


Keywords: chronic kidney disease, epoxyeicosatrienoic acids, renal fibrogenesis, soluble epoxide hydrolase, unilateral ureteral obstruction
Abstract
Epoxyeicosatrienoic acids (EETs) are arachidonic acid metabolites with biological effects, including antiapoptotic, anti-inflammatory, and antifibrotic functions. Soluble epoxide hydrolase (sEH)-mediated hydrolysis of EETs to dihydroxyeicosatrienoic acids (DHETs) attenuates these effects. Recent studies have demonstrated that inhibition of sEH prevents renal tubulointerstitial fibrosis and inflammation in the chronic kidney disease model. Given the pathophysiological role of the EET pathway in chronic kidney disease, we investigated if administration of EET regioisomers and/or sEH inhibition will promote antifibrotic and renoprotective effects in renal fibrosis following unilateral ureteral obstruction (UUO). EETs administration abolished tubulointerstitial fibrogenesis, as demonstrated by reduced fibroblast activation and collagen deposition after UUO. The inflammatory response was prevented as demonstrated by decreased neutrophil and macrophage infiltration and expression of cytokines in EET-administered UUO kidneys. EET administration and/or sEH inhibition significantly reduced M1 macrophage markers, whereas M2 macrophage markers were highly upregulated. Furthermore, UUO-induced oxidative stress, tubular injury, and apoptosis were all downregulated following EET administration. Combined EET administration and sEH inhibition, however, had no additive effect in attenuating inflammation and renal interstitial fibrogenesis after UUO. Taken together, our findings provide a mechanistic understanding of how EETs prevent kidney fibrogenesis during obstructive nephropathy and suggest EET treatment as a potential therapeutic strategy to treat fibrotic diseases.
NEW & NOTEWORTHY Epoxyeicosatrienoic acids (EETs) are cytochrome P-450‐dependent antihypertensive and anti-inflammatory derivatives of arachidonic acid, which are highly abundant in the kidney and considered renoprotective. We found that EET administration and/or soluble epoxide hydrolase inhibition significantly attenuates oxidative stress, renal cell death, inflammation, macrophage differentiation, and fibrogenesis following unilateral ureteral obstruction. Our findings provide a mechanistic understanding of how EETs prevent kidney fibrogenesis during obstructive nephropathy and suggest that EET treatment may be a potential therapeutic strategy to treat fibrotic diseases.
INTRODUCTION
Chronic kidney disease (CKD) is a major public health concern and is highly prevalent due to an increase in the incidence of hypertension, obesity, diabetes, and other cardiovascular diseases (1–3). Current therapies inhibiting the renin-angiotensin-aldosterone system can slow CKD progression and prevent CKD-related complications, but their therapeutic efficacy is quite limited (4). Therefore, the development of novel approaches to treat CKD is urgently required. Renal fibrosis is characterized by tubular cell injury, interstitial infiltration of inflammatory cells, excessive accumulation of fibroblasts, and interstitial matrix deposition, leading to CKD progression and, ultimately, end-stage kidney disease (5–7). Damaged tubular cells secrete proinflammatory cytokines, such as interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α, which constitute the major receptor-triggered fibrotic and apoptotic signals in the injured kidney (6, 8). Interstitial infiltration of inflammatory cells also produces cytokines responsible for tubular apoptosis and fibroblast proliferation and activation, which leads to renal fibrosis (6, 8).
Epoxygenase cytochrome P-450 (CYP) enzymes generate epoxyeicosatrienoic acids (EETs) by catalyzing the epoxidation of arachidonic acid olefin bonds, resulting in the production of four regioisomeric EETs: 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET (9). Once formed, EETs act in an autocrine or paracrine manner to elicit biological responses (9). Soluble epoxide hydrolase (sEH), a key enzyme responsible for the conversion of EETs to their corresponding less potent diols dihydroxyeicosatrienoic acids (DHETs), is widely distributed in mammalian tissues, including the kidney (9, 10). Increased kidney sEH expression and decreased EET levels contribute to kidney disease through vascular dysfunction, inflammation, and fibrosis (11, 12). These findings have led to the suggestion that increase of EET levels and inhibition of sEH activity could have protective effects in the treatment of kidney disease. We have previously demonstrated that increased EETs by genetic or pharmacological inhibition of sEH promotes anti-inflammatory and fibroprotective effects in unilateral ureteral obstruction (UUO) kidneys via activation of peroxisome proliferator-activated receptor isoforms and downregulation of NF-κB, transforming growth factor (TGF)-β1/Smad3, and inflammatory signaling pathways (13, 14). Recent studies have also reported that increased EET levels by either genetic sEH deficiency or its pharmacological inhibition enhance anti-inflammatory, antiapoptotic, and antifibrotic effects in the kidney, supporting our previous findings (15–17). Yang et al. (18) demonstrated that EET stabilization through the sEH inhibitor 12(3-adamantan-1-yl-ureido)-dodecanoic acid can decrease myofibroblast formation by attenuating endothelial-mesenchymal transition in the UUO model. Moreover, 12(3-adamantan-1-yl-ureido)-dodecanoic acid inhibits proinflammatory M1 polarization by suppressing the NF-κB pathway, leading to prevent the progression of renal interstitial fibrosis in the IgA nephropathy model (19). Treatment with 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl)urea, an sEH inhibitor, significantly attenuated high-fat diet-induced renal injury (20). To test the efficacy of EET administration and to examine if combined EET and sEH inhibition will exhibit additive or synergistic antifibrotic and renoprotective effects in renal fibrosis induced by UUO, we used genetic or pharmacological sEH inhibition and exogenous EET administration.
MATERIALS AND METHODS
Animal Experiments
Male C57BL/6J mice aged 8–10 wk were purchased from Jackson Laboratories. Male sEH-knockout (Ephx2−/−) mice were generated as previously reported (14). All mouse experi ments were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of University of Nebraska Medical Center. UUO was conducted as previously reported (21, 22). Briefly, mice were anesthetized with an intraperitoneal injection of a cocktail containing ketamine (100 mg/kg body wt) and xylazine (16 mg/kg body wt). After exposure of the left kidney through a left flank incision, the left ureter was ligated completely near the kidney pelvis using a 5-0 silk tie, as previously described (21, 22). Sham-operated mice were subjected to the same surgical procedure except for the ureter ligation. Kidneys were harvested at 7 days after UUO. Kidneys were either snap frozen in liquid nitrogen for Western blot analysis or fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for histological experiments. For the pharmacological inhibition of sEH, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid (t-TUCB; 0.4 mg/mouse/day) or vehicle (0.5% methylcellulose) was administered by oral gavage beginning 24 h before UUO as previously described until the time of euthanasia (13). Mice were administered with isomers 11,12‐EET and 14,15‐EET (15 μg/kg/day, respectively, Cayman, Ann Arbor, MI) or vehicle (30% ethanol) through an intraperitoneally implanted osmotic pump (Alzet, Cupertino, CA) beginning at the time of UUO until the time of euthanasia. Briefly, mice were anesthetized, and an osmotic pump (model 1007 D, Alzet) was implanted in the peritoneal cavity. The osmotic pumps are designed to deliver a flow rate of 0.5 µL/h for 7 days and were filled with vehicle or 11,12-EET and 14,15-EET (23).
Western Blot Analysis
We performed electrophoresis of protein extracts using Tris-glycine buffer systems and subsequent blotting as previously described (24). Membranes were incubated with antibody against α-smooth muscle actin (α-SMA; 1:10,000, Sigma, St. Louis, MO), 4-hydroxynonenal (4-HNE; 1:5,000, Abcam, Cambridge, MA), and sEH (1:5,000, Cayman). Anti-GAPDH antibody (1:5,000, NOVUS, Littleton, CO) was used for loading controls on stripped membranes. Membranes were incubated with the appropriate secondary antibody against horseradish peroxidase (HRP) horse anti-mouse IgG (1:5,000, Vector Laboratories, Burlingame, CA) and HRP rabbit anti-goat IgG (1:5,000, Vector Laboratories). Bands were quantified using Lab Works analysis software (Ultra-Violet Products, Cambridge, UK).
Sirius Red Staining
Kidney paraffin-embedded sections were stained with picrosirius red staining according to the manufacturer’s instructions, as previously described (25). Digital images were acquired using a Leica THUNDER Imager microscope with a ×20 objective (Leica, Wetzlar, Germany). Sirius red staining was measured in five randomly chosen fields per kidney using ImageJ software (National Institutes of Health).
Histology
Kidney paraffin-embedded sections were stained with periodic acid-Schiff stain, as previously described (26). Histological damage of tubular injury was scored by the percentage of tubules that displayed tubular necrosis, cast formation, and tubular dilation as follows: 0 = normal, 1 = <10%, 2 = 10–25%, 3 = 26–50%, 4 = 51–75%, and 5 = >75%. Ten randomly chosen high-power (×200 magnification) fields per kidney were used for the counting in a blinded manner.
Immunohistochemical and Immunofluorescence Staining
Immunohistochemical and immunofluorescence staining of the kidneys was performed on paraffin-embedded sections as previously described (27). Briefly, 4% paraformaldehyde-fixed kidney sections were rehydrated and labeled with antibodies against α-SMA (1:200, Sigma), collagen type I (1:100, UNLB, Birmingham, AL), aquaporin-1 (AQP1; 1:100, Sigma), F4/80 (1:100, Bio-Rad, Hercules, CA), polymorphonuclear neutrophils (PMN; 1:100, Accurate Chemical & Scientific, Carle Place, NY), or CD31 (Developmental Studies Hybridoma Bank, Iowa City, IA). Sections were then incubated with the appropriate secondary antibody against HRP horse anti-mouse IgG (1:100, Vector Laboratories), HRP rabbit anti-goat IgG (1:100, Vector Laboratories), HRP horse anti-rat IgG (1:100, Vector Laboratories), HRP goat anti-rabbit IgG (1:100, Vector Laboratories), or Texas red anti-rabbit (1:100, Vector Laboratories). The respective α-SMA positive and collagen type I-positive areas were measured in 10 randomly chosen high-power (×200 magnification) fields per kidney using ImageJ software. The number of F4/80-positive and PMN-positive cells was counted in 10 randomly chosen high-power fields per kidney.
TUNEL Assay
TUNEL assay was performed using an in situ cell death detection kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions (24). In brief, 4-μm kidney sections were deparaffinized and rehydrated. Sections were then incubated with TUNEL reagent mixture for 30 min and then washed with PBS three times for 5 min each; the nuclei were stained with DAPI. Images were observed under a microscope (Leica). TUNEL-positive cells were counted in 10 fields per kidney.
Quantitative Real-Time PCR Analysis
RNA was extracted using TRIzol reagent (Invitrogen, Waltham, MA) from homogenized kidneys, as previously described (28). Next, 1 μg of RNA was used for cDNA synthesis using iScript Reverse Transcription Supermix (Bio-Rad). Quantitative real-time PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and the CFX connect real-time PCR system (Bio-Rad). Quantitative real-time PCR was carried out for 40 cycles of denaturation at 95°C for 15 s, annealing at 58°C for 15 s, and extension at 72°C for 15 s. Target gene expression was quantified relative to that of an internal control gene (GAPDH) based on a comparison of the threshold cycle (CT) at constant fluorescence intensity. The amount of transcript was inversely related to the observed CT, and CT was expected to increase by one for every twofold dilution of the transcript. Relative expression (R) was calculated using the following equation: R = 2−ΔΔCT. All data were normalized relative to GAPDH and to the respective controls. The murine quantitative real-time PCR primer sequences were as follows: 5′- TTCAGGACATCCTGCAAAAG-3′ (sense) and 5′- GGCTCTTGAGCTGGAAGAAA-3′ (antisense) for inducible nitric oxide synthase (iNOS), 5′- CCCAAGCAATACCCAAAGAA-3′ (sense) and 5′- ACGGATTCCATGGTGAAGTC-3′ (antisense) for IL-1β, 5′- CGTCAGCCGATTTGCTATCT-3′ (sense) and 5′- CTTGGGCAGATTGACCTCAG-3′ (antisense) for TNF-α, 5′- CTCCAAGCCAAAGTCCTTAGAG-3′ (sense) and 5′- AGGAGCTGTCATTAGGGACATC-3′ (antisense) for arginase-1 (Arg-1), 5′- GCTCTTACTGACTGGCATGAG-3′ (sense) and 5′- CGCAGCTCTAGGAGCATGTG-3′ (antisense) for IL-10, and 5′- CATCACTGCCACCCAGAAGACTG-3′ (sense) and 5′- ATGCCAGTGAGCTTCCCGTTCAG-3′ (antisense) for GAPDH.
Cell Culture and Treatment
Raw 264.7 cells, murine macrophage cell line cells (American Type Culture Collection, Manassas, VA), were maintained in DMEM (GIBCO, Grand Island, NY) supplemented with 10% FBS (GIBCO) and 100 U/mL streptomycin-penicillin (GIBCO) at 37°C in a humidified atmosphere containing 5% CO2 (21). Raw 264.7 cells were grown until 80% confluence on culture plates and then treated with 1 μM t-TUCB, 1 μM of 11,12-EET and 14,15-EET (Cayman), or 10 ng/mL TGF-β (R&D Systems, Minneapolis, MN) for 18 h.
In Vitro Migration Assay
Migration was assayed as previously described (29) using a modified Boyden chamber (Corning Costar, Cambridge, MA) that contains a polycarbonate Transwell membrane filter (8-μm pore size). The number of 104 cells were plated on the upper chamber. The lower chamber contained 10 ng/mL TGF-β (R&D Systems). Cells were incubated for 18 h at 37°C in a humidified atmosphere containing 5% CO2. Cells were stained with hematoxylin and eosin, and migrated cells that remained on the bottom surface were counted after nonmigrated cells were scraped from the upper surface of the membrane with a cotton swab. Pictures were taken using a microscope (Leica).
Measurement of EETs
An ELISA kit (MyBioSource, San Diego, CA) was used to measure the concentrations of 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET in kidney tissues, according to the manufacturer’s instructions.
Statistical Analyses
All data were analyzed using GraphPad Prism 7 software (San Diego, CA). Results are expressed as means ± SD. Two-group comparison was performed using an unpaired Student t test. Multiple-group comparisons were performed by one-way or two-way ANOVA followed by a Tukey post hoc test as appropriate. Differences were regarded as statistically significant when they had P values of <0.05.
RESULTS
Administration of EETs Attenuates Renal Interstitial Inflammation and Fibrogenesis and Morphological Impairments Induced by UUO
To assess the role of EETs during renal interstitial fibrosis and inflammation, 8-wk-old male mice were administered vehicle or a combination of EET regioisomers (11,12-EET and 14,15-EET) at one of several different concentrations (3, 6, 7.5, or 15 µg/kg/day) using an osmotic pump beginning at the time they underwent UUO or sham surgery until the time of euthanasia. Based on our previous report showing that sEH deletion enhanced the ratio of both 11,12-EET/DHET and 14,15-EET/DHET in UUO compared with wild type, we explored the roles of 11,12-EET and 14,15-EET regioisomers in this study (14). After UUO, more abundance of α-SMA, a marker of myofibroblasts, was observed in the kidneys of both vehicle- and smaller-dose EET-administered mice. Significantly less abundance of α-SMA was observed using combination of 11,12-EET and 14,15-EET at a concentration of 15 µg/kg/day (Fig. 1A). In addition, there was no significant difference of α-SMA abundance after single treatment with either 11,12-EET or 14,15-EET (15 µg/kg/day) compared with vehicle treatment (Fig. 1B).
Figure 1.

Effect of 11,12-epoxyeicosatrienoic acid (EET) and 14,15-EET on kidney fibrosis following unilateral ureteral obstruction (UUO). A: male C57BL/6 mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (3, 6, 7.5, or 15 µg/kg/day of each, using an osmotic pump) for 7 days. Kidneys were subjected to Western blot analysis using anti-α-smooth muscle actin (α-SMA) antibody. B: mice were subjected to either UUO or sham operation and then administered with a single treatment of either 11,12-EET or 14,15-EET (15 µg/kg/day). The kidneys were subjected to Western blot analysis using anti-α-SMA antibody. GAPDH was used as a loading control. Band density was measured using ImageJ software. Data are presented as means ± SD; n = 4. ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. NS, not significant; V, vehicle.
More α-SMA abundance was observed in 15 µg/kg/day EET-administered mice compared with vehicle-administered mice following UUO (Supplemental Fig. S1). Collagen deposition, as assessed by Sirius red staining, was increased mainly in the interstitium after UUO, and this increase was attenuated in EET-administrated mice compared with vehicle-treated mice (Fig. 1B). Consistent with this result, the interstitial collagen I-positive area in the kidney was lower in EET-administered mice compared with vehicle-treated mice (Supplemental Fig. S1C). It has been previously demonstrated that EETs have anti-inflammatory effects in various organs (30–33). Thus, we tested whether administration of EETs affects immune cell infiltration into the injured site after UUO. The increased F4/80 (a marker of macrophages)-positive cells were less in EET-administered mice compared with control mice after UUO (Supplemental Fig. S1D). Taken together, these data suggest that EETs mitigate the infiltration of immune cells into the renal interstitium during fibrogenesis. UUO can induce loss of brush borders of tubular cells and disruption, congestion, dilation, and flattening of tubules (Supplemental Fig. S1E). UUO-induced tubular cell damages were lesser in EET-administered mice than in control mice. There were no significant differences in plasma creatinine and blood urea nitrogen concentrations between the two mouse groups (Supplemental Fig. S1, G and H), as expected.
Inhibition of sEH Prevents the EET-to-DHET Conversion in UUO Kidneys
After UUO, significantly more abundance of sEH protein was observed (Supplemental Fig. S2, A and B). However, there was no difference of sEH abundance between vehicle-treated and EET-treated kidneys in both sham and UUO groups (Supplemental Fig. S2, A and B). In addition, there was no difference of sEH abundance between vehicle-treated and t-TUCB-treated kidneys in both sham and UUO groups (Supplemental Fig. S2A). sEH was not observed in sEH-deficient mice (Supplemental Fig. S2B). Levels of all four EETs (5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET) were assessed by immunoassay (Supplemental Fig. S2, C and D). All four EET levels decreased in UUO-operated mice compared with sham-operated mice. Levels of 11,12-EET and 14,15-EET were increased by exogenous EET administration or sEH inhibition in both sham and UUO mice. EET administration inhibited UUO-induced decreases of 11,12-EET and 14,15-EET levels in the kidneys of both Ephx2−/− and Ephx2+/+ mice and both control and t-TUCB-treated mice, whereas the levels of 5,6-EET and 8,9-EET were not significantly different. These data indicate that t-TUCB treatment or Ephx2 deficiency are effective at inhibiting sEH activity. Our data also demonstrate that administration of 11,12-EET and 14,15-EET increases their levels in UUO kidneys but has no effect on levels of 5,6-EET and 8,9-EET.
Inhibition of sEH and/or Administration of EETs Prevent Renal Fibrosis Induced by UUO
To assess the additive effect of EET administration and sEH inhibition on fibrogenesis, we tested renal fibrosis in sEH-inhibited mice with or without EET administration following UUO. To avoid experimental variation, we first tested the effect of EET administration only, as shown in Fig. 1, in a different group of mice; at the same time, we tested the combinatorial effect of EET administration and sEH inhibition. Consistent with the data provided in Supplemental Fig. S1, the percentage of α-SMA expression and collagen deposition, as assessed by immunohistochemistry and ImageJ analysis, was decreased by 34% and 45%, respectively (Fig. 2A) after EET treatment. Western blot analysis also showed that α-SMA expression was decreased by 44% (Fig. 2B). For pharmacological inhibition of sEH, t-TUCB (0.4 mg/mouse/day) was administered by oral gavage beginning 24 h before UUO. For the combined inhibition experiments, mice were administered t-TUCB 24 h before UUO and 11,12-EET and 14,15-EET beginning at the time they underwent UUO. Collagen deposition was assessed by Sirius red staining and immunostaining using anti-collagen type I antibody. At 7 days after UUO, the increase of collagen deposition in the interstitium was significantly lower in t-TUCB-treated mice compared with vehicle-treated mice (Fig. 2A). After EET administration, collagen deposition was reduced in both sEH-inhibited and EET-treated mice compared with vehicle-treated mice (Fig. 2A). Less α-SMA abundance was also observed in t-TUCB-treated mice than in vehicle-treated mice (Fig. 2, A and B). After combined t-TUCB and EET administration, less α-SMA abundance and collagen were observed compared with vehicle-treated mice; however, there was no significant difference between t-TUCB and EET treatments alone. We also examined whether t-TUCB and/or EET administration affect TGF-β expression in UUO kidneys. TGF-β mRNA expression was significantly reduced in EET- and/or t-TUCB-treated kidneys after UUO compared with vehicle-treated kidneys (Fig. 2C), but the combination treatment had no difference in TGF-β mRNA expression compared with single treatments.
Figure 2.
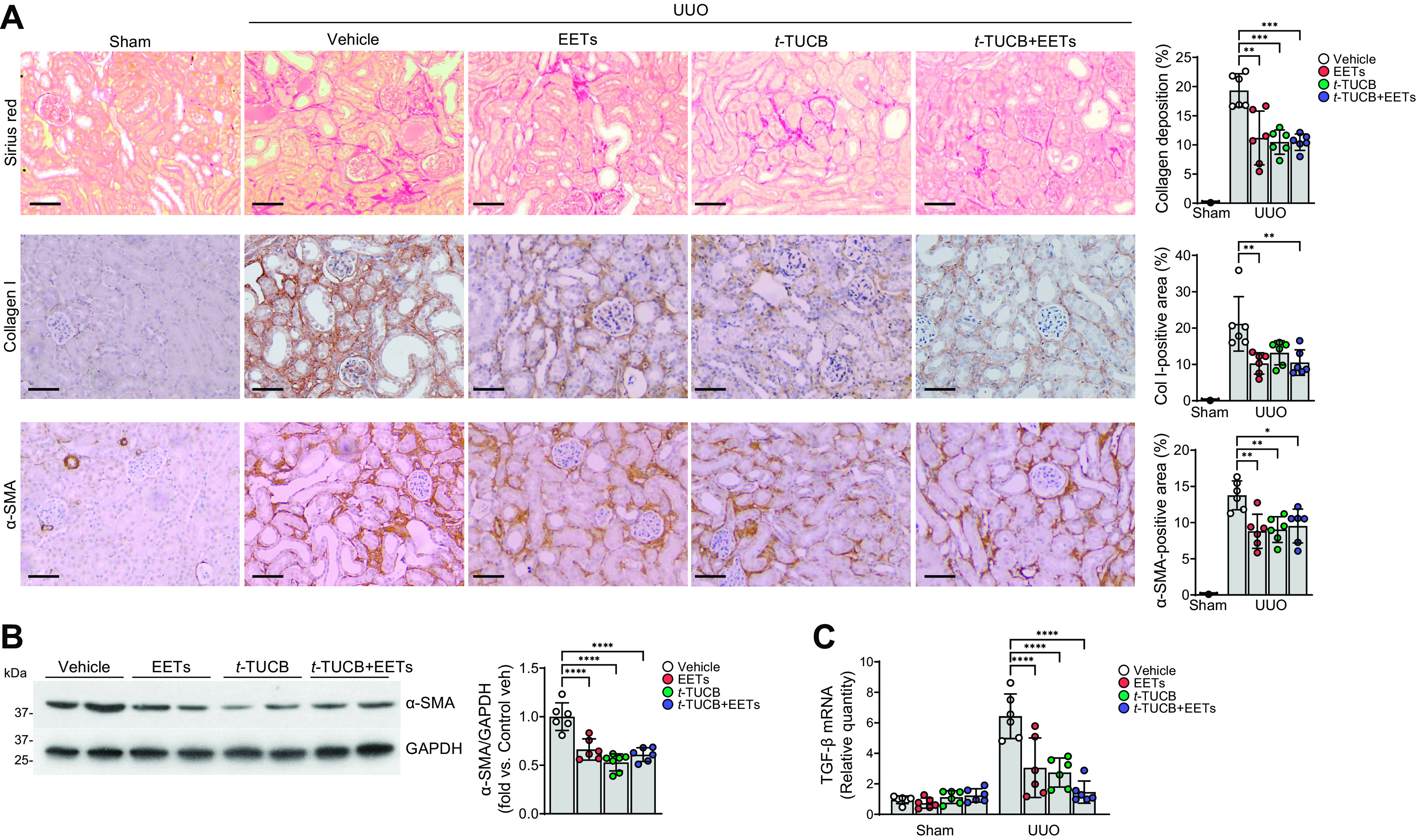
Epoxyeicosatrienoic acid (EET) administration or pharmacological inhibition of soluble epoxide hydrolase attenuates renal interstitial fibrogenesis during unilateral ureteral obstruction (UUO). Male C57BL/6 mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (15 µg/kg/day, using an osmotic pump) for 7 days. For the pharmacological inhibition of soluble epoxide hydrolase, t-TUCB (0.4 mg/mouse/day) or vehicle was administered by oral gavage beginning 24 h before UUO. The kidneys were harvested at 7 days after the operation. A: kidney sections were subjected to Sirius red staining and immunohistochemical staining using anti-collagen type I (Col I; brown) and anti-α-smooth muscle actin (α-SMA) antibodies. Hematoxylin stain (blue color) was used for counterstaining. Pictures of the cortex were taken. Scale bars = 50 μm. B: kidneys were subjected to Western blot analysis using anti-α-SMA antibody. GAPDH was used as a loading control. Band density was measured using ImageJ software. C: mRNAs were extracted from kidney tissues using RNA extraction solution as described in materials and methods. The mRNA level of transforming growth factor (TGF)-β was measured and normalized using the GAPDH level. Data are presented as means ± SD; n = 6. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. t-TUCB, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid.
Next, we examined the levels of renal fibrosis in Ephx2−/− mice at 7 days after UUO. Consistent with the results of pharmacological inhibition of sEH mice, collagen deposition, α-SMA abundance, and TGF-β mRNA expression were significantly lower in Ephx2−/− mice with or without EET treatment (Fig. 3, A–C). However, there was no difference of collagen deposition, α-SMA abundance, and TGF-β mRNA expression between single treatments with either EETs or sEH inhibition and combined EET and sEH inhibition. These results suggest that sEH inhibition and/or EET administration attenuated tubulointerstitial fibrogenesis; however, an additive protective effect was not observed. Since peritubular capillary rarefaction is a cause for interstitial fibrosis and tubular atrophy (34), we determined capillary density using CD31 stain (Supplemental Fig. S3, A and B). Loss of peritubular capillaries was observed following UUO, but no significant differences were observed in she-inhibited and/or EET-treated kidneys after UUO. These results suggest that increased EETs might not affect renal vascular density in UUO.
Figure 3.
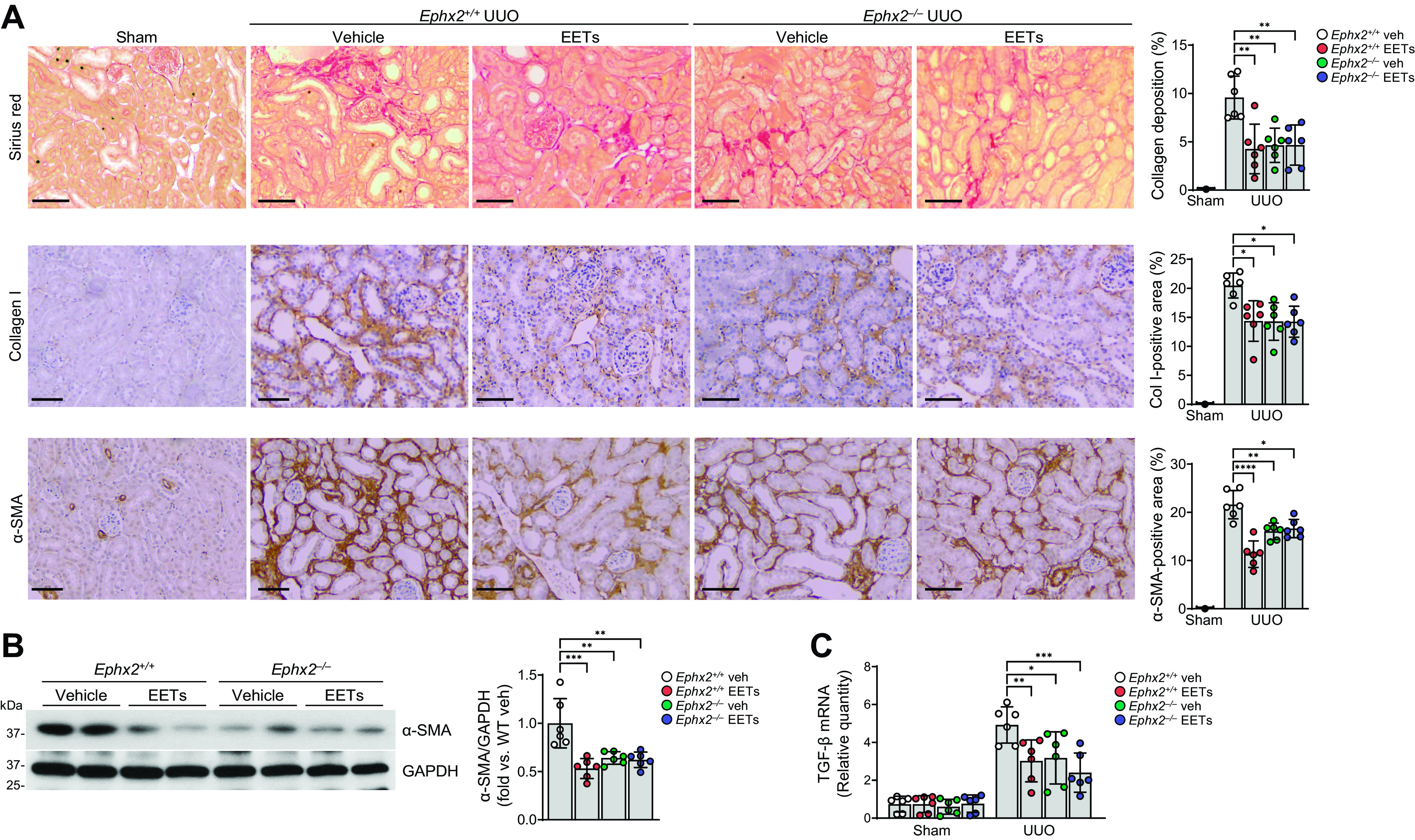
Epoxyeicosatrienoic acid (EET) administration or genetic inhibition of soluble epoxide hydrolase attenuates renal interstitial fibrogenesis during unilateral ureteral obstruction (UUO). Male Ephx2+/+ and Ephx2–/– mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (15 µg/kg/day, using an osmotic pump) for 7 days. The kidneys were harvested at 7 days after the operation. A: kidney sections were subjected to Sirius red staining (red) and immunohistochemical staining using anti-collagen type I (Col I; brown) and anti-α-smooth muscle actin (α-SMA; brown) antibodies. Hematoxylin stain (blue color) was used for counterstaining. Pictures of the cortex were taken. Scale bars = 50 μm. B: kidneys were subjected to Western blot analysis using anti-α-SMA antibody. GAPDH was used as a loading control. Band density was measured using ImageJ software. C: the mRNA level of transforming growth factor (TGF)-β was measured in the kidneys. mRNA levels were normalized by GAPDH levels. Data are presented as means ± SD; n = 6. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. Veh, vehicle; WT, wild type.
Inhibition of sEH or Administration of EETs Attenuates Renal Interstitial Inflammation Induced by UUO
To determine whether EETs contribute to renal inflammation in obstructive nephropathy, we next examined the infiltration of inflammatory cells in UUO kidneys. After UUO, infiltration of both F4/80-positive macrophages and PMN-positive neutrophils occurred in vehicle-treated kidneys, but EET administration greatly inhibited the infiltration in UUO kidneys (Fig. 4, A and B). Infiltration of F4/80-positive and PMN-positive cells was also reduced after sEH inhibition compared with that in vehicle-treated mice after UUO (Fig. 4, A and B). In addition, the infiltration of macrophages and neutrophils decreased in Ephx2−/− mice compared with Ephx2+/+ mice after UUO (Fig. 4, C and D). In a combination treatment approach, after EET administration in Ephx2+/+ mice, the numbers of macrophages and neutrophils in both Ephx2−/− and Ephx2+/+ mice were also decreased compared with vehicle-treated mice (Fig. 4, C and D). However, there was no difference in the number of infiltrated cells between EET-administrated or sEH-inhibited mice and combined EET-administrated and sEH-deficient mice.
Figure 4.
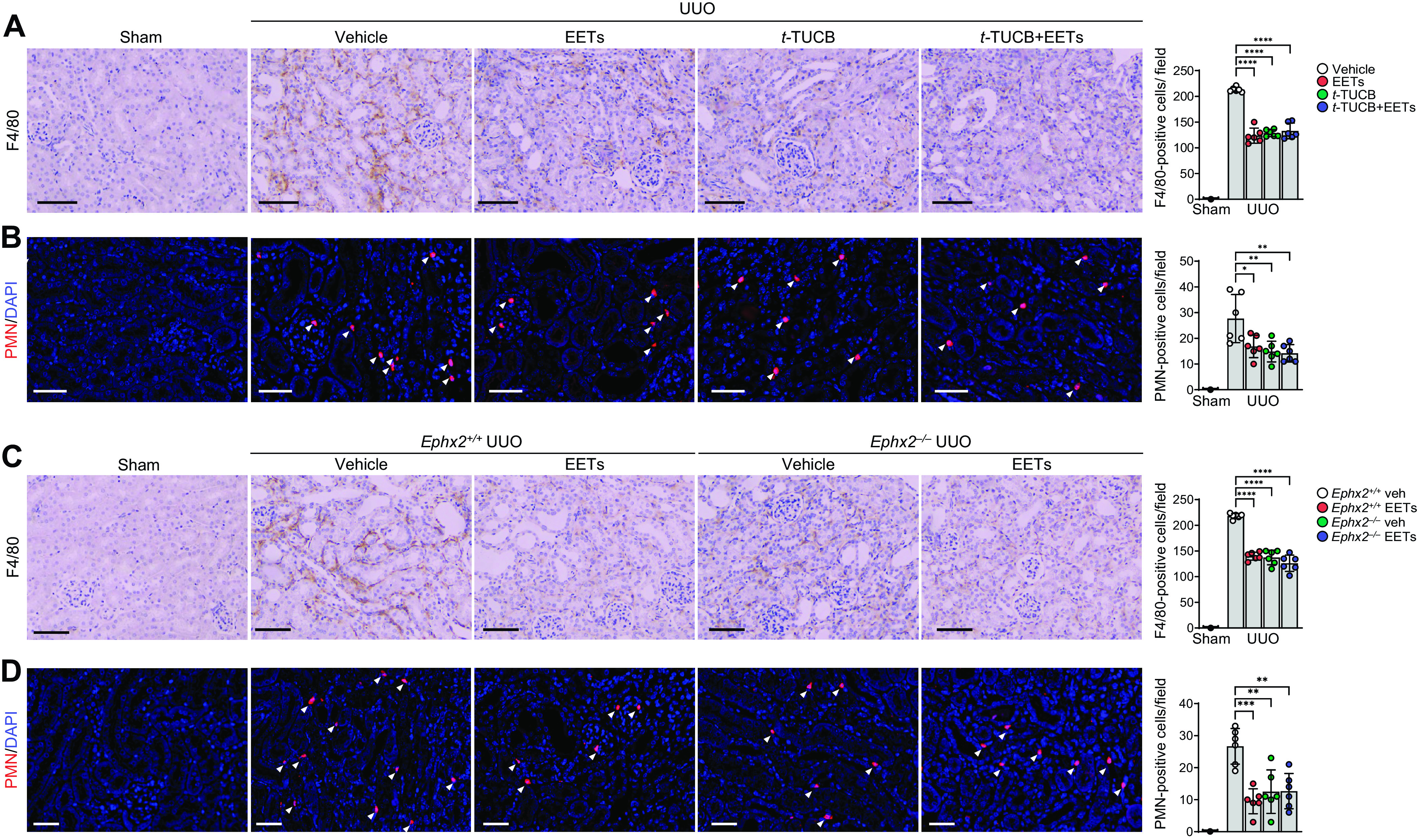
Epoxyeicosatrienoic acid (EET) administration or soluble epoxide hydrolase inhibition reduces renal interstitial inflammation during unilateral ureteral obstruction (UUO). Male C57BL/6 mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (15 µg/kg/day, using an osmotic pump) for 7 days. A and B: for the pharmacological inhibition of soluble epoxide hydrolase, t-TUCB (0.4 mg/mouse/day) or vehicle (Veh) was administered by oral gavage beginning 24 h before UUO. The kidneys were harvested at 7 days after the operation. Kidney sections were stained by immunohistochemical staining using anti-F4/80 (brown; A) antibody and antipolymorphonuclear neutrophil (PMN; red; B) antibodies. C and D: Ephx2+/+ and Ephx2–/– mice were subjected to UUO or sham operation and then administered with the combination of EETs. Kidney sections were stained by immunohistochemical staining using anti-F4/80 (brown; C) and anti-PMN (red; D) antibodies. Hematoxylin and DAPI stain (both blue) were used for counterstaining. Pictures of the cortex were taken. Scale bars = 50 μm. Data are presented as means ± SD; n = 6. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. t-TUCB, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid.
Macrophages have been thought to be key immune response effector cells and are categorized into phenotypically, classically activated M1 macrophages and alternatively activated M2 macrophages (35). To determine whether EETs can induce macrophage M2 activation and suppress UUO-induced M1 activation, we assessed the expression of M1- and M2-associated markers in the kidneys after UUO. UUO upregulated the expression of both M1 and M2 macrophage marker genes (Fig. 5, A–J). EET administration and/or sEH inhibition significantly reduced the expression of M1 macrophage markers, including iNOS, IL-1β, and TNF-α, compared with vehicle-treated mice after UUO (Fig. 5, A–C and F–H). Expression of M2 macrophage markers (Arg-1 and IL-10) was highly upregulated in EET-treated and/or sEH-inhibited mice with UUO (Fig. 5, D, E, I, and J).
Figure 5.
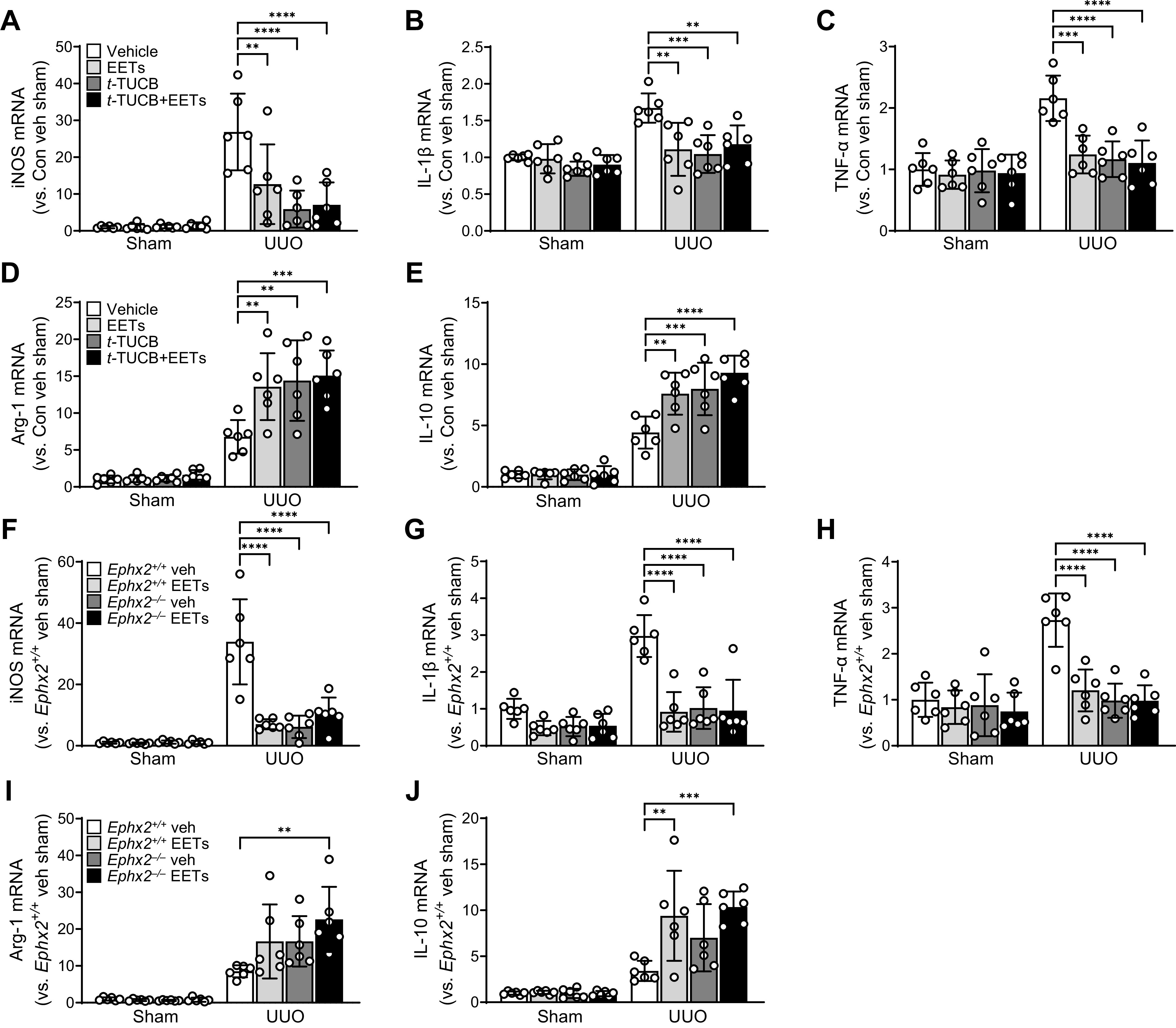
Epoxyeicosatrienoic acid (EET) regulate macrophage polarization in the kidney during unilateral ureteral obstruction (UUO). Male C57BL/6 mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (15 µg/kg/day, using an osmotic pump) for 7 days. A–E: for the pharmacological inhibition of soluble epoxide hydrolase, t-TUCB (0.4 mg/mouse/day) or vehicle (Veh) was administered by oral gavage beginning 24 h before UUO. mRNA levels of inducible nitric oxide synthase (iNOS; A), interleukin (IL)-1β (B), tumor necrosis factor (TNF)-α (C), arginase-1 (Arg-1; D), and IL-10 (E) were measured in the kidneys. F–J: Ephx2+/+ and Ephx2–/– mice were subjected to UUO or sham operation and then administered with the combination of EETs. mRNA levels of iNOS (F), IL-1β (G), TNF-α (H), Arg-1 (I), and IL-10 (J) were measured in the kidneys. mRNA levels were normalized by GAPDH levels. Data are presented as means ± SD; n = 6. **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. t-TUCB, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid.
To elucidate the EET-mediated mechanism underlying the increased infiltration of inflammatory cells into the kidney, we investigated the effect of EETs on macrophage migration and polarization. Using a Transwell system, we assessed the effect of EETs on TGF-β-induced macrophage migration. The number of migrated macrophages significantly decreased in EET-treated and/or sEH-inhibited groups compared with vehicle treatment (Fig. 6A). After TGF-β treatment, the number of migrated cells increased compared with control but significantly decreased in EET-treated and/or sEH-inhibited groups. TGF-β treatment upregulated the expression of both M1 and M2 macrophage marker genes (Fig. 6, B–E). EET and/or t-TUCB treatment significantly decreased expression of iNOS and IL-1β compared with vehicle-treated macrophages (Fig. 6, B and C), whereas expression of Arg-1 and IL-10 was highly upregulated in EET- and/or t-TUCB-treated macrophages (Fig. 6, D and E). These data suggested an important role for EETs in the regulation of macrophage-mediated renal inflammation following UUO.
Figure 6.
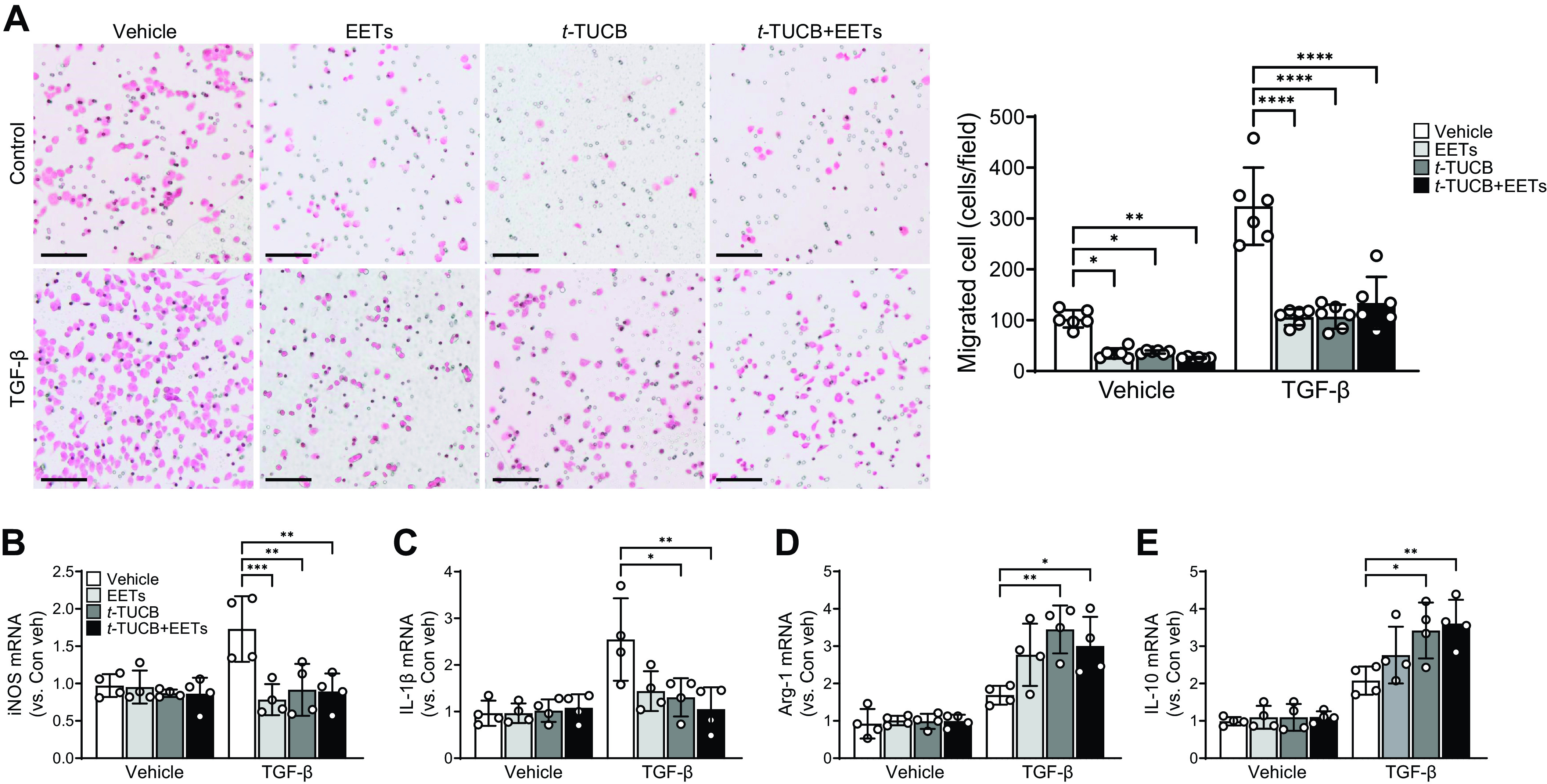
Epoxyeicosatrienoic acid (EET) administration or soluble epoxide hydrolase inhibition reduces macrophage migration and M1/M2 polarization. Raw 264.7 cells were incubated with 1 µM of t-TUCB and/or 1 µM of 11,12-EET and 14,15-EET in the absence or presence of 10 μg/mL of transforming growth factor (TGF)-β for 18 h. A: representative images of bright-field microscopy of hematoxylin and eosin-stained migrated cells. Macrophages were stained pink. Small circles are pores of the membrane filters. The graph summarizes the number of migrated macrophages. Scale bars = 200 μm. mRNA levels of inducible nitric oxide synthase (iNOS; B), interleukin (IL)-1β (C), arginase-1 (Arg-1; D), and IL-10 (E) were measured in the kidneys. mRNA levels were normalized by GAPDH levels. Data are presented as means ± SD; n = 4. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. Con, control; t-TUCB, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid; Veh, vehicle.
Inhibition of sEH or Administration of EETs Diminishes Renal Cell Injury and Oxidative Stress Induced by UUO
Since tubular injury can lead to inflammation and fibrosis (36), we evaluated the effects of EETs on tubular injury and apoptotic cell death in UUO-subjected kidneys. UUO increased tubular atrophy, dilation, and apoptosis in nonproximal tubule cells (37). EET administration or inhibition of sEH using t-TUCB significantly prevented this tubular injury and apoptotic cell death (Fig. 7, A and B). In addition, gene ablation of sEH significantly prevented tubular injury and apoptosis in UUO kidneys (Fig. 7, C and D). EET administration prevented tubular injury and cell death in both Ephx2+/+ and Ephx2−/− mice compared with vehicle-injected mice, but the combination had no additive protective effect in preventing tubular injury and cell death (Fig. 7, C and D).
Figure 7.
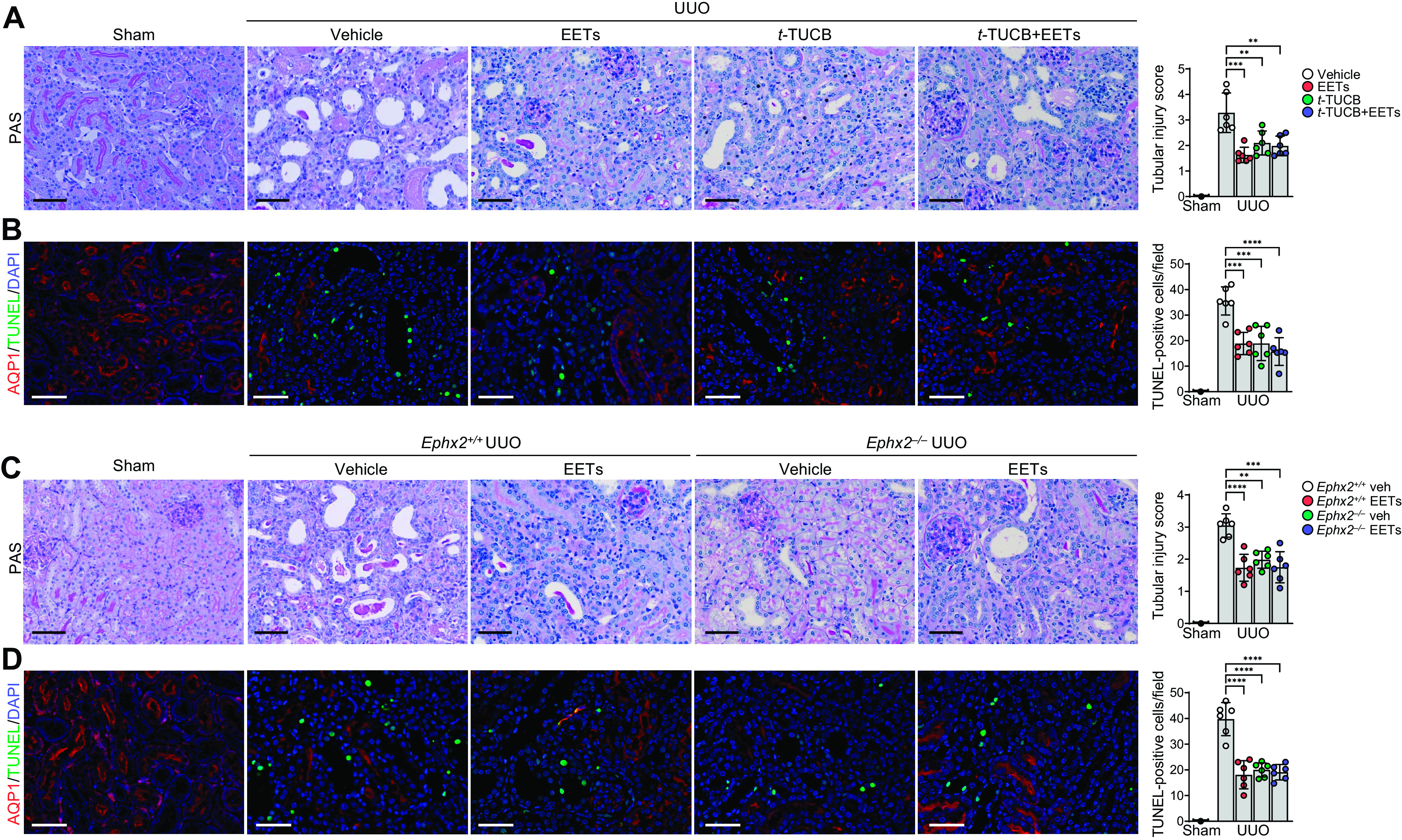
Epoxyeicosatrienoic acid (EET) administration or pharmacological or genetic inhibition of soluble epoxide hydrolase suppresses tubular cell damages and cell death during unilateral ureteral obstruction (UUO). Male C57BL/6 mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (15 µg/kg/day, using an osmotic pump) for 7 days. The kidneys were harvested at 7 days after the operation. A and B: for the pharmacological inhibition of soluble epoxide hydrolase, t-TUCB (0.4 mg/mouse/day) or vehicle (Veh) was administered by oral gavage beginning 24 h before UUO. A: kidney damage was evaluated by periodic acid-Schiff (PAS) staining and damage scoring. B: kidney sections were stained using a TUNEL assay kit and aquaporin-1 (AQP1) antibody (red). TUNEL-positive cells (green) were counted by microscopy. C and D: Ephx2+/+ and Ephx2–/– mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET. C: kidney damage was evaluated by PAS staining and damage scoring. D: kidney sections were stained using a TUNEL assay kit and AQP1 antibody (red). Hematoxylin and DAPI stain (both blue) were used for counterstaining. Pictures of the cortex were taken. Scale bars = 50 μm. Data are presented as means ± SD; n = 6. **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. t-TUCB, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid.
To explore whether the administration of EETs or inhibition of sEH using t-TUCB has an antioxidative effect during UUO, we next evaluated lipid peroxidation in UUO kidneys. UUO significantly increased lipid peroxidation, an indicator for oxidative stress, as represented by increased levels of 4-HNE (a product of lipid peroxidation) and lipid hydroperoxide in vehicle-treated kidneys, whereas either pharmacological or genetic inhibition of sEH ameliorated lipid peroxidation induced by UUO (Fig. 7, A–F). EET administration reduced the UUO-induced increase of lipid peroxidation in both control and she-inhibited mice (Fig. 8, A–F) but the combination had no additive protective effect in preventing lipid peroxidation. Taken together, these results indicate that EET administration and/or either pharmacological or genetic inhibition of sEH attenuate UUO-induced tubular cell injury, apoptosis, and oxidative stress.
Figure 8.
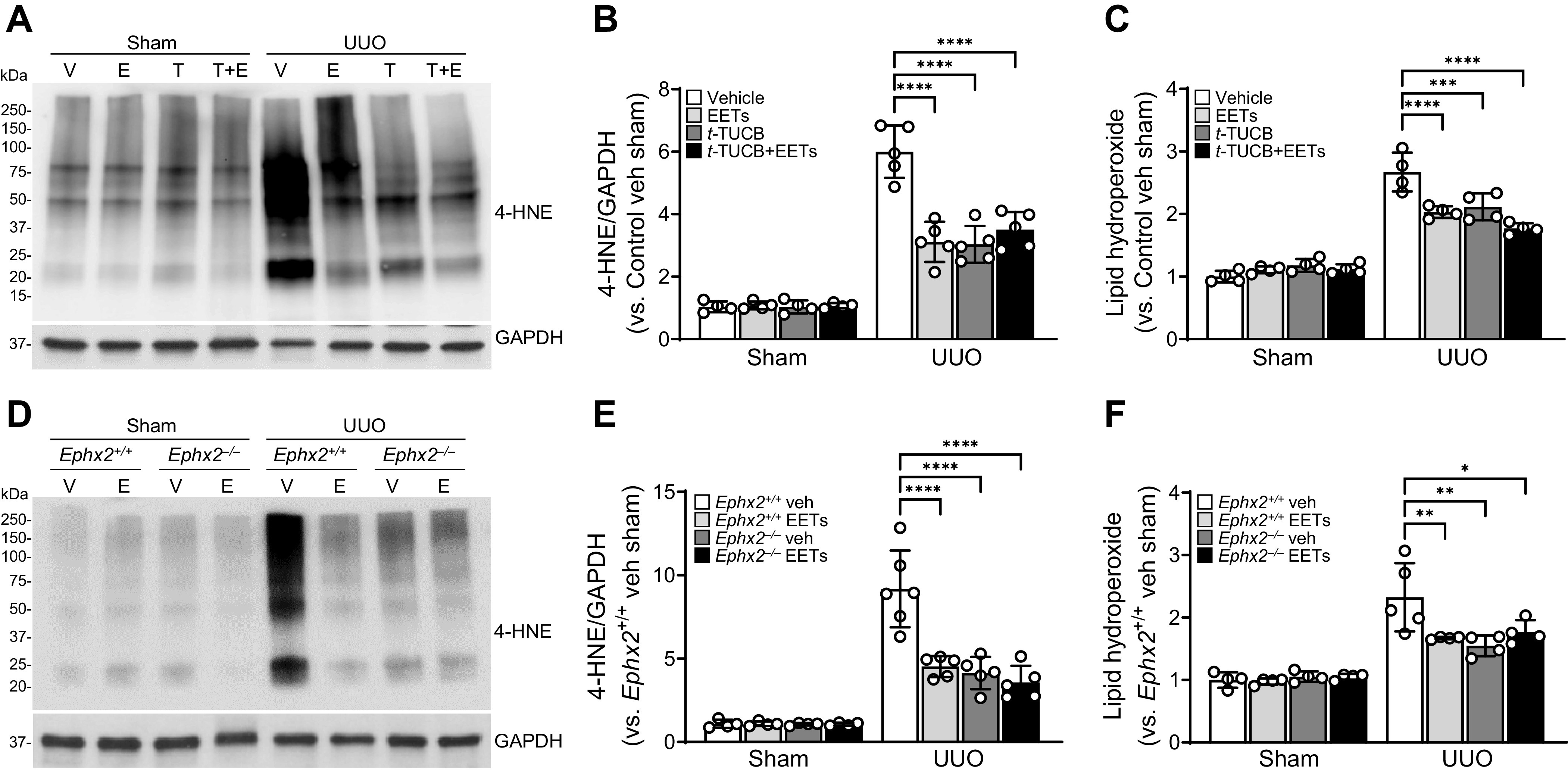
Epoxyeicosatrienoic acid (EET) administration or pharmacological or genetic inhibition of soluble epoxide hydrolase (sEH) mitigates unilateral ureteral obstruction (UUO)-induced oxidative stress. Male C57BL/6 mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET (E; 15 µg/kg/day, using an osmotic pump) for 7 days. Kidneys were harvested at 7 days after the operation. A–C: for the pharmacological inhibition of soluble epoxide hydrolase, t-TUCB (T; 0.4 mg/mouse/day) or vehicle (V) was administered by oral gavage beginning 24 h before UUO. A and B: the 4-hydroxynonenal (4-HNE) level was evaluated by Western blot analysis using anti-4-HNE antibody. GAPDH was used as a loading control. Band density was measured using ImageJ software. C: lipid peroxidation as indicated by the lipid hydroperoxide level in kidneys using a lipid hydroperoxide assay kit. C and D: Ephx2+/+ and Ephx2–/– mice were subjected to either UUO or sham operation and then administered with the combination of 11,12-EET + 14,15-EET. D and E: the 4-HNE level was evaluated by Western blot analysis using anti-4-HNE antibody. F: lipid peroxidation as indicated by the lipid hydroperoxide level in kidneys. Data are presented as means ± SD; n = 4–6. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way or two-way ANOVA followed by a Tukey’s post hoc multiple comparison test was used to determine significance. TUCB, 4-[[trans-4-[[[[4-(trifluoromethoxy)phenyl]amino]carbonyl]amino]cyclohexyl]oxy]benzoic acid.
DISCUSSION
In this study, we tested the effects of EET administration, sEH inhibition, and the combination of EET administration and sEH inhibition in CKD progression in mice subjected to UUO. Our data indicated that increasing levels of EETs by 11,12-EET and 14,15-EET administration and/or sEH inhibition significantly reduced the infiltration of neutrophils and macrophages into the renal interstitium and activation of fibroblasts and downregulated M1 while upregulated M2 macrophage numbers. Increased EET levels prevented collagen and α-SMA deposition and prevented histological damage, apoptosis, and oxidative stress in UUO-induced kidneys. Interestingly, the combination of EET administration and sEH inhibition did not have an additive effect in protecting kidneys from renal tubular injury, inflammation, and fibrosis, suggesting that the threshold level of EET protection may have been reached by either EET administration or sEH inhibition alone. Taken together, our data suggest that either sEH inhibition and/or EET administration have a beneficial effect in attenuating renal injury, inflammation, and interstitial fibrogenesis after UUO.
CKD is a growing worldwide health problem with high morbidity and mortality and is a major unmet medical need. Tubulointerstitial fibrogenesis is a hallmark of CKD progression and eventually end-stage kidney disease (38). Accumulating evidence suggests that inflammation plays a critical role in the initiation and progression of renal fibrosis (6, 8, 39). Inflammation initiated in the kidneys can lead to the release of cytokines and chemokines, resulting in the accumulation and activation of myofibroblasts, and consequent production and deposition of the extracellular matrix (6, 8, 39). Various cytokines (IL-13, IL-21, and TGF-β1), chemokines (monocyte chemoattractant protein-1 and macrophage inflammatory protein-1β), angiogenic factors (VEGF), growth factors (platelet-derived growth factor), and the renin-angiotensin-aldosterone system contribute to renal fibrosis (39). Treatment strategies targeting single cytokine functions are not effective, possibly due to compensation by other cytokines (40). Despite being a major unmet medical need, current efforts are restricted to the control of blood pressure and optimization of renin-angiotensin-aldosterone system blockade. These therapies, at best, may reduce proteinuria, a surrogate marker of renal disease, but they only partially reduce progression of CKD (41).
sEH converts EETs to less active DHETs, attenuating anti-inflammatory and antifibrotic functions (9, 10). We have previously reported that increasing endogenous EET bioavailability by genetic or pharmacological sEH inhibition prevented the progression of renal fibrogenesis in two experimental models of CKD, UUO and glomerulonephropathy, by suppressing the influx of F4/80-positive macrophages with decreased levels of inflammatory cytokines and chemotactic factors (13, 14). Similar to our reports showing that sEH inhibition prevents disease progression in obstructive nephropathy, numerous studies have reported beneficial effects in IgA nephropathy, high-fat diet-mediated renal injury, and diabetic or hypertensive renal injury. These studies also demonstrated a significant reduction of macrophage infiltration, release of inflammatory factors, and attenuation of renal tubulointerstitial fibrosis (17–20, 42–44). Studies in CKD models have demonstrated the ability for EET analogs to slow or arrest progression to end-stage kidney disease through antiapoptotic, anti-inflammatory, and antifibrotic mechanisms (11, 12). EET-A, an EET analog, treatment of UUO mice reduced kidney collagen and α-SMA and prevented increased epithelial-to-mesenchymal transducers such as the zinc finger transcription factor Snail1 and ZEB-1 while reducing myofibroblast markers such as fibroblast-specific protein 1, E-cadherin, and fibronectin (45). In addition, EET-B treatment in Dahl salt-sensitive hypertensive rats markedly reduced urinary albumin and nephrin excretion along with reductions in glomerular injury, intratubular cast formation, and kidney fibrosis (46). However, whether direct administration of EETs or combined EET administration and/or sEH inhibition have protective effect in UUO has not been studied yet. In the present study, we showed that EET administration and/or sEH inhibition attenuated UUO-induced fibrosis, as shown by markedly diminished inflammatory cell infiltration, collagen and α-SMA deposition, and renal tubular cell damage and death. Collectively, these data suggest that increasing levels of EETs inhibit malignant signaling events to prevent renal interstitial fibrogenesis in UUO. Reduced capillary density is a common feature in CKD, which correlates with interstitial fibrosis (34). In the present study, capillary density was decreased in UUO-subjected kidneys compared with sham-operated kidneys. However, no difference in capillary density was observed between vehicle and EETs in either control or sEH-inhibited kidneys following UUO. Thus, we suggest that EETs attenuate renal injury and fibrogenesis apart from a direct effect on endothelial cell viability.
Macrophages are key cellular mediators of inflammation (47). In general, M1 macrophages promote proinflammatory reactions, whereas M2 macrophages produce anti-inflammatory cytokines (47). In the present study, we observed that increased EET levels by EET administration or sEH inhibition attenuated macrophage infiltration to the injured site following UUO. Moreover, increased EET levels reduced mRNA levels of iNOS, IL-1β, and TNF-α, whose expression is associated with M1 polarization. On the other hand, increased EET levels increased mRNA levels of Arg-1 and IL-10, whose expression is associated with M2 polarization in UUO-induced mice. Several studies have shown that increased EET levels by EET analog administration or sEH inhibition significantly diminish macrophage infiltration and regulate macrophage polarization (19, 30, 31, 48, 49). Treatment with t-TUCB, an sEH inhibitor, shifted macrophage polarization toward the anti-inflammatory M2 phenotype in hepatic macrophage infiltration induced by high-fat diet (48). EETs inhibited the recruitment of macrophages to adipose tissue and their switch to the M1 phenotype, whereas their M2 phenotype remained preserved, reducing inflammation, and the associated insulin resistance in high fat-induced obesity (49). Furthermore, EETs inhibit proinflammatory M1 polarization through the NF-κB pathway and stimulating M2 polarization through the phosphatidylinositol 3-kinase pathway in the IgA nephropathy model (19). In UUO-induced rats, reduced EET generation by epoxygenase Cyp2j4 deletion results in a profibrotic macrophage transcriptome and upregulation of genes belonging to the collagen, ECM, collagen-associated gene families, and TGF-β signaling pathway-related genes [connective tissue growth factor, Vegfa, Gremlin-1, and chemokine (C-X-C motif) ligand 12; 50]. Furthermore, in the present study, EET treatment and/or sEH inhibition effectively inhibited macrophage migration in the absence or presence of TGF-β. Consistent with our in vitro data, Dai et al. (30) showed that 11,12-EET effectively inhibited macrophage migration induced by LPS and significantly inhibited LPS-induced M1 macrophage polarization and diminished proinflammatory cytokines. These findings are consistent with our data and indicate that increased EET levels suppressed inflammation in tubular cells by inhibiting M1 polarization while maintaining or increasing M2 polarization after UUO, suggesting that EET has antifibrotic effects in different fibrosis-related diseases, including CKD. A role for TGF-β in the transition of the M1 into M2 phenotype has been well established. In our study, TGF-β treatment along with inhibition of sEH or EET administration induced expression of both M1 and M2 macrophage markers. In our experimental setup, it is possible that additional pathways are regulated following inhibition of sEH or EET administration, in the presence of TGF-β. Modulation of several factors, including peroxisome proliferator-activated receptor-γ transcription activity (30, 51), SMAD2/3 and phosphatidylinositol 3-kinase/AKT signaling pathways regulating SNAIL, have been implicated in M1-M2 polarization (52). We speculate that it is likely that one or more of these potential pathways may contribute to the activation of both M1 and M2 macrophages in our study. Further studies are needed to determine what may have caused the activation of both M1 and M2 macrophages.
Increased reactive oxygen species (ROS) generation and oxidative stress stimulate fibrogenic intracellular signaling, resulting in an increase in the formation of ECM components, leukocyte infiltration in the interstitium, and interstitial fibroblast proliferation (53). Previous studies have demonstrated that Ephx2 deficiency and sEH inhibition decrease oxidative stress in diabetic nephropathy by inhibiting excessive ROS production (42). Moreover, the EET analogs EET-F01 and EET-A decreased oxidative stress in cisplatin nephrotoxicity by reducing lipid peroxidation and expression of the oxidative marker genes NADPH oxidase subunits p47phox, p67phox, gp91phox, and NOX4 (54). Thus, we tested the hypothesis that the protective effect of sEH inhibition or EET supplementation may also be related to reduced oxidative stress in renal tubular cells after UUO. Consistent with previous studies, exogenous EETs and/or sEH inhibition suppressed oxidative stress. ROS, including free radicals such as superoxide, hydroxyl radical, and hydrogen peroxide, are produced at high levels under oxidative stress, causing cellular damage and cell death (55). Our data indicate that sEH inhibition and exogenous EETs significantly protect renal tubular epithelial cells from apoptotic cell death and cell damage induced by UUO. EETs inhibit renal tubular apoptosis by activating the phosphatidylinositol 3-kinase/Akt signaling pathway in streptozotocin-induced diabetic nephropathy (56), lung ischemia-reperfusion injury (57), and pulmonary arterial hypertension (58), suggesting a plausible similar mechanism in UUO. Together, these data suggest that EETs can play as a key regulator of renal tubular cell apoptosis and a potent antiapoptotic factor in the kidney.
In this study, Ephx2−/− mice and the pharmacological sEH inhibitor t-TUCB were used to increase endogenous EET levels. Several reports have demonstrated that either genetic or pharmacological inhibition of sEH increases EET levels in various organs, including the kidney, liver, and heart (9, 13, 14, 48). Based on our data and previous reports, we hypothesized that combined exogenous EET administration and sEH inhibition may have a synergistic or additive renoprotective effect after UUO. In contrast to our expectations, combined sEH inhibition and EET administration did not provide additive or synergic effects in renal fibrogenesis following UUO. Consistent with our findings, Červenka et al. (59) demonstrated that combined treatment with sEH inhibitor and EET-A exerts distinct antihypertensive and antiarrhythmic actions in the angiotensin II-dependent hypertension model but were not more effective than single treatment. Our data, however, did not demonstrate the activation of distinct pathways after EET administration or sEH inhibition. Although it remains unclear why the combination of sEH inhibition and EET treatment was not more effective, the results imply that this might be due to already maximally increased EET levels by either administration of EETs or by sEH inhibition and thus exogenous EET administration in Ephx2−/− mice did not additively augment renoprotective effects in UUO-induced disease progression. Unlike our results, in a previous report, Skibba et al. (45) demonstrated that administration of 14,15-EET alone attenuated fibrosis by reducing collagen content and α-SMA expression. The discrepancies between the results of Skibba et al. (45) and the result presented here may be owing to the differences in the concentration of EET administration; they administrated 10 mg/kg/day of 14,15-EET alone, whereas we administrated 15 µg/kg/day of either 11,12-EET or 14,15-EET, which may have less efficacy in reducing renal fibrosis.
In conclusion, we demonstrate that increasing EET levels by 11,12-EET and 14,15-EETs administration and genetic or pharmacological inhibition of sEH significantly attenuates oxidative stress, renal cell death, renal interstitial inflammation, and fibrogenesis following UUO. Furthermore, increased EETs reduced inflammatory responses in UUO kidneys, such as M1 macrophage polarization and inflammatory cell infiltration. Our data, for the first time, provide a mechanistic understanding of how EETs prevent kidney fibrogenesis during obstructive nephropathy. Collectively, our findings may pave the way to design therapeutic strategies focusing on EETs or EET derivatives to prevent fibroproliferative diseases not only in UUO but also in other chronic diseases including those of the kidney, liver, lung, and heart.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21539082.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.21539118.v1.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.21539139.v1.
GRANTS
B.J.P. is supported by National Institutes of Health Grants DK116987, DK120533, and DK120846.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.J.P. conceived and designed research; M.R.N. performed experiments; M.R.N. analyzed data; M.R.N., H.-S.J., F.E.S., F.A.F., J.K., and B.J.P. interpreted results of experiments; M.R.N. prepared figures; M.R.N. drafted manuscript; H.-S.J., F.A.F., J.K., and B.J.P. edited and revised manuscript; M.R.N., H.-S.J., F.E.S., F.A.F., J.K., and B.J.P. approved final version of manuscript.
REFERENCES
- 1. Bakris G, Vassalotti J, Ritz E, Wanner C, Stergiou G, Molitch M, Nesto R, Kaysen GA, Sowers JR; CKD Consensus Working Group. National Kidney Foundation consensus conference on cardiovascular and kidney diseases and diabetes risk: an integrated therapeutic approach to reduce events. Kidney Int 78: 726–736, 2010. doi: 10.1038/ki.2010.292. [DOI] [PubMed] [Google Scholar]
- 2. Narkiewicz K. Obesity and hypertension–the issue is more complex than we thought. Nephrol Dial Transplant 21: 264–267, 2006. doi: 10.1093/ndt/gfi290. [DOI] [PubMed] [Google Scholar]
- 3.GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990—2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395: 709–733, 2020. doi: 10.1016/S0140-6736(20)30045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Declèves AE, Sharma K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol 10: 257–267, 2014. doi: 10.1038/nrneph.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boor P, Ostendorf T, Floege J. Renal fibROSis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol 6: 643–656, 2010. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- 6. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7: 684–696, 2011. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 124: 2299–2306, 2014. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Black LM, Lever JM, Agarwal A. Renal inflammation and fibrosis: a double-edged sword. J Histochem Cytochem 67: 663–681, 2019. doi: 10.1369/0022155419852932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8: 794–805, 2009. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 526: 61–74, 2013. doi: 10.1016/j.gene.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan F, Roman RJ. Effect of cytochrome P450 metabolites of arachidonic acid in nephrology. J Am Soc Nephrol 28: 2845–2855, 2017. doi: 10.1681/ASN.2017030252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imig JD. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol Ther 192: 1–19, 2018. doi: 10.1016/j.pharmthera.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim J, Yoon SP, Toews ML, Imig JD, Hwang SH, Hammock BD, Padanilam BJ. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol 308: F131–F139, 2015. doi: 10.1152/ajprenal.00531.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol 307: F971–F980, 2014. doi: 10.1152/ajprenal.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stavniichuk A, Hye Khan MA, Yeboah MM, Chesnik MA, Jankiewicz WK, Hartmann M, Blöcher R, Kircher T, Savchuk O, Proschak E, Imig JD. Dual soluble epoxide hydrolase inhibitor/PPAR-γ agonist attenuates renal fibrosis. Prostaglandins Other Lipid Mediat 150: 106472, 2020. doi: 10.1016/j.prostaglandins.2020.106472. [DOI] [PubMed] [Google Scholar]
- 16. Zhu Y, Blum M, Hoff U, Wesser T, Fechner M, Westphal C, Gürgen D, Catar RA, Philippe A, Wu K, Bubalo G, Rothe M, Weldon SM, Dragun D, Schunck WH. Renal ischemia/reperfusion injury in soluble epoxide hydrolase-deficient mice. PLoS One 11: e0145645, 2016. doi: 10.1371/journal.pone.0145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiang CW, Lee HT, Tarng DC, Kuo KL, Cheng LC, Lee TS. Genetic deletion of soluble epoxide hydrolase attenuates inflammation and fibrosis in experimental obstructive nephropathy. Mediators Inflamm 2015: 693260, 2015. doi: 10.1155/2015/693260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang SH, Kim YC, An JN, Kim JH, Lee J, Lee HY, Cho JY, Paik JH, Oh YK, Lim CS, Kim YS, Lee JP. Active maintenance of endothelial cells prevents kidney fibrosis. Kidney Res Clin Pract 36: 329–341, 2017. doi: 10.23876/j.krcp.2017.36.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Q, Liang Y, Qiao Y, Zhao X, Yang Y, Yang S, Li B, Zhao Q, Dong L, Quan S, Tian R, Liu Z. Expression of soluble epoxide hydrolase in renal tubular epithelial cells regulates macrophage infiltration and polarization in IgA nephropathy. Am J Physiol Renal Physiol 315: F915–F926, 2018. doi: 10.1152/ajprenal.00534.2017. [DOI] [PubMed] [Google Scholar]
- 20. Luo Y, Wu MY, Deng BQ, Huang J, Hwang SH, Li MY, Zhou CY, Zhang QY, Yu HB, Zhao DK, Zhang G, Qin L, Peng A, Hammock BD, Liu JY. Inhibition of soluble epoxide hydrolase attenuates a high-fat diet-mediated renal injury by activating PAX2 and AMPK. Proc Natl Acad Sci USA 116: 5154–5159, 2019. doi: 10.1073/pnas.1815746116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jang HS, Kim JI, Noh M, Rhee MH, Park KM. Regulator of G protein signaling 2 (RGS2) deficiency accelerates the progression of kidney fibrosis. Biochim Biophys Acta 1842: 1733–1741, 2014. doi: 10.1016/j.bbadis.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 22. Noh MR, Woo CH, Park MJ, In Kim J, Park KM. Ablation of C/EBP homologous protein attenuates renal fibrosis after ureteral obstruction by reducing autophagy and microtubule disruption. Biochim Biophys Acta Mol Basis Dis 1864: 1634–1641, 2018. doi: 10.1016/j.bbadis.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 23. Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol 24: 229–242, 2013. doi: 10.1681/ASN.2012070678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang HS, Noh MR, Jung EM, Kim WY, Southekal S, Guda C, Foster KW, Oupicky D, Ferrer FA, Padanilam BJ. Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int 97: 327–339, 2020. doi: 10.1016/j.kint.2019.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jang HS, Padanilam BJ. Simultaneous deletion of Bax and Bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 309: F540–F550, 2015. doi: 10.1152/ajprenal.00170.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noh MR, Kim KY, Han SJ, Kim JI, Kim HY, Park KM. Methionine sulfoxide reductase A deficiency exacerbates cisplatin-induced nephrotoxicity via increased mitochondrial damage and renal cell death. Antioxid Redox Signal 27: 727–741, 2017. doi: 10.1089/ars.2016.6874. [DOI] [PubMed] [Google Scholar]
- 27. Jang HS, Kim JI, Kim J, Park JW, Park KM. Angiotensin II removes kidney resistance conferred by ischemic preconditioning. Biomed Res Int 2014: 602149, 2014. doi: 10.1155/2014/602149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Noh MR, Kong MJ, Han SJ, Kim JI, Park KM. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol 34: 101548, 2020. doi: 10.1016/j.redox.2020.101548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Noh MR, Jang HS, Song DK, Lee SR, Lipschutz JH, Park KM, Kim JI. Downregulation of exocyst Sec10 accelerates kidney tubule cell recovery through enhanced cell migration. Biochem Biophys Res Commun 496: 309–315, 2018. doi: 10.1016/j.bbrc.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 30. Dai M, Wu L, He Z, Zhang S, Chen C, Xu X, Wang P, Gruzdev A, Zeldin DC, Wang DW. Epoxyeicosatrienoic acids regulate macrophage polarization and prevent LPS-induced cardiac dysfunction. J Cell Physiol 230: 2108–2119, 2015. doi: 10.1002/jcp.24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hye Khan MA, Stavniichuk A, Sattar MA, Falck JR, Imig JD. Epoxyeicosatrienoic acid analog EET-A blunts development of lupus nephritis in mice. Front Pharmacol 10: 512, 2019. doi: 10.3389/fphar.2019.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khan AH, Falck JR, Manthati VL, Campbell WB, Imig JD. Epoxyeicosatrienoic acid analog attenuates angiotensin II hypertension and kidney injury. Front Pharmacol 5: 216, 2014. [Erratum in Front Pharmacol 5: 250, 2014]. doi: 10.3389/fphar.2014.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thomson SJ, Askari A, Bishop-Bailey D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int J Vasc Med 2012: 605101, 2012. doi: 10.1155/2012/605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kida Y, Tchao BN, Yamaguchi I. Peritubular capillary rarefaction: a new therapeutic target in chronic kidney disease. Pediatr Nephrol 29: 333–342, 2014. doi: 10.1007/s00467-013-2430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 5: 953–964, 2005. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 36. Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hiatt MJ, Ivanova L, Trnka P, Solomon M, Matsell DG. Urinary tract obstruction in the mouse: the kinetics of distal nephron injury. Lab Invest 93: 1012–1023, 2013. doi: 10.1038/labinvest.2013.90. [DOI] [PubMed] [Google Scholar]
- 38. Glassock RJ, Warnock DG, Delanaye P. The global burden of chronic kidney disease: estimates, variability and pitfalls. Nat Rev Nephrol 13: 104–114, 2017. doi: 10.1038/nrneph.2016.163. [DOI] [PubMed] [Google Scholar]
- 39. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199–210, 2008. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ruggenenti P, Cravedi P, Remuzzi G. Mechanisms and treatment of CKD. J Am Soc Nephrol 23: 1917–1928, 2012. doi: 10.1681/ASN.2012040390. [DOI] [PubMed] [Google Scholar]
- 41. Ohtake T, Oka M, Maesato K, Mano T, Ikee R, Moriya H, Kobayashi S. Pathological regression by angiotensin II Type 1 receptor blockade in patients with mesangial proliferative glomerulonephritis. Hypertens Res 31: 387–394, 2008. doi: 10.1291/hypres.31.387. [DOI] [PubMed] [Google Scholar]
- 42. Jiang XS, Xiang XY, Chen XM, He JL, Liu T, Gan H, Du XG. Inhibition of soluble epoxide hydrolase attenuates renal tubular mitochondrial dysfunction and ER stress by restoring autophagic flux in diabetic nephropathy. Cell Death Dis 11: 385, 2020. doi: 10.1038/s41419-020-2594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol 15: 1244–1253, 2004. [PubMed] [Google Scholar]
- 44. Hye Khan MA, Hwang SH, Sharma A, Corbett JA, Hammock BD, Imig JD. A dual COX-2/sEH inhibitor improves the metabolic profile and reduces kidney injury in Zucker diabetic fatty rat. Prostaglandins Other Lipid Mediat 125: 40–47, 2016. doi: 10.1016/j.prostaglandins.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Skibba M, Hye Khan MA, Kolb LL, Yeboah MM, Falck JR, Amaradhi R, Imig JD. Epoxyeicosatrienoic acid analog decreases renal fibrosis by reducing epithelial-to-mesenchymal transition. Front Pharmacol 8: 406, 2017. doi: 10.3389/fphar.2017.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hye Khan MA, Neckár J, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, Falck JR, Imig JD. Orally active epoxyeicosatrienoic acid analog attenuates kidney injury in hypertensive Dahl salt-sensitive rat. Hypertension 62: 905–913, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest 129: 2619–2628, 2019. doi: 10.1172/JCI124615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. López-Vicario C, Alcaraz-Quiles J, García-Alonso V, Rius B, Hwang SH, Titos E, Lopategi A, Hammock BD, Arroyo V, Clària J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc Natl Acad Sci USA 112: 536–541, 2015. doi: 10.1073/pnas.1422590112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dai M, Wu L, Wang P, Wen Z, Xu X, Wang DW. CYP2J2 and its metabolites EETs attenuate insulin resistance via regulating macrophage polarization in adipose tissue. Sci Rep 7: 46743, 2017. doi: 10.1038/srep46743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Behmoaras J, Diaz AG, Venda L, Ko JH, Srivastava P, Montoya A, Faull P, Webster Z, Moyon B, Pusey CD, Abraham DJ, Petretto E, Cook TH, Aitman TJ. Macrophage epoxygenase determines a profibrotic transcriptome signature. J Immunol 194: 4705–4716, 2015. doi: 10.4049/jimmunol.1402979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARγ, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci USA 102: 16747–16752, 2005. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heming M, Gran S, Jauch SL, Fischer-Riepe L, Russo A, Klotz L, Hermann S, Schafers M, Roth J, Barczyk-Kahlert K. Peroxisome proliferator-activated receptor-γ modulates the response of macrophages to lipopolysaccharide and glucocorticoids. Front Immunol 9: 893, 2018. doi: 10.3389/fimmu.2018.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dendooven A, Ishola DA Jr, Nguyen TQ, Van der Giezen DM, Kok RJ, Goldschmeding R, Joles JA. Oxidative stress in obstructive nephropathy. Int J Exp Pathol 92: 202–210, 2011. doi: 10.1111/j.1365-2613.2010.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Imig JD, Hye Khan MA, Burkhan A, Chen G, Adebesin AM, Falck JR. Kidney-targeted epoxyeicosatrienoic acid analog, EET-F01, reduces inflammation, oxidative stress, and cisplatin-induced nephrotoxicity. Int J Mol Sci 22: 2793, 2021. doi: 10.3390/ijms22062793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 9: 49–89, 2007. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 56. Chen G, Xu R, Wang Y, Wang P, Zhao G, Xu X, Gruzdev A, Zeldin DC, Wang DW. Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am J Physiol Endocrinol Physiol 303: E563–E575, 2012. doi: 10.1152/ajpendo.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen W, Zheng G, Yang S, Ping W, Fu X, Zhang N, Wang DW, Wang J. CYP2J2 and EETs protect against oxidative stress and apoptosis in vivo and in vitro following lung ischemia/reperfusion. Cell Physiol Biochem 33: 1663–1680, 2014. doi: 10.1159/000362950. [DOI] [PubMed] [Google Scholar]
- 58. Feng W, Xu X, Zhao G, Li G, Liu T, Zhao J, Dong R, Wang DW, Tu L. EETs and CYP2J2 inhibit TNF-α-induced apoptosis in pulmonary artery endothelial cells and TGF-β1-induced migration in pulmonary artery smooth muscle cells. Int J Mol Med 32: 685–693, 2013. doi: 10.3892/ijmm.2013.1435. [DOI] [PubMed] [Google Scholar]
- 59. Červenka L, Husková Z, Kopkan L, Kikerlová S, Sedláková L, Vaňourková Z, Alánová P, Kolář F, Hammock BD, Hwang SH, Imig JD, Falck JR, Sadowski J, Kompanowska-Jezierska E, Neckář J. Two pharmacological epoxyeicosatrienoic acid-enhancing therapies are effectively antihypertensive and reduce the severity of ischemic arrhythmias in rats with angiotensin II-dependent hypertension. J Hypertens 36: 1326–1341, 2018. doi: 10.1097/HJH.0000000000001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21539082.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.21539118.v1.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.21539139.v1.
Data Availability Statement
Data will be made available upon reasonable request.