 10 mice), 2 independent experiments. d,e, Mice were injected with anti-CD3 antibody every 48 h for 100 h. Frequency of CD4+TCRβ+eYFP+ cells (n = 5 mice) (d) and IL-17A expression in restimulated CD4+TCRβ+eYFP+ cells (e) (control: n = 7, Opa1Il17a-cre: n = 8 mice, 2 independent experiments). f, Mice were immunized with myelin oligodendrocyte glycoprotein (MOG) peptide and monitored for clinical disease score (n = 7 mice per group). g, Frequency of CD4+TCRβ+eYFP+ cells from brain, spinal cord (SC) and draining lymph nodes (dLNs) 20 days after immunization (n = 8 mice per genotype, 2 independent experiments). h, Expression of IL-17A and IFNγ in brain CD4+TCRβ+eYFP+ cells (control: n = 13, Opa1Il17a-cre: n = 9 mice, 2 independent experiments). i–k, Expression of RORγt and TBET (i; n = 6 mice per genotype), Ki67 (j) and active caspase-3 (k) (n= 8 mice per genotype, 2 independent experiments) in CD4+TCRβ+eYFP+ cells from brain (i) and spinal cord (j ,k). Representative plots and graphs summarize results of at least two independent experiments, except where noted otherwise. Data are mean ± s.e.m. Two-sided šidák’s test (c,f‘h) or unpaired two-tailed t-test (d,e,i‘k). Exact P-values are indicated.
TH17 cells expand in the small intestine lamina propria upon treatment with anti-CD3 antibody20. In this model, Opa1Il17a-cre mice had reduced numbers of TH17 cells in the lamina propria compared with control mice (Fig. 2d and Extended Data Fig. 4j). Opa1Il17a-cre TH17 cells expressed less IL-17A and RORγt (Fig. 2e and Extended Data Fig. 4k) but similar levels of activation, migration and proliferation markers compared with control TH17 cells (Extended Data Fig. 4l,m), supporting a requirement for OPA1 for effector function and expansion. Following immunization with myelin oligodendrocyte glycoprotein, Opa1Il17a-cre mice were protected from EAE (Fig. 2f and Extended Data Fig. 4n), exhibiting decreased T cell and neutrophil infiltration to the spinal cord (Fig. 2g and Extended Data Fig. 4o,p). Opa1Il17a-cre TH17 cells showed defective IL-17A production (Fig. 2h), with similar expression of transcription factors (Fig. 2i), chemokine receptors CCR6 and CXCR3 and the mTOR targets pS6 and p4E-BP1 (Extended Data Fig. 4q,r). Moreover, levels of Ki67 and active caspase-3 showed that Opa1Il-17a-cre TH17 cells proliferated normally, but were less viable than control TH17 cells (Fig. 2j,k). Thus, OPA1 is required for the maintenance of tissue-resident intestinal and pathogenic TH17 cells.
TH17 cells need OPA1 independent of energetics
Cells depend on respiration in low-glucose conditions21. We explored whether differences in T cell metabolism drive the selective reliance on respiration and OPA1. The robust glycolytic and energetic metabolism in TH1 and TH2 cells contrasted with that in TH17 and Treg cells, which had more quiescent and oxidative metabolism with reduced glucose and leucine uptake and mTORC1 activity (Extended Data Fig. 5a,b). TH17 cells exhibited lower rates of protein and RNA synthesis and increased dependence on mitochondrial metabolism for protein translation22 (Extended Data Fig. 5c,d). To lower metabolic activity in an energetic cell type, we cultured TH1 cells at different glucose concentrations (Extended Data Fig. 5e). At low glucose concentration, control TH1 cells expressed IFNγ, whereas IFNγ expression was severely impaired in OPA1-deficient TH1 cells (Extended Data Fig. 5f). We next explored whether low metabolic activity in TH17 cells makes them dependent on OPA1 for IL-17 expression. It is known that pTH17 and npTH17 cells have distinct metabolic profiles23. pTH17 cells had higher metabolic activity than npTH17 cells, with increased oxidative metabolism and protein and RNA synthesis, as well as reduced dependence on mitochondrial metabolism for protein translation (Extended Data Fig. 5g–i), but still required OPA1 for IL-17 expression (Extended Data Fig. 4b,d). Thus, whereas low metabolic activity confers OPA1 dependence, TH17 cells require OPA1 for cytokine expression independent of their metabolic state.
OPA1 affects glutamine flux and 2-HG
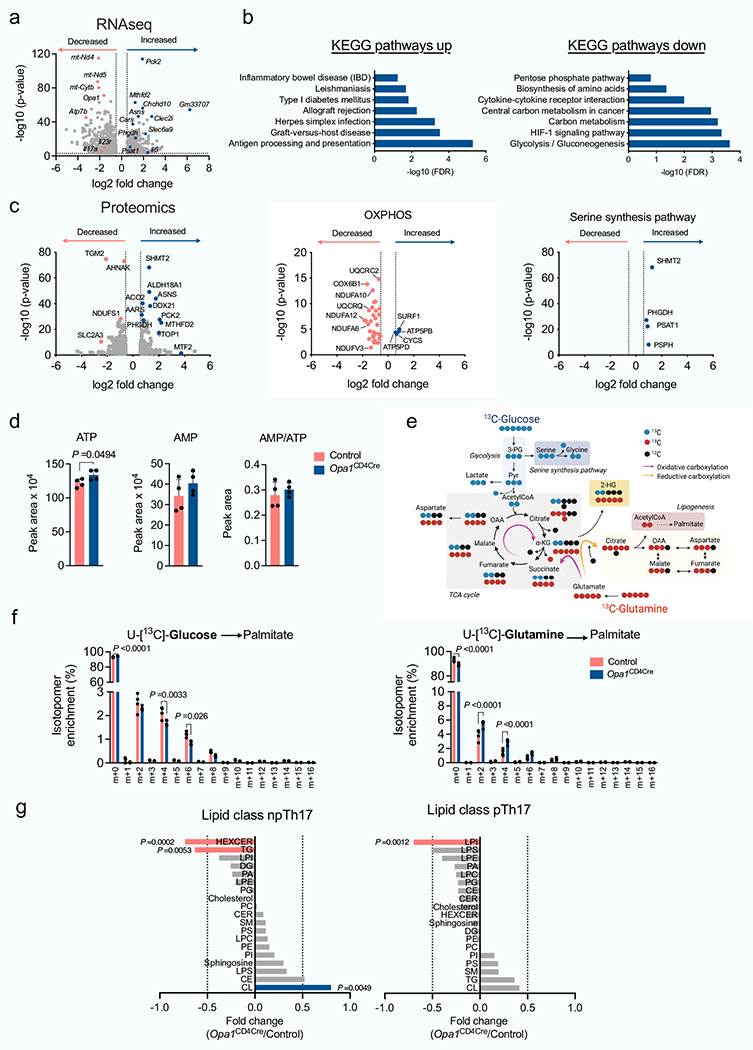
Opa1Cd4-cre TH17 cells exhibited transcriptional and proteomic changes associated with immune function, infection, immune-mediated diseases and metabolic pathways compared with control TH17 cells (Fig. 3a and Extended Data Fig. 6a–c). Proteomic analysis revealed reduced expression of electron transport chain (ETC) complex subunits and increased expression of serine and glycine biosynthesis enzymes in these cells (Extended Data Fig. 6c), suggesting mitochondrial dysfunction and a stress response24,25, and metabolomics showed accumulation of TCA cycle metabolites, with the α-ketoglutarate (α-KG)-derived metabolite 2-hydroxyglutarate (2-HG) being highly upregulated (Fig. 3b,c). Increased NADH/NAD+ and reduced aspartate (Fig. 3d)–the production of which depends on a functional ETC26,27–further indicated disrupted oxidative phosphorylation (OXPHOS) and TCA cycle metabolism in OPA1-deficient cells. Similar ATP and AMP levels, glucose flux to fatty acids and lipid accumulation across genotypes (Extended Data Fig. 6d–g), ruled out a bioenergetic failure28 or decreased de novo fatty acid synthesis29 underlying impaired IL-17A expression in OPA1-deficient cells.
Fig. 3 |. OPA1 modulates the TCA cycle and 2-HG for epigenetic control of TH17 cell function.
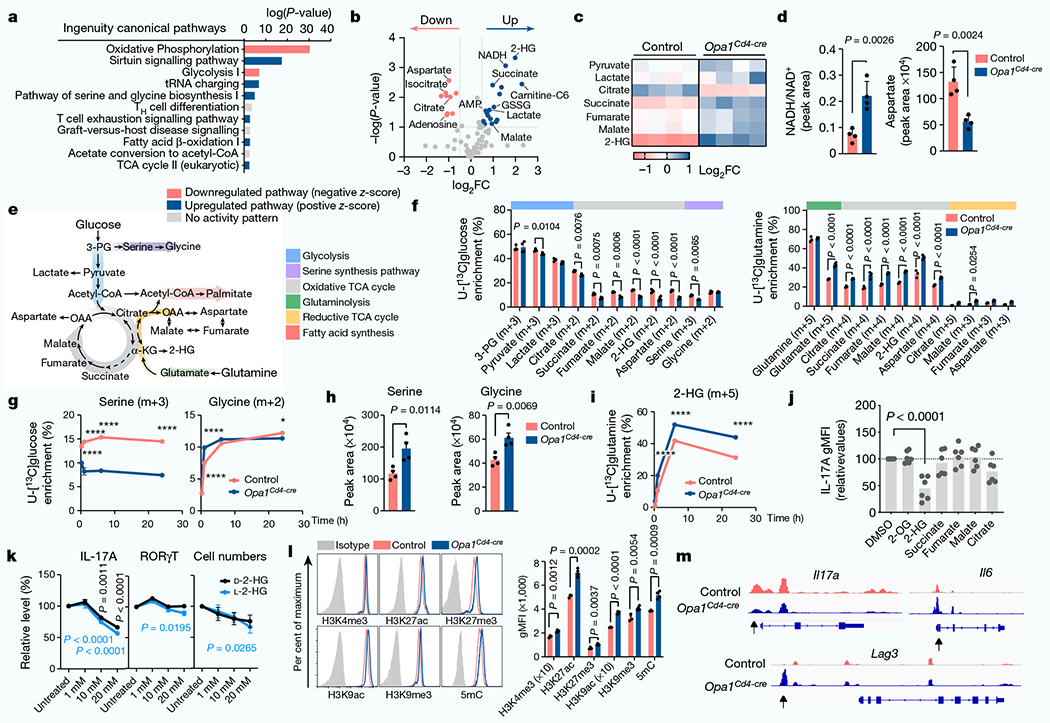
a, ingenuity pathway analysis of differentially regulated proteins (Opa1Cd4-creversus control TH17 cells; log10(P-value<1.3, log2FC >0.58)). b, Volcano plot of metabolite levels determined by liquid chromatography–mass spectrometry (LC-MS) analysis. The plot shows metabolites that are increased (blue) or decreased (red) in Opa1Cd4-cre versus control TH17 cells; FC>0.5 and log10(P-value) > 1. GSSG, oxidized glutathione. c, Heat map showing metabolite levels (log2FC) in control and Opa1Cd4-cre TH17 cells. d, Cellullar NADH/NAD+(left) and aspartate (right), determined by LC–MS. e, Schematic of central carbon metabolism. 3-PG, 3-phosphoglycerate; OAA, oxaloacetate. f, Fractional labelling of metabolites in TH17 cells cultured for 20 h with fully-labelled U-[13C]glucose on day 3 (left, n = 4 biological replicates per group) or U-[13C]glutamine (right, control: n = 3, Opa1Cd4-cre: n = 4 biological replicates). Colours along the top of the graph correspond to the scheme in e. g, Fractional labelling of serine and glycine from U-[13C]glucose. h, intracellular serine and glycine levels, determined by LC–MS. i, Fractional labelling of 2-HG from U-[13C]glutamine. j, IL-17A expression in CD4+ T cells cultured for 4 days in TH17 conditions after overnight treatment with metabolites (n = 6 biological replicates, 2 independent experiments). 2-OG, 2-oxoglutarate. k, Cell numbers (n = 7 biological replicates) and cytokine and transcription factor expression (n = 8 biological replicates) in cells cultured for 3 days in TH17 conditions with D-2-HG (black) or L-2-HG (blue) for the last 48 h, 2 independent experiments. I, Acetylation and methylation status of histone H3 lysine 4 (H3K4), lysine 9 (H3K9) and lysine 27 (H3K27) and DNA methylation. 5mC, 5-methylcytosine. m, Accessible chromatin regions as indicated by ATAC-seq. Representative plots and graphs summarize results of at least two independent experiments. Arrows indicate differentially accessible chromatin regions. a,g,i, n = 3 biological replicates. b–d,h,l, n = 4 biological replicates. Data in d,f–l are mean±s.e.m. Two-sided šidák’s test (f,g,i,k), Dunnett’s test (j) or unpaired two-tailed t-test (d,h,l). Exact P-values are indicated. *P<0.05, ****P<0.0001.
Opa1Cd4-cre TH17 cells exhibited lower glucose flux to glycolysis, TCA cycle and other associated pathways (Fig. 3e,f). Notably, glucose flux to serine biosynthesis30 decreased upon OPA1 loss (Fig. 3g), although it did not impede intracellular serine and glycine levels, which were increased in Opa1Cd4-cre TH17 cells (Fig. 3h). OPA1-deficient TH17 cells robustly incorporated glutamine-derived carbons into glutamate, TCA cycle intermediates and 2-HG (Fig. 3f,i). To test the effect of accumulated TCA cycle metabolites and 2-HG on IL-17 production, we cultured CD4+ TN cells in TH17 conditions with cell-permeable forms of these metabolites. IL-17A production and cell numbers were markedly reduced by 2-HG (Fig. 3j), independently of transcription factor expression or 2-HG enantiomer configuration (l-2-HG or d-2-HG) (Fig. 3k). Thus, OPA1 deletion increases glutamine oxidation and 2-HG generation, which negatively affects IL-17A expression.
Altered metabolism affects chromatin
Epigenetic and transcriptional programmes are regulated by 2-HG5; 2-HG inhibits α-KG-dependent DNA and histone dioxygenases and demethylases, increasing histone and/or DNA methylation7. Permissive (H3K4me3, H3K27ac and H3K9ac) and repressive (H3K9me3 and H3K27me3) histone modifications and global DNA methylation, increased in Opa1Cd4-cre TH17 cells (Fig. 3l), suggesting altered chromatin organization. The DNA and histone methyltransferase inhibitors 5-azacytidine (5-Aza) and UNC1999 (UNC), but not histone acetyltransferase inhibitors, restored IL-17A expression (Extended Data Fig. 7a), indicating that DNA and histone methylation changes caused altered IL-17A activity. Using assay for transposase-accessible chromatin with high throughput sequencing (ATAC–seq) we found that OPA1 deficiency altered chromatin accessibility in regions with recognition motifs for T cell differentiation, transcription factors and stress response elements (Extended Data Fig. 7b–d). Pathway annotation analysis showed that these regions were involved in transcriptional regulation, signalling and T cell responses, including IL-17A, IL-6 and LAG3 (Fig. 3m and Extended Data Fig. 7e). These data suggest that mitochondrial disturbances (that is, OPA1 deficiency) augment glutamine oxidation with 2-HG accumulation, which affects TH17 effector function by altering the chromatin accessibility of immune response genes.
LKB1 responds to OPA deficiency
To investigate signals coordinating the cellular response to OPA1 deficiency in TH17 cells, we performed Ingenuity pathway analysis and identified LKB1 (encoded by Stk11) as the top potential upstream regulator (Fig. 4a). LKB1 has roles in mitochondrial function, metabolism and energy homeostasis31–34. Notably, LKB1 also coordinates serine metabolism and DNA methylation with cancer transformation35. LKB1 activity, as determined by Ser428 phosphorylation36, was increased in Opa1Cd4-cre TH17 cells (Fig. 4b). Increased LKB1 phosphorylation occurred 48 h after activation in TH17 conditions, but was absent in TH1 and TH2 conditions, and only appeared at later times in Treg cells (Extended Data Fig. 8a,b), demonstrating differential LKB1 signalling in Opa1Cd4-cre TH17 cells.
Fig. 4 |. LKB1 couples TCA cycle and de novo serine metabolism for 2-HG production.
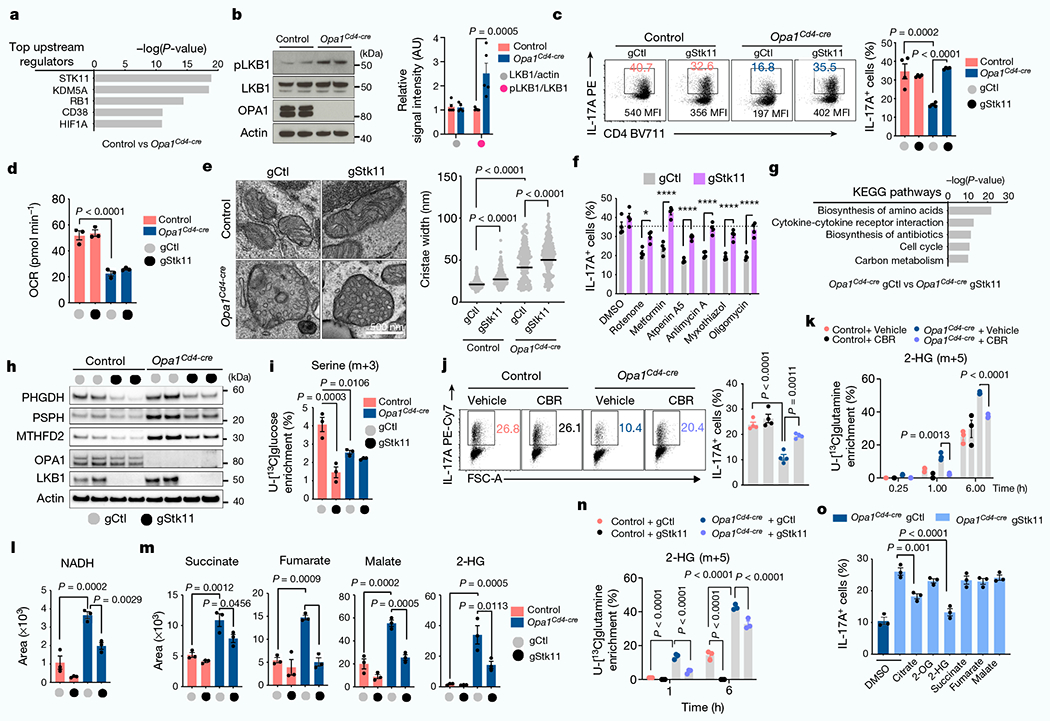
a, Top upstream regulators of differentially regulated proteins (Opa1Cd4-cre versus control TH17 cells; log10(P-value < 1.3, log2FC > 0.58, Ingenuity pathway analysis)). b, Immunoblot (left) and quantification (right) of LKB1 phosphorylation and expression (n = 5 biological replicates, 2 independent experiments). Actin is used as a sample processing control. c, 1L-17A expression in T cells transfected with Cas9 and indicated guide RNAs (gRNAs). gCtl, control guide RNA; gStk11, Stk11-targeting guide RNA. d, Basal OCR. e, Electron microscopy image (left) and width of mitochondrial cristae (right) (control gCtl: n = 214; control gStk11: n = 271, Opa1Cd4-cre gCtl: n = 236; Opa1Cd4-cre gStk11: n = 257 cristae, 3 biological replicates). Data are shown as individual measurements and geometric mean (black bar). f, 1L-17A expression in T cells transfected with Cas9 and indicated gRNAs, cultured in TH17 conditions for four days and treated overnight as indicated. g, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation of differentially expressed genes (Opa1Cd4-cre gStkll versus Opa1Cd4-cre gCtl, P <0.001, log2FC > 1). h, Immunoblot in TH17 cells transfected with Cas9 and indicated gRNAs. Two replicate transfections shown . i, Detection of serine (m+3) in cells cultured for 6 h with U-[13C]glucose. j, 1L-17A expression in cells treated for 20 h with CBR-5884 on day 2. k, Detection of 2-HG (m+5) fractional labelling from U-[13C]glutamine. l,m, Intracellular NADH (l), succinate, fumarate, malate and 2-HG (m) detection by LC-MS. n, 2-HG (m+5) in cells cultured with U-[13C]glutamine. o, 1L-17A expression in Opa1Cd4-cre T cells transfected with Cas9 and indicated gRNAs, cultured in TH17 conditions for four days and treated overnight as indicated. Representative plots and graphs summarize results of at least two independent experiments, except where noted otherwise. Data a,d,e,g,i,k-o: n = 3 biological replicates per group; c,f,j: n = 4 biological replicates per group. Data in b–d,f,i–o are mean ± s.e.m. Two-sided šidák’s test (b,f), Tukey’s test (c‘e,i‘n) or Dunnett’s test (o). Exact P-values are indicated.
LKB1 deletion restores IL-17
Deletion of LKB1 in OPA1-deficient TH17 cells restored IL-17A expression (Fig. 4c and Extended Data Fig. 8c,d). LKB1 deletion did not affect early activation markers, but reduced RORγt expression, proliferation and cell cycle progression36,37 (Extended Data Fig. 8e–h). Heterozygous LKB1 loss in Opa1Il17a-cre mice (Opa1Stk11hetIl17a-cre) mildy restored disease and T cell infiltration compared with Opa1Il17a-cre mice (Extended Data Fig. 8i,j). However, full LKB1 deletion (Opa1Stk11Il17a-cre) restricted disease and T cell infiltration (Extended Data Fig. 8k–m), probably owing to proliferation defects caused by LKB1 loss (Extended Data Fig. 8g,h), but re-established IL-17A production (Extended Data Fig. 8n), demonstrating that LKB1 senses mitochondrial integrity in vivo to control Th17 effector function. LKB1 deletion corrected IL-17A expression in Opa1Cd4-cre TH17 cells without restoring respiration or cristae morphology defects (Fig. 4d,e), and in an AMPK- and mTOR-independent manner (Extended Data Fig. 9a–e). Remarkably, LKB1 loss preserved IL-17A expression in cells treated acutely with mitochondrial inhibitors (Fig. 4f). Overall, LKB1 signalling restrains IL-17A expression following mitochondrial disturbances, and its loss unleashes cytokine expression during mitochondrial stress.
Several kinases regulate LKB1 in response to signals such as DNA damage, NADH/NAD+ imbalance, cAMP, calcium levels or reactive oxygen species38 (ROS). OPA1-deficient cells showed alterations in many of these signals (Extended Data Fig. 9f–i), indicating that multiple factors may converge on LKB1 during mitochondrial stress. Pharmacological inhibition of LKB1-activating kinases, or restoration of the NADH/NAD+ balance or ROS pointed to ataxia telangiectasia mutated (ATM) kinase as a driver of LKB1 signalling upon OPA1 deficiency (Extended Data Fig. 9j). Thus, TH17 cells are sensitive to perturbed mitochondrial membrane integrity and function, and this information is relayed through LKB1 to control IL-17 expression.
The OPA1–LKB1 axis regulates TCA and 2-HG
We analysed the transcriptome of LKB1-deleted control and Opa1Cd4-cre TH17 cells (Extended Data Fig. 10a). Analysis of genes differentially regulated by LKB1 revealed pathways associated with amino acid biosynthesis, cytokine–cytokine receptor interaction, cell cycle and carbon metabolism (Fig. 4g and Extended Data Fig. 10b–d), illustrating processes by which LKB1 may control IL-17A expression upon OPA1 deficiency. Among them, LKB1 deletion reduced the expression of serine biosynthesis enzymes (Fig. 4h), including PHGDH, the first enzyme in serine biosynthesis, which converts the glycolytic intermediate 3-phosphoglycerate to phosphohydroxypyruvate. We measured glucose flux into serine30 in control and Opa1Cd4-cre TH17 cells in which LKB1 was deleted. OPA1-deficient cells diverted less glucose to serine–as previously observed (Fig. 3g)–and LKB1 deletion reduced this further (Fig. 4i). Although serine biosynthesis was decreased, Opa1Cd4-cre TH17 cells had high intracellular serine compared with controls, and these levels increased upon acute LKB1 deletion (Extended Data Fig. 10e), probably reflecting differences in extracellular serine uptake and/or catabolism. The disparity between the expression of serine biosynthesis pathway enzymes and the activity of the pathway led us question how this might affect the cellular response to OPA1 deficiency.
LKB1 coordinates TCA, NADH and 2-HG
In hypoxic or acidic conditions, NADH-dependent dehydrogenases, including PHGDH and lactate dehydrogenase (LDHA) engage in promiscuous activity and catalyse the NADH-dependent reduction of α-KG to l-2-HG and d-2-HG in addition to their primary enzymatic reactions39–41. We speculated that PHGDH, which was expressed in OPA1-deficient T cells, but not fully engaged in serine biosynthesis (Fig. 4h,i), produced 2-HG. We therefore pharmacologically targeted PHGDH and other dehydrogenases, including LDHA and TCA cycle enzymes that produce 2-HG, isocitrate dehydrogenase 1 and 2 (IDH1/2) and malate dehydrogenase 1 and 27 (MDH1/2) (Extended Data Fig. 10f). Out of these interventions, only PHGDH inhibition restored IL-17A in OPA1-deficient TH17 cells (Fig. 4j and Extended Data Fig. 10f). Furthermore, PHGDH inhibition reduced glutamine flux to 2-HG in OPA1-deficient cells (Fig. 4k), indicating that PHGDH contributes to 2-HG production and decreases IL-17A expression.
ETC inhibition, genetic mitochondrial deficiencies and hypoxia impair respiration and cause NADH accumulation through mitochondrial serine catabolism and–specifically–methylenetetrahy-drofolate dehydrogenase42 (MTHFD2). We tested whether LKB1 loss, which reduced serine biosynthesis enzymes (Fig. 4h), would similarly re-establish NADH levels in OPA1-deficient cells. Indeed, LKB1 deletion restored NADH in Opa1Cd4-cre cells to control levels (Fig. 4l) as well as TCA cycle metabolites and 2-HG (Fig. 4m). Notably, LKB1 loss decreased the exacerbated glutamine carbon allocation to TCA metabolites and 2-HG in Opa1Cd4-cre cells (Fig. 4n and Extended Data Fig. 10g). Consistent with LKB1 restraining IL-17A expression by regulating TCA cycle metabolites and 2-HG, single metabolite treatment–and most significantly 2-HG–dampened IL-17A expression in LKB1-deficient Opa1Cd4-cre TH17 cells (Fig. 4o). Overall, our results indicate that LKB1, by coordinating glutamine oxidation and PHGDH activity, generates 2-HG that modulates the cell’s epigenetic state and restrains IL-17A expression.
Discussion
Here we show that mitochondrial disturbances in OPA1-deficient cells trigger a response that increases the expression of serine biosynthesis enzymes and 2-HG production. Loose cristae in OPA1-deficient cells alter the electron flow through the disorganized ETC, leading to increased NADH/NAD+ and TCA cycle inhibition43, as it requires NAD+ for enzymatic reactions5. Our findings indicate that NADH accumulation pushes PHGDH to generate 2-HG from α-KG39–reinforcing glutamine oxidation in the TCA cycle as cells need to replenish α-KG. We show that LKB1 responds to OPA1 deficiency by controlling glutamine oxidation and TCA cycle metabolism. LKB1 signalling exacerbates mitochondrial deficiencies in OPA1-deficient TH17 cells, and its deletion is sufficient to restore intracellular metabolites and IL-17A expression. Although it is linked to glutamine oxidation44, how LKB1 regulates glutamine usage remains unclear, but during mitochondrial stress it could affect redox shuttles, glutamine intake and hydrolysis or serine biosynthesis enzyme expression. Nevertheless, our findings are consistent with a role for glutamine metabolism in TH17 cells45. We do not exclude the possibility that TCA cycle metabolites and 2-HG–which can regulate gene expression by epigenetic and transcriptional remodelling or protein function via posttranslational modifications46–may control TH17 effector function upon mitochondrial stress through other mechanisms.
During immune responses, T cells must adapt to fluctuating environments to meet their metabolic demands and tune effector function47. How mitochondrial membrane rearrangements modulate cell metabolism and immune function in inflammatory environments in vivo is currently unknown. However, our data suggest that an LKB1–mitochondrial axis sustains T cell metabolic adaptations to distinct inflammatory and tissue conditions, and illustrate metabolic and mitochondrial vulnerabilities in TH17 cells that might be therapeutically exploited to control autoimmune disease.
Methods
Animal models
C57BL/6J (000664), PhAM (018397), Cd4-cre (022071), Il17a-cre (016879), Rosa26-eYFP (006148), Great (IFNγ eYFP reporter, 017581)48, 4Get (IL-4 eGFP reporter, 004190)49, FIR (FOXP3-RFP reporter, 008374)50 and conditional floxed Stk11 (Stk11fl, 014143) mice were purchased from The Jackson Laboratory. Opa1 and Drp1 conditional floxed mice were obtained from H. Sesaki. Mfn1 and Mfn2 conditional floxed mice were obtained from D. C. Chan. Conditional floxed mice were crossed with Cd4-cre to generate Opa1Cd4-cre, Mfn1Cd4-cre, Mfn2Cd4-cre, Drp1Cd4-cre and Stk11Cd4-cre mice. For conditional deletion of OPA1 or LKB1 in IL-17A-expressing cells, Il17a reporter animals (Il17a-cre × Rosa26e-YFP, which harbour a loxP-site-flanked STOP cassette followed by the eYFP-encoding sequence inserted into the Rosa26 locus19) were crossed with Opa1fl/fl mice (Il17-cre+/−Rosa26e-YFP+/−Opa1fl/fl (Opa1Il17a-cre)) or Stk11fl/fl mice (IL17Cre+/−R26eYFP+/− Stk11fl/fl (Stk11Il17Cre)). For conditional deletion of OPA1 and LKB1, Cd4-cre or Il17a reporter mice were crossed with Opa1fl/fl and Stk11fl/fl mice (Cd4-cre+/−, Opa1fl/flStk11fl/fl, Opa1Stk11Cd4-cre; Il17Cre+/−Rosa26-eYFP+/− Opa1fl/flStk11fl/+, Opa1Stk11hetIl17-cre and Il17-cre+/−Rosa26-eYFP+/− Opa1fl/fl Stk11fl/fl (Opa1Stk11Il17-cre)). Age- and sex-matched Il17a reporter animals (Il17-cre+/−Rosa26-eYFP+/− (control)) were used as controls. All mice were used at 8–12 weeks of age unless otherwise noted. All mice required for this study were bred in specific-pathogen-free conditions in the animal facility of the Max Planck Institute for Immunobiology and Epigenetics (Freiburg, Germany), and all corresponding animal protocols were approved by the animal care committee of the Regierungspraesidium Freiburg. All cells used in this study were collected from male and female mice at 8–12 weeks of age.
T cell assays
CD4+ TN cells were isolated from cell suspensions from spleen and peripheral lymph nodes from mice from the indicated genotypes. Pooled spleen and lymph node single cell suspensions were incubated with biotinylated antibodies against mouse CD44, CD25, CD8, IgM, B220, CD19, Gr-1, MHC-II, F4/80, CD11c, CD11b and DX5 (Biolegend), followed by negative selection using magnetic beads (Stem cell Technology). Approximately 0.3 × 106 CD4+ TN cells were plated into 48-well-plates coated with 5 μg ml−1 anti-mouse CD3 (clone 17A2, BioXcell), 2 μg ml−1 soluble anti-mouse CD28 (clone 37.51, BioXcell) in RPMI 1640 (Gibco) supplemented with 10% fetal calf serum (Gibco), 1% (v/v) penicillin–streptomycin (Invitrogen), 55 μM β-mercaptoethanol (Sigma), 1 mM sodium pyruvate, 2 mM glutamine and 5 mM Hepes. For TH1 conditions, culture was supplemented with IL-12 (10 ng ml−1) and anti-mouse IL-4 (4 μg ml−1, clone 11B11, BioXcell). For TH2 conditions, IL-4 (10 ng ml−1) and anti-mouse IFNγ (4 μg ml−1, clone XMG1.2, BioXcell) were added. For TH17 conditions, TGFβ (5 ng ml−1), IL-6 (10 ng ml−1), IL-1β (10 ng ml−1), anti-mouse IFNγ (10 μg ml−1) and anti-mouse IL-4 (10 μg ml−1) were included in cell cultures. For regulatory T cell conditions, TGFβ (10 ng ml−1), anti-mouse IFNγ (4 μg ml−1) and anti-mouse IL-4 (4 μg ml−1) were added. Where specificied, cells were polarized towards non-pathogenic (np) and pathogenic (p) TH17 cell conditions as follows. For npTH17 conditions, TGFβ (5 ng ml−1), IL-6 (10 ng ml−1), anti-mouse IFNγ (10 μg ml−1) and anti-mouse IL-4 (10 μg ml−1) were included in cell cultures. For pTH17 conditions, IL-6 (20 ng ml−1), IL-1β (20 ng ml−1), IL-23 (20 ng ml−1), anti-mouse IFNγ (10 μg ml−1) and anti-mouse IL-4 (10 μg ml−1) were included in cell cultures. Human IL-2 (10 ng ml−1) was added to all cultures except in TH17 conditions. All cytokines were from PrepoTech. The cells were supplemented with fresh medium at day 3 and analyses performed at day 4 unless otherwise stated.
For analysis of intracellular cytokine production and transcription factor expression, cells were collected on day 4 and restimulated 6 h with phorbol myristate acetate (50 ng ml−1) (PMA) and ionomycin (1 μg ml−1) in the presence of brefeldin A (Biolegend) for the last 4 h. After restimulation, cells were stained with cell-surface marker antibodies and LIVE/DEAD Fixable dye Blue (Thermo) to exclude dead cells, fixed and permeabilized with a FOXP3/transcription factor staining kit (eBioscience), followed by staining with cytokine- and/or transcription-factor-specific antibodies. All flow cytometry analyses were performed on an LSR III flow cytometer (BD) and data were analysed with FlowJo software (BD).
For analysis of cytokines in cell culture supernatants, equal numbers of cells were collected and cultured in fresh medium containing PMA and ionomycin for 16 h on day 4. After restimulation, supernatant was collected and concentration of cytokines determined by a multiplexed assay (LEGENDplex, Biolegend), as per manufacturer instructions.
Mitochondrial ETC inhibitors were added overnight on day 3 of culture with the following doses: 1 μM rotenone (Sigma), 5 mM metformin (Sigma), 1 μM atpenin A5 (Hellobio), 1 μM antimycin A (Sigma), 1 μM myxothiazol (Sigma), 1 μM oligomycin (Sigma). AMPK inhibitor Compound C (Merk) was added at 1 mM from the initial activation (day 0) or 16 h at day 3. Cells were treated with the following TCA metabolites as indicated; 1 mM dimethyl oxoglutarate, 5 mM diethyl succinate, 100 nM diethyl fumarate, 0.25 mM octyl-2-HG, l-and d-α-hydroxyglutaric acid disodium salt, 1 mM diethyl malate, and 1 mM citrate (all from Sigma).
The following compounds were added for 48 h in cells at day 1 of culture at 10 μM unless otherwise indicated: CBR-5884 (Sigma), BAY 1436032 (MedChemExpress), LW6 (TCI Deutschland), AGI-5198 (Sigma), NHI-2 (Sigma), GSK-864 (Sigma), CPTH2 (Cayman), Anacardic acid (Cayman), A485 (Tocris), 1 μM C6464 (Sigma), Sinefungin (Abcam), 1 μM 5-aza-2′-deoxycytidine (Sigma), 1 μM UNC1999 (Abcam), KDM5-C70 (Xcess Bio), 1 μM GSK-J4 (Xcess Bio), KU-55933 (Sigma), 0.5 μM BI-D1870 (Abcam), 1 μM Ex-527 (Abcam), 1 μM sirtinol (Fischer Scientific), 1 μM PKCζ pseudosubstrate inhibitor (Cayman), 1 μM PKCζ pseudosubstrate inhibitor, myristoylated (sc-397537, Santa Cruz Biotechnology), 1 μM PKA Inhibitor 14-22 Amide, Myristoylated (Sigma), 0.5 mM nicotinamide riboside (Cayman), 1 μM Tempol (Santa Cruz), 5 mM N-acetylcysteine (NAC) from Sigma. Cells were processed for flow cytometry and acquired with CytExpert software on a CytoFLEX (Beckman Coulter) cytometer and data analysed with FlowJo software v10.7.2. (BD).
CRISPR–Cas9 delivery
CRISPR–Cas9 deletion of LKB1, DRP1 and AMPKα1 was performed with Alt-R CRISPR–Cas9 System (IDT) and 4D-Nucleofector technology (Lonza) in freshly isolated CD4+ T cells. gRNA duplexes were prepared by mixing equimolar concentrations of CRISPR RNAs (crRNAs) and transactivating crRNAs (tracrRNAs), annealing 5 min at 98 °C, mixing gRNA complexes (crRNA–tracrRNA) with Alt-R Streptococcus pyogenes Cas9 Nuclease V3 (IDT), and incubating for 20 min at room temperature to form ribonucleoprotein (RNP) complexes (crRNA–tracrRNA–Cas9). For the delivery of RNP complexes, cells were extensively washed in PBS, pelleted, and the RNP complexes and P4 Primary Cell buffer added before transferring into a nucleofection chamber. The complexes were delivered by electroporation using DS137 pulse on Amaxa 4D-Nucleofector (Lonza). Electroporated cells were recovered in T cell medium for 120 min at 37 °C before activation in TH conditions, as described. Two gRNAs were used to target LKB1 (Mm.Cas9.STK11.1.AA: 5′-GGGCTCGTACGGCAAGGTGA-3′; Mm.Cas9.STK11.1.AC: 5′-TTGACGTTGGCCTCTCCATT-3′), DRP1 (Mm. Cas9.DNM1L.1.AA: 5′-GTTCCCACTACGACGATCTG-3′; Mm.Cas9. DNM1L.1.AB: 5′-AAATAGCTATGGTGAACCGG-3′) and AMPKα subunit 1 (Mm.Cas9.PRKAA1.1.AA: 5′-CGAGTTGACCGGACATAAAG-3′; Mm.Cas9. PRKAA1.1.AB: 5′-CCGGTGTGGATTATTGTCAC-3′), and a non-targeting gRNA (Alt-R CRISPR–Cas9 negative control crRNA) was used as control (all from IDT) at same concentration of RNP complexes between conditions.
Metabolic assays
OCR (in pmol min−1) and ECAR (in mpH min−1) were measured in a 96-well XF Extracellular Flux Analyzer (Seahorse Bioscience) on cells at day 4 of activation. Cells (105 per well; at least 3 injections per condition) were plated in complete XF medium (non-buffered RPMI 1640 containing 25 mM glucose, 2 mM l-glutamine, and 1 mM sodium pyruvate) and spun onto poly-d-lysine-coated Seahorse 96-well plates. Cells were equilibrated for 1 h at 37 °C in the absence of CO2 before the assay. OCR and ECAR were measured in basal conditions and in response to 1 μM oligomycin, 1.5 μM FCCP and 100 nM rotenone + 1 μM antimycin A (all from Sigma). Basal mitochondrial respiration was determined from baseline measurements. ATP-coupled respiration was determined by the subtraction of oligomycin A values from basal respiration. Maximal mitochondrial respiration was determined by subtracting the OCR after treatment with rotenone and antimycin A from the OCR measured following treatment with FCCP. For glycolysis characterization, cells were plated in complete XF medium without glucose and ECAR (in mpH min−1) was measured in basal conditions and in response to 10 mM glucose (Sigma), 1 μM oligomycin and 25 mM 2-deoxyglucose (2-DG, Sigma). Basal glycolytic rate was determined by subtracting ECAR measurement following 2-DG treatment from ECAR measurements following glucose addition. Total glycolytic capacity was determined by subtracting the ECAR measured after 2-DG treatment from the ECAR measured after oligomycin treatment.
Glucose uptake was analysed by flow cytometry in CD4+ T cells cultured in complete RPMI medium without glucose (Gibco) containing 100 mM 2-NBDG (Sigma) for 20 min at 37 °C.
Kynurenine uptake was analysed by flow cytometry as described51. CD4+ T cells were stained with anti-CD4 and plated in HBSS at 37 °C in a water bath. 200 μM kynurenine (Sigma) was added to the cells and uptake was stopped after 4 min by fixing cells in 1% paraformaldehyde (PFA, Sigma) for 30 min at room temperature. The 405 nm laser and 450/50 BP filter were used for kynurenine fluorescence detection.
Protein and RNA synthesis were analysed by flow cytometry in CD4+ T cells incubated 30 min at 37 °C in complete RPMI medium containing 20 μM Click-iT O-propargyl-puromycin (OPP, Thermo) or 1 mM Click-iT 5-ethynyl uridine (EU, Thermo), respectively. Incorporation of protein and RNA Click-iT analogues was detected with a standard Click-it reaction (Thermo) following manufacturer’s instructions. Negative controls for protein and RNA synthesis were included in the assay by incubation of cells with 100 μg ml−1 cycloheximide (Sigma) 30 min or 5 μg ml−1 actinomycin D (Sigma) 45 min, respectively.
For the analysis of protein translation dependence on mitochondrial function and glycolysis, protein synthesis rates in vehicle-treated cells was subtracted to protein synthesis rates of cells treated with oligomycin or 2-deoxyglucose, as described22.
Immunoblot analysis
Cells were washed with ice-cold PBS and lysed in 1× cell lysis buffer (Cell Signaling). Cleared protein lysates were denatured (LDS loading buffer, 10 min, 70 °C), and equal number of cells or equal protein amount resolved on precast 4% to 12% NuPAGE Bis-Tris protein gels (Life Technologies), and transferred onto nitrocellulose membranes (iBLOT2, Life Technologies). To resolve long and short OPA1 isoforms, equal amounts of protein lysates were run for at least 100 min in precast 8% Bis-Tris protein gels (Life Technologies), and transferred onto nitrocellulose membranes (iBLOT2, Life Technologies).
Membranes were blocked in TBS-Tween 5% milk for 1 h at room temperature and incubated overnight at 4 °C with the following antibodies (dilution; clone; catalogue number); anti-β-actin (1:5,000; 13E5; 4970S), anti-LKB1 (1:2,000; D60C5; 3047S), anti-phosphorylated LKB1 Ser428 (1:2,000; C67A3, 3482S), anti-AMPKα (1:2,000; D5A2; 5831S), anti-phosphorylated AMPKα Thr172 (1:2,000; 40H9; 2535), anti-phosphorylated ACC Ser79 (1:2,000; 3661), anti-ACC (1:2,000; C83B10; 3676), anti-DRP1(1:2,000; D6C7; 8570), anti-TOM20 (1:2,000; D8T4N; 42406) and anti-MFN2 (1:2,000; D2D10; 9482) from Cell Signaling, anti-OPA1 (1:2,000; 18/OPA1; 612606) from BD Biosciences, anti-MFN1 (1:1,000; EPR21953-74; ab221661) from Abcam, anti-tubulin (1:4,000; DM1A; T9026) and anti-PHGDH (1:2,000; HPA021241) from Sigma, anti-PSPH (1:2,000; PA5-96863) from Thermo and anti-MTHFD2 (1:2,000; 12270-1-AP) from Proteintech.
All primary antibody incubations were followed by three TBS-Tween washes (10, 5 and 5 min, respectively) and 1 h incubation with secondary HRP-conjugated antibodies (1:10,000 for rabbit anti-mouse IgG (H+L) secondary antibody, HRP (31450) or 1:20,000 for goat anti-rabbit IgG (H+L) secondary antibody, HRP (31460)) from Thermo Fisher in TBS-Tween with 5% milk. Membranes were washed with TBS-Tween (10, 5 and 5 min, respectively) and visualized using SuperSignal West-Pico or Femto Chemiluminescent Substrate (Pierce) on Biomax MR films (Kodak) or the ChemiDoc Imaging System (Bio-Rad). Optical intensity of the signals was quantified using ImageJ software (v1.53c, NIH). Tubulin or β-actin were used as loading controls when samples were run in same gels or as sample processing controls, when run on parallel gels.
Uncropped and unprocessed scans of all blots are provided in the Source data files.
Imaging analysis of mitochondrial structure
Spinning disk confocal microscopy was performed as described43. For live imaging of mitochondria in cells from PhAM mice, CD4+ TN cells were purified and activated as indicated for four days, transferred to glass bottom dishes (MatTek) pre-coated with poly-d-lysine (Sigma) in complete RPMI medium, and acquired using a Zeiss spinning disk confocal with an Evolve (EMCCD) camera with a 100× objective. Cells were kept in a humidified incubation chamber at 37 °C with 5% CO2 during acquisition. For nuclei visualization, Hoechst 42 (Thermo) was added prior to acquisition. For the analysis of mitochondrial morphology in fixed cells, cells from the indicated genotypes were activated as indicated, stained with Mitotracker Deepred 30 min at 37 °C, allowed to settle for 30 min at 37 °C into poly-d-lysine-coated coverslips, fixed 10 min with 2% PFA in PBS, and mounted with Prolong antifade medium containing DAPI (Invitrogen). At least 15 fields were randomly acquired per condition and biological replicate. Confocal images were analysed using Imaris analysis software (v9.5, Bitplane).
For electron microscopy imaging 0.5–1.0 × 106 T cells were fixed in 2.5% glutaraldehyde in 100 mM sodium cocodylate, washed in cocodylate buffer. After dehydration samples were embedded in Eponate 12 resin (Ted Pella) and sections were cut. Images were acquired using a JOEL 1200 EX trans-mission electron microscope with an ATMP digital camera. Cristae width was measured using ImageJ software (v1.53c, NIH) and averaged over 40 independent images in 3 biologically independent samples per condition. Acquisition of electron microscopy micrographs and measurements of max cristae width displayed were performed blinded.
For electron microscopy analysis of mitochondrial structure in ex vivo T cells: great mice (which express an IRES-enhanced YFP construct after the Ifng stop codon that allows expression of both IFNγ and eYFP from the same mRNA and then analysis of IFNγ-expressing cells) were injected intravenously with a sublethal dose 1 × 104 colony-forming units (CFU) of wild-type L. monocytogenes. T cells were sorted by fluorescence-activated cell sorting (FACS) (CD4+IFNγ-eYFP+ live cells) from cell suspensions from spleen and peripheral lymph nodes obtained at day 7 post-infection and processed for electron microscopy as indicated. For H. polygyrus infection, mice expressing IL-4-eGFP (which express an IRES-enhanced EGFP construct between the translational stop codon and the 3′ UTR of the interleukin 4 gene, and allow the analysis of IL-4 expressing cells) and FOXP3-RFP (which express a bicistronic reporter expressing a red fluorescent protein knocked into the endogenous Foxp3 locus, and allow the analysis of FOXP3-expressing cells) were gavaged with 200 infectious L3 stage H. polygyrus larvae in PBS. On day 14, MLNs were collected and CD4 T cells were FACS-sorted (CD4+IL-4-eYFP+ live cells, for TH2 CD4 T cells and CD4+FOXP3-RFP+ live cells, for Treg cells) and processed for electron microscopy as indicated. TN cells were FACS-sorted from the spleen and lymph nodes from steady-state C57BL/6J mice.
Mouse EAE model
EAE was induced by immunizing mice subcutaneously with 200 μg of myelin oligodendrocyte glycoprotein peptide (MOG35–55: MEVGWYRSPFSRVVHLYRNGK) emulsified in complete Freund’s adjuvant (supplemented with killed Mycobacterium tuberculosis strain H37RLa) and intraperitoneal injections of 200 ng pertussis toxin (Hooke Labs) at the time of immunization and 24 h later. The disease was scored daily on a scale of 0–5 as follows: 0, no overt signs of disease; 1, limp tail; 2, limp tail plus hindlimb weakness; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis; 5, moribund. At the end of experiment, the mice were euthanized for analysis of the T cells infiltrated into the brain and spinal cord. Mice that did not develop symptoms of EAE were not excluded from the analysis. When indicated, the statistical significance was determined by Student’s t-test or one-way ANOVA (exact P-values are presented on each graph). Sample size is indicated on each graph. All animal work was performed in compliance with all relevant ethical regulations and approved by the animal care committee of the Regierungspraesidium Freiburg and Max Planck Institute.
Mouse CD3 model
Mice were injected intra-peritoneally every 48 h during 100 h with 50 μg anti-CD3 monoclonal antibody (2C11, BioXcell) and euthanized 4 h after the last injection. Lymphocytes from small intestine lamina propria were isolated by removing Peyer’s patches from the small intestine and then cutting into 1 cm pieces. Tissue was washed in RPMI 3% FCS supplemented with 5 mM EDTA (Thermo) and 0.15 mg ml−1 DTT by shaking 25 min at 37 °C for removal of epithelial and intraepithelial cells, washed 4 times with RPMI containing 2 mM EDTA, digested with 0.1 mg ml−1 Liberase TL (Roche) and 50 μg ml−1 DNase I (Roche) 35 min at 37 °C, followed by separation on a Percoll (GE Healthcare) gradient.
Flow cytometry
To assess cytokine production in in vitro experiments, cells were activated for the indicated times, collected and restimulated 6 h with 50 ng ml−1 PMA and 1 μg ml−1 ionomycin in the presence of brefeldin A (Biolegend) for the last 4 h. Intracellular cytokine staining was performed using BD CytoFix/CytoPerm kit (BD Biosciences) and nuclear staining of transcription factors using the FOXP3 Permeabilisation kit (eBioscience). Cells were stained with Live/Dead viability dye (Thermo) prior to antibody staining. Cells were collected on LSR III Fortessa flow cytometers (BD Bioscience) and analysed using FlowJo (BD Biosciences) software. The following antibodies were used: anti-CD4 (A20), anti-IL17A (TC11-18H10.1), anti-IFNγ (XMG1.2) and anti-TBET (4B10) from Biolegend; anti-GATA-3 (TWAJ) from eBioscience and anti-FOXP3 (FJK-16s, R16-715) and anti-RORγt (Q31-378) from BD Biosciences.
To determine eYFP+ TH17 cells (CD4+/TCRβ+) in the small intestine lamina propria, Peyer’s patches and MLNs from Il17a reporter mice, lymphocytes were cell-surface stained with anti-CD45 (30-F11), anti-CD4 (A20), anti-TCRβ (H57-597) from Biolegend and LIVE/DEAD Fixable dye Blue (Thermo) and identified as live CD45+CD4+TCRβ+eYFP+ cells. For detection of TH17 cells (CD4+TCRβ+) expressing eYFP in the central nervous system of EAE mice, lymphocyte fraction from the brain and spinal cord was obtained by cutting tissues in small pieces, followed by digestion with 2.4 μg ml−1 collagenase II (Sigma) and 4 μg ml−1 DNAse I (Sigma), 45 min at 37 °C and separation on a Percoll gradient. Lymphocytes were cell-surface stained with CD45 (30-F11), CD11c (N418), CD4 (A20), anti-TCRα (H57-597) from Biolegend and LIVE/DEAD Fixable dye Blue (Thermo) and TH17 cells identified as live CD45+CD4+TCRβ+eYFP+. For intracellular detection of cytokines in cells from the small intestine or the central nervous system, lymphocytes were restimulated 4–5 h at 37 °C in complete RPMI with PMA (50 ng ml−1, Sigma) and ionomycin (1 μg ml−1, Sigma) in the presence of Brefeldin A (1 μg ml−1, Sigma), cell-surface stained, fixed with 2 % PFA 10 min at room temperature, and stained intracellularly with IL-17A-PE (TC11-18H10.1) using the BD CytoFix/CytoPerm kit (BD Biosciences).
Analysis of cell cycle was performed by flow cytometry in cells stained with anti-CD4 and LIVE/DEAD Fixable dye for 30 min at 4 °C, fixed with FOXP3 Permeabilisation kit (eBioscience) 60 min at room temperature and stained for 40 min with Ki67 (Biolegend). DNA content was assessed by FxCycle staining (Thermo) in the live CD4+ T cell gate.
Analysis of phospho-S6 ribosomal protein (S235/236) and phospho-4E-BP1 (Thr37/46) was performed by flow cytometry in CD4 T cells stained with anti-CD4, fixed with 2% PFA, permeabilized for 1 h with 90% methanol at −20 °C and stained with anti-pS6 (D57.2.2E) and p4EB-P1 (2365B4) fluorescent conjugated antibodies from Cell Signaling Technology.
Analysis of active caspase-3 was performed by flow cytometry in cells stained with anti-CD45, anti-CD4, anti-TCRβ (Biolegend) and LIVE/DEAD Fixable dye (Thermo) for 30 min at 4 °C, fixed with BD CytoFix/CytoPerm kit (BD Biosciences) and stained for 45 min with anti-active caspase-3 antibody (1:100, clone C92-605, BD Biosciences).
Analysis of the histone methylation and acetylation marks was performed by flow cytometry in CD4+ T cells fixed 60 min at room temperature with FOXP3 Permeabilisation kit (eBioscience) and stained for 90 min with fluorescently labelled antibodies against H3K4me3 (clone C42D8), H3K27ac (clone D5E4), H3K27me3 (clone C36B11), H3K9ac (clone C5B11), H3K9me3 (clone D4W1U), and rabbit monoclonal antibody IgG isotype control (DA1E) (all from Cell Signaling). Cells were gated on diploid cells with ‘single’ DNA content based on FxCycle staining (Thermo) in the live-cell gate.
Analysis of the DNA methylation mark 5-methylcytosine (5mC) was performed by flow cytometry in CD4+ TN cells activated in TH17 cell culture conditions for 4 days. Cells were collected, washed in PBS and stained with anti-CD4 and LIVE/DEAD Fixable dye (Thermo) for 30 min at 4 °C, followed by 60 min fixation at room temperature with FOXP3 Permeabilisation kit (eBioscience). Cells were washed, permeabilized (0.5% Triton X-100 in PBS) 15 min at room temperature and treated with 2 N HCl 30 min at 37 °C. Neutralization was performed with 100 mM Tris-HCl pH 8.8 for 10 min, followed by extensive washes with 0.05% Tween-20 in PBS. After blocking (1% BSA and 0.05% Tween-20 in PBS) for 2 h, cells were incubated with a monoclonal antibody against 5 mC (33D3, AbD Serotec) at a final concentration of 10 μg ml−1 or with an isotype control antibody (mouse IgG) for 90 min at room temperature. Samples were washed in 1% BSA, 0.05% Tween-20 in PBS and incubated with the secondary antibody for 35 min at room temperature. Cells were washed and incubated with FxCycle staining (Thermo) for DNA content analysis prior to flow cytometry acquisition.
Analysis of mitochondrial membrane potential was performed by flow cytometry in cells labelled 30 min with 50 nM tetramethylrhodamine methyl ester (Thermo) in complete medium and LIVE/DEAD Fixable dye (Thermo). Cells were gated on live cells and results expressed as geometric mean fluorescence intensity.
Analysis of mitochondrial content by flow cytometry was performed in CD4+ TN cells and CD4+ TN cells activated in TH and regulatory T cell conditions for the indicated times, staining with anti-CD4 and LIVE/DEAD Fixable dye (Thermo) for 30 min at 4 °C, followed by 60 min fixation at room temperature with FOXP3 Permeabilisation kit (eBioscience) and 60 min staining at room temperature with fluorescently labelled antibodies against TOMM20 (clone EPR15581-54), ATP5A (clone 15H4C4) and COX IV (clone EPR9442(ABC)) from Abcam and Mitofilin (clone D-3) from Santa Cruz. Cells were gated on live CD4+ cells and results expressed as geometric mean fluorescence intensity relative to CD4+ TN cells.
Intracellular calcium content was determined by flow cytometry with the ratiometric calcium indicator Indo-1 (Thermo) in cells incubated with Indo-1-AM (1 μM,) 30 min at 37 °C in complete RPMI medium without FCS. Cells were washed, resuspended in complete medium and the mean fluorescence intensity of the Calcium-bound and unbound Indo-1 signal acquired on a linear scale on a LSR III Fortessa flow cytometer (BD Bioscience) equipped with 37 °C temperature-controlled tube holder. Fluorescence intensity was analysed in FlowJo (BD Biosciences) software and represented as the bound/unbound Indo-1 ratio.
Quantitative PCR analysis
Total RNA was extracted using the RNeasy mini kit (Qiagen) with an additional on-column DNase treatment using the RNAse-Free DNase set (Qiagen), and quantified with Nanodrop (Thermo Fisher). cDNA was prepared using 200-300 ng total RNA by a reverse transcription PCR (RT–PCR) using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems), per manufacturer’s instructions. Quantitative PCR for the assessment of Il17a mRNA expression was performed on cDNA using TaqMan Gene Expression Assays (Assay ID:Mm00439619_ m1 IL17A, Thermo Fisher) on an Applied Biosystems 7000 sequence detection system, using iTaq Universal SYBR Green Supermix (Bio-Rad). Fold changes in expression were calculated by the ΔΔCt method, using Hprt or 18s ribosomal RNA (Assay ID: Mm03024075_m1 Gene: Hprt, Assay ID: Mm03928990_g1 18s Ribosomal RNA, Thermo Fisher) as endogenous controls for mRNA expression.
RNA-sequencing analysis
RNA was extracted using RNeasy Kit (QIAGEN) following the manufacturer’s instructions with an additional on-column DNase treatment using the RNAse-Free DNase set (Qiagen). RNA was quantified using Qubit 2.0 (Thermo Fisher Scientific). Libraries were prepared using the TruSeq stranded mRNA kit (Illumina) and sequenced in a HISeq 3000 (Illumina) with a read depth of approximately 30 million aligned reads per sample by the Deep-sequencing Facility at the Max Planck Institute for Immunobiology and Epigenetics (Freiburg). Sequenced libraries were processed with deepTools52 v_2.0, using STAR53 v_2.7.10, for trimming and mapping, and feature Counts54 v_2.0.3 to quantify mapped reads. Raw mapped reads were processed in R (Lucent Technologies) with DESeq255 v_1.36 to generate normalized read counts to visualize as heatmaps using Morpheus (Broad Institute) and determine differentially expressed genes with greater than 2-fold change and lower than 0.05 adjusted P-value. Gene ontology analysis was performed with DAVID56 (v_2016 and v_2021) and Ingenuity pathway analysis (QIAGEN).
ATAC–seq
Libraries were prepared using the Nextera DNA library Prep Kit (Illumina) adapting a published protocol57. In vitro-polarized TH17 cells were washed in PBS and then lysed for 3 min in 10 mM Tris-HCl, pH 7.4,10 mM NaCl, 3 mM MgCl2 and 0.1% Igepal CA-630, 0.1% Tween-20, 0.01% digitonin, 1% BSA (all from Sigma). Cold wash buffer (same as lysis buffer without Igepal and digitonin) was added to the lysate and mixed three times by inverting the tubes and immediately spun at 500g for 5 min, 4 °C. Supernatant was discarded and nuclei resuspended in 2.5 μl TDE1 (Nextera Tn5, 100 nM final), 0.5 μl 1% digitonin (0.01% final), 0.5ul 10% Tween-20 (1% final) 16.5 μl PBS and 5 μl water and incubated for 30 min at 37 °C with gentle mixing (1,000 rpm). DNA was purified with the QIAGEN MinElute PCR Purification Kit (Thermo Fisher Scientific). PCR amplification was performed with the NEBNext High-Fidelity 2× PCR Master Mix (New England Labs) using custom Nextera PCR primers containing barcodes. Adapters were removed with AMPure XP beads according to manufacturer’s protocol. Libraries were quantified with the Qubit and sequenced with a HISeq 3000 (Illumina) at the Deep-sequencing Facility at the Max Planck Institute for Immunobiology and Epigenetics (Freiburg).
Sequenced samples were trimmed with Trimmomatic58 v_0.4, mapped using Bowtie259 v_2.4.5 and replicate mapped files merged with SAM tools60 v_1.4.1. Coverage files were generated with deepTools52 v_2.0. Open chromatin and differentially regulated chromatin regions were detected with MACS261 v_2.2.7.1 using a P-value <1 × 10−7 and a q-value of less than 0.1 and differentially regulated regions identified with CSAW62,63 v_1.30.1 using a 2-fold enrichment threshold and P-value lower than 0.01. Bed files were analysed with Bedtools64 v_2.28.0, and visualized alongside coverage files on IGV v_2.4.0. Transcription factor enrichment was done with HOMER65 v_4.11.
Metabolite extraction
Equal number of cells were pelleted, medium removed by aspiration and washed once with 500 μl PBS. Metabolome extraction was performed by the addition of 10 μl ice-cold solvent (40:40:20 acetonitrile: methanol: water) every 100,000 cells. After centrifugation for 10 min at 16,000g at 4 °C, clean supernatant was transferred to a clean tube, frozen on dry ice and kept at −80 °C until LC–MS analysis.
LC–MS analysis
Targeted metabolite quantification by LC–MS was carried out using an Agilent 1290 Infinity II UHPLC in line with an Agilent 6495 QQQ-MS operating in MRM mode. MRM settings were optimized separately for all compounds using pure standards. LC separation was on a Phenomenex Luna propylamine column (50 × 2 mm, 3 μm particles) using a solvent gradient of 100% buffer B (5 mM ammonium carbonate in 90% acetonitrile) to 90% buffer A (10 mM NH4 in water). Flow rate was from 1,000 to 750 μl min−1. Autosampler temperature was 5 °C and injection volume 2 μl. Peak areas were measured using MassHunter B.07.01 (Agilent).
GC–MS analysis
To measure [13C]glucose and [13C]glutamine tracing, CD4+ TN cells were cultured and activated as described followed by incubation with fresh RPMI medium containing 11 mM [13C]glucose or 2 mM [13C]glutamine for the indicated periods of time. Cells were collected, processed for metabolome extraction and dried by SpeedVac. Dried metabolite extracts were resuspended in pyridine and derivatized with methoxyamine (sc-263468 Santa Cruz Bio) for 60 min at 37 °C and subsequently with N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamid, with 1% tert-butyldimethylchlorosilane (375934 Sigma-Aldrich) for 30 min at 80 °C. Isotopomer distributions were measured using a DB5-MS GC column in a 7890 GC system (Agilent Technologies) combined with a 5977 MS system (Agilent Technologies). Data processing, including correction for natural isotope abundance was performed by an in-house R script (https://gitlab.gwdg.de/joerg.buescher/metabolomics_scripts).
Lipidomics
Lipids were extracted using a biphasic methyl tert-butyl ether (MTBE) extraction protocol. The same number of cells per condition were resuspended in 100 μl cold PBS in glass vials. Cold methanol (750 μl), MTBE (2 ml) and water (625 μl) were added sequentially with vortexing. Samples were centrifuged to separate phases, and the upper organic phase was dried using a Genevac EZ2 speed vac. Samples were resuspended in 2:1:1 isopropanol: acetonitrile: water prior to analysis. LC–MS was carried out using an Agilent Zorbax Eclipse Plus C18 column using an Agilent 1290 Infinity II UHPLC in line with an Agilent 6495 Triple Quad QQQ-MS. Lipids were identified by fragmentation and retention time, and quantified using MassHunter B.07.01 (Agilent).
Proteomics
Sample preparation.
Protein sample preparation was carried out using 5 × 106 cells using an iST 8X kit (PreOmics), according to the manufacturer’s recommendation. All samples used for data-dependent acquisition (DDA) and data-independent acquisition (DIA) analyses were spiked with index retention time (iRT) kit peptides (Biognosys), according to the manufacturer’s instructions.
Construction of DIA spectral library.
Spectral libraries were generated by Spectronaut version 10.0 using MaxQuant results as an input66. Seventyshotgun (DDA) runs (using 2 or 3 biological replicates from each biological conditions) were acquired using a Q Exactive Plus instrument, and data were searched using MaxQuant (version 1.6.1.0). The spectral library was constructed using an FDR cut-off of 1% and a minimum and maximum of 3 and 6 fragment ions, respectively, and protein grouping was performed according to MaxQuant search results.
Mass spectrometric acquisition.
The general nanoLC–MS setup was similar to that previously described66, with minor modifications. A Q Exactive Plus mass spectrometer (Thermo Fisher) and an Easy nanoLC-1200 (Thermo Fisher) were used for both DDA and DIA experiments. For the chromatographic separation of peptides, 4 μg peptide digest was analysed at 50 °C (controlled by Sonation column oven) on a 50-cm in-house-packed fused-silica emitter microcolumn (75 μm inner diameter × 360 μm outer diameter SilicaTip PicoTip; New Objective) packed with 1.9-μm reverse-phase ReproSilPur C18-AQ beads (Dr Maisch). Peptides were separated by a 4-h linear gradient of 5–80% (80% acetonitrile, 0.1% formic acid) at a constant flow rate of 300 nl min−1.
Data analysis.
The MS2-based label-free quantification was carried out by analysing DIA raw data using Biognosys Spectronaut (version 10.0) software using default parameters as previously described66, with minor modifications. In brief, the decoy method was set to ‘mutated’, data extraction and extraction window were set to ‘dynamic’ with correction factor 1, identification was set to ‘normal distribution P-value estimator’ with q-value cut-off of 0.1, and the profiling strategy was set to ‘iRT profiling’ with q-value cut-off of 0.01. Ultimately, protein quantity was set to ‘average precursor quantity’ and smallest quantitative unit was set to ‘precursor ion’ (summed fragment ions). For statistical testing and identification of deregulated proteins in all approaches, a two-sample Student’s t-test was used to identify differentially expressed proteins filtered to 1% FDR.
Statistical analysis
Statistical analysis was performed using Prism 9 software (GraphPad) and Microsoft Excel (v16.46). Data are expressed as mean ± s.e.m. (standard error of the mean), unless otherwise indicated. Comparisons for two groups were calculated using unpaired two-tailed Student’s t-tests (for two groups meeting the normal distribution criteria, according to the Shapiro-Wilk normality test). Comparisons of more than two groups and grouped data were calculated using one-way or two-way ANOVA, and corrected for multiple comparisions between groups or to a reference group using Tukey’s, šidák’s or Dunnett’s tests. Exact P-values and details of statistical testing can be found in the Figure legends and in the Source data file. Unless otherwise specified, n represents the number of individual biological replicates and is represented in graphs as one dot per sample. Flow cytometry plots are representative of at least three replicates. Immunoblots are representative of at least two independent experiments. Confocal and electron microscopy images are representative of at least three independent biological replicates. No statistical method was used to predetermine sample size, but a minimum of three samples were used per experimental group and condition. Experiments were not randomized.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Mitochondrial membrane remodeling in CD4+TN cells under T-helper (Th) and regulatory (Treg) conditions in vitro and in vivo.
a) Mitochondrial sphericity In live cell spinning disk microscopy images In CD4+ TN cells (PhAM mice) cultured in Th and Treg conditions (TN: n = 1673, Th1: n = 8406, Th2: n = 5485, Th17: n = 7672 and Treg: n = 4824 segmented objects (mitochondria), 2 independent experiments. Violin plot shows median with quartiles. b) EM images (left) and cristae width (right) in TN cells cultured in Th and Treg conditions (TN: n = 362, Th1: n = 479, Th2: n = 369, Th17: n = 319, and Treg: n = 294 cristae, 3 biological replicates). (c) Intracellular expression in TN, Th and Treg cells (n = 4 biological replicates). Representative graph summarize results of two independent experiments. d) Immunoblot of CD4+ TN, Th and Treg cells, day 4, representative experiment out of two. ACTIN: sample processing control (e) Quantification of protein levels from immunoblots as in Fig. 1b (n = 4 biological replicates, 2 independent experiments). f) Immunoblot of proteins that control mitochondrial dynamics in CD4+ TN cells cultured for the indicated times in Th and Treg conditions. TUBULIN: sample processing control. Right, protein levels (n = 4 biological replicates, 2 independent experiments). g) EM images (left) and cristae width (right) in CD4+ TN and effector T cells from reporter mice subjected to immune challenges (naive: n = 410, Th1: n = 262, Th2: n= 675, Th17: n = 461 and Treg: n = 402 cristae, 3 mice per condition). Data b, g are geometric mean, c mean and e, f means ± s.e.m. Two-sided Tukey’s test (b) or Dunnett’s test (a, c, e, f, g). Exact P values are indicated.
Extended Data Fig. 2 |. Mitochondrial dysfunction and proliferation defects in Opa1CD4cre T cells.
CD4+ TN cells from control and Opa1cd4cre mice were cultured 4 days in Th and Treg conditions. a) Immunoblot for OPA1 at the end of the culture (2 biological replicates per condition shown). ACTIN: loading control. b) Spinning disk microscopy images from mitochondria (MitoTracker Deep Red, purple) and nuclei (Hoechst, blue). Scale bar: 2 μm. Representative images from 3 biological replicates. c) Baseline OCR (left) and ECAR (right) (n = 4 biological replicates). d) Mitochondrial membrane potential determined by Tetramethylrhodamine methyl ester (TMRM) (n = 3 biological replicates) e) Cell proliferation, day 3 (n = 8 biological replicates, 2 independent experiments). f) Cell viability determined by 7-aminoactinomycin D (7-AAD) and annexin-V staining, day 4 (n = 7 biological replicates, 2 independent experiments). g) Cell-cycle analysis determined by Ki-67 and DAPI staining (n = 3 biological replicates). h) Surface expression of CD25, CD44 and CD69, and intracellular expression of Nur77 in CD4+ TN cells from control and Opa1CD4Cre mice cultured 3 days in Th and Treg conditions (n = 4 biological replicates). i) IL-17A expression and proliferation in control and Opa1CD4Cre Th17 cells, day 3. Graph shows IL-17A expression per each cell division state gated on live CD4+ T cells (n = 4 biological replicates).j) IL-17A expression in CD4+ TN cells cultured in Th17 conditions with increasing concentration of plate bound anti-CD3 or IL-6 for three days and restimulated (n = 4 biological replicates). Representative plots and a graph summarizing the results of at least two independent experiments are shown, except where noted otherwise. Data are means ± s.e.m. Two-sided šidák’s test (d, e, f, h, i, j) or unpaired two-tailed t-test (c). Exact P values are indicated.
Extended Data Fig. 3 |. Role of inner and outer MM remodeling in IL-17A production.
Mfn1, Mfn2 or Dnml1 floxed mice were crossed with CD4Cre mice (Mfn1CD4Cre, Mfn2CD4Cre and Drp1CD4Cre). a, b, c, d, e, CD4+ TN cells across genotypes were cultured in Th and Treg conditions. a) Immunoblot of MFN1, MFN2 and DRP1 in a representative experiment out of two. ACTIN: loading control. b) Cytokine and TF expression in Th- and Treg-reestimulated cells across genotypes (Th1 conditions: Control n=15; Mfn1, Mfn2 n =11; Drp1 n =8; Th2, Th17 and Treg conditions; Control n =11; Mfn1, Mfn2 n =7; Drp1 n =4, biological replicates). c) Intracellular levels of T-bet and ROR-γt (Control n = 15; Mfn1CD4Cre n =11; Mfn2CD4Cre n =11; Drp1CD4Cre n =8, biological replicates). d) Proliferation as determined by Cell Trace Violet dilution staining (n = 4 biological replicates, except control; n = 6). e) Relative baseline OCR, ATP-coupled respiration, and maximal respiration in Th17 cells across genotypes (n = 4 biological replicates per group).f, g, h, i, Control and Opa1CD4Cre CD4+ TN cells were transfected with Cas9 and indicated gRNAs. f) Immunoblot for DRP1 and OPA1. ACTIN: loading control. g) Representative spinning disk confocal images from mitochondria (Mitotracker Deepred, green) and nuclei (Hoechst, blue) from 3 biological and transfection replicates. h) OCR upon oligomycin (Oligo), fccp and rotenone + Antimycin A (ROT+AA) addition (left) and relative baseline OCR (right) (n =4 biological and transfection replicates). i) Intracellular IL-17A expression (n =7 biological and transfection replicates). Representative plots and a graph summarizing the results of at least two independent experiments are shown, except where noted otherwise. Data are means ± s.e.m. Two-sided Tukey’s test (b, c, d, e, h, i). Exact P values are indicated.
Extended Data Fig. 4 |. OPA1-requirement for IL-17A expression across cell culture and in vivo conditions.
TN cells cultured in Th17 non-pathogenic (np) and pathogenic (p) conditions. a) EM images and cristae width (Control np: n = 528, p: n = 343; Opa1CD4Cre np: n = 321, p: n = 267 cristae, 3 biological replicates). Graph shows geometric mean, black. (b) Cytokine (c) TF expression, d) supernatant cytokines, and e) proliferation, n = 4 biological replicates. f) Cell viability (n = 8, 2 independent experiments). g) Gating strategy to identify CD4+TCRβ+eYFP+ (Th17) cells. h) Ki-67 expression in LP Th17 cells (Control: n = 6; Opa1IL17aCre: n = 3), PP (Control: n = 7; Opa1IL17aCre: n = 4) and MLN (Control: n = 6; Opa1IL17aCre: n = 3), 2 independent experiments. i) Th17 cell numbers (LP: n = 3; PP; Control: n = 3; Opa1IL17aCre: n = 4, MLN: n = 4). Mice were anti-CD3 immunized.j) LP Th17 cell numbers (n = 4), k) ROR-γT expression (n = 4), l) surface expression (CCR6, CXCR3: Control: n = 4; Opa1IL17aCre: n = 3; ICOS, CD27, CD103: n = 4) and m) Ki-67 levels (Control: n = 9; Opa1IL17aCre: n =8, 2 independent experiments). Mice were MOG immunized. n) Disease incidence, onset and peak disease (days, controlIL17aCre: n = 16; Opa1IL17aCre: n = 14, 2 independent experiments. o) Cell numbers (n = 8). p) Neutrophil percentage (CD45+CD11b+Ly6G+, n = 8). q) CCR6 and CXCR3 levels (n = 8). r) Phosphorylated S6 and 4E-BP1 (CD4+TCRβ+eYFP+cells). Plots and graphs summarize results of at least two independent experiments, except noted otherwise. Data are means ± s.e.m. Two-sided Tukey’s test (a) or šidák’s test (b, c, e, i, l, q) or unpaired two-tailed t-test (h, j, k, m, o, p). Exact P values are indicated.
Extended Data Fig. 5 |. Th17 cells rely on OPA1 independent of metabolic activity.
a, b, c, d, CD4+ TN cells were cultured in Th and Treg conditions.
a) Bioenergetic profile (baseline OCR/ECAR, n = 4 independent experiments). b) Glucose and kynurenine uptake, phosphorylated S6 (n = 4) and 4E-BP1 (Th1: n = 3, Th2,Th17,Treg: n = 4 biological replicates). c) de novo protein and nascent RNA synthesis by O-propargyl puromycin (OPP) and ribonucleoside ethynyl uridine (EU) incorporation, respectively (n = 4 biological replicates). Cyclohexamide (CHX) and actinomycin D (ActD): negative controls. d) Protein translation dependence on mitochondria (left, Th1, Th2: n = 6; Th17,Treg: n = 7 biological replicates) or glycolysis (right, Th1, Th2: n = 7: Th17, Treg: n = 8 biological replicates), 2 independent experiments. (e) OCR and ECAR (10 mM: n = 8; 1 mM and 0 mM n = 4 biological replicates) and (f) IFN-γ expression (n = 3 biological replicates) in cells cultured in Th1 conditions 3 days before overnight culture in RPMI with 10% non-dialyzed FBS and decreasing glucose concentrations. g, h, i, CD4+ TN cells cultured in Th17 np/p conditions. g) Bioenergetic profile (n = 4 biological replicates). h) de novo protein synthesis (left, n = 7 biological replicates, 2 independent experiments) and nascent RNA synthesis (right, np/p: n = 4, ActD: n = 2 biological replicates). i) Protein translation dependence on mitochondria (left, np: n = 8: p: n = 3 biological replicates) or glycolysis (right, np: n = 8: p: n = 7 biological replicates, 2 independent experiments). Representative plots and graphs summarize results of at least two independent experiments, except where noted otherwise. Data are means ± s.e.m. Two-sided Tukey’s test (b–e), šidák’s test (f) or unpaired two-tailed t-test (h, i). Exact P values are indicated.
Extended Data Fig. 6 |. Multi-OMICS analysis of Th17 cells upon OPA1 deletion.
Control and Opa1CD4CTe CD4+ TN cells were cultured in Th17 conditions and the RNA extracted and sequenced. a) Volcano plot shows differentially expressed genes (1518: log10 P value < 3, log2 FC > 0.5). b) DAVID KEGG pathway analysis from differentially up- (779) or downregulated (739) genes (FDR, false discovery rate). c) Control and Opa1CD4Cre CD4+ TN cells were cultured in Th17 conditions and proteins from whole cell lysates extracted and analyzed (LC-MS). Left, volcano plot from differentially expressed proteins (562: log10 P value<1.3, log2 FC > 0.58). Middle and right volcano plots, protein expression in relation to OXPHOS and serine and glycine biosynthesis pathways, respectively. d) Cellular ATP, AMP and AMP/ATP ratio by LC-MS (n= 4 per genotype, day 4). e) Schematic of [U-13C]-Glucose and [U-13C]-Glutamine carbon allocation into central carbon metabolites. f) Fractional labelling of palmitate with [U-13C]-Glucose (left) or [U-13C]-Glutamine (right) in control and Opa1CD4Cre Th17 cells cultured 20 h on day 3 with fully labelled subsrates (n = 4 per group). g) Control and Opa1CD4ae CD4+ TN cells were cultured in Th17 conditions 4 days and lipids extracted and analyzed (n = 4 biological replicates). Graph, FC of distinct lipid classes in Opa1CD4Cre vs. control (HEXCER: hexosylceramide, TG: triacylglycerades, LPI: lysophosphatidylinositol, DG: diacylglycerol, PA: phosphatidic acid, LPE: lysophosphatidylethanolamine, PG: phosphatidylglycerol, Cholesterol, PC: phosphatidylcholines, CER: ceramides, SM: sphingomyelin, PS: phosphatidylserine, LPC: lysophosphatidylcholines, PE: phosphatidylethanolamine, PI: phosphatidylinositol, Sphingosine, LPS: lipopolysaccharide, CE: cholesterol esters, CL: cardiolipins). Transcriptomics, proteomics and lipidomics were performed once (a,b: n = 3; g: n = 4 biological replicates). Graphs summarize results of at least two independent experiments in d, f. Data are means ± s.e.m. Unpaired two-tailed t-test (a, c, d, f, g). Exact P values are indicated.
Extended Data Fig. 7 |. OPA1 controls chromatin accessibility in Th17 cells. a) CD4+ TN cells.
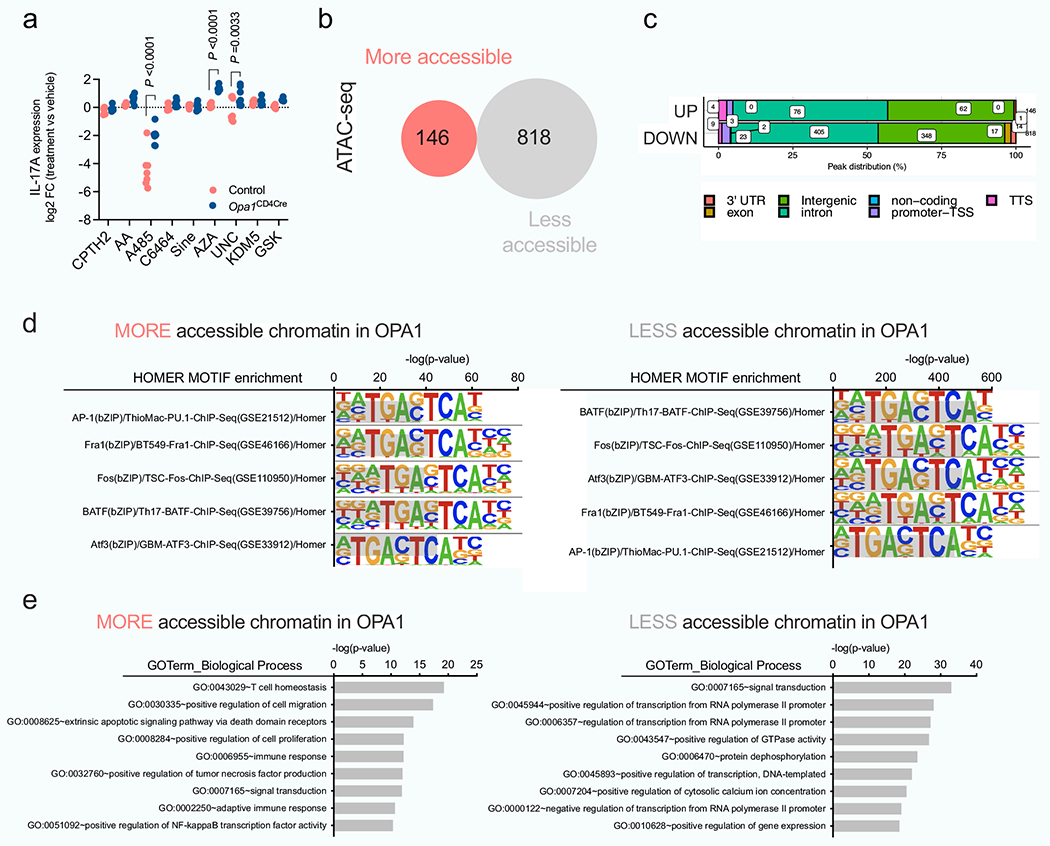
a) CD4+ TN cells from control and Opa1CD4Cre mice were cultured in Th17 conditions for 1 day followed by 48 h incubation with inhibitors for histone acetylation and methylation, and DNA methylation. IL-17A expression was analyzed by flow cytometry and results show log2 FC of IL-17A MFI in inhibitor-treated vs vehicle-treated cells (n = 7 biological replicates for all conditions except GSK: n = 4 biological replicates). Graph combines data from two independent experiments. Data are mean ± s.e.m. Two sided šidák’s test. Exact P values are indicated. b, c, d, e, CD4+ TN cells from control and Opa1CD4Cre mice were cultured in Th17 conditions 4 days and the DNA extracted and processed for ATAC-seq (n = 3 biological replicates). b) Number of differentially accessible chromatin regions (FC >2, p < 0.01). c) Genomic localization of the differentially regulated accessible chromatin regions. d) HOMER motif TF analysis on regulated chromatin regions. e) GO term pathway enrichment analysis from the differentially regulated accessible chromatin regions between Opa1CD4Cre and control Th17 cells (FC >2, p < 0.01).
Extended Data Fig. 8 |. LKB1 deletion in OPA1 reestablishes IL-17A expression.
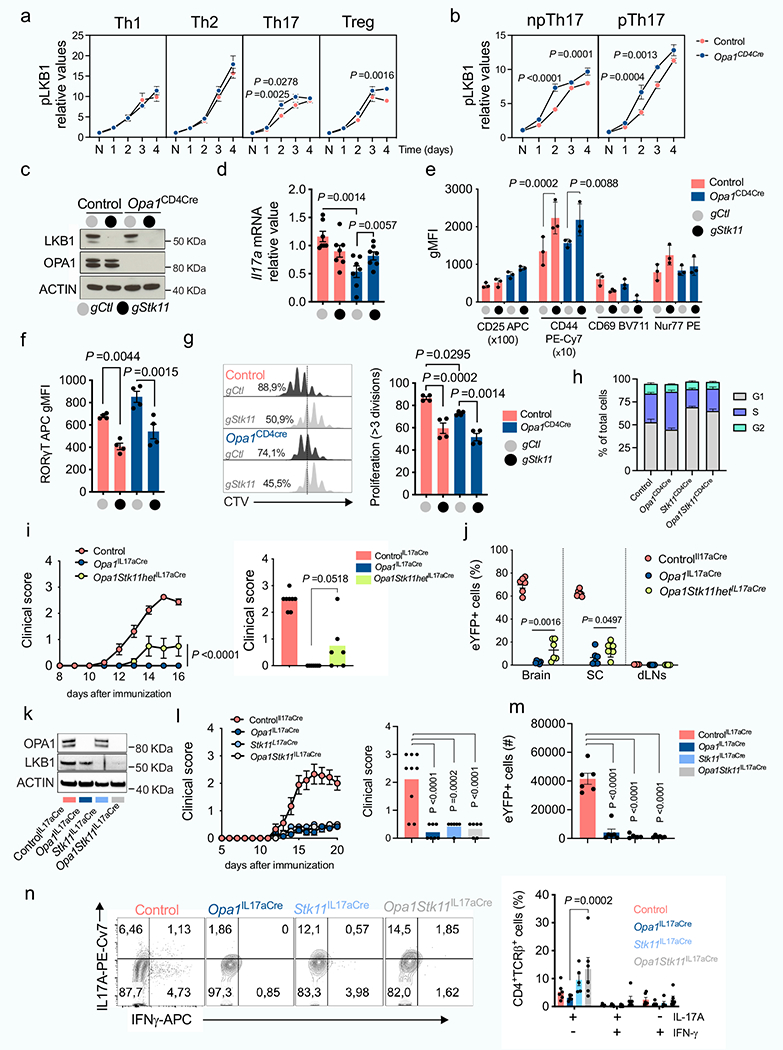
LKB1 phosphorylation kinetics in CD4+ TN cells cultured in Th and Treg (a) and np/p Th17 (b) conditions (Control: n = 4; Opa1CD4Cre: n = 3 biological replicates). c, d, e,f g, h Control and Opa1CD4Cre CD4+ TN cells transfected with Cas9 and indicated gRNAs. (c) Immunoblot for LKB1 and OPA1. ACTIN: loading control, (d) IL-17A mRNA expression (RT-PCR, n = 7), (e) CD25, CD44,CD69 and Nur77 expression (n = 3), (f) ROR-γT expression (n = 4), (g) cell proliferation (n = 4), (h) and cell-cycle analysis (n = 4, biological and transfection replicates). i) Left, clinical disease score upon MOG immunization and at day 16 (Right), (control: n = 8; Opa1IL17aCre: n = 7; and Opa1Stk11hetIL17aCre: n = 6 mice).j) CD4+TCRβ+eYFP+cell frequencies, day 20 post-immunization (n = 6 mice). k) Immunoblot for LKB1 and OPA1 in CD4+TCRβ+eYFP+ cells from control, Opa1IL17aCre, Stk11IL17aCre and Opa1Stk11IL17aCre mice. ACTIN: loading control. I) Left, clinical disease score upon MOG immunization and at day 16 (Right) (control: n = 9; Opa1IL17aCre: n = 7; Stk11IL17aCre: n = 6 and Opa1Stk11IL17aCre: n = 6 mice). m) CD4+TCRβ+eYFP+ SC cell numbers (n = 6 mice per genotype except Stk11IL17aCre: n = 5 mice) and n) IL-17A and IFN-γ expression, day 20 postimmunization (control: n = 6 mice; Opa1IL17Cre: n = 5 mice; Stk11IL17Cre: n = 5 mice; Opa1Stk11IL17Cre: n = 6 mice). Representative plots and graphs summarize results of at least two independent experiments, except noted otherwise. Data are means ± s.e.m. Two-sided šidák’s test (a, b, h, n) or Tukey’s test (d, e, f, g, i, j, l, m) . Exact P values are indicated.
Extended Data Fig. 9 |. LKB1 antagonizes OPA1 deficiency independent of mTOR and AMPK.
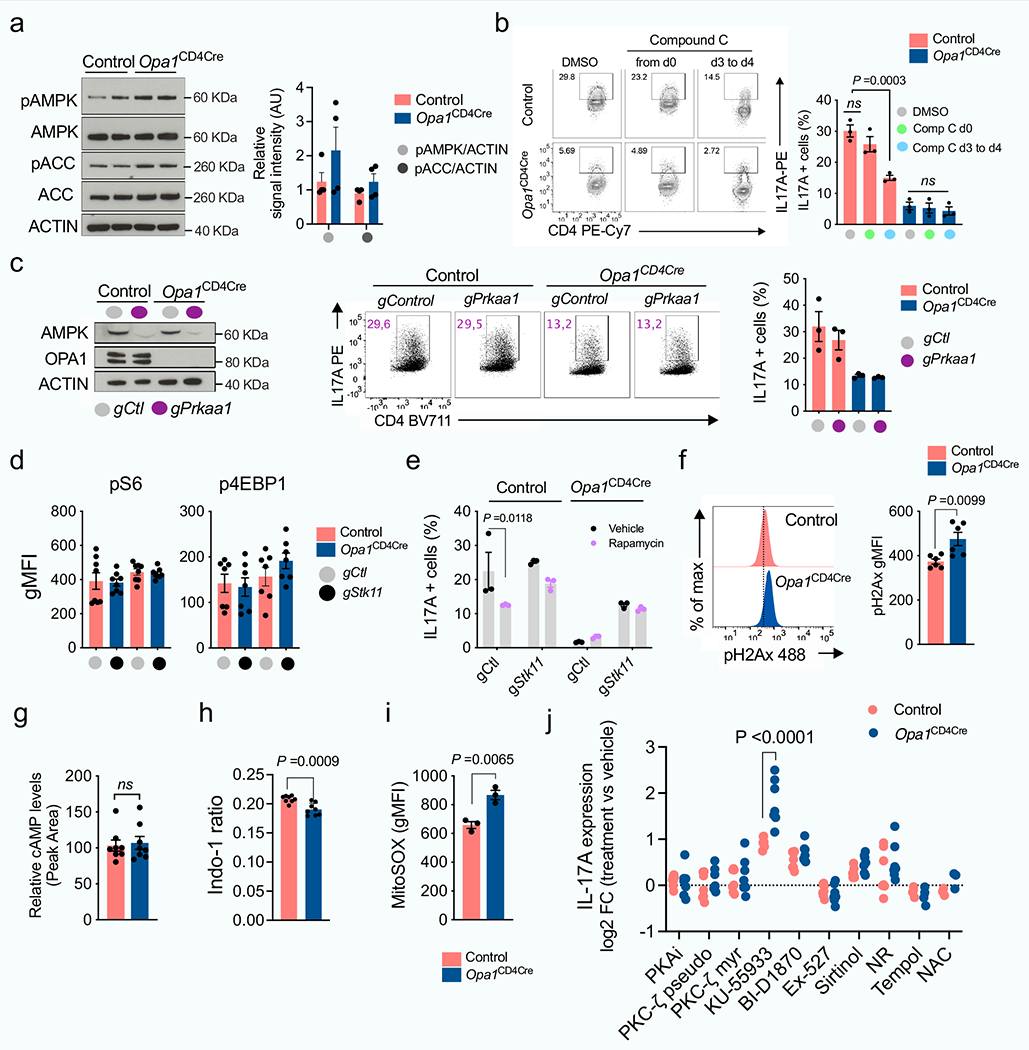
a) Immunoblot for phosphorylated and total AMPK and ACC in control and Opa1CD4Cre Th17 cells. ACTIN: sample processing control. Graph, relative signal intensity (n = 4, 2 independent experiments). b) IL-17A expression in control and Opa1CD4Cre CD4+ TN cells cultured in Th17 conditions and Compound C from day 0 (d0) or from day 3 to 4 (d3) and restimulated (n = 3). c) Left, immunoblot for AMPK and OPA1 in T cells transfected with Cas9 and indicated gRNAs and cultured 5 days in Th17 conditions. ACTIN: sample processing control. Right, IL-17A expression in restimulated cells (n = 3 biological and transfection replicates). d) Expression of phosphorylated S6 (n = 8) and 4E-BP1 (n = 7 biological and transfection replicates), 2 independent experiments. e) IL-17A expression in T cells transfected with Cas9 and indicated gRNAs and cultured in Th17 conditions 2 days followed by 48 h rapamycin treatment (n = 3 biological and transfection replicates). f) Phosphorylated H2Ax expression (n = 6, 2 independent experiments). g) Cyclic AMP (cAMP) levels (LC-MS, n = 8, 2 independent experiments). h) Basal calcium levels (n = 8, 2 independent experiments). i) Mitochondrial ROS (mitoSOX, n = 3). j) Control and Opa1CD4Cre CD4+ TN cells cultured in Th17 conditions 1 day followed by 2 day inhibitor-treatment. Graph, IL-17A MFI log2 FC inhibitor-treated vs. vehicle-treated cells (n = 7 biological replicates, except Tempol: n = 4 and NAC: n = 3, 2 independent experiments). Representative plots and graphs summarize results of at least two independent experiments, except noted otherwise. Data (a–i) are means ± s.e.m. Unpaired two-tailed t-test (a, f, g, h, i), two-sided Tukey’s test (b, c, d) or šidák’s test (e, j). Exact P values are indicated.
Extended Data Fig. 10 |. LKB1 couples TCA cycle and serine metabolism to Th17 effector function.
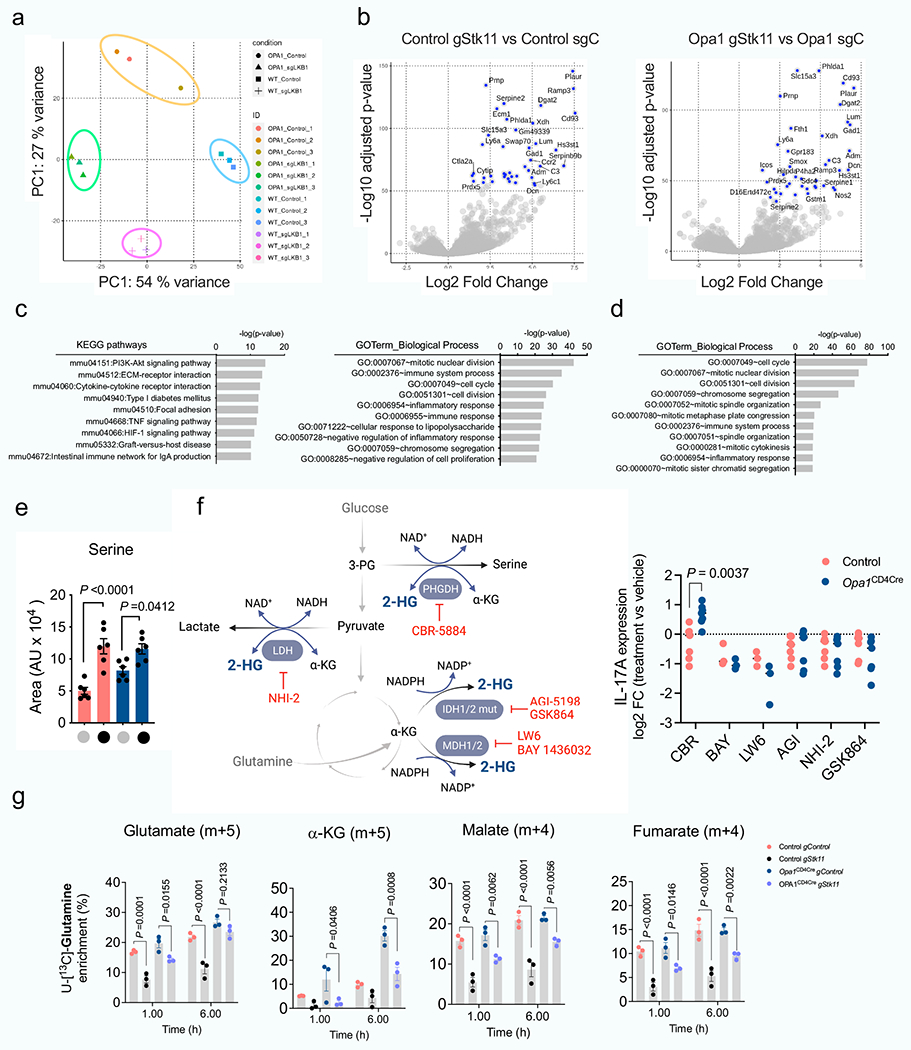
a) Principal component analysis (PCA) of the transcriptome of control and Opa1CD4Cre CD4+ TN cells transfected with Cas9 and indicated gRNAs, cultured 4 days in Th17 conditions (n = 3 biological and transfection replicates). b) Volcano plots of the differentially expressed genes (DEG, FC > 1, p-value < 0.001) for the comparisons: control gStk11 vs. control gCtl (left) and Opa1CD4CregStk11 vs. Opa1CD4CregCtl (right). Blue dots annotate the highest 40 DEG. c, d, KEGG pathway and Gene Ontology (GO) annotation analysis from DEG (FC>1, p-value<0.001) between (c) control gStk11 vs. control gCtl or (d) Opa1CD4CregStk11 vs. Opa1CD4CregCtl. e) Intracellular serine in control and Opa1CD4Cre CD4+ TN cells transfected with Cas9 and indicated gRNAs (n = 6 transfection replicates, 2 independent experiments) by LC-MS. f) Left, schematic shows 2-HG production from primary and promiscuous enzymatic reactions and the inhibitors used for targeting its production. Right, CD4+ TN cells from control and Opa1CD4Cre mice were cultured under Th17 conditions for 1 day followed by 48 h inhibitor treatment. IL-17A expression was analyzed by flow cytometry in restimulated cells and expressed as log2 FC inhibitor-treated vs. vehicle-treated cells (CBR, AGI, NHI-2, GSK864: n = 7 per group; BAY, LW6: n = 3 per group). Graph summarizes data from 2 experiments. g) Kinetics of fractional labelling of glutamate (m + 5), α-ketoglutarate (m + 4), malate (m + 4) and fumarate (m+4) in control and Opa1CD4Cre CD4+ T cells transfected with Cas9 and indicated gRNAs cultured for 4 days in Th17 conditions and incubated with [U-13C]-Glutamine for the indicated times (n = 3 biological and transfection replicates). Data are means ± s.e.m. Two-sided šidák’s test (f) or Tukey’s test (e, g). Exact P values are indicated.
Supplementary Material
Acknowledgements
We thank the members of the Immunometabolism Department at the MPI-IE for helpful discussion; the Electron Microscopy Laboratory at the University of Padova for the invaluable help; and the Imaging, Metabolomics, FACS, Sequencing and Animal facilities at the MPI-IE for their excellent technical support. This work was supported in part by the Max Planck Society, the Leibniz Prize (to E.L.P.), two Bloomberg Distinguished Professorships from Johns Hopkins University (E.L.P. and E.J.P.), the National Institutes of Health R01AI156274 (to E.L.P.) and R35GM144103 (to H.S.), a Marie Sklodowska-Curie actions Individual Fellowship (MSCA-IF) (to F.B.), a Sir Henry Wellcome Fellowship (to D.J.P.) and an Alexander von Humboldt Postdoctoral Fellowship (to M.V.).
Footnotes
Code availability
LC–MS and GC–MS metabolomics data were analysed using R code developed in-house, which is publicly available at https://gitlab.gwdg.de/joerg.buescher/metabolomics_scripts.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-05264-1.
Competing interests E.L.P. is member of the scientific advisory boards of ImmunoMet Therapeutics. E.L.P. and E.J.P. are founders of and scientific advisors to Rheos Medicines.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-05264-1.
Peer review information Nature thanks Navdeep Chandel, Luca Scorrano and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Data availability
RNA-sequencing and ATAC–seq data have been deposited in the Gene Expression Omnibus (GEO) as the superseries GSE207603. This super-series contains RNA-sequencing datasets with accession numbers GSE156742 and GSE207601 and a single ATAC–seq dataset with accession number GSE207602. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository, with the dataset identifier PXD036162. Source data are provided with this paper.
References
- 1.Buck MD, O’Sullivan D & Pearce EL T cell metabolism drives immunity. J. Exp. Med 212, 1345–1360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailis W et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu T et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 548, 228–233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giacomello M, Pyakurel A, Glytsou C & Scorrano L The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol 21, 204–224 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Reyes I & Chandel NS Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun 11, 102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cogliati S et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye D, Guan KL & Xiong Y Metabolism, activity, and targeting of d- and l-2-hydroxyglutarates. Trends Cancer 4, 151–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan DC Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol 15, 235–259 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Youle RJ & van der Bliek AM Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matilainen O, Quiros PM & Auwerx J Mitochondria and epigenetics—crosstalk in homeostasis and stress. Trends Cell Biol. 27, 453–463 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Herkenne S et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab. 31, 987–1003.e1008 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Song Z, Chen H, Fiket M, Alexander C & Chan DC OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol 178, 749–755 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishihara N, Fujita Y, Oka T & Mihara K Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacVicar T & Langer T OPA1 processing in cell death and disease—the long and short of it. J. Cell Sci 129, 2297–2306 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Korn T, Bettelli E, Oukka M & Kuchroo VK IL-17 and Th17 Cells. Annu. Rev. Immunol 27, 485–517 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wagner A et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 184, 4168–4185.e4121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanov II et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sano T et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirota K et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol 12, 255–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esplugues E et al. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birsoy K et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arguello RJ et al. SCENITH: a flow cytometry-based method to functionally profile energy metabolism with single-cell resolution. Cell Metab. 32, 1063–1075.e1067 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaublomme JT et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao XR et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, e10575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikkanen J et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Birsoy K et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sullivan LB et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L et al. Niche-selective inhibition of pathogenic Th17 cells by targeting metabolic redundancy. Cell 182, 641–654.e620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berod L et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med 20, 1327–1333 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Locasale JW et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet 43, 869–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blagih J, Krawczyk CM & Jones RG LKB1 and AMPK: central regulators of lymphocyte metabolism and function. Immunol Rev 249, 59–71 (2012). [DOI] [PubMed] [Google Scholar]
- 32.MacIver NJ et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J. Immunol 187, 4187–4198 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He N et al. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc. Natl Acad. Sci. USA 114, 12542–12547 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang K et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature 548, 602–606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kottakis F et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shackelford DB & Shaw RJ The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tiainen M, Ylikorkala A & Makela TP Growth suppression by Lkb1 is mediated by a G1 cell cycle arrest. Proc. Natl Acad. Sci. USA 96, 9248–9251 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kullmann L & Krahn MP Controlling the master-upstream regulation of the tumor suppressor LKB1. Oncogene 37, 3045–3057 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Fan J et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol 10, 510–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Intlekofer AM et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 22, 304–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Intlekofer AM et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol 13, 494–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang L et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 31, 809–821.e806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buck MD et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faubert B et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl Acad. Sci. USA 111, 2554–2559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson MO et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795.e1719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baksh SC & Finley LWS Metabolic coordination of cell fate by α-ketoglutarate-dependent dioxygenases. Trends Cell Biol. 31, 24–36 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buck MD, Sowell RT, Kaech SM & Pearce EL Metabolic instruction of immunity. Cell 169, 570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reinhardt RL, Liang HE & Locksley RM Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol 10, 385–393 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohrs K, Wakil AE, Killeen N, Locksley RM & Mohrs M A two-step process for cytokine production revealed by IL-4 dual-reporter mice. Immunity 23, 419–429 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wan YY & Flavell RA Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl Acad. Sci. USA 102, 5126–5131 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinclair LV, Neyens D, Ramsay G, Taylor PM & Cantrell DA Single cell analysis of kynurenine and system L amino acid transport in T cells. Nat. Commun 9, 1981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC–seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.21–21.29.29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lun AT & Smyth GK De novo detection of differentially bound regions for ChIP-seq data using peaks and windows: controlling error rates correctly. Nucleic Acids Res. 42, e95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lun AT & Smyth GK csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 44, e45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Musa YR, Boller S, Puchalska M, Grosschedl R & Mittler G Comprehensive proteomic investigation of Ebf1 heterozygosity in pro-B lymphocytes utlizing data independent acquisition. J. Proteome Res 17, 76–85 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing and ATAC–seq data have been deposited in the Gene Expression Omnibus (GEO) as the superseries GSE207603. This super-series contains RNA-sequencing datasets with accession numbers GSE156742 and GSE207601 and a single ATAC–seq dataset with accession number GSE207602. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository, with the dataset identifier PXD036162. Source data are provided with this paper.