

Abstract
The generation of organoids is one of the biggest scientific advances in regenerative medicine. Here, by lengthening the time that human pluripotent stem cells (hPSCs) were exposed to a three-dimensional microenvironment, and by applying defined renal inductive signals, we generated kidney organoids that transcriptomically matched second-trimester human fetal kidneys. We validated these results using ex vivo and in vitro assays that model renal development. Furthermore, we developed a transplantation method that utilizes the chick chorioallantoic membrane. This approach created a soft in vivo microenvironment that promoted the growth and differentiation of implanted kidney organoids, as well as providing a vascular component. The stiffness of the in ovo chorioallantoic membrane microenvironment was recapitulated in vitro by fabricating compliant hydrogels. These biomaterials promoted the efficient generation of renal vesicles and nephron structures, demonstrating that a soft environment accelerates the differentiation of hPSC-derived kidney organoids.
Kidney organoids have been produced from human pluripotent stem cells (hPSCs) by specific induction of the metanephric mesenchyme (MM) lineage (including nephron progenitor cells, NPCs)1–7, or by the simultaneous induction of MM- and ureteric bud (UB)-like progenitors8,9, the two progenitor cell populations that give rise to the adult kidney during development. Recently, NPCs and UB progenitors were separately induced and then aggregated together into three-dimensional (3D) spheroids that generated kidney organoids with higher-order architecture10. For kidney organoids generated from human embryonic stem cells (hESCs), CRISPR/Cas9 technology can be used to recapitulate the molecular features of kidney diseases4. Human kidney organoids can also be used as unprecedented in vitro models to screen for nephrotoxicity3,4,9. Besides the importance of these findings, major concerns related to the lack of vascularization and insufficient maturation still require further investigation to advance the field of hPSC-derived kidney organoids (kidney organoids). Biophysical cues have been shown to regulate cell behaviour, including the stemness and differentiation of different stem cell populations. Recently, application of fluid flow enhanced hPSC-derived podocyte-like cell differentiation in monolayer culture11, and the modulation of adherent forces in kidney organoids, resulted in changes in the functional performance of proximal tubular epithelial-like cells within kidney organoids12.
Efficient generation of kidney organoids in 3D culture
During mammalian kidney development, the posterior primitive streak (PPS) and anterior primitive streak (APS) give rise to the intermediate mesoderm (IM) and definitive endoderm, respectively. The posterior IM generates the MM, whereas the anterior IM forms the UB. The PPS can be generated from hPSCs using a combination of growth factors (including BMP4)1,7,8, or by exposing undifferentiated cells to varying doses and durations of the Wnt signalling agonist CHIR99021 (CHIR), a widely used inhibitor of glycogen synthase kinase 3 (GSK3β)1–5,8,9. Building upon these observations, we asked whether PPS cells could be generated by exposing hPSCs to a high dose of CHIR (8 μM) in two-dimensional monolayer culture over three consecutive days (Supplementary Fig. 1a; Methods). This treatment regimen was sufficient to induce PPS-committed cells that were positive for the PPS marker BRACHYURY (referred to as T), at 82.2 ± 2.6% efficiency (Supplementary Fig. 1b,c). PPS-committed cells upregulated the expression of PPS genes in comparison with APS genes (Supplementary Fig. 1d). Subsequent exposure of PPS-committed cells to a combination of FGF9 and activin A (20:1) for an additional day resulted in acquisition of the early IM marker PAX2 at 85.0 ± 1.4% efficiency (Supplementary Fig. 2a–c; Methods). Accordingly, messenger RNA levels for the posterior IM markers OSR1 and HOXD11 and the anterior IM marker GATA3 were also upregulated at this stage (Supplementary Fig. 2d).
We next reasoned that increasing the time that IM-committed cells are exposed to 3D culture, which increase cell-to-cell and cell-to-extracellular matrix interactions, would generate kidney organoids at higher efficiencies than previously reported3,8,9. Therefore, 3D spheroids were generated by the self-aggregation of IM cells and maintained under 3D organotypic culture until day 16 (Fig. 1a; see Methods). Treatment of IM-committed 3D spheroids (day 0) with CHIR (3 μM) for 3 d (from day 0 to day 3), while maintaining FGF9 signalling (from day 0 to day 7), resulted in the formation of numerous renal vesicles (RVs) on day 8 (Supplementary Fig. 3a,b), which were analysed by immunofluorescence for RV-associated markers, including PAX2, WT1, LHX1, PAX8, HNF1β, ECAD and BRN1 (Supplementary Fig. 3a,c,d). Remarkably, SIX2-positive cells were absent on day 8, indicating the lack of MM progenitors at this stage (Supplementary Fig. 3d). In addition, downregulation of the epithelial-to-mesenchymal markers TWIST and SNAIL and upregulation of WNT4 and ECAD coincided with initiation of the mesenchymal-to-epithelial transition, which is essential for nephrogenesis (Supplementary Fig. 3e). RV-stage organoids were then differentiated in the absence of growth factors (from day 7 to day 16). This resulted in the derivation of kidney organoids with multiple nephron-like structures that were segmented into typical nephron components, including proximal tubules (LTL+ AQP1+/SLC3A1+), loops of Henle (ECAD+ UMOD+), distal tubules (UMOD−ECAD+), and glomeruli (PODXL+/PODOCIN+/NEPHRIN+/NEPH1+/WT1+ PODXL+ LTL−/PODOCIN+ LTL−) (Fig. 1b, Supplementary Fig. 4a–g). In addition, quantitative PCR (qPCR) and immunofluorescence analyses confirmed that isolated LTL− and LTL+ cell fractions from day 16 organoids expressed markers of glomerular and proximal tubular identity, respectively (Supplementary Fig. 5a–f). Similarly, markers representative of the major steps of differentiation were analysed by qPCR (Supplementary Fig. 6). Our methodology was quite robust, as kidney organoids from two commercial hESC lines and one human induced pluripotent stem cell (hiPSC) line were also generated (Supplementary Fig. 7a–d, Supplementary Fig. 8).
Fig. 1 |. Efficient generation of kidney organoids in 3D culture.
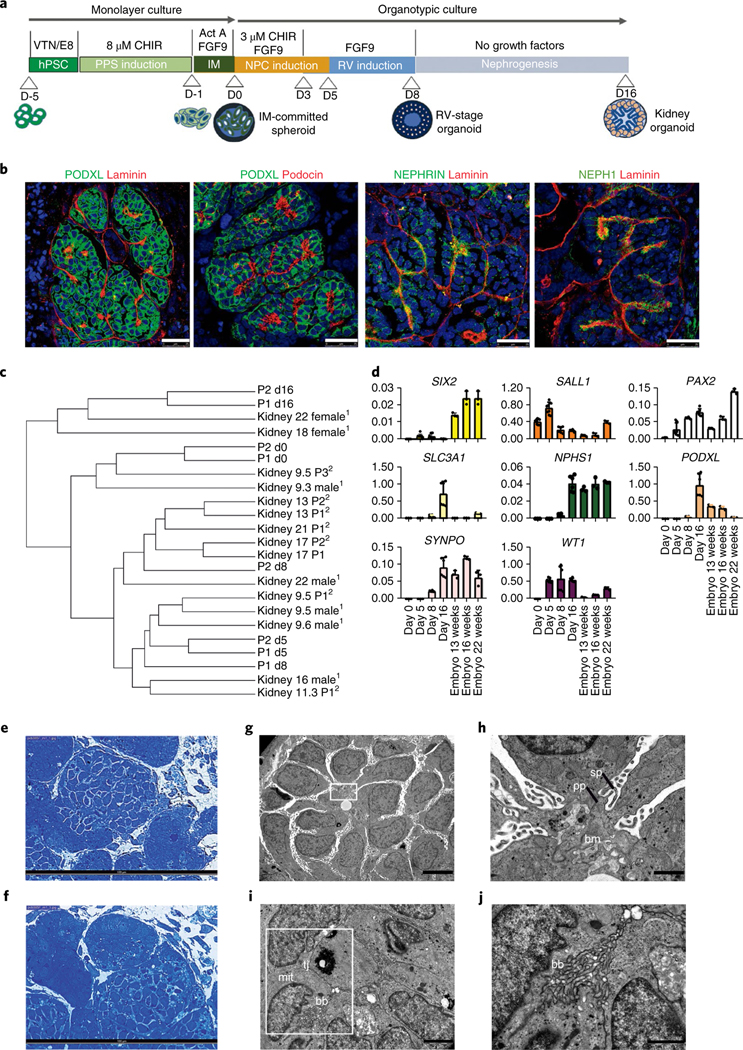
a, Schematic of the stepwise differentiation methodology for generating kidney organoids from hPSCs. b, Confocal microscopy images of glomerular structures in day 16 kidney organoids showing podocyte-like cells positive for PODXL, nephrin, NEPH1 and podocin, and the basement membrane protein laminin. Scale bars, 25 μm. c, Dendrogram representing the hierarchical clustering of day 0,5,8 and 16 kidney organoids with human fetal kidneys from 9, 13, 17 and 18 weeks of gestation (first trimester) and 22 weeks of gestation (second trimester). Data from Chuva de Sousa Lopes (SRP055513)13 (1) and McMahon (SRP111183)14 (2) are included in the analysis. d, qPCR analysis during kidney organoid differentiation and 13-, 16-, and 22-week human fetal kidneys (genes are indicated). Data are mean ± s.d. For SIX2, WT1, SALL1 and PAX2, day 0, day 5, n = 3; day 8, day 16, n = 2. For PODXL, SLC3A1, SYNPO and NPHS1, day 0, day 5, n = 1; day 8, day 16, n = 2. Each sample is a pool of six organoids. Three technical replicates are shown per sample. e,f, Semithin sections of day 16 kidney organoids showing glomerular (e) and tubular-like (f) structures. Scale bars, 100 μm. g–j, TEM of day 16 kidney organoids. g, Immature podocytes. Scale bar, 5 μm. h, A magnified view of the boxed region in g showing a detail of podocyte-related structures including the deposition of a basement membrane (bm), and primary (pp) and secondary cell processes (sp). Scale bar, 1 μm. i, Epithelial tubular-like cells with brush borders (bb), high mitochondrial (mit) content and tight junctions (tj). Scale bar, 2 μm. j, A magnified view of the boxed region in i showing a detail of brush borders. Scale bar, 1 μm.
To gain insight into the sequence of transcription regulatory events necessary to promote renal differentiation from hPSCs, we performed RNA sequencing (RNA-Seq) analysis at major stages during the differentiation process. We compared our results with transcriptional data from human fetal organs/tissues from the first and second trimesters of gestation13,14 (Supplementary Fig. 9, Supplementary Table 1; Methods), demonstrating that our technique specifically generated renal lineages. Importantly, RNA-Seq analysis showed that day 8 RV-stage organoids transcriptionally matched human fetal kidneys at 16 weeks of gestation, whereas day 16 kidney organoids matched human fetal kidneys at 22 weeks of gestation (Fig. 1c, Supplementary Table 2; Methods). We validated these findings by analysing markers of nephron progenitors (SIX2, SALL1, PAX2), the proximal tubular segment (SLC3A1) and the glomerular compartment (NPHS1, PODXL, SYNPO, WT1) via qPCR (Fig. 1d). Additionally, immunofluorescence analysis showed that localization of late-stage nephron markers was comparable between kidney organoids and human fetal kidney samples (Supplementary Fig. 4a–g). Transmission electron microscopy (TEM) analysis was performed on day 16 kidney organoids (Fig. 1e–j). Ultrathin sections revealed the presence of primitive podocyte-like cells with deposition of a basement membrane (Fig. 1g) and developing primary and secondary cell processes (Fig. 1h). Epithelial tubular-like cells with brush borders and high mitochondrial content were also detected (Fig. 1i,j).
Kidney organoids recapitulate human kidney development
The formation of kidney organoids with segmented nephrons may depend on the existence of a transient population of NPCs responsible for the generation of nephron structures in vitro. We analysed by immunofluorescence the expression of OSR1, WT1, PAX2 and SIX2, confirming that cells exhibiting a NPC signature were present in day 5 spheroids (Supplementary Fig. 10a,b). The posterior origin of NPCs was also confirmed by the detection of HOXD11, OSR1, and WT1 mRNA by qPCR (Supplementary Fig. 10c). Interestingly, the anterior IM fate was also generated at this stage, as GATA3 mRNA was detected (Supplementary Fig. 10c). We next evaluated the ability of day 5 NPCs to form kidney chimaeric structures ex vivo, taking advantage of a faithful reaggregation assay with mouse embryonic kidney cells15,16. After 6 d of culture, differentiated NPCs, identified by the expression of human nuclear antigen (HuNu), integrated into nascent nephron structures that expressed WT1 in the glomerular segment and PAX8 in the nascent nephron (Fig. 2a–f; Methods). Thus, day 5 NPCs exhibited the capacity to integrate into mouse nascent nephron structures, but not into the UB compartment, suggesting that the induction of UB derivatives from hPSCs may depend on additional exogenous signals. These results challenge previous findings9 and agreed with a recent study that identified optimal time windows and exogenous signals for selectively inducing NPC and UB lineages from mouse and human PSCs10.
Fig. 2 |. Kidney organoids model human kidney organogenesis in vitro.
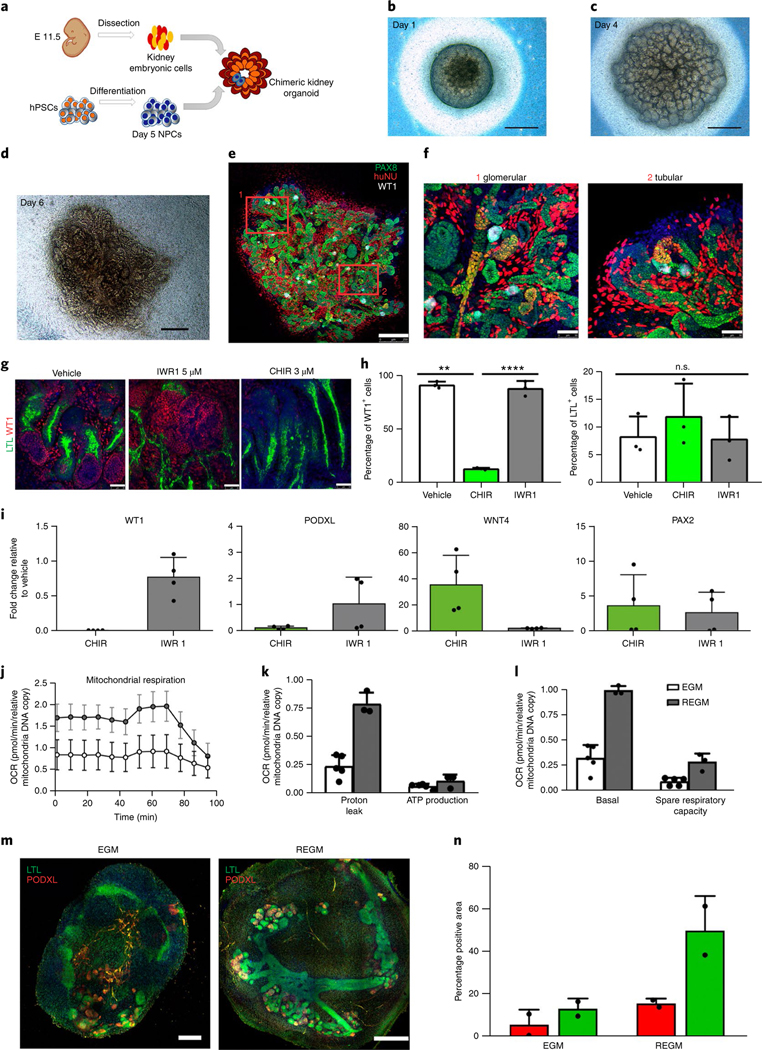
a, Representation of the coculture of day 5 NPCs with mouse embryonic kidney cells. b–d, Bright-field images of reaggregates after 1 d (b), 4 d (c) and 6 d (d) in culture. Scale bars, 500 μm. e, Immunocytochemistry for PAX8, WT1 and HuNu of the reaggregate in d. Scale bar, 250 μm. f, Magnified views of d. Scale bars, 50 μm. g–i, Modulation of β-catenin signalling in kidney organoids with IWR1 and CHIR inhibitors. g, Immunocytochemistry for WT1 and LTL in day 16 kidney organoids with the indicated regimens. Scale bars, 50 μm. h, Corresponding quantification of the percentage of WT1+ cells and LTL+ structures. Data are mean ± s.d. n = 3 organoids per treatment. One-way analysis of variance with Tukeýs post hoc test. For % WT1, F (1.009, 2.017) = 213.6, P = 0.0045; vehicle versus CHIR, **P = 0.0082; CHIR versus IWR1, ****P = 0.000034; vehicle versus IWR1, n.s., not significant, P = 0.9995. For % LTL, F (1.002, 2.004) = 0.9976, P = 0.4232, not significant. i, Corresponding qPCR analysis (genes are indicated). Data are mean ± s.d. (three technical replicates). j–l, Energy metabolism profile of kidney organoids maintained in EGM or REGM: kinetic oxygen consumption rate (OCR) response (j), inner mitochondrial membrane proton leak and cellular ATP production (k) and basal respiration and spare respiratory capacity (l). Data are normalized to mitochondrial DNA copy number/sample. Data are mean ± s.d. n = 3 (EGM) and n = 2 (REGM) organoids. m, Immunocytochemistry for LTL and PODXL in day 16 kidney organoids under EGM or REGM regimen. Scale bars, 200 μm (EGM) and 400 μm (REGM). n, Corresponding quantification of the percentage of PODXL+ and LTL+ structures. Data are mean ± s.d. n = 2 organoids per condition.
Next, we investigated the capacity of human kidney organoids to faithfully recapitulate complex nephron patterning events that have been mainly studied in the mouse model17. Day 8 RV-stage organoids were exposed to inhibitors of tankynase (IWR1) and GSK3β (CHIR) to decrease or increase β-catenin signalling, respectively. CHIR treatment reduced the number of WT1+ glomerulus-like structures when compared with vehicle (control) and IWR1. In contrast, the percentage of LTL+ proximal tubule-like structures was unchanged (Fig. 2g,h). qPCR analysis confirmed a decrease in the expression of WT1 and PODXL (proximal segment), and the induction of WNT4 (a β-catenin target gene) in CHIR-treated organoids relative to control, whereas PAX2 (whole nephron) remained unchanged (Fig. 2i). We next determined the effect of disrupting Notch signalling by treating day 8 RV-stage organoids with the γ-secretase inhibitor DAPT. Inhibition of Notch resulted in a severe loss of proximal tubule-like structures (LTL+), together with a reduction in PODXL+ glomerulus-like structures when compared with control (Supplementary Fig. 11a,b). qPCR analysis confirmed the downregulation of proximal (WT1) and medial (SLC3A1) nephron segment markers (Supplementary Fig. 11c). These findings agree with the role of Notch signalling in specifying proximal and medial identity during nephron patterning in the mouse17, and expand previous knowledge about the effect of Notch signalling on kidney organoids3.
The kidney is a highly metabolic organ that generates ATP through oxidative phosphorylation. Into the light of this knowledge, we hypothesized that the energy metabolism profile of cells should be taken into account to promote the differentiation of hPSCs into renal subtypes. Therefore, we exposed day 8 RV-stage organoids to either cell culture medium that promotes glycolysis in stem cells18 (endothelial cell growth medium, EGM) or cell culture medium favouring oxidative phosphorylation (renal epithelial cell growth medium (REGM) with insulin) for 8 d. Seahorse analysis revealed that REGM increased mitochondrial respiration in kidney organoids when compared with EGM (Fig. 2j–l), promoting an oxidative phosphorylation bioenergetic phenotype. Kidney organoids under REGM conditions enhanced tubule differentiation, as shown by the development of prominent proximal tubular structures (LTL+), more than with EGM (Fig. 2m,n), in agreement with previous findings19.
Vascularization of kidney organoids
Kidney organoids have shown the presence of nascent vascular endothelial cells surrounding renal structures, but lack a proper vascular pattern4,9. Of note, only two independent studies have reported in vivo vascularization of either hiPSC-derived NPCs20 or kidney organoids21 when transplanted under the kidney capsule of immunodeficient mice, identifying in both cases host-derived vascularization. We decided to explore an alternative approach for providing a vascular environment to kidney organoids. We made use of the chick chorioallantoic membrane (CAM), a highly vascularized extraembryonic tissue that has been used in tumour angiogenesis research22,23 and for the grafting of biomaterials24. More so than other in vivo models, such as the mouse, CAM represents a naturally immunodeficient environment that offers direct, minimally invasive access to the assay site, thereby facilitating the monitoring of the experiments in situ. We implanted day 16 kidney organoids into the CAM of 7-day-old chick embryos, and then maintained them in ovo for 5 d (Fig. 3a; Methods). On day 3 of implantation, multiple blood vessels from the CAM were macroscopically distinguished throughout kidney organoids (Fig. 3b, Supplementary Video 1). The circulation of chick blood within kidney organoids was clearly observed after 5 d (Fig. 3c, Supplementary Video 2). At this stage, in vivo injection of dextran–FITC (fluorescein isothiocyanate) into the CAM allowed for live imaging of the vasculature, confirming the grafting of the organoids into the CAM (Fig. 3d, Supplementary Video 3). Compared with in vitro counterparts (Supplementary Fig. 12a), CAM-implanted kidney organoids (implanted organoids) exhibited glomeruli with an enlarged Bowman’s space and tubule-like structures with enlarged lumens (Supplementary Fig. 12b,c, magnified views). Furthermore, CAM blood vessels (indicated with asterisks in Supplementary Fig. 12b,c,d) were found in close vicinity to glomerulus structures. Immunofluorescence analysis of consecutive sections confirmed the presence of chick blood vessels (labelled with Lens culinaris agglutinin) within implanted organoids. The latter were identified by the expression of the human marker HuNu and the presence of glomerulus-like structures (WT1+) (Supplementary Fig. 12e). Next, we tested the ability of implanted organoids to respond to the well known nephrotoxic agent cisplatin. Twenty-four hours after injecting cisplatin into the chick vasculature, levels of KIM-1 (a marker of renal tubule toxicity) and cleaved CASPASE 3 (a classical apoptotic marker) were upregulated in proximal tubular structures (LTL+), compared with specimens injected with a control solution (Supplementary Fig. 12f,g).
Fig. 3 |. In vivo vascularization of kidney organoids using chick CAM.
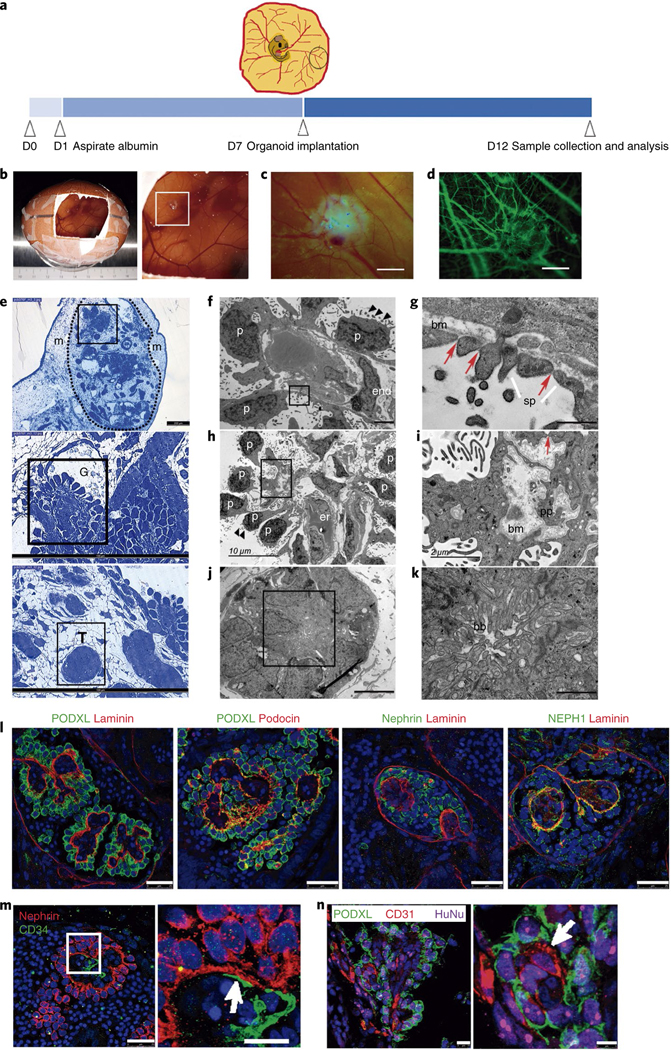
a, Methodology for the implantation of day 16 kidney organoids into chick CAM. b,c, Macroscopic views of implanted organoids maintained in ovo for 3 d (b) and 5 d (c). d, The implanted organoid in c after intravital injection of dextran–FITC through the chick vasculature. Scale bars, 1000 μm (c,d). e, Semithin sections of a kidney organoid (dashed line) implanted in the CAM mesenchyme (m) for 5 d. Magnified views of glomerular (G) and tubular (T) cells are shown. Scale bars, 200 μm, 100 μm (magnified views). f–k, TEM of implanted organoids. Magnified views of the boxed regions in f,h,j are shown in g,i,k, respectively). f, Differentiated podocytes (p) extending primary cell processes and apical microvilli (black triangles) are located on one side of the basement membrane and a vascular endothelial cell (end) is found on the opposite side. g, Slit diaphragm-like structures (red arrows) between secondary cell processes (sp). bm, basement membrane. h, Aligned podocytes showing primary cell processes and apical microvilli (black triangles). er, chicken erythrocytes. i, A detail of the basement membrane (bm), primary cell processes (pp) and a slit diaphragm-like structure (red arrow). j, Tubular-like cells. k, A detail of brush borders (bb). Scale bars, 2 μm (f), 500 nm (g), 10 μm (h), 2 μm (i), 5 μm (j), 1 μm (k). l–n, Confocal microscopy images of glomerular structures in implanted organoids. l, Immunohistochemistry for PODXL, nephrin, NEPH1, podocin and laminin. Scale bars, 25 μm. m, Immunohistochemistry for nephrin and CD34. Scale bars, 25 μm, 10 μm (magnified view). n, Immunohistochemistry for PODXL, CD31 and the human marker HuNu. Scale bars, 10 μm, 5 μm (magnified view). White arrows indicate endothelial-like cells in close contact with podocyte-like cells (m,n).
We next analysed semithin (Fig. 3e) and ultrathin sections (Fig. 3f–k) of implanted organoids. TEM images revealed the presence of aligned podocyte-like cells on one side of a linear basement membrane (Fig. 3f–h). Endothelial-like cells were found on the opposite side of the basal lamina (Fig. 3f) and, occasionally, chicken erythrocytes were observed within the glomerular-like structures (Fig. 3h). Podocyte-like cells exhibited multiple microvilli on the apical surface and extended primary and secondary cell processes on the basal side (Fig. 3f–i, Supplementary Fig. 12h–j). Secondary cell processes were bridged by slit diaphragm-like structures (Fig. 3g–i, Supplementary Fig. 12h–j). These features, which reflect functional differentiation, were not detected in the organoids cultured in vitro (Fig. 1g, h). Furthermore, tubular-like cells with thick brush borders and high mitochondrial content were observed (Fig. 3j,k). Immunofluorescence analysis showed apical localization of PODXL in aligned podocyte-like cells situated on the basement membrane (LAMININ+). Conversely, podocin and NEPH1 localized on the podocyte basal side (Fig. 3l). Likewise, CD34+ endothelial-like cells (stained with an antihuman specific CD34 antibody) were closely associated with NEPHRIN+/PODXL+ podocyte-like cells within glomerulus-like structures in implanted organoids (Fig. 3m, Supplementary Fig. 12k). Moreover, endothelial-like cells (CD31+) within glomerulus-like structures (PODXL+) coexpressed the human marker HuNu (Fig. 3n). In contrast, for kidney organoids cultured in vitro, CD34+ endothelial-like cells were not found within glomerulus-like structures (PODXL+) (Supplementary Fig. 12l).
Soft hydrogels enhance the formation of kidney organoids
Mirroring the exact biochemical (for example, site-specific bioactive ligands) and biophysical (for example, extracellular matrix stiffness, fluid flow, oxygen tension) properties of a physiological environment represents an as yet unaffordable technical approach in tissue engineering. By contrast, fabrication of hydrogels with mechanical properties (for example, Young’s modulus) similar to native tissues is a key methodology for guiding cellular responses and differentiation25. Therefore, we decided to characterize the specific mechanical properties of the CAM (by measuring the Young’s modulus, Supplementary Fig. 13a; Methods), which exhibited a stiffness value of about 1 kPa (Supplementary Fig. 13b), representative of an early embryonic microenvironment26 in which undifferentiated cells are primed for lineage commitment27. We next explored whether substrates mimicking a soft microenvironment may favour the generation of kidney organoids, compared with stiffer substrates. Thus, we fabricated functionalized polyacrylamide hydrogels of tunable stiffness (ranging from soft, 1 kPa, to very rigid, 60 kPa) as substrates for hPSC differentiation (Supplementary Fig. 14a; Methods). In comparison with rigid hydrogels, hPSCs grown on soft hydrogels under undifferentiated conditions formed tightly compacted hPSC colonies (ECAD+) (Supplementary Fig. 14b), showing reduced nuclear localization of the mechanotransduction marker Yes-associated protein (YAP) (Supplementary Fig. 14c). RNA-Seq analysis of hPSCs revealed that soft hydrogels promoted the expression of genes related to embryo and mesodermal differentiation (Supplementary Table 3), suggesting that a soft milieu may better replicate early stages of embryonic development, during which time counteracting gene regulatory networks control both pluripotency and differentiation ground states28. Based on these observations, we hypothesized that using soft hydrogels during the first steps of monolayer differentiation (including PPS and IM induction) may help guide hPSCs toward renal commitment. PPS induction of hPSCs differentiated on soft hydrogels resulted in higher mRNA levels of T and SALL1 markers when compared with rigid conditions (Supplementary Fig. 15a,b). PPS differentiation was also analysed by RNA-Seq, showing that soft hydrogels induced the expression of genes related to transcription regulation and downregulated genes related to extracellular matrix and basement membrane (Supplementary Table 4, Supplementary Fig. 15c). Induction of PPS-committed cells into IM-committed cells (Supplementary Fig. 16a) showed that soft hydrogels promoted increased mRNA levels of the early IM marker PAX2, the posterior IM marker HOXD11, the anterior IM marker LHX1, and SALL1 when compared with rigid conditions (Supplementary Fig. 16b). Upon differentiation under 3D organotypic culture, IM-committed cells derived on soft hydrogels began to develop RVs one day earlier (at day 7: Supplementary Fig. 16c), and resulted in the generation of more RVs than those derived on rigid hydrogels, as shown by quantitative analysis of PAX2+ RVs (Fig. 4a,b). Moreover, day 16 kidney organoids from soft conditions developed more WT1+ glomerulus-like and LTL+ tubule-like structures than those derived from rigid conditions (Fig. 4c,d), and expressed increased mRNA levels of late-stage nephron (NPHS1, SCNN1B) and vascularization (ENDOGLIN, VEGFR) markers (Fig. 4e). TEM of day 16 kidney organoids showed the presence of tubule-like structures containing epithelial cells with prominent brush borders in both soft and rigid conditions (Fig. 4f, Supplementary Fig. 17). Interestingly, soft hydrogels induced the differentiation of podocyte-like cells containing slit diaphragm-like structures between the cell processes (Fig. 4g–i), a podocyte differentiation feature that was absent in rigid conditions (Fig. 1g,h, Supplementary Fig. 17). Considering these findings, day 16 kidney organoids derived from soft hydrogels were then implanted into the CAM for 5 days (Fig. 4j). TEM revealed the presence of tubular epithelial-like cells with brush borders (Fig. 4k) and numerous glomerular structures containing podocyte-like cells above a dense basement membrane and in close vicinity to endothelial cells and chicken erythrocytes (Fig. 4l). Furthermore, induced podocyte-like cells exhibited secondary cell processes with slit diaphragm-like structures (Fig. 4m,n).
Fig. 4 |. Soft hydrogels accelerate the differentiation of kidney organoids.
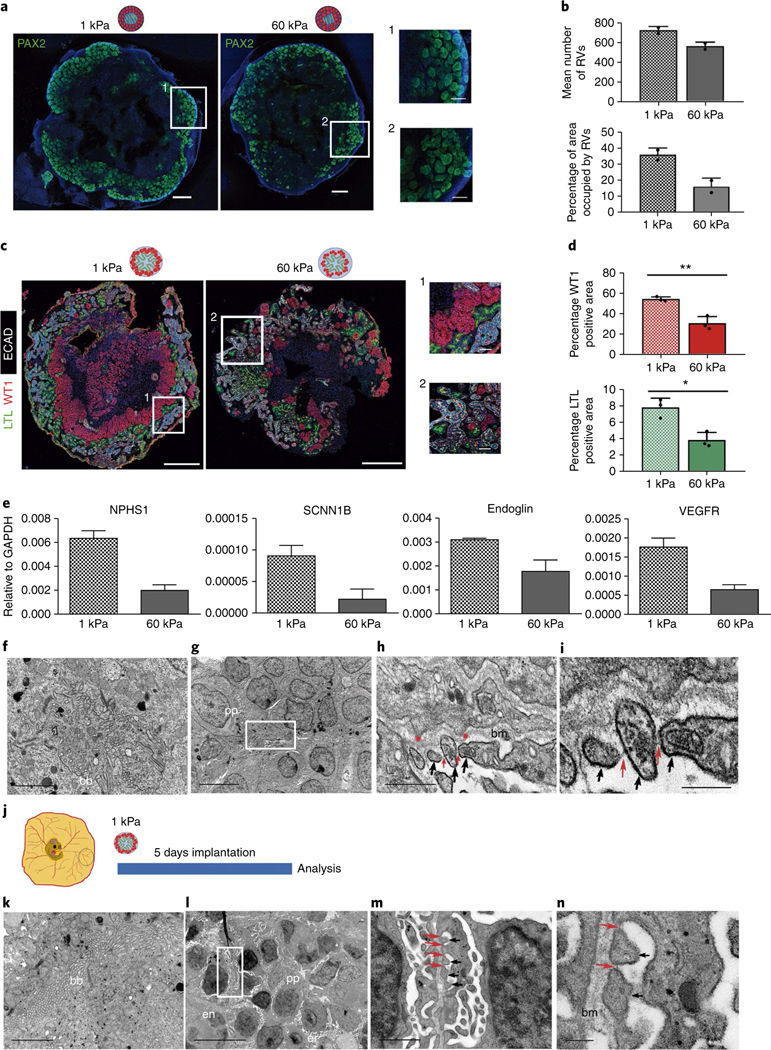
a, Immunocytochemistry for PAX2 in RV-stage organoids generated using 1 kPa or 60 kPa hydrogels. Scale bars, 500 μm, 150 μm (magnified views). b, Quantification of a. The mean number of RVs and area percentage occupied by RVs were quantified. Data are mean ± s.d. n = 2 organoids per condition. c, Immunohistochemistry for LTL, WT1 and ECAD in day 16 kidney organoids from 1 kPa or 60 kPa. Scale bars, 500 μm and 50 μm (magnified views). d, Quantification of c. The percentages of WT1+ and LTL+ area were quantified. Data are mean ± s.d. n = 3 organoids per condition. For WT1+, t(4) = 5.8057, **P = 0.0044. For LTL+, t(4) = 4.6023, *P = 0.0100. Two-tailed Student’s t-test. e, qPCR analysis of day 16 kidney organoids from 1 kPa or 60 kPa (genes are indicated). Data are mean ± s.d. (technical replicates). f–i, TEM of day 16 kidney organoids from 1 kPa. f, Epithelial tubular-like cells with brush borders (bb). g, Podocyte-like cells with primary cell processes (pp). h,i, Magnified views of g. Secondary cell processes (black arrows) with slit diaphragm-like structures (red arrows). Red asterisks, podocyte membrane protrusions. bm, basement membrane. Scale bars, 2 μm (f), 5 μm (g), 500 nm (h), 200 nm (i). j, Day 16 kidney organoids from 1 kPa were implanted into the CAM. k–n, TEM of implanted kidney organoids from 1 kPa. k, Tubular-like cells with brush borders. l, Aligned podocyte-like cells extending primary cell processes near endothelial (en) cells and chicken erythrocytes (er). m,n, Magnified views of l. Secondary cell processes with slit diaphragm-like structures. Scale bars, 2 μm (k), 10 μm (l), 1 μm (m), 200 nm (n).
Outlook
The methodology described here reduced the time needed to generate kidney organoids when compared with previous protocols by about 30%1,3–5,9, leading to the generation of kidney organoids that transcriptionally resembled second-trimester human fetal kidneys. This is an improvement over previous findings, in which kidney organoids clustered with trimester 1 human fetal kidneys9. Furthermore, here we have shown that kidney organoids implanted into chick CAM successfully engrafted and were vascularized in ovo, providing a straightforward model for nephrotoxicity and kidney disease modelling applications. Importantly, CAM-implanted kidney organoids showed morphological features that reflect functional differentiation compared with in vitro conditions. When CAM stiffness was mimicked in vitro via compliant hydrogels, hPSCs differentiated on soft substrates (CAM-like) generated IM-committed cells that showed an accelerated formation of more RVs and nephron structures than those produced on rigid substrates. Furthermore, kidney organoids generated from soft hydrogels exhibited improved differentiation characteristics when compared with those found under stiffer conditions. These differentiation features were also enhanced after CAM transplantation. Overall, the methodology described here paves the way toward further developing biomimetic approaches that will enhance organoid differentiation (either in vitro or in vivo). These advances will enable future studies of kidney development and disease.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41563-019-0287-6.
Methods
Culture of hPSCs.
All hPSC lines were obtained after the approval of the Ethics Committee of the Center of Regenerative Medicine in Barcelona and the Comisión de Seguimiento y Control de la Donación de Células y Tejidos Humanos del Instituto de Salud Carlos III (project numbers: 0336E/7564/2016; 0336E/5311/2015; 0336E/15986/2016; 0336E/79489/2015; 00336E/20031/2014). ES[4] hESC and CBiPSsv-4F-40 were obtained from The National Bank of Stem Cells (ISCIII, Madrid). H1 and H9 hESC lines were purchased at Wicell. All the lines were maintained in Essential 8 medium (A1517001, Life Technologies) in cell culture plates coated with 5 μg ml−1 vitronectin (A14700, Fisher Scientific) with 5% CO2 at 37 °C. Cells were passaged every 4–6 d.
hPSC differentiation into renal progenitor cells and generation of 3D kidney organoids.
hPSCs grown on vitronectin-coated plates were rinsed twice with PBS (1001–015, Life Technologies) and disaggregated into small cell clusters with 0.5 mM EDTA (E9884, Sigma). Cells were then seeded onto vitronectin-coated culture plates at a density of 5 × 104–1.5 × 104 cells per cm2 in Essential 8 medium (day −5). After overnight culture, the differentiation was initiated by treating hPSCs with 8 μM CHIR (SML1046, Sigma) in advanced RPMI 1640 basal medium (12633–012, Life Technologies) supplemented with 2 mM L-GlutaMAX (35050–038, Life Technologies) and penicillin/streptomycin (penicillin 10,000 U ml−1:streptomycin 10,000 μg ml−1; 15140122, Life Technologies) for 3 d (from day −4 to day −1). Next, cultures were treated with 200 ng ml−1 FGF9 (100–23B, Peprotech), 1 μ ml−1 heparin (H3149–10KU, Sigma) and 10 ng ml−1 activin A (338-AC-050, Vitro) for 1 d (from day −1 to day 0). Media changes were performed every day. On day 0, single-cell suspensions were obtained by dissociating cells with Accumax (07921, Stem Cell Technologies). Cells were then resuspended in advanced RPMI 1640 basal medium containing 3 μM CHIR, 200 ng ml−1 FGF9 and 1 μg ml−1 heparin, placed in 96-well plates (V bottom) at 5 × 105 cells per well, spun down (300 g for 3 min) and maintained in culture for 2 d without medium change. On day 2, cell spheroids were placed onto Transwells (CLS3460, Sigma) and cultured in advanced RPMI 1640 basal medium containing 3 μM CHIR, 200 ng ml−1 FGF9 and 1 μg ml−1 heparin for another 1 d. On day 3, CHIR was removed and organoids were maintained in advanced RPMI 1640 basal medium with 200 ng ml−1 FGF9 and 1 μg ml−1 heparin for another 4 d. From day 7 organoids were maintained in advanced RPMI 1640 basal medium until day 16 unless otherwise indicated, changing the medium every second day.
Immunocytochemistry.
After a single wash with PBS, samples were fixed with 4% paraformaldehyde (153799, Aname) for 20 min at room temperature. Next, samples were washed twice with PBS and further blocked using Tris-buffered saline (TBS) with 6% donkey serum (S30, Millipore) and 1% Triton X-100 (T8787, Sigma) for 1 h at room temperature. Samples were then treated overnight at 4 °C with primary antibodies diluted in antibody dilution buffer consisting of TBS with 6% donkey serum and 0.5% Triton X-100. After three rinses with antibody dilution buffer, samples were treated for 4 h at room temperature with fluorescent-conjugated secondary antibodies (Alexa Fluor (A) 488-, Cy3- or A647-; 1:200). A previous blocking step with a streptavidin/biotin blocking kit (SP-2002, Vector Labs) was performed when samples were assayed for biotinylated LTL (B-1325, Vector Labs) and Alexa Fluor 488-conjugated streptavidin (SA5488, Vector Labs) was used to detect LTL+ cells. Nuclei were detected using 4,6-diamidino-2-phenylindole (DAPI; 1:5000, D1306, Life Technologies) for 30 min. For mounting, samples were immersed in Fluoromount-G (0100–01, Southern Biotech). Image acquisition was carried out using an SP5 Leica microscope or a Zeiss LSM780 confocal microscope. Primary antibodies and associated information are provided in Supplementary Table 5.
Electron microscopy.
After fixation of samples with 2.5% glutaraldehyde containing 1% tannic acid in 0.1 M phosphate buffer (pH 7.4), samples were postfixed for 1 h at 4 °C with 1% OsO4 in 0.1 M phosphate buffer. Graded ethanol series were performed followed by epoxy resin embedding. Toluidine blue staining was performed in semithin sections before examination using a light microscope. Then, ultrathin sections were obtained using an EM UC7 ultramicrotome (Leica Microsystems) and collected on copper grids. 4% uranyl acetate and lead citrate were then used for staining. Samples were subsequently analysed with a JEM 1230 electron microscope (JEOL).
Total RNA isolation and qPCR with reverse transcription.
TRI Reagent was employed for total RNA purification following the manufacturer’s recommendations (T9424, Sigma). TURBO DNase inhibitor (AM1907, Ambion) was used in order to eliminate any residual genomic DNA. Complementary DNA was synthesized from 1 μg of RNA using a Cloned AMV First-Strand cDNA synthesis kit (12328, Invitrogen). Quantitative PCR with reverse transcription (QUANTSTUDIO 5 Applied Biosystems, Thermo Fisher Scientific) was used to quantify gene expression from cDNAs (25 ng/well) using PowerUp Sybr Green Master Mix (A25742, Thermo Fisher Scientific). GAPDH or Rplp0 were used for data normalization. Primer sequences used in this study are listed in Supplementary Table 6.
Next-generation RNA sequencing.
Sequencing libraries were prepared from 1 μg of total RNA (previously isolated using TRI Reagent—T9424, Sigma) using an Illumina TruSeq RNA Sample Prep Kit (catalogue no. FC-122–1001). Sequencing was carried out to produce between 50 and 60 million paired end reads/sample using the Illumina Hiseq 2500 platform. Raw sequences were inspected for their quality using FastQC (version v0.11.5)29 and trimmed using Skewer (version 0.2.2)30. STAR mapper (version 2.5.3a)31 was used to align the data to the human reference genome (GRCh38). The option ‘–quantMode’ was employed to count the number of mapped tags within genes (annotation Gencode v26). On average, 90% of tags were univocally mapped either to the genomic or to a splice junction. Data from Chuva de Sousa Lopes (SRP055513)13 were analysed in the same way. Read counts per genes were finally analysed using the R statistical package DESeq232. Since the two experiments were performed using different techniques (RNA-Seq and DeepSAGE), we used the ComBat function from the svaseq R package33 on rlog-transformed read counts in order to mitigate the batch effect. Scaled rlog values were then used to calculate the sample-to-sample distance and plotted as a dendrogram. Data from Little (SRP059518)9 and McMahon (SRP111183)14 were analysed using the same procedure. The package Keygenes13 was used to classify every sample according to its similarity to a tissue. Keygenes compares the transcriptional profiles of test samples with that from organs or cell types from a training set. In this study, the ‘fetal wo’ training set was used. This training set contains transcriptional data from17 fetal organs.
Flow cytometry.
Cells were dissociated using Accumax (07921, Stem Cell Technologies) for 5 min at 37 °C. Next, cells were resuspended in PBS and incubated with LIVE/DEAD Fixable Violet stain reagent (L23105, Life Technologies) (1:1000) for 30 min in the dark. For intracellular staining, a Foxp3/Transcription Factor Staining Buffer Set (00–5523–00, Labclinics) was used according to manufacturer’s instructions. Briefly, cell suspensions were fixed in the dark for 30–60 min at room temperature with Foxp3 fixation/permeabilization working solution. Permeabilization of samples was performed using the permeabilization buffer for 5 min at room temperature. Blocking was performed using 2% fetal bovine serum for 15 min. Incubations with conjugated antibodies were performed for 30 min. The antibodies used were OCT4 conjugated to Alexa Fluor 488 (560253, BD Pharmigen), brachyury conjugated to allophycocyanin (IC2085A, R&D Systems) and PAX2 (AF3364, R&D Systems) conjugated to A488 using a Lightning-Link® Rapid conjugation kit (322–0010, Innova Biosciences) following the manufacturer’s instructions. Samples were then washed with permeabilization buffer and resuspended in PBS + 2% fetal bovine serum. For cell sorting experiments, kidney organoids were stained with fluorescein-conjugated LTL (FL-1321, Vector Laboratories) as described elsewhere12. Kidney organoids were then dissociated to single cells using Accumax (07921, Stem Cell Technologies) for 15 min followed by 0.25% (wt/vol) trypsin (25300–054, Life Technologies) for 15 min at 37 °C. SA3800 software version 2.0.4 (SONY) was used to acquire flow cytometry samples in the Sony SA3800 spectral cell analyser (SONY). FACSDiva software version 8.0.1 (BD Biosciences) was used in the FACS Aria Fusion instrument (BD Biosciences) for cell sorting experiments. FlowJo software version 10 was used to analyse the data.
Reaggregation of mouse embryonic kidney cells with hPSC-derived NPCs.
These experiments were performed following approval by the Ethics Committee on Animal Research of the University of Barcelona, Spain (protocol no. OB 391/18). Reaggregation experiments were carried out as previously described in ref. 16. In brief, embryonic kidneys from 11.5–12.5 d post conception were collected from time-mated pregnant C57BL/6J mice. Kidney rudiments were removed from mouse embryos by manual dissection under a dissecting microscope. Dissociation of kidney rudiments into single cells was performed by incubating kidneys with 0.25% (wt/vol) trypsin (25300–054, Life Technologies) for 1–2 min at 37 °C, followed by quenching of trypsin using complete medium (MEM + 10% fetal bovine serum + penicillin/streptomycin) and pipetting up and down vigorously for 30 s to disaggregate kidneys. Day 5 organoids were dissociated into single cells with Accumax (07921, Stem Cell Technologies). The resultant cell suspensions were sieved through a 40 μm pore cell strainer. Next, 7.2 × 105 mouse kidney cells were combined with 8 × 103 cells from day 5 organoids, placed in a 96-well plate (V bottom) in complete medium with 10 μM ROCK inhibitor (72304, Stem Cell Technologies), spun down (300 g for 3 min) and incubated at 37 °C, 5% CO2 to allow aggregate formation. After overnight culture, aggregates were placed onto Transwells (CLS3460, Sigma) and maintained in complete medium at 37 °C and 5% CO2 for 4–6 d. Medium changes were performed every 48 h.
Nephron patterning assays.
For nephron patterning assays, samples were cultured in basic differentiation media supplemented with 10 μM DAPT (565770, Sigma), 3 μM CHIR (SML1046, Sigma) or 5 μM IWR1 (681669, Sigma) from day 8 to day 16. Organoids were then collected for RNA isolation and fixed with 4% paraformaldehyde for immunocytochemistry.
Seahorse analysis.
Kidney organoids at day 16 of differentiation were resuspended in warm Seahorse XF Assay Medium (Seahorse Bioscience). Individual organoids were transferred to an islet plate (one organoid per well) containing 400 μl of medium per well. After 1 h of incubation at 37 °C, plates were loaded into an XF24 respirometry machine (Seahorse Bioscience). Uncoupled and maximum OCR were assayed with oligomycin (1 μM) and FCCP (1.5 μM). To inhibit complex I- and III-dependent respiration, rotenone (1 μM) and antimycin A (1 μM) were used, respectively. OCR represents the oxygen tension and acidification of the medium as a function of time (pmol min−1).
Implantation of kidney organoids onto chick CAM.
Following animal care guidelines in Spain, no approval was required to perform the experiments described here. Briefly, fertilized white Leghorn chicken eggs were supplied by Granja Gibert. Eggs were placed horizontally in a humidified atmosphere at 38 °C in a Javier Masalles 240 N incubator. After 24 h, 3 ml of albumin was evacuated from the egg using a 18-gauge syringe. At embryonic day 7 (ED 7), a small window was created by cutting the egg shell using a sterile scalpel. Then, day 16 kidney organoids were implanted onto the surface of the CAM (one kidney organoid per egg) by gently scraping the upper CAM layer (avoiding bleeding or visible rupture of capillaries) at the desired implantation site. Egg windows were sealed with conventional plastic tape and incubated for 3–5 additional days (until ED 10–12).
Intravital imaging of the CAM vasculature.
Under a dissecting microscope, superficial CAM veins were injected with 1 mg ml−1 FITC–dextran (2 MDa) (FD-2000S, Sigma) in PBS using a 30-gauge Hamilton syringe, allowing solutions to circulate for 5 min. Injected volumes were kept at 50 μl. Live imaging was performed using a MZ10 F Leica stereomicroscope equipped with a MC170 HD Leica camera.
Nephrotoxicity assay.
Chick embryos (ED 14) that contained kidney organoids implanted into the CAM were intravenously injected with desired dosages of cisplatin (P4394, Sigma) using a 30-gauge Hamilton syringe, as previously reported34. Specimens injected with control solution (without cisplatin) were used as controls. Eggs were then sealed with conventional plastic tape and incubated overnight at 38 °C. After 24 h, CAM-implanted kidney organoids were collected and analysed.
Histological analysis and immunohistochemistry on CAM-implanted kidney organoids.
CAM-implanted kidney organoids were harvested at day 5 of the implantation period, fixed in 4% paraformaldehyde at 4 °C overnight and embedded in paraffin. For histological analysis, 5 μm thick sections were stained with haematoxylin and eosin. For immunohistochemistry, antigen retrieval consisting of citrate buffer (pH 6) at 95 °C for 30 min was performed. Samples were then blocked with TBS containing 3% donkey serum and 1% Triton X-100 for 1 h at room temperature. Subsequently, primary antibodies were used overnight at 4 °C in TBS with 3% donkey serum and 0.5% Triton X-100. After three washing steps with TBS containing 3% donkey serum and 0.5% Triton X-100, samples were treated with the appropriate conjugated secondary antibodies (Alexa Fluor 488-, Cy3- or A647-; all 1:200) for 2 h at room temperature. Nuclei were stained with DAPI (1:5000, D1306, Life Technologies) for 10 min. Samples were immersed in Fluoromount-G (0100–01, Southern Biotech). Image acquisition was carried out using a SP5 Leica microscope or a Zeiss LSM780 confocal microscope. Primary antibodies and associated information are provided in Supplementary Table 5.
Determination of the Young’s modulus of the chick CAM.
The ball indentation method was used to assess the Young’s modulus (E) of the chick CAM, as described in ref. 35. The indentation depth (d) was calculated based on the derivative of fluorescence intensity profile using a custom-made MATLAB code. E was calculated from the indentation force (F), d and the radius of the ball indentor (R). For d < 0.3 R, the Hertz contact mechanics model was used to calculate E as follows: E = [3(1 −2)F]/4R0.5d1.5, where ν is the Poisson’s ratio of the CAM.
Fabrication of functionalized polyacrylamide hydrogels.
Glass-bottom dishes were loaded with a solution of acetic acid, 3-(trimethoxysilyl)propyl methacrylate (Sigma) and ethanol (1/1/14). Wells were next rinsed three times with 96% ethanol. Different concentrations of acrylamide and bis-acrylamide were combined with a solution containing 0.5% ammonium persulfate, 0.05% tetramethylethylenediamine (Sigma) and 2% fluorescent 200 nm far-red carboxylated nanobeads (Invitrogen). Specifically, concentrations of 5 and 0.04% of acrylamide and bis-acrylamide were used for the softer hydrogels, and 12% and 0.25% of acrylamide and bis-acrylamide for the stiffer hydrogels, resulting in a nominal Young’s modulus of 1 kPa and 60 kPa, respectively, according to ref. 36. The substrates were functionalized as previously described in ref. 37. Briefly, a drop containing 1 mg ml−1 acrylic acid NHS (A8060, Sigma), 0.2% bis-acrylamide, 0.2% tetramethacrylate (408360, Sigma) and 0.05% (w/v) Irgacure 2959 was added on the surface of the hydrogel and photoactivated under exposure to ultraviolet light for 10 min. Afterwards, functionalized hydrogels were washed with HEPES and PBS and incubated overnight with 50 μg ml−1 vitronectin (A14700, Fisher Scientific) at 4 °C.
Quantification of immunofluorescence images.
A custom-made MATLAB code was used to perform the quantification of immunofluorescence images. For quantification of number and area of RVs, the DAPI image was first smoothed with a mean filter to homogenize the intensity values of the nuclei within an RV. This image was converted into a binary image after applying an intensity threshold. The binary image was used to segment the RVs by applying a watershed algorithm. From this segmentation, a list containing the area of each RV and the number of RVs was obtained. The entire area of the organoid was identified using the same principle but reducing the threshold for the binary conversion. The percentage of area occupied by RVs was calculated by adding the area of all RVs identified divided by the entire area of the organoid. For quantification of LTL+ and PODXL+ structures, the percentage of area occupied by LTL+ and PODXL+ structures was calculated using the same procedure as described above. For quantification of WT1+ cells, the DAPI image was converted into a binary image after applying an intensity threshold. The binary image was used to segment the nuclei by applying a watershed algorithm. All nuclei found were counted. The same procedure was used to identify WT1+ nuclei. The positive nuclei in WT1 images were divided by the number of DAPI nuclei, giving a percentage of WT1+ cells for each condition.
Human kidney material.
Primary human proximal tubular cells were obtained from collaborators at Hospital Clinic de Barcelona, Spain. The procedure was approved by the ethics committee of Hospital Clinic de Barcelona (project no. 2009/5023). Primary renal proximal tubular epithelial cells were obtained in the laboratory of origin as previously described38. Human fetal kidney samples included in this study were provided by the Fetal Tissue Bank of Vall d’Hebron University Hospital Biobank (PT13/0010/0021), part of the Spanish National Biobanks Network, and they were processed following standard operating procedures with the appropriate approval of the Ethical and Scientific Committees (project no. 0336E/9934/2015). The gestational age of human fetal kidney samples was determined using ultrasound heel-to-toe and crown-to-rump measurements39. Human fetal kidney samples from 13, 16 and 22 weeks of gestation were supplied as whole tissues embedded in OCT (at −80 °C) and as frozen tissue samples for RNA extraction.
Statistics and reproducibility.
Data are mean ± standard deviation (s.d.). Statistical differences between two groups were tested with a two-tailed Student’s t-test or one-way analysis of variance followed by Tukey’s post hoc test. Data were statistically significant if P < 0.05. Number of replicates (n = x), P values and degrees of freedom are included in the figure legends. GraphPad Prism version 6.01 software was used for statistical analysis. A table summarizing sample size, number of experiments and statistical test results (when applicable) for each figure panel is also provided as Supplementary Table 7.
For in vitro experiments, two to six organoids were analysed at the times and conditions indicated in each experiment. For ex vivo reaggregation assay, we used one to three pregnant mice (to collect embryonic kidneys from mouse embryos) per experiment. Two or three reaggregates were analysed per experiment. For implantation of organoids into chick CAM in ovo, about 6–22 chicken eggs were used per experiment and about 2–10 implanted kidney organoids were analysed per experiment.
RNA-Seq of kidney organoids during the time course differentiation was performed on six pooled kidney organoids at each time analysed from two independent experiments (Fig. 1c and Supplementary Fig. 9).
For main figures where representative immunofluorescence images are shown, at least n = 2 biologically independent kidney organoids were analysed from independent experiments showing similar results (Fig. 1b, n = 2 organoids; Fig. 2g, n = 3 organoids per treatment; Fig. 2m, n = 4 organoids per culture condition; Fig. 3l,m, n = 2 implanted organoids; Fig. 3n, n = 3 implanted organoids; Fig. 4a, n = 2 organoids per stiffness condition; Fig. 4c, n = 3 organoids per stiffness condition).
For main figures where representative TEM images are shown, n = 2 biologically independent kidney organoids were analysed from independent experiments showing similar results (Fig. 1e–j, Fig. 3e–k, Fig. 4f–i and Fig. 4k–n).
Macroscopic images of kidney organoids after implantation into chick CAM are representative of three independent CAM implantation experiments (Fig. 3b, n = 6 implanted organoids; Fig. 3c, n = 10 implanted organoids; Fig. 3d, n = 3 implanted organoids after dextran–FITC injection).
For supplementary figures, complete information on the number of independent experiments and samples analysed is provided in the corresponding figure legends.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
MATLAB codes can be requested from the corresponding author.
Data availability
RNA-Seq data are publicly available in Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) under the accession numbers GSE108349, GSE108350 and GSE108351. All remaining datasets supporting the findings described here are available within the article and its supplementary information files. Additionally, data are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgements
We are grateful to members of the N. Montserrat laboratory for insightful discussions and critical reading of the manuscript. We thank D. O’Keefe and M. Schwarz for administrative help, L. Bardia, A. Lladó and J. Colombelli from the Advanced Digital Microscopy facility at the Institute for Research in Biomedicine for assistance in confocal microscopy imaging and the Electron Cryo-Microscopy Unit at the Scientific and Technological Centers of the University of Barcelona for their technical assistance. We would particularly like to acknowledge the patients and the Fetal Tissue Bank of Vall d’Hebron University Hospital Biobank (PT13/0010/0021), part of the Spanish National Biobanks Network, for its collaboration. This work has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (StG-2014-640525_REGMAMKID to E.G., P.P., C.T. and N.M. and CoG-616480 to X.T.), the European Commission (project H2020-FETPROACT-01-2016-731957 to X.T. and P.R.-C.), the Spanish Ministry of Economy and Competitiveness/FEDER (BFU2016-77498-P to L.F. and E.M., BFU2015-65074 to X.T., BFU2016-79916-P to P.R.-C., SAF2015-72617-EXP to N.M., SAF2017-89782-R to N.M. and RYC-2014-16242 to N.M.), the Generalitat de Catalunya and CERCA programme (2014-SGR-927 to X.T. and 2017 SGR 1306 to N.M.), Asociación Española contra el Cáncer (AECC CI2016 to L.F. and E.M., LABAE16006 to N.M.). R.O. is supported by an FI fellowship (Generalitat de Catalunya). P.R.-C. is also supported by Obra Social La Caixa. J.C.I.B. is supported by the G. Harold and Leila Y. Mathers Charitable Foundation, the Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED002), the Moxie Foundation, the National Institutes of Health (5R21AG055938), the Universidad Católica San Antonio de Murcia and Fundación Dr. Pedro Guillén. C.H.P. is supported by the Bioengineering Excellence of Scientific Training project, cofunded from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 712754 and from the Spanish Ministry of Economy and Competitiveness under the Severo Ochoa grant SEV-2014-0425 (2015-2019). N.M. is also supported by CardioCel (TerCel, Instituto de Salud Carlos III). IBEC is the recipient of a Severo Ochoa Award of Excellence from MINECO.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41563-019-0287-6.
References
- 1.Taguchi A. et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14, 53–67 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Lam AQ et al. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J Am Soc Nephrol. 25, 1211–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morizane R. et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol 33, 1193–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freedman BS et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun 6, 8715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toyohara T. et al. Cell therapy using human induced pluripotent stem cell-derived tenal progenitors ameliorates acute kidney injury in mice. Stem Cells Transl. Med 4, 980–992 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imberti B. et al. Renal progenitors derived from human iPSCs engraft and restore function in a mouse model of acute kidney injury. Sci. Rep 5, 8826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia Y. et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat. Cell Biol 15, 1507–1515 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Takasato M. et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol 16, 118–126 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Takasato M. et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Taguchi A, & Nishinakamura R. Higher-order kidney organogenesis from pluripotent stem cells. Cell Stem Cell 21, 730–746 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Musah S. et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip.Nat. Biomed. Eng 1, 0069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz NM et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater 16, 1112–1119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roost MS et al. KeyGenes, a tool to probe tissue differentiation using a human fetal transcriptional atlas. Stem Cell Rep. 4, 1112–1124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindström NO et al. Conserved and divergent features of human and mouse kidney organogenesis. J. Am. Soc. Nephrol 29(3), 785–805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Unbekandt M. & Davies JA Dissociation of embryonic kidneys followed by reaggregation allows the formation of renal tissues. Kidney Int.77, 407–416 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Davies JA, Unbekandt M, Ineson J, Lusis M. & Little MH Dissociation of embryonic kidney followed by re-aggregation as a method for chimeric analysis. Methods Mol. Biol 886, 135–146 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Lindström NO et al. Integrated β-catenin, BMP, PTEN, and Notch signalling patterns the nephron. eLife 3, e04000 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Bock K. et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Narayanan K. et al. Human embryonic stem cells differentiate into functional renal proximal tubular-like cells. Kidney Int. 83, 593–603 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Sharmin S. et al. Human induced pluripotent stem cell-derived podocytes mature into vascularized glomeruli upon experimental transplantation. J. Am. Soc. Nephrol 27, 1778–1791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van den Berg CW et al. Renal subcapsular transplantation of PSC-derived kidney organoids induces neo-vasculogenesis and significant glomerular and tubular maturation in vivo. Stem Cell Rep. 10, 751–765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribatti D. Chick embryo chorioallantoic membrane as a useful tool to study angiogenesis. Int. Rev. Cell Mol. Biol 270, 181–224 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Cimpean AM, Ribatti D. & Raica M. The chick embryo chorioallantoic membrane as a model to study tumor metastasis. Angiogenesis 11, 311–319 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Baiguera S, Macchiarini P. & Ribatti D. Chorioallantoic membrane for in vivo investigation of tissue-engineered construct biocompatibility. J. Biomed. Mater. Res. B 100, 1425–1434 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Vining KH & Mooney DJ Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol 18, 728–742 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Przybyla L, Lakins JN & Weaver VM Tissue mechanics orchestrate Wnt-dependent human embryonic stem cell differentiation. Cell Stem Cell 19, 462–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed K. et al. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PLoS One 5, e10531 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theunissen TW & Jaenisch R. Mechanisms of gene regulation in human embryos and pluripotent stem cells. Development 144, 4496–4509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrews S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
- 30.Jiang H, & Lei R. & Ding SW & Zhu S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 15, 182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love MI, Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leek JT Svaseq: removing batch effects and other unwanted noise from sequencing data. Nucl. Acids Res 42, e161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kue CS, Tan KY, Lam ML & Lee HB Chick embryo chorioallantoic membrane (CAM): an alternative predictive model in acute toxicological studies for anti-cancer drugs. Exp. Anim 64, 129–138 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee D, Rahman MM, Zhou Y. & Ryu S. Three-dimensional confocal microscopy indentation method for hydrogel elasticity measurement. Langmuir 31, 9684–9693 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Yeung T. et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskelet 60, 24–34 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Przybyla L, Lakins JN, Sunyer R, Trepat X. & Weaver VM Monitoring developmental force distributions in reconstituted embryonic epithelia. Methods 94, 101–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montserrat N. et al. Generation of induced pluripotent stem cells from human renal proximal tubular cells with only two transcription factors, OCT4 and SOX2. J. Biol. Chem 287, 24131–24138 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Rahilly R. & Müller F. Developmental stages in human embryos: revised and new measurements. Cells Tissues Organs 192, 73–84 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data are publicly available in Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) under the accession numbers GSE108349, GSE108350 and GSE108351. All remaining datasets supporting the findings described here are available within the article and its supplementary information files. Additionally, data are available from the corresponding author upon reasonable request.