

Abstract
Painful peripheral neuropathy is a common neurological complication associated with human immunodeficiency virus (HIV) infection and anti-retroviral therapy. We characterized the impact of two CB2 cannabinoid agonists (AM1710 and LY2828360 – ligands differing in signaling bias and CNS penetration) on neuropathic nociception induced by the antiretroviral agent Zalcitabine (2’-3’-dideoxycitidine; ddC). We also used a conditional knockout approach to identify cell types mediating CB2 agonist-induced antinociceptive efficacy and sparing of morphine tolerance. AM1710 and LY2828360 alleviated ddC-induced neuropathic nociception in mice of both sexes. These benefits were absent in global CB2 knockout mice, which exhibited robust morphine antinociception. Like morphine, AM1710 blunted ddC-induced increases in proinflammatory cytokine (IL-1β, TNF-α) and chemokine (CCL2) mRNA expression levels. We generated advillinCre/+;CB2f/f conditional knockout mice to ascertain the role of CB2 localized to primary sensory neurons in CB2-mediated therapeutic effects. Antinociceptive efficacy of both AM1710 and LY2828360, but not reference analgesics, were absent in advillinCre/+;CB2f/f mice, which exhibited robust ddC-induced neuropathy. In ddC-treated CB2f/f mice, LY2828360 suppressed development of morphine tolerance and reversed established morphine tolerance, albeit with greater efficacy in male compared to female mice. LY2828360 failed to block or reverse morphine tolerance in advillinCre/+;CB2f/f mice. The present studies indicate that CB2 activation may alleviate HIV-associated antiretroviral neuropathy and identify a previously unreported mechanism through which CB2 activation produces antinociceptive efficacy. Our results also provide the first evidence that a CB2 agonist can reverse established morphine tolerance and demonstrate that CB2 localized to peripheral sensory neurons mediates the opioid tolerance sparing efficacy of CB2 agonists.
Keywords: CB2, dorsal root ganglia, neuropathic pain, HIV, opioid tolerance
Graphical Abstract
1. Introduction
In humans living with human immunodeficiency virus (HIV) infection, the development of a painful peripheral neuropathy is the most common neurological complication, with prevalence rates ranging from 20-90% (1). HIV-associated distal sensory polyneuropathy is thought to arise from two primary causes, neuropathic pain associated with toxic viral protein byproducts secreted during viral replication, or antiretroviral toxic neuropathy (ATN) caused by drugs such as the nucleoside reverse transcriptase inhibitor Zalcitabine (2’-3’-dideoxycitidine; ddC) that have been widely used to combat HIV infection. Clinically, neuropathic pain syndromes originating from HIV infection and the use of antiretroviral drugs are indistinguishable from one another (2). The pathological mechanisms surrounding the development of peripheral neuropathy as a sequela to HIV-infection remain poorly understood and are likely to arise from a complex array of factors acting in concert (2). Perineural application of toxic viral protein byproducts like envelope glycoprotein 120 (gp120) (3) fail to recapitulate the pathological signs observed in human peripheral sensory neurons including demyelination, axonal degeneration and apoptosis of dorsal root ganglia (DRG) cells (4-6), possibly due to the localized nature of gp120 exposure in rodent models versus chronic, systemic distribution of gp120 in human HIV patients. However, preclinical models of antiretroviral-induced neuropathy produce pathological signs observed clinically such as a loss of intraepidermal nerve fibers (7), along a time course that parallels the development of symptoms observed clinically (8). Therefore, due to the complexity of modeling an essentially human disease in rodents, and the correspondence between rodent and clinical antiretroviral-induced neuropathies, the present studies relied on toxic neuropathy induced by ddC to model HIV-associated antiretroviral induced neuropathic pain. Currently available pharmacotherapies used for the management of HIV-associated sensory neuropathy fail to provide adequate symptom relief (9), indicating a need for novel treatment strategies in managing HIV-associated painful neuropathy.
The endocannabinoid system has emerged as a therapeutic target for the management of HIV-associated sensory neuropathy, with several reports suggesting that cannabis alleviates pain symptoms in HIV-affected individuals (10-12). Components of cannabis which target cannabinoid type 1 receptors (CB1), such as Δ9-tetrahydrocannabinol (THC), promote pain relief but also produce problematic side effects (13). Cannabinoid type 2 receptor (CB2) agonists alleviate various types of pathological pain without producing unwanted side effects associated with CB1 receptor activation (14, 15). However, therapeutic efficacy of targeting CB2 to alleviate anti-retroviral toxic neuropathy remains largely unexplored. To date, a single report has suggested that the phytocannabinoid β-caryophyllene, which acts as a CB2 agonist, among other targets, effectively alleviates antiretroviral-associated neuropathic pain (16). However, the mechanisms by which CB2 agonists produce antinociception remain poorly understood.
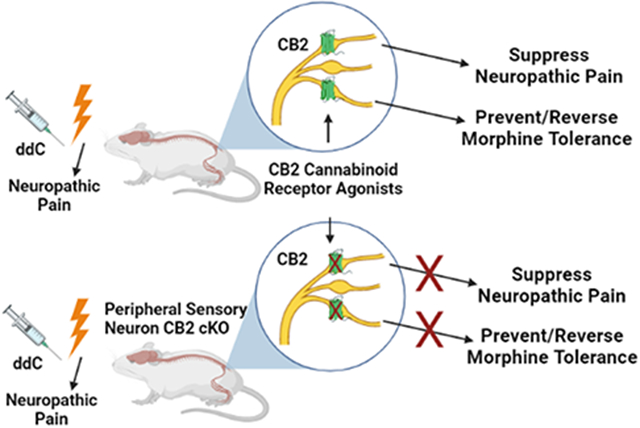
Identification of cell types expressing CB2 that mediate the therapeutic benefits of CB2 ligands has remained problematic given the lack of reliable antibodies for CB2 and low levels of CB2 expression in central nervous system (17-19). Therefore, the present studies sought to characterize the impact of CB2 receptor agonists on ddC-evoked neuropathic nociception and probe the cell types governing the antinociceptive effects of CB2 receptor agonists using mice of both sexes. To accomplish this objective, we used two structurally distinct CB2 agonists (LY2828360 and AM1710) that differ in their signaling profiles and their ability to penetrate the CNS; LY2828360 is a G-protein biased agonist that signals slowly through adenyl cyclase and readily penetrates the CNS (20, 21), whereas AM1710 is a balanced CB2 agonist that does not couple to calcium channels and exhibits limited CNS availability (22-25). To circumvent CB2 antibody specificity issues, the current studies employed a recently developed CB2 green fluorescent reporter mouse (GFP) reporter mouse (CB2f/f) together with a ddC-induced toxic neuropathy model to aid in identification of cell types within nociceptive circuitry which express CB2 (26, 27). In this mouse line, the entire cnr2 coding region is flanked by loxP sites, allowing for cell-type specific deletion of CB2 from cells expressing cre recombinase. To examine the contribution of peripheral sensory neuronal populations to the antinociceptive efficacy of CB2 agonists in ddC neuropathy, we used an advillinCre driver mouse line (28) to generate conditional knockout (cKO) mice in which CB2 was selectively removed from peripheral sensory neurons (advillinCre/+;CB2f/f). As μ opioid receptors (MOR) present on primary afferent nociceptors are responsible for the development of tolerance to the antinociceptive efficacy of morphine (29), the present studies also sought to determine whether primary sensory CB2 expression contributes to the tolerance sparing effects of CB2 agonists.
2. Materials and Methods
2.1. Subjects
Adult (approximately 3 months old at the start of experiments) male and female C57Bl6/J mice were purchased from Jackson labs (Bar Harbor, ME). Adult (3-5 months old at start of experiments) CB2 knockout mice, advillinCre/+;CB2f/f and CB2f/f GFP reporter mice were bred at Indiana University on a C57Bl6/j background.
2.2. Drugs
2’-3’-dideoxycitidine (ddC) was purchased from Sigma Aldrich (St. Louis, MO). AM1710 was synthesized by the lab of Alexandros Makriyannis (Northeastern University, Boston, MA). LY2828360 was synthesized by Sai Life Sciences Ltd. (Hyderabad, India) for use in all experiments with the exception of experiment 1. LY2828360 used in experiment 1 was provided by Eli Lilly (Indianapolis, IN) and used to validate results of LY2828360 synthesis from Sai Life Sciences Ltd. Morphine sulfate was provided by the NIDA Drug Supply Program (Bethesda, MD). ddC was dissolved in 0.9% sterile saline (Baxter Healthcare Corporation, Deerfield, IL). All other compounds were dissolved in a vehicle consisting of 3% DMSO, and the remaining 97% consisting of ethanol, emulphor, and 0.9% saline at a ratio of 1:1:18 respectively. All compounds were delivered at a volume of 5 ml/kg via intraperitoneal (i.p.) injection 30 minutes before behavioral testing occurred.
2.3. Behavioral hypersensitivity testing
Paw withdrawal thresholds to mechanical stimulation were measured using an electronic von Frey anesthesiometer (IITC, Woodland Hills, CA) attached to a semiflexible plastic tip as described previously (20, 27, 30-32). Animals were placed on a mesh table under Plexiglas chambers and allowed to habituate to the environment for 30 minutes prior to the onset of behavioral testing. Mechanical stimulation was applied to the midplantar surface of the hind paw and the amount of force in grams necessary to provoke a withdrawal response was recorded in duplicate for each paw. Cold allodynia was assessed by applying a droplet of acetone to the midplantar surface of the hind paw and measuring the amount of time spent attending to the stimulated paw (i.e. licking, shaking, and lifting of the paw) in triplicate for each paw (20, 27, 30-32).
2.4. General experimental procedures
ddC was administered 3 times per week for a total of 3 weeks at 25 mg/kg, i.p (cumulative dose: 225 mg/kg i.p. over 3 weeks) as described previously (3, 7). For all experiments, pharmacological testing began on day 14 after the initiation of ddC treatments when mechanical and cold allodynia were maximal and stable. Within-subjects ascending dose response curves were generated allowing four days to pass between each pharmacological treatment. In experiment 1, the development of ddC-induced mechanical and cold allodynia was measured three times per week (Monday, Wednesday, Friday). In all subsequent experiments, behavioral responsiveness during the development of ddC-induced pain was measured every 4 days. In the experiment utilizing CB2 KO mice, male and female CB2 KO mice were treated with a single injection of AM1710 or LY2828360. Then, 24 h later, the same subjects received a single i.p. injection of morphine. In chronic dosing experiments, mice received once daily injections of AM1710, LY2828360 or morphine beginning on day 14 of ddC treatment. The time course of acute drug treatments was determined on day one of chronic dosing. In experiments examining the ability of LY2828360 to prevent or reverse morphine tolerance, CB2f/f and advillinCre/+;CB2f/f mice received once daily injections of morphine (10mg/kg, i.p.) or vehicle, in combination with LY2828360 (0.03 mg/kg or 0.1 mg/kg, i.p.) or vehicle for the co-administration dosing schedule. For the reversal dosing schedule, mice received once daily injections of morphine (10 mg/kg, i.p.) or vehicle for all 12 days of chronic dosing, in combination with vehicle for days 1-6, followed by LY2828360 (0.03 mg/kg or 0.1 mg/kg) or vehicle, on days 7-12 of administration. For all conditions, mice were tested every three days to evaluate the development of tolerance, and behavioral testing began 30 minutes after the injection of the pharmacological treatments. For all experiments, experimenters were blinded to genotype and treatment conditions.
2.5. Tissue preparation for immunohistochemistry/methods for immunohistochemistry
Male and female CB2f/f GFP reporter mice used for immunohistochemical experiments received ddC (25 mg/kg i.p./day 3x/week) for a total of 2 weeks and were sacrificed on day 14 following the onset of treatments to coincide with the time point used to initiate pharmacological treatments in all other experiments. Tissue was harvested approximately 48 hours after last ddC injection. Mice were deeply anesthetized with isoflurane and then transcardially perfused with 0.1% heparinized 0.1M phosphate-buffered saline (PBS) followed by ice cold 4% paraformaldehyde. Spleens, lumbar spinal cords, and brains were extracted and kept in the same fixative for 24 hours and then cryoprotected in 30% sucrose for 3 days prior to sectioning. Lumbar L3-L6 DRG were extracted, kept in the same fixative for 4 hours and cryoprotected in 30% sucrose overnight prior to sectioning. Brains, spleens, and spinal cords were cryosectioned at 30 μm and maintained in an antifreeze solution (50% sucrose in ethylene glycol and 0.1M PBS) prior to immunostaining. Free floating sections were washed in 0.1 M PBS, blocked with buffer consisting of 5% donkey serum and 0.1% Triton X-100 in 0.1 M PBS, then incubated overnight in goat polyclonal antibody to GFP (AB0020-200, 1:1000, Sicgen, Cantenhede, Portugal) at 4° C. Sections were then incubated in donkey anti-goat AlexaFluor 594 (1:500, ThermoFisher, Waltham, MA) counterstained with DAPI (0.1 μg/mL) and treated with Sudan black B (0.2% weight/volume, Sigma Aldrich, St Louis, MO). DRG were cryosectioned at 12 μm, mounted on gelatin-subbed slides, and kept at −80° C prior to immunostaining. Slide-mounted immunostaining was carried out using identical procedures as free-floating immunostaining.
2.6. Digital imaging and microscopy
Images were captured using a Leica DM6 B microscope equipped with a Leica DFC9000 GT camera and Leica Application Suite X software (Leica Microsystems, Wetzlar, Germany). No images underwent post-processing or alterations of any sort.
2.7. RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Naïve mice were deeply anesthetized with isoflurane, and lumbar spinal cord, DRG, and spleen were dissected and flash frozen in isopentane and stored at −80 °C until use. Total RNA was purified using TRIzol™ (Thermofisher, Waltham, MA) and RNeasy mini kit from lumbar spinal cord, spleen, and DRG according to manufacturer’s manual. Quantification of total mRNA was assessed by using a NanoDrop 2000C UV-Vis spectrophotometer at 260 nm.
One-step RT-qPCR was performed using the Luna universal One-Step RT-qPCR kit (New England BioLabs) in a total volume of 20 μl and a template concentration of 50 ng/μl according to the manufacturer’s recommendations. Thermal cycle conditions were 55 °C for 10 minutes (RT step), 95 °C for 1 minute, followed by 40 cycles of 95 °C for 10 seconds and 60 °C for 1 minute. A melting curve analysis was performed at 95 °C for 15 seconds, 60 °C for 1 minute, 95 °C for 15 seconds following every run to ensure a single amplified product for each reaction. All reactions were performed in duplicates in 96 well reaction plates (Thermofisher, Waltham, MA). Primer sequences are listed as follows:
-
Forward GAPDH/Reverse GAPDH:
GGGAAGCTCACTGGCATGGC/GGTCCACCACCCTGTTGCT
-
Forward CB2/Reverse CB2:
CTCGGTTACAGAAACAGAGGCTGATGTG/TCTCTCTTCGAGGGAGTGAACTGAACG
-
Forward GFP/Reverse GFP:
ACATGGTCCTGCTGGAGTTCGTGAC/CTCTTCGAGGGAGTGAACTGAACGG
For experiments evaluating impact of ddC and pharmacological treatments of cytokine/chemokine mRNA expression levels, RT-qPCR was performed using Taqman RNA-to-CT 1-step kit (Thermo fisher scientific, Waltham, MA) in a total volume of 10 μl and a template concentration of 50 ng/μl according to manufacturer’s recommendations. Thermal cycle conditions were 48 °C for 30 minutes (RT step), 95 °C for 10 minutes, followed 40 cycles of by 95 °C for 15 seconds, and 60C °C of 1 minute. A melting curve analysis was performed at 95 °C for 15 second, 60 °C for 1 minute, 95 °C for 15 seconds following every run to ensure a single amplified product for every reaction. Taqman gene expression assays used in the experiments were: GAPDH mm9999995-g1; CNR2 m02620087_s1; IL10 mm00439616_m1; IL-1beta mm00434228_m1; CXCL12 mm00445553_m1; CXCR4 mm01996749_s1; TNFα mm00443258_m1; CCL2 mm00441242_m1.
All reactions were performed in duplicate in 394 well reaction plates (Thermofisher Scientific, Waltham, MA). The mRNA expression levels were expressed as relative quantities to control and calculated by 2(−ΔΔCt) method where GAPDH is used as in house reference gene.
2.8. Statistical analyses
Paw withdrawal thresholds and duration of time spent attending to the acetone-stimulated paw were averaged across paws for each modality (mechanical, cold) for each subject on each day to obtain a single data point per subject per observation interval. The time course of the development of ddC-induced neuropathic pain, as well as within-subjects dose-response curves, were analyzed by two-way repeated measures ANOVA followed by Tukey multiple comparisons post-hoc tests to examine differences between groups. Unpaired sample two-tailed t-tests were used for two group comparisons to analyze qPCR data. Statistical analyses of data presented in each figure was performed using Graphpad prism 7 (Graphpad prism software, San Diego, CA) and are summarized in Table 1.
Table 1.
Summary of Statistical Analyses of Data Presented in Figure Panels
| Figure | Time | Drug | Interaction |
|---|---|---|---|
| 1A | mechanical: F1,44 = 386.3, p<0.0001 | mechanical: F6,44 = 94.68, p<0.0001 | mechanical: F6,44 = 31.26, p<0.0001 |
| 1B | cold: F1,44 = 541.6, p<0.0001 | cold: F6,44 = 121, p<0.0001 | cold: F6,44 = 80.17, p<0.0001 |
| 1C | mechanical: F3,18 = 31.59, p<0.0001 | mechanical: F4,18 = 56.66, p<0.0001 | mechanical: F12,18 = 11.51, p<0.0001 |
| 1D | cold: F3,18 = 89.55, p<0.0001 | cold: F4,18 = 80.45, p<0.0001 | cold: F12,18 = 26.57, p<0.0001 |
| 1E | mechanical: F3,18 = 57.32, p<0.0001 | mechanical: F4,18 = 73.81, p<0.0001 | mechanical: F12,18 = 11.45, p<0.0001 |
| 1F | cold: F3,18 = 79.81, p<0.0001 | cold: F4,18 = 60.14, p<0.0001 | cold: F4,19 = 63.68, p<0.0001 |
| 1G | mechanical: F3,19 = 57.44, p<0.0001 | mechanical: F4,19 = 73.71, p<0.0001 | mechanical: F12,19 = 14.15, p<0.0001 |
| 1H | cold: F3,19 = 107.9, p<0.0001 | cold: F4,19 = 63.68, p<0.0001 | cold: F12,19 = 26.22, p<0.0001 |
| Time | Drug | Interaction | |
| 2A | mechanical: F2,66 = 439.8, p<0.0001 | mechanical: F6,66 = 213.8, p<0.0001 | mechanical: F12,66 = 22.75, p<0.0001 |
| 2B | cold: F6,66 = 480.4, p<0.0001 | cold: F6,66 = 418.5, p<0.0001 | cold: F12,66 = 83.78, p<0.0001 |
| AM1710 | LY2828360 | Vehicle | |
| 2C | mechanical: F3,7=252.5, p<0.0001 | mechanical: F3,7=109.1, p<0.0001 | Mechanical: F3,6=297.3, p<0.0001 |
| 2D | cold: F3,7=768.9, p<0.0001 | cold: F3,7=814.4, p<0.0001 | cold: F3,6=967.8, p<0.0001 |
| Figure | Time | Drug | Interaction |
| 3A | mechanical: F5,60 = 186.3, p<0.0001 | mechanical: F3,12 = 38.82, p<0.0001 | mechanical: F15,60 = 16.54, p<0.0001 |
| 3B | cold: F5,60 = 296.4, p<0.0001 | cold: F3,12 = 117, p<0.0001 | cold: F15,60 = 50.94, p<0.0001 |
| 3C | mechanical: F5,60 = 77.09, p<0.0001 | mechanical: F3,12 = 28.09, p<0.0001 | mechanical: F15,60 = 8.735, p<0.0001 |
| 3D | cold: F5,60 = 89.54, p<0.0001 | cold: F3,12 = 46.43, p<0.0001 | cold: F15,60 = 15.45, p<0.0001 |
| 3E | mechanical: F5,60 = 123.4, p<0.0001 | mechanical: F3,12 = 49.93, p<0.0001 | mechanical: F15,60 = 15.48 p<0.0001 |
| 3F | cold: F5,60 = 138.9, p<0.0001 | cold: F3,12 = 87.59, p<0.0001 | cold: F15,60 = 25.93, p<0.0001 |
| 4A | mechanical: F6,72 = 124.6, p<0.0001 | mechanical: F3,12 = 108, p<0.0001 | mechanical: F18,72 = 15.69, p<0.0001 |
| 4B | cold: F6,72 = 247.3, p<0.0001 | cold: F3,12 = 504.6, p<0.0001 | cold: F18,72 = 62.47, p<0.0001 |
| 4C | mechanical: F6,72 = 86.43, p<0.0001 | mechanical: F3,12 = 61.48, p<0.0001 | mechanical: F18,72 = 11.13, p<0.0001 |
| 4D | cold: F6,72 =89.94, p<0.0001 | cold: F3,12 = 407.6, p<0.0001 | cold: F18,72 = 21.65, p<0.0001 |
| 4E | mechanical: F6,72 = 274.6, p<0.0001 | mechanical: F3,12 =33.51, p<0.0001 | mechanical: F18,72 = 18,36, p<0.0001 |
| 4F | cold: F6,72 = 215.9, p<0.0001 | cold: F3,12 = 61, p<0.0001 | cold: F18,72 = 24.04, p<0.0001 |
| Figure 5 | |||
| IL-1β: T5=2.8, p=0.04 | |||
| TNF-α: T5=3.52, p=0.016 | |||
| CCL2: T6=2.5, p=0.047 | |||
| Figure 6 | |||
| 6A | F2,8=5.11, p=0.04 | ||
| 6B | F2,7=8.07, p=0.02 | ||
| 6C | F2,8=4.87, p=0.04 | ||
| 6D | F2,8=5.52, p=0.03 | ||
| 6E | F2,8=7.05, p=0.02 | ||
| 6F | F2,9=2.12, p=0.18 | ||
| 6G | F2,9=4.22, p=0.051 | ||
| 6H | F2,9=2.86, p=0.12 | ||
| 6I | F2,9=3.53, p=0.07 | ||
| 8A | T14=1.6, p=0.13 | ||
| 8B | T13=1.6, p=0.13 | ||
| 8C | T13=2.13, p=0.053 | ||
| 8D | T13=4.73, p=0.0004 | ||
| 8E | T14=10.17, p<0.0001 | ||
| 8F | T12=2.08, p=0.05 | ||
| 9A | T13=4.041, p=0.001 | ||
| 9B | T14=1.611, p=0.13 | ||
| 9C | T13= 0.76, p=0.46 | ||
| 9D | T14=0.82, p=0.43 | ||
| 9E | T13=3.31, p=0.006 | ||
| 9F | T14=1.62, p=0.13 | ||
| 9G | T13=0.44, p=0.67 | ||
| 9H | T14=2.03, p=0.062 | ||
| Figure | Drug | Genotype | Interaction |
| 10A | mechanical: F7,112 = 245.9, p<0.0001 | mechanical: F3,16 = 39.38, p<0.0001 | mechanical: F21,112 = 17.2, p<0.0001 |
| 10B | cold: F7,112 = 384.7, p<0.0001 | cold: F3,16 = 46.6, p<0.0001 | cold: F21,112 = 37.66, p<0.0001 |
| 10C | mechanical: F7,112 = 286.5, p<0.0001 | mechanical: F3,16 = 71.86, p<0.0001 | mechanical: F21,112 = 19.17, p<0.0001 |
| 10D | cold: F7,112 = 414.9, p<0.0001 | cold: F3,16 = 54.81, p<0.0001 | cold: F21,112 = 36.35, p<0.0001 |
| Figure | Within | Between | Interaction |
| 11A | mechanical: F6,192 = 162.2, p<0.0001 | mechanical: F3,32 = 159.2, p<0.0001 | mechanical: F18,192 = 32.51, p<0.0001 |
| 11B | cold: F6,192 = 131.8, p<0.0001 | cold: F3,32 = 174.2, p<0.0001 | cold: F18,192 = 24.9, p<0.0001 |
| 11C | mechanical: F6,126 = 243.3, p<0.0001 | mechanical: F3,27 = 78.5, p<0.0001 | mechanical: F18,162 = 22.11, p<0.0001 |
| 11D | cold: F6,162 = 212.9, p<0.0001 | cold: F3,27 = 139.5, p<0.0001 | cold: F18,162 = 20.42, p<0.0001 |
| 11E | mechanical: F6,168 = 210.6, p<0.0001 | mechanical: F3,28 = 52.67, p<0.0001 | mechanical: F18,168 = 14.68, p<0.0001 |
| 11F | cold: F1,168 = 403.3, p<0.0001 | cold: F3,28 = 111.4, p<0.0001 | cold: F18,168 = 31.51, p<0.0001 |
| 11G | mechanical: F6,156 = 322.4, p<0.0001 | mechanical: F3,26 = 9.351, p<0.001 | mechanical: F18,156 = 4.47, p<0.0001 |
| 11H | cold: F6,156 = 618.3, p<0.0001 | cold: F3,26 = 37.18, p<0.0001 | cold: F18,156 = 15.85, p<0.0001 |
| Figure | Within | Between | Interaction |
| 12A | mechanical: F6,180 = 201, p<0.0001 | mechanical: F3,30 = 90.69, p<0.0001 | mechanical: F18,180 = 32.5, p<0.0001 |
| 12B | cold: F6,180 = 147, p<0.0001 | cold: F3,30 = 124.5, p<0.0001 | cold: F18,180 = 23.47, p<0.0001 |
| 12C | mechanical: F6,156 = 243.6, p<0.0001 | mechanical: F3,26 = 20.89, p<0.0001 | mechanical: F18,156 = 20.93, p<0.0001 |
| 12D | cold: F6,156 = 221.7, p<0.0001 | cold: F3,26 = 43.37, p<0.0001 | cold: F18,156 = 18.11, p<0.0001 |
| 12E | mechanical: F6,162 = 178.5, p<0.0001 | mechanical: F3,27 = 60.03, p<0.0001 | mechanical: F18,162 = 15.06, p<0.0001 |
| 12F | cold: F6,162 = 362.7, p<0.0001 | cold: F3,27 = 96.56, p<0.0001 | cold: F18,162 = 29.34, p<0.0001 |
| 12G | mechanical: F6,156 = 377.4, p<0.0001 | mechanical: F3,26 = 6.8, p<0.001 | mechanical: F18,156 = 6.281, p<0.0001 |
| 12H | cold: F6,156 = 617.3, p<0.0001 | cold: F3,26 = 26.18, p<0.0001 | cold: F18,156 = 14.87, p<0.0001 |
3. Results
3.1. CB2 Activation alleviates ddC-evoked mechanical and cold allodynia.
Anti-retroviral treatment with ddC (25 mg/kg, i.p. x 3 injections/week x 3 weeks) induced hypersensitivity to mechanical and cold stimulation; this sensitization emerged by the second day after initiation of ddC administration and was stable for at least 14 days (p<0.001 for each time point). Morphine treatment dose dependently alleviated ddC-induced mechanical and cold hypersensitivity. The lowest dose of morphine tested (1 mg/kg, i.p.) produced a partial reversal of ddC-induced mechanical (p=0.02 vs. non-ddC treated mice, Figure 1A) and cold (p<0.001 vs. non-ddC-treated mice, Figure 1B) hypersensitivity, whereas the two highest doses (3 and 10 mg/kg i.p.) completely reversed ddC-induced mechanical and cold hypersensitivity. Treatment with the CB2 agonist AM1710 also alleviated ddC-induced mechanical and cold hypersensitivity. The lowest dose of AM1710 partially reversed ddC-induced mechanical hypersensitivity (1 mg/kg, i.p., p=0.015 vs. non ddC-treated mice, Figure 1C), whereas the highest doses (5 and 10 mg/kg, i.p., p<0.001 vs. ddC-vehicle treated mice, Figure 1C) completely reversed ddC-induced mechanical hypersensitivity. AM1710 produced a full reversal of ddC-induced cold hypersensitivity at all doses evaluated (1, 5 and 10 mg/kg i.p., p<0.001 for all doses vs. ddC-vehicle treated mice). Similarly, treatment with a structurally distinct CB2 agonist, LY2828360, dose-dependently alleviated ddC-induced mechanical hypersensitivity. The lowest dose of LY2828360 (0.3 mg/kg, i.p.) produced a partial reversal of ddC-induced mechanical (p=0.004 vs. non ddC-treated mice) and cold (p=0.048 vs. non-ddC treated mice) allodynia, whereas the higher doses (1 and 3 mg/kg, i.p.; Figure 1G) produced complete reversal of ddC-induced mechanical and cold hypersensitivity (p<0.001 vs. ddC-vehicle treated mice; Figure 1H).
Figure 1. Morphine, AM1710, and LY2828360 alleviate ddC-evoked mechanical and cold allodynia.

ddC induces mechanical and cold hypersensitivity. C57Bl6/J mice treated with ddC displayed mechanical (p<0.001 for all time points, A) and cold (p<0.001 for all time points, B) allodynia on days 2-14 following onset of ddC treatments. N=23 per group. Morphine (A, B), AM1710 (C, D), and LY2828360 (E, F) suppress mechanical and cold allodynia induced by ddC treatment without altering responding in C57Bl6/J mice not treated with ddC. ***p<0.001, **p<0.01, *p<0.05 vs. non ddC-treated mice. $$$p<0.001, $$p<0.01 vs. all other groups. Two-way ANOVA followed by Tukey multiple comparisons test post-hoc N= 5-6 per group. BL: baseline, ddC: 2’-3’-dideoxycitidine, LY: LY02828360, MOR: morphine.
3.2. AM1710 and LY2828360 fail to alleviate ddC-evoked mechanical and cold allodynia in mice lacking CB2 receptors.
CB2 knockout mice (KO) developed ddC-induced mechanical and cold hypersensitivity at levels largely equivalent to wild type (WT) mice. At baseline, CB2 KO mice displayed modestly lower paw withdrawal thresholds relative to WT mice treated with ddC and vehicle (p<0.01; Figure 2A). ddC reduced mechanical paw withdrawal thresholds in both genotypes on days 2-14 (p<0.001 for each time point) following initiation of ddC dosing relative to WT mice treated with vehicle (Figure 2A), consistent with development of mechanical allodynia. CB2 KO mice displayed lower mechanical paw withdrawal thresholds than WT mice treated with ddC at days 2 (p=0.003), 9 (p=0.03), 11 (p=0.002) and 14 (p=0.003) following initiation of ddC dosing (Figure 2A). ddC also elevated acetone-evoked nocifensive behavior in both genotypes on days 2-14 (p<0.001 for each time point; Figure 2B). However, CB2 KO mice displayed elevated levels of cold hypersensitivity on days 2 (p=0.02), 7 (p<0.001) and 14 (p<0.001) relative to WT mice treated with ddC. ddC produced mechanical and cold allodynia in all CB2 KO groups (p<0.0001 for all treatment groups). Most relevant to our studies, in CB2 KO mice, neither LY2828360 (3 mg/kg, i.p. Mechanical: p=0.85, cold: p=0.99) nor AM1710 (10 mg/kg, i.p. Mechanical: p=0.93; cold: p=0.96) altered mechanical or cold responding relative to vehicle (mechanical: p=0.81; cold: p=0.92) treatment (Figure 2C and 2D). By contrast, morphine (10 mg/kg, i.p.) treatment completely reversed ddC evoked mechanical and cold allodynia in all CB2 KO groups (p<0.0001 for all groups; Figure 2C and D).
Figure 2. The antinociceptive effects of AM1710 and LY2828360 are absent in CB2 KO mice.

Neither AM1710 nor LY2828360 alleviate ddC-evoked neuropathic nociception in CB2 KO mice. CB2 KO mice displayed slightly lower mechanical paw withdrawal thresholds at baseline relative to WT mice (p=0.0004). CB2 KO and WT mice treated with ddC displayed lower mechanical paw withdrawal thresholds on days 2-14 post initiation of ddC dosing relative to vehicle-treated mice (p<0.001 for all observations, A). CB2 KO mice displayed lower paw withdrawal thresholds relative to WT mice treated with vehicle on days 2 (p=0.003), 9 (p=0.03), 11 (p=0.002) and 14 (p=0.003). CB2 KO and WT mice treated with ddC display elevated levels of cold hypersensitivity from day 2-14 following onset of ddC treatments (p<0.001 for all observations, B). CB2 KO mice display elevated levels of cold hypersensitivity on days 2 (p=0.02), 7 (p<0.001) and 14 (p<0.001) following onset of ddC treatments. ##p<0.01 vs. CB2 KO, $$$p<0.001, $$p<0.01, $p<0.05 vs. WT ddC, ***p<0.001 vs. all other groups. Two-way ANOVA followed by Tukey multiple comparisons test post-hoc. N= 23 per group. WT: wild type, CB2 KO: CB2 knockout, ddC: 2’-3’-dideoxycitidine. LY2828360 (3 mg/kg, i.p.) and AM1710 (10 mg/kg, i.p.) fail to alleviate ddC-induced mechanical (C) and cold allodynia (D) in CB2 KO mice, whereas morphine (10 mg/kg, i.p.) restores mechanical (C) and cold (D) allodynia to pre-ddC levels in the same mice. +++p<0.001, +p<0.05 vs. baseline. One-way repeated measures ANOVA performed on each drug condition to examine within-subjects effects, Tukey multiple comparisons test post-hoc. N= 7-8 per group. BL: baseline, ddC: 2’-3’-dideoxycitidine, LY: LY2828360, MOR: morphine.
3.3. CB2 agonists produce comparable levels of antinociception in male and female mice.
Male and female C57BlJ/6 mice exhibited comparable levels of ddC-evoked mechanical and cold allodynia on days 2-14 of the testing period (data not shown). All compounds tested showed a similar time course of anti-allodynic effects. AM1710 (10 mg/kg, i.p. Figure 3A,B), LY2828360 (3 mg/kg, i.p. Figure 3C,D) and morphine (10 mg/kg, i.p. Figure 3E,F) all reversed ddC-evoked mechanical and cold allodynia from 0.5-4.5 hours post-administration (p<0.0001 for all comparisons (Figure 3B). In female mice, AM1710 lowered cold responsiveness 24 hours-post injection relative to vehicle (p=0.02; Figure 3B). Male mice showed slightly elevated cold responsiveness at 0.5 (p=0.007), 2.5 (p=0.04), 4.5 (p=0.04), and 24 hours (p=0.03) post vehicle treatment relative to female mice similarly treated with vehicle (Figure 3D). Male mice treated with LY2828360 displayed slightly lower cold responsiveness 24 hours post-administration (p=0.002) relative to female mice treated with vehicle (Figure 3D). Male mice treated with morphine displayed higher mechanical paw withdrawal thresholds 24 hours post-administration relative to female mice similarly treated with morphine (p=0.04; Figure 3E). At 24 hours post-injection, female mice treated with morphine showed lower cold responsiveness relative to female mice treated with vehicle (p=0.0004; Figure 3F). AM1710, LY2828360 and morphine did not alter mechanical or cold responsiveness in the absence of ddC (data not shown).
Figure 3. Time course and comparison of the anti-allodynic efficacy of CB2 agonists in male and female mice.

AM1710 (10 mg/kg, i.p., A,B), LY2828360 (3 mg/kg i.p., C,D) and morphine (10 mg/kg, i.p., E,F) alleviated ddC evoked mechanical and cold hypersensitivity at comparable levels in male and female C57BlJ/6 mice at 0.5, 2.5 and 4.5 hours post-administration relative to ddC-treated mice receiving vehicle. ###p<0.0001 all groups vs. baseline. ***p<0.0001 vs. veh-treated male and female, $$p<0.01, $p<0.05 vs. female veh, &p<0.05 female AM1710 vs. female veh, ~p<0.05 male MOR vs. female MOR, %%%p<0.001 female MOR vs. female veh. Two-way ANOVA, Tukey multiple comparisons test post-hoc. (N=4 per group).
3.4. CB2 agonists, but not morphine, display sustained anti-allodynic efficacy with chronic dosing.
AM1710 (10mg/kg, i.p. A,B) and LY2828360 (3 mg/kg, i.p. C,D) produced sustained antinociceptive efficacy, at similar levels between male and female C57BlJ/6 mice, alleviating ddC-induced mechanical and cold hypersensitivity throughout the 12 day chronic dosing period (p<0.0001 for all time points) relative to same sex mice treated with vehicle. In comparison, the antinociceptive effects of morphine were largely absent after 6 days of repeated injections (Figure 4E,F). (A) Male mice treated with AM1710 displayed higher mechanical paw withdrawal thresholds on day 3 of administration (p=0.01) relative to female mice treated with AM1710. (Figure 4B) Female mice treated with vehicle displayed transient increases in cold responsiveness relative to male mice similarly treated with vehicle on days 1 (p=0.0007) and 12 of injection (p=0.02). Male mice receiving LY2828360 displayed higher mechanical paw withdrawal thresholds on day 9 of chronic dosing (p=0.002) relative to female mice similarly treated with LY2828360 (Figure 4C). ddC-treated male mice displayed lower cold responsiveness relative to ddC-treated female mice receiving either vehicle or LY2828360 (p=0.02) (Figure 4D). Morphine alleviated ddC-induced mechanical hypersensitivity on day 1 (p<0.0001), 3 (p<0.0001) and 6 (p=0.03) of administration relative to vehicle treatment (Figure 4E). ddC-treated male mice treated with morphine displayed slightly higher mechanical withdrawal thresholds on day 9 of daily dosing relative to female ddC-treated mice receiving vehicle (p=0.02, Figure 4E). Morphine alleviated ddC-evoked cold hypersensitivity, at comparable rates between male and female mice, on days 1 (p<0.0001), 3 (p<0.0001) and 6 (p=0.001) of administration. Male mice showed lower cold responsiveness relative to male and female mice treated with vehicle on day 9 (p=0.03) and female mice treated with vehicle (p=0.006) on day 12 of administration. AM1710, LY2828360 and morphine did not alter mechanical withdrawal thresholds or cold responsiveness in mice treated with vehicle in lieu of ddC (data not shown).
Figure 4. CB2 agonists display sustained antinociceptive efficacy with repeated dosing whereas opioid analgesics do not.

AM1710 (10 mg/kg/day, i.p., A,B) and LY2828360 (3 mg/kg/day, i.p., C,D) produce sustained efficacy following chronic administration at comparable levels in male and female C57BlJ/6 mice. Tolerance develops to the pain relieving effects of morphine (10 mg/kg/day, i.p., E,F). ###p<0.0001 vs. baseline, ***p<0.0001 vs. veh-treated male and female, &&&p<0.0001, &p<0.05 female veh vs. male veh, ^^p<0.01 male LY2828360 vs. female LY2828360, $p<0.05 male MOR vs. female veh, ~p<0.05 male MOR vs. veh, @@p<0.01 male MOR vs. female veh. Two-way ANOVA, Tukey multiple comparisons test post-hoc. (N=4 per group).
3.5. Impact of AM1710, Morphine and LY on mRNA expression levels of cytokines and chemokines in lumbar spinal cord.
To identify the effect of toxic challenge with ddC (25 mg/kg i.p., administered 3 times a week for 3 weeks) at the molecular level at the level of the central nervous system, we compared mRNA expression levels of a battery of genes in lumbar spinal cords derived from C57BlJ/6 mice receiving either ddC or saline at the same times. Lumbar spinal cord mRNA expression levels of IL-1β (p=0.04), TNF-α (p=0.016) and CCL2 (i.e. MCP-1; p=0.047) were increased in mice that received ddC relative to vehicle injection (Figure 5). By contrast, CNR2 (p=0.8768), IL-10 (p=0.310), CXCL12 (p=0.991) and CXCR4 (p=0.599) mRNA expression levels were not altered by ddC treatment. We further analyzed IL-1β, TNF-α, and CCL2 mRNA expression levels in lumbar spinal cords derived from ddC-treated mice chronically treated (1x/ day x 12 days, i.p.) with AM1710 (10 mg/kg), morphine (10 mg/kg) or LY2828360 (3 mg/kg). AM1710 decreased mRNA expression levels of IL-1β (p=0.057, Figure 6A), TNF-α (p=0.016, Figure 6B) and CCL2 (p=0.031, Figure 6C), cytokines and chemokines that were elevated by ddC treatment. Morphine reduced mRNA expression levels of IL-1β (p=0.0278, Figure 6D) and TNF-α (p=0.0349, Figure 6E) but not CCL2 (Figure 6F) in ddC-treated mice. LY2828360 did not alter IL-1β (p=0.963, Figure 6G), TNF-α (p=0.363, Figure 6H) or CCL2 (p=0.085, Figure 6I) mRNA expression levels in ddC-treated mice at the same timepoint.
Figure 5. Impact of toxic challenge with ddC on CB2, cytokine and chemokine mRNA expression levels in lumbar spinal cord derived from male wildtype mice.

ddC treatment produced an upregulation of IL-1β, TNF-α, and CCL2 in the lumbar spinal cord compared with the saline group similarly injected with vehicle (i.p.). Values were calculated using the 2(−ΔΔCt) method. GAPDH was used as reference gene. Data are expressed as mean ± S.E.M. (n=4 per group). * p<0.05 vs. vehicle, unpaired sample two tailed t-test.
Figure 6. Impact of ddC and systemic (i.p.) administration of AM1710, morphine and LY2828360 on expression levels of IL-1β, TNF-α and CCL2 mRNAs in lumbar spinal cord derived from male wildtype mice.

(A, B, C) AM1710 blunts ddC-evoked increases in IL-1β, TNF-α and CCL2 mRNAs, morphine blunts ddC-induced increases in IL-1β, TNF-α mRNA’s (Mor; D,E,F), whereas LY2828360 fails to alter ddC-induced increases in IL-1β, TNF-α and CCL2 mRNAs (LY; G, H, I). ddC treatment (Veh ddC) induced upregulation of IL-1β, TNF-α and CCL2 mRNAs compared to saline treated groups that were similarly injected with vehicle (Veh-Veh). Values were calculated using the 2(−ΔΔCt) method with GAPDH was used as reference gene and all groups compared to the Veh Veh group. Data are expressed as mean ± S.E.M. (n=4 per group). * p<0.05, one-way ANOVA.
3.6. Immunohistochemical mapping of CB2/GFP receptor expression in CB2f/f GFP reporter mice.
Next, we performed immunohistochemical experiments to localize GFP protein expression throughout the brain, spinal cord, DRG, and spleen of male and female CB2f/f GFP mice; these mice were sacrificed 14 days after initiation of treatment with ddC or vehicle. As expected, robust anti-GFP immunoreactivity was observed in the spleens of male and female CB2f/f GFP mice treated with ddC or vehicle (Figure 7 C,F). Direct GFP fluorescence was below detection threshold in all tissues under conditions in which Sudan Black treatment was used to quench autofluorescence (Figure 7 B,E). In contrast to the robust anti-GFP immunoreactivity we observed in spleen, GFP and anti-GFP immunoreactivity were below detection threshold in the lumbar spinal cord, brain, and lumbar L3-6 DRG of mice treated with vehicle or ddC (data not shown). Anti-GFP immunostaining was also carried out using wild type C57BL6/J mice as negative controls. As expected, anti-GFP immunoreactivity was absent in the spleens, spinal cord, and brains of WT mice processed under identical conditions (data not shown).
Figure 7. Anti-GFP-like immunoreactivity is detectable in the spleens of CB2f/f mice.

Robust anti-GFP-like immunoreactivity was located in the spleens of CB2f/f mice treated with ddC (A,C) or vehicle (D,F). Sudan black treatment quenched native GFP expression (B, E). Scale bar equal to 1 mm.
3.7. Identification of CB2 and GFP mRNA within nociceptive circuitry.
To circumvent possible concerns that limited sensitivity of IHC may mask low levels of protein expression and that chemical methods used to quench (auto)fluorescence in the tissues examined, we conducted qRT-PCR experiments to determine whether CB2 and GFP mRNA was present in spleen, spinal cord and DRG of CB2f/f and wild type C57BL6/j mice. CB2 mRNA was detectable in the spleens, lumbar spinal cord and DRG at comparable levels in CB2f/f and WT C57BlJ/6 mice. Moreover, GFP mRNA was present at very high levels in the spleen, and at lower levels in both lumbar spinal cord and DRG of CB2f/f mice (Figure 8 D-F). However, as expected, GFP mRNA was not reliably detected in spleen, spinal cord and DRG of WT mice lacking GFP (Figure 8 D-F).
Figure 8. CB2 and GFP mRNA are detectable within nociceptive circuitry.

CB2 and GFP mRNAs are detectable in tissues implicated in nociceptive processing. CB2 mRNA was present at comparable levels between CB2f/f and wild type C57BlJ/6 mice in the spleen, spinal cord and DRG (A-C). GFP mRNA was detectable in the spleen, spinal cord and DRG of CB2f/f mice (D-F). (N=6 for wildtype group, N=11 for CB2f/f, animals used for this study were age matched between 90 days 150 days. Values were calculated using the 2(−ΔΔCt) method. GAPDH was used as reference gene. In WT tissues, CT values for GFP were typically undefined and assigned an arbitrary CT value of 37 for comparison with values in reporter mice. Data are expressed as mean ± S.E.M. **p < 0.01, ***p<0.001, unpaired sample two tailed t-test.
3.8. Comparison of mRNA levels of CB2 and GFP in DRG, paw skin, lumbar spinal cord and spleen derived from advillinCre/+;CB2f/f and advillinCre/−;CB2f/f mice.
To ascertain whether primary sensory neuron CB2 receptors mediated anti-allodynic effects of CB2 agonists, we generated advillinCre/+;CB2f/f conditional knockout mice. CB2 receptors were selectively deleted from peripheral sensory neurons by crossing CB2f/f mice with advillin+/cre mice (28). In DRG, CB2 mRNA expression levels were lower in advillinCre/+;CB2f/f compared to advillinCre/−;CB2f/f mice (p=0.0014,Figure 9A). GFP mRNA was still detectable in DRG of advillinCre/+;CB2f/f mice albeit at lower levels than that observed in advillinCre/−;CB2f/f mice (p=0.0056, Figure 9E). By contrast, CB2 mRNA expression levels did not differ between advillinCre/+;CB2f/f and advillinCre/−;CB2f/f mice in paw skin (Figure 9B), lumbar spinal cord (Figure 9C) or spleen (Figure 9D), indicating that insertion of GFP and deletion of CB2 gene in DRG did not alter the basal expression of CB2 in these tissues. As expected, GFP mRNA was detected in the paw skin, lumbar spinal cord and spleen of both advillinCre/−;CB2f/f and advillinCre/+;CB2f/f mice (Figure 9F, 9G, 9H). In addition, GFP mRNA was expressed at comparable level in paw skin, lumbar spinal cord and spleen of the advillinCre/−;CB2f/f and advillinCre/+;CB2f/f mice. Thus, advillinCre/+;CB2f/f mice showed a selective deletion of CB2 and its EGFP reporter in DRG, but not in other tissues (spleen, spinal cord, paw skin), documenting the selectivity of our cKO mouse for deletion of primary sensory neuron CB2 expression levels.
Figure 9. CB2 and GFP mRNA expression patterns in various tissues derived from advillinCre/+;CB2f/f mice and CB2f/f mice.

Comparisons of (A, B, C, D) CB2 mRNA and (E, F, G,H) GFP mRNA in (A, B) DRG, (C, D) paw skin, (E, F) lumbar spinal cord and (G,H) spleen between advillinCre/+;CB2f/f mice and CB2f/f mice (n=6-10/group). AdvillinCre/+;CB2f/f mice display lower levels of CB2 and GFP mRNA in lumbar DRG relative to CB2f/f mice, but no difference in expression levels between genotypes for either mRNA species in paw skin, spinal cord and spleen. Values were calculated using the 2(−ΔΔCt) method. GAPDH was used as reference gene. Data are expressed as mean ± S.E.M. **p < 0.01, unpaired sample two tailed t-test.
3.9. CB2 agonists fail to alleviate ddC-evoked mechanical and cold allodynia in advillinCre/+;CB2f/f mice.
Next, we assessed the impact of removing CB2 receptors from peripheral sensory neurons on the antinociceptive efficacy of LY2828360 and AM1710. Both AM1710 (Figure 10A,B) and LY2828360 (Figure 10C,D) alleviate ddC-evoked mechanical and cold hypersensitivity in male and female CB2f/f GFP mice. Specifically, AM1710 decreased ddC-evoked mechanical (Figure 10A) and cold (Figure 10B) hypersensitivity at 1 mg/kg (p<0.002 for all comparisons), 5 mg/kg (p<0.0001 for all comparisons), and 10 mg/kg (p<0.0001 for all comparisons), but not at 0.1 mg/kg (p>0.9 for all comparison). By contrast, AM1710 failed to alter mechanical or cold hypersensitivity in advillinCre/+;CB2f/f mice at any dose (p>0.2 for all comparisons; Figure 10A,B). In the same subjects, morphine (10 mg/kg, i.p.) and gabapentin (50 mg/kg, i.p.) reversed ddC-induced mechanical and cold allodynia in all groups (p<0.0001 for all comparisons; Figure 10A,B). Similarly, LY2828360 decreased ddC-evoked mechanical (Figure 10C) and cold (Figure 10D) allodynia in CB2f/f mice. In male and female CB2f/f mice, LY2828360 attenuated ddC-evoked mechanical and cold hypersensitivity at 0.3 mg/kg (p<0.01 for all comparisons), 1 mg/kg (p<0.0001 for all comparisons) and 3 mg/kg (p<0.0001 for all comparisons), but not at 0.03 mg/kg (p>0.9 for all comparisons; Figure 10C). By contrast, LY2828360 did not alter ddC-evoked mechanical hypersensitivity in advillinCre/+;CB2f/f mice, at any dose (p>0.6 for all comparisons) (Figure 10C). Morphine (10 mg/kg, i.p.) and gabapentin (50 mg/kg, i.p.) reversed ddC-evoked mechanical hypersensitivity in both sexes and genotypes (p<0.0001 for all comparisons; Figure 10A-D). By contrast, vehicle treatment did not alter levels of mechanical or cold responsiveness in either sex or genotype (data not shown).
Figure 10. Removal of CB2 receptors from peripheral sensory neurons eliminates the antinociceptive efficacy of CB2 receptor agonists.

AM1710 (A,B) and LY2828360 (C,D) dose dependently alleviate ddC-evoked mechanical and cold allodynia in CB2f/f GFP mice, but fail to alter ddC-induced behavioral hypersensitivities in advillinCre/+;CB2f/f mice. The reference analgesics morphine and gabapentin reverse ddC-evoked mechanical (A) and cold (B) hypersensitivity in CB2f/f and advillinCre/+;CB2f/f mice. ###p<0.0001 vs. pre-ddC baseline, **p<0.01, ***p<0.0001 male and female CB2f/f vs. ddC, $$$p<0.0001 all groups vs. ddC. Two-way ANOVA, Tukey multiple comparisons test post-hoc. (N= 5 per group).
3.10. LY2828360 prevents the development of tolerance to morphine’s antinociceptive efficacy in male CB2f/f mice, and to a lesser extent in female CB2f/f mice, whereas removal of CB2 receptors from peripheral sensory neurons eliminates these effects.
In male CB2f/f GFP mice, morphine (10 mg/kg i.p.) alleviated ddC-evoked mechanical (Figure 11A) and cold allodynia (Figure 11B) relative to vehicle treatment on days 1 (p<0.001) and 3 (p<0.01) of chronic dosing, but efficacy was absent on days 6-12 of repeated dosing (p>0.3 for all comparisons); these observations are consistent with development of morphine tolerance. LY2828360 co-administration (0.03 or 0.1 mg/kg, i.p.) with morphine (10 mg/kg, i.p.) prevented the development of tolerance to morphine’s antinociceptive efficacy in CB2f/f GFP mice (Figure 11A). Mechanical paw withdrawal thresholds were higher in mice treated with either dose of LY2828360 in combination with morphine on day 1-12 (p<0.0001) relative to mice receiving vehicle and on days 3-12 of administration relative to mice receiving morphine alone (p<0.001 for each comparison). Cold responsiveness varied slightly between the two doses of LY2828360 (Figure 11B). CB2f/f GFP mice treated with the high dose of LY2828360 (0.1 mg/kg) in conjunction with morphine displayed lower levels of cold responsiveness (p<0.01) on day 1 of administration relative to mice treated with the low dose of LY2828360 (0.03 mg/kg). Both doses of LY2828360 suppressed the development of morphine tolerance, producing sustained relief of ddC-evoked cold hypersensitivity on days 3 (p<0.01) and 6-12 of administration (p<0.001) relative to CB2f/f GFP mice treated with either vehicle or morphine alone (Figure 11B).
Figure 11. Co-administration of LY2828360 with morphine prevents the development of tolerance to morphine’s antinociceptive effects in male, and to a lesser extent in female CB2f/f mice but not in advillinCre+/;CB2f/f mice.

Morphine (10 mg/kg i.p.) alleviated ddC-evoked mechanical (A, C, E, G) and cold (B, D, F, H) responsiveness on days 1 and 3 of administration. Co-administration of LY2828360 (0.03 mg/kg or 0.1 mg/kg i.p.) with morphine (10 mg/kg i.p.) prevented the development of tolerance, producing sustained suppressions of ddC-evoked mechanical (A,C) or cold (B,D) allodynia on days 1-12 of administration in male CB2f/f GFP mice but not male advillincre+/;CB2f/f mice. Co-administration of LY2828360 (0.03 mg/kg or 0.1 mg/kg i.p.) with morphine (10 mg/kg i.p.) lessened the development of tolerance in CB2f/f GFP mice, though to a lesser extant than that observed in male CB2f/f GFP mice (E-H). The tolerance sparing effects of LY2828360 were absent in female advillincre+/;CB2f/f mice (G,H). ###p<0.001 vs. baseline, ***p<0.001 all groups vs. veh+veh, $$$p<0.001, $$p<0.01 MOR+LY0.1 mg/kg and MOR+ LY0.03 mg/kg vs. all other groups, +++p<0.001, ++p<0.01 MOR+Veh vs. Veh+Veh, &&p<0.01, &p<0.05 MOR+LY0.1 mg/kg vs. MOR+LY0.03 mg/kg, @@p<0.01, @p<0.05 MOR+LY0.1mg/kg vs. MOR+Veh, ^^p<0.01 MOR+LY0.1 mg/kg and MOR+LY0.03 mg/kg vs. Veh+Veh, ===p<0.001, =p<0.05 MOR+LY0.1 mg/kg vs. all other groups, %p<0.05 MOR+LY0.03 mg/kg vs. Veh, −p<0.01 MOR+LY0.03 mg/kg vs. Veh+Veh and MOR+LY0.1 mg/kg. :::p>0.001, ::p>0.01, :p>0.05 CB2f/f MOR+LY vs. all other groups, >p<0.05 CB2f/f MOR+LY vs. CB2f/f MOR+Veh, !p<0.05 CB2f/f MOR+Veh vs. Adv cre+;CB2 f/f MOR+LY, <<p<0.01, <p<0.05 CB2f/f MOR+Veh vs. all other groups, \p<0.05 CB2f/f MOR+LY vs. vs. advillincre+/;CB2f/f MOR+VEH, −p<0.05 CB2f/f vs. advillincre+/;CB2f/f MOR+LY, “p<0.05 CB2f/f vs. advillincre+/;CB2f/f, ~p<0.05 CB2f/f MOR+LY vs. advillincre+/;CB2f/f MOR+LY, /p<0.05 CB2f/f MOR+Veh vs. advillincre+/;CB2f/f MOR+LY. Two-way ANOVA, Tukey Multiple comparisons test post-hoc. (N=6-10 per group). Black bars indicate period of LY and morphine co-administration. MOR: morphine, LY: LY2828360, Veh: vehicle.
In female CB2f/f GFP mice, morphine alleviated ddC-evoked mechanical and cold allodynia on days 1 and 3 of administration (p<0.001 for all comparisons), but not on days 6-12 of administration (p>0.05 for all comparisons) (Figure 11E, F), consistent with development of tolerance. LY2828360 (0.03 mg/kg or 0.1 mg/kg i.p.) co-administration prolonged the antinociceptive efficacy of morphine in female CB2f/f GFP mice. Female CB2f/f GFP mice treated with either dose of LY2828360 displayed higher mechanical withdrawal thresholds on day 6 and 9 of co-administration (p<0.01) relative to female mice receiving vehicle, and relative to all groups on days and 12 (p<0.0001 for each comparison) (Figure 11E). The high dose of LY2828360 in conjunction with morphine also elevated paw withdrawal thresholds on days 6-12 (p<0.05 for all comparisons) relative to female CB2f/f GFP mice receiving morphine alone. Similarly, both doses of LY2828360 reduced cold responsiveness on day 6 relative to vehicle (p<0.001 for each comparison) and on days 9 and 12 relative to all other groups (p<0.001 for each comparison, Figure 11F). LY2828360 dose-dependently lessened the development of tolerance to morphine’s ability to suppress cold hypersensitivity. Female CB2f/f GFP mice treated with the high dose of LY2828360 in conjunction with morphine displayed lower levels of cold responsiveness on days 6-12 of administration relative to female CB2f/f mice receiving the low dose of LY2828360 in conjunction with morphine (p<0.05 for all comparisons, Figure 11F).
In both genotypes, ddC induced hypersensitivity to mechanical and cold stimulation (p<0.0001 for all comparisons) (Figure 11C, D, G, H). Morphine (10mg/kg, i.p.) co-administered with LY2828360 (0.1 mg/kg, i.p.) increased mechanical paw withdrawal thresholds (Figure 11C) and lowered cold responsiveness (Figure 11D) on days 3-12 (p<0.001 for all comparisons) in male (Figure 11C, D) and female (Figure 11G, H) CB2f/f mice, but not in advillincre+/;CB2f/f mice (Figure 11C, D, G, H). Thus, removal of CB2 receptors from peripheral sensory neurons eliminated the efficacy of CB2 agonists in sparing morphine tolerance. Transient differences in mechanical and cold allodynia were observed between groups throughout the testing period. Male CB2f/f GFP mice treated with morphine alone displayed modestly higher mechanical withdrawal thresholds at baseline (p=0.003), and lower mechanical withdrawal thresholds following acute drug treatment (dose 1; p=0.04) relative to CB2f/f GFP mice treated with morphine in combination with LY2828360. Male CB2f/f GFP mice treated with morphine alone also displayed higher mechanical withdrawal thresholds following dose 3 (p=0.04) relative to male advillincre+/;CB2f/f mice treated with morphine in combination with LY2828360. Male CB2f/f GFP mice treated with morphine displayed higher levels of cold responsiveness following dose 1 relative to all other groups (p<0.01 for all comparisons).
In female CB2f/f GFP mice, morphine (10 mg/kg, i.p.) in combination with LY2828360 (0.1 mg/kg, i.p.) increased mechanical withdrawal thresholds relative to female advillincre+/;CB2f/f mice receiving morphine alone on day 3 (p=0.02), and relative to all other groups on days 6-12 (p<0.01 for all comparisons). Female CB2f/f GFP mice treated with morphine alone displayed higher mechanical paw withdrawal thresholds on day 3 of administration relative to female advillincre+/;CB2f/f mice treated with morphine alone or in combination with LY2828360 (p<0.05 for each comparison; Figure 11G).
3.11. LY2828369 reverses established morphine tolerance in male, and to a lesser extent in female CB2f/f GFP mice, whereas removal of CB2 receptors from peripheral sensory neurons eliminates the tolerance reversing effects LY2828360.
In male CB2f/f GFP mice receiving the reversal dosing schedule, prior to initiation of LY2828360 dosing (0.1 mg/kg i.p., days 7-12 of administration), morphine (10 mg/kg i.p.) produced initial antinociceptive efficacy, alleviating ddC-evoked mechanical (Figure 12A) and cold (Figure 12B) allodynia on days 1 and 3 of administration (p<0.0001 for each comparison). By day 6 of administration, morphine’s antinociceptive efficacy was no longer observed (p>0.3 for all comparisons), consistent with development of tolerance. Treatment with LY2828360 (0.03 or 0.1 mg/kg i.p.) beginning on day 7 of morphine administration reversed established morphine tolerance, alleviating ddC-evoked mechanical (Figure 12A) and cold (Figure 12B) hypersensitivity on days 9 and 12 of administration relative to male CB2f/f GFP mice treated with morphine alone or vehicle (p<0.0001 for all comparisons). Male CB2f/f GFP mice treated with LY2828360 (0.1 mg/kg i.p. on days 7-12 of administration), however, displayed modestly lower levels of mechanical hypersensitivity on days 6 (p<0.05) and 9 (p<0.01) (Figure 12A) and lower levels of cold allodynia on day 9 (p<0.05) of administration (Figure 12B) relative to male CB2f/f GFP mice receiving the low dose of LY2828360. Neither dose of LY2828360 alone altered mechanical or cold responsiveness in the absence of morphine (data not shown).
Figure 12. LY2828360 reverses established tolerance to the anti-allodynic effects of morphine in male, and to a lesser extent in female CB2f/f GFP mice, but not in advillinCre+/;CB2f/f mice.

In all groups receiving morphine (10 mg/kg i.p.) alone on days 1-6 of administration, tolerance rapidly develops to morphine’s antinociceptive efficacy (A-H). In CB2f/f GFP mice, following initiation of LY2828360 dosing (0.03 mg/kg or 0.1 mg/kg i.p.) in conjunction with morphine on days 7-12, the antinociceptive efficacy of morphine returned, alleviating ddC-evoked mechanical (A, C, E, G) and cold (B, D, F, H) hypersensitivity on days 9-12 of administration. LY2828360 produced a reversal of established morphine tolerance in male, and to a lesser extent in female CB2f/f mice (A-H), but not in advillincre+/;CB2f/f mice of either sex (C,D,G,H). ###p<0.001 vs. baseline, :::p<0.001, ::p<0.01, :p<0.05 CB2f/f MOR+LY vs. all other groups, >p<0.05 CB2f/f MOR+LY vs. CB2f/f MOR+Veh, !p<0.05 CB2f/f MOR+Veh vs. Adv cre+;CB2 f/f MOR+LY, <<p<0.01, <p<0.05 CB2f/f MOR+Veh vs. all other groups, \p<0.05 CB2f/f MOR+LY vs. vs. advillincre+/;CB2f/f MOR+VEH, −p<0.05 CB2f/f vs. advillincre+/;CB2f/f MOR+LY, “p<0.05 CB2f/f vs. advillincre+/;CB2f/f, ~p<0.05 CB2f/f MOR+LY vs. advillincre+/;CB2f/f MOR+LY, /p<0.05 CB2f/f MOR+Veh vs. advillincre+/;CB2f/f MOR+LY. Two-way ANOVA, Tukey Multiple comparisons test post-hoc. (N=6-10 per group). Black bars indicate period of LY and morphine co-administration. For reversal condition, grey bar indicates period of vehicle+morphine administration (days 1-6), black bar indicates period of LY+morphine co-administration (days 7-12). MOR: morphine, LY: LY2828360, Adv: advillin, Veh: vehicle.
In female CB2f/f GFP mice receiving the dosing schedule used to reverse established tolerance, morphine alleviated ddC-evoked mechanical and cold allodynia on days 1 and 3 (p<0.0001 for all comparisons) of injections in all morphine-treated groups (Figure 12E). By day 6 of repeated dosing, the antinociceptive effects of morphine were largely absent, consistent with the development of tolerance. Strikingly, initiation of LY2828360 treatment on day 7 of morphine administration reversed established tolerance to morphine’s antinociceptive efficacy. Both doses of LY2828360 (0.03 mg/kg or 0.1 mg/kg i.p. on day 7-12) in conjunction with morphine increased mechanical paw withdrawal thresholds (Figure 12E), and lowered levels of cold responsiveness (Figure 12F) on days 9 and 12 relative to all other groups (p<0.001 for all comparisons). Additionally, the high dose of LY2828360 was more efficacious than the low dose in female CB2f/f GFP mice, producing reduced cold responsiveness on days 9 and 12 of administration relative to all other groups (p<0.05 for each comparison; Figure 12F). In female CB2f/f GFP mice, LY2828360 did not reliably alter ddC-evoked allodynia in the absence of morphine (data not shown).
In male and female CB2f/f GFP mice receiving LY2828360 (0.1 mg/kg, i.p.) co-administered with morphine on days 7-12, mechanical withdrawal thresholds were higher and cold responsiveness was lower on days 9 and 12 (p<0.001 for each comparison) relative to CB2f/f GFP mice receiving morphine alone or advillincre+/;CB2f/f mice receiving either treatment (Figure 12C-D, G-H). In male CB2f/f GFP mice, prior to initiation of reversal dosing with LY2828360 all groups displayed similar levels of mechanical and cold allodynia with a few exceptions. Male CB2f/f GFP mice treated with morphine alone displayed higher mechanical withdrawal thresholds (Figure 12A,C) at baseline (p=0.04) and after dose 3 (p=0.01) relative to male CB2f/f GFP mice treated with the reversal dosing schedule of LY2828360, and lower mechanical withdrawal thresholds following the first injection (p=0.02) relative to male CB2f/f GFP mice treated with the reversal dosing schedule of LY2828360. Male CB2f/f GFP mice treated with morphine alone also displayed higher levels of cold responsiveness following dose 1 relative to all other groups (p<0.05 for all comparisons; Figure 12B, D). Female CB2f/f and advillincre+/;CB2f/f mice treated with morphine alone or with the reversal dosing schedule displayed roughly equivalent mechanical (Figure 12G) and cold (Figure 12H) responses prior to the initiation of reversal dosing with LY2828360 on day 7. On day 3 of administration female CB2f/f GFP mice treated with morphine alone displayed higher mechanical withdrawal thresholds relative to all other groups (p<0.05 for each comparison). Prior to initiating the reversal dosing regimen of LY2828360 and morphine, morphine increased mechanical withdrawal thresholds (Figure 12G) and reduced cold responsiveness (Figure 12H) in female CB2f/f GFP mice relative to female advillincre+/;CB2f/f mice in the same treatment group on day 3 (p=0.03). On day 6, female CB2f/f GFP mice treated with morphine alone displayed lower levels of cold responsiveness relative to both groups of female advillincre+/;CB2f/f mice (p<0.05 for all comparisons). In male and female CB2f/f and advillincre+/;CB2f/f mice treated with vehicle in lieu of morphine, LY2828360 did not alter mechanical or cold responsiveness (p>0.1 for all comparisons, data not shown). These results indicate that, in male and female mice, removal of CB2 from peripheral sensory neurons eliminates the ability of LY2828360 to reverse established morphine tolerance.
4. Discussion
To our knowledge, the present studies provide the first evidence that CB2 receptors on peripheral sensory neurons are necessary for the antinociceptive effects of CB2 agonists. They also support a previously unrecognized role for primary sensory CB2 receptors in mediating the ability of CB2 agonists to both spare and reverse opioid antinociceptive tolerance. These studies further validate CB2 receptors as a viable therapeutic target for the management of ATN-associated pain. In our studies, two structurally distinct CB2 agonists, AM1710 and LY2828360, alleviated ddC-evoked mechanical and cold hypersensitivity at a wide range of doses, without altering mechanical or cold responsiveness in the absence of ddC. The antinociceptive efficacy of AM1710 and LY2828360 was absent in mice globally lacking CB2, indicating the pain-relieving effects of both compounds are mediated by CB2 receptors. In these same CB2 KO mice, morphine completely reversed ddC-evoked mechanical and cold hypersensitivity, further indicating that the lack of antinociceptive efficacy of LY2828360 and AM1710 is not due to some gross alteration of nociceptive circuitry resulting from CB2 deletion. The present studies add to the previous body of literature from preclinical to clinical research suggesting that the cannabinoid system may be targeted to efficaciously alleviate HIV-associated sensory neuropathy. Cannabis consumption in HIV-infected patient populations provides a range of beneficial effects, from alleviating HIV-associated pain (10-12, 33), to increasing appetite and reducing nausea and psychiatric symptoms such as anxiety and depression (12). Evidence from preclinical research also supports the therapeutic potential of targeting the cannabinoid system for alleviating both primary sources implicated in the development of HIV-associated pain. Mixed CB1/CB2 agonists (3), and selective CB2 agonists have demonstrated efficacy in alleviating pain produced by administration of the HIV-associated toxic viral protein gp120 (34). Mixed CB1/CB2 agonists (7), exogenously administered endocannabinoids (35), and the Cannabis sativa terpene β-caryophyllene which is thought to exert its pharmacological effects via CB2 receptors, similarly have proven efficacious in alleviated ATN-associated pain (16). ATN-associated pain is specifically modeled with ddC in a mouse model in the present report; it should be recognized that HIV remains an essentially human disease that cannot be transmitted to mice, and a complex milieu of factors contributes to HIV-associated neuropathic pain in people (2).
CB2 receptor stimulation alleviates allodynia in multiple preclinical pain models without producing the problematic side effects associated with CB1 receptor stimulation (14), including the development of tolerance (15, 20, 25), cannabimimetic side effects or physical dependence (15). In the current study, AM1710 and LY2828360 produced sustained efficacy in reducing ddC-evoked mechanical and cold hypersensitivity following 12 days of chronic dosing, at similar levels between male and female mice, whereas mice rapidly developed tolerance to the antinociceptive effects of morphine. The results of the current study support the possibility that targeting CB2 represents a superior therapeutic target associated with fewer side effects. In addition to development of antinociceptive tolerance and the high risk for abuse and addiction associated with opioids such as morphine, opioids may exacerbate HIV-induced neuroinflammation and infectivity (36). Morphine administration upregulates the co-receptors HIV uses to enter and infect immune cells, such as CCR5 (37-39) and CXCR4 (40), which is implicated in the development of HIV-associated neuropathic pain (41, 42). MOR agonist administration in HIV-infected patient populations therefore has the potential to worsen HIV-induced neuroinflammation, HIV-infectivity and HIV-associated neuropathic pain. In contrast, CB2 stimulation may be protective against HIV-induced neurotoxicity/neuroinflammation (43, 44).
The present studies provide the first evidence that CB2 expression on peripheral sensory neurons may be responsible for the antinociceptive efficacy of CB2 agonists. Selective deletion of CB2 from excitatory neurons or peripheral sensory neurons completely eliminated the antinociceptive efficacy of two structurally distinct CB2 receptor agonists with different in vitro signaling properties (20, 25). Moreover, these effects were observed in both male and female mice. On the other hand, in advillinCre/+;CB2f/f mice, two mechanistically distinct reference analgesics (morphine and gabapentin) still effectively eliminated ddC-induced mechanical and cold hypersensitivity. Additionally, the two CB2 receptor agonists used in the current studies exhibit differential levels of CNS availability and signaling properties. LY2828360 is a slowly signaling G-protein biased agonist that readily crosses the blood brain barrier, whereas AM1710 is a balanced agonist that does not couple to calcium channels that exhibits limited CNS penetration (22, 45). Given that both compounds displayed a similar ability to decrease ddC-induced mechanical and cold hypersensitivity suggests CB2 stimulation exerting antinociceptive effects via a peripheral site of action. There exists considerable controversy regarding the distribution of CB2 receptors within the central nervous system (CNS) (19). A growing number of reports indicate CB2 receptors may be present within the CNS, though the types of cells, and under what circumstances they may be expressed are still debated (26, 46-62). CB2 expression has also been suggested to increase in primary afferent neurons under pathological conditions (63-65). In the present studies, native GFP fluorescence and anti-GFP immunoreactivity were below the threshold for detection throughout the brain and sensory neurocircuitry including in the cell bodies of primary afferent neurons as well as the dorsal horn of the lumbar spinal cord, where primary afferent neurons terminate in the central nervous system. However, CB2 mRNA was detected in both lumbar DRG and lumbar spinal cord, albeit at low levels, suggesting immunohistochemical approaches may have insufficient sensitivity to detect protein expressed at such low levels. Both CB2 and GFP mRNA were decreased in the DRG of advillincre+;CB2f/f mice, but not in the spleen or spinal cord relative to CB2f/f mice, indicating deletion of CB2 receptors by advillin-Cre produced a selective decrease in the mRNA for both markers of CB2 receptor transcription in the cell bodies of peripheral sensory neurons. Additionally, local administration of CB2 receptor agonists in the paw decrease mechanical and cold allodynia induced by the chemotherapeutic drug paclitaxel, and CB2EGFP is detected in keratinocytes, Langerhans cells and dendritic cells within the epidermis (27). In fact, using the same CB2f/f GFP mouse used here, we recently showed that CB2EGFP in Langerhans cells and keratinocytes are dynamically regulated by paclitaxel in peripheral paw tissue under conditions in which only scattered axonal labeling of large diameter fibers were observed (27). These latter effects are consistent with likely localization of CB2 on Aβ fibers and Merkel cell endings whereas thinly myelinated and unmyelinated epidermal nerve fibers lacked CB2EGFP labeling (27). CB2 agonist administered locally in the paw also suppressed paclitaxel-induced allodynia and increases spinal IL-10 mRNA levels (27). These observations raise the possibility that CB2 on the peripheral terminals of primary afferents may contribute to the antinociceptive effects of CB2 receptor agonists in ddC-induced neuropathic pain. Future studies, beyond the scope of the present report, are needed to determine whether GFP/CB2 can be detected in other areas on primary afferent neurons such as peripheral terminals in the paw skin, or whether more sensitive techniques such as RNAscope may provide insight into the tissues and cells expressing GFP and/or CB2 mRNA.
Recently, it was reported that global deletion of CB2 receptors or selective removal of CB2 from myeloid cells exacerbated the development of neuropathic pain induced by partial ligation of the sciatic nerve (66), whereas removal of CB2 from neuronal populations had no effect on the development of pain. However, this study did not examine the in vivo efficacy of CB2 agonists in ameliorating established pain in neuronal CB2-knockout mice. Furthermore, given previous reports indicating CB2 protein expression is induced following peripheral nerve injury (63, 65), these results do not preclude the possibility that CB2 in neuronal populations might contribute to antinociception once neuropathic pain has been established. In a subsequent report, removal of CB2 from neurons in mice expressing Cre recombinase under the Synapsin I promoter (Syn-Cre+:Cnr2fl/fl), but not Nav1.8+ primary afferents (Nav1.8− Cre+:-Cnr2fl/fl), modestly attenuated the ability of the CB2 receptor agonist JWH133 to alleviate mechanical hypersensitivity in a partial sciatic nerve injury model (67). This slight decrease in the antinociceptive efficacy of JWH133 was accompanied by an increase in JWH133 self-administration, suggesting an increase in spontaneous pain and indicating that despite increased intake of a CB2 agonist the ability of JWH133 to alleviate established neuropathic pain was blunted (but not eliminated) by removal of CB2 receptors from neurons. The sustained efficacy of JWH133 following removal of CB2 from Nav1.8+ nociceptors is interesting given the robust loss of CB2 agonist efficacy observed in our conditional knockout mice in the present studies. Discrepancies may be explained by the fact that in the present study, CB2 was removed from all peripheral sensory neurons, not just Nav1.8+ nociceptive primary afferents. Additionally, we used a model of toxic neuropathy whereas a surgically-induced peripheral nerve injury model was used in the previous report. The presence of CB2 on peripheral sensory that in vitro CB2 receptor agonists produce an inhibitory effect on the activity of guinea pig and/or human vagus nerve preparations to stimulation via prostaglandin E2 (guinea pig), hypertonic saline (guinea pig) or capsaicin (both) (68). Additionally, in cultured human DRG, CB2 agonists prevent capsaicin-induced Ca2+ influx indicating CB2 receptors are indeed present on cultured nociceptive primary afferent neurons (69). Future studies are needed to probe the specific subtypes of peripheral sensory afferents that contribute to the antinociceptive effects of CB2 receptor stimulation. Future studies will determine whether CB2 is dynamically altered, induced or upregulated on certain primary afferent subtypes by different toxic challenges. Such studies can be expected to provide further insight into particular pain conditions that may, or may not, benefit from CB2 receptor agonist therapy.
In the present study, the CB2 agonist LY2828360 robustly prevented the development of morphine tolerance in male CB2f/f GFP mice, and to a lesser extent in female CB2f/f GFP mice. To our knowledge, our study is the first demonstration of tolerance sparing effects of a CB2 agonist in female mice. Mechanisms underlying differences between the tolerance sparing effects of LY2828360 in male and female mice in the present studies requires additional examination.
The present study is the first report to indicate that treatment with a CB2 agonist can reverse established morphine tolerance. The ability of a pharmacological intervention to reverse tolerance once established may prove critical in a clinical setting, as there are great individual differences regulating the development of tolerance in clinical populations (70). Once again, LY2828360 more robustly reversed established morphine tolerance in male versus female CB2f/f GFP mice. Although many studies have reported the existence of sex differences in opioid analgesia and tolerance ((71) for review), the current study failed to find any reliable differences in opioid analgesia or opioid tolerance between male and female mice. The difference in LY2828360’s ability to prevent or reverse morphine tolerance is unlikely to be attributed to differences in the kinetics of the development or extent of morphine tolerance between male and female mice; in the absence of LY2828360, mice of both sexes developed tolerance to morphine’s ability to alleviate ddC-evoked mechanical and cold hypersensitivity at equivalent rates and to a similar extent. Further experiments are necessary to examine whether differences in CB2 receptor expression between male and female mice can be detected in regions of nociceptive circuitry controlling the development of morphine tolerance (such as primary afferent neurons (29)).
The present results build on previous work from our laboratory (20) indicating that administration of LY2828360 prior to or co-administered with morphine prevents the development of tolerance to morphine’s antinociceptive efficacy in a model of chemotherapy-induced peripheral neuropathy (27). Previous studies from other laboratories have suggested CB2 stimulation prevents the development of morphine tolerance by upregulating MOR mRNA levels in DRG and spinal cord (72), or by reducing tolerance produced by the release of proinflammatory cytokines via stimulation of CB2 receptors on microglia (73). LY2828360 may more effectively engage either of these pathways in male mice. Further experiments are required to determine whether CB2 receptor stimulation produces greater upregulations of MOR mRNA in males relative to female mice or suppresses opioid induced increases in proinflammatory cytokines more effectively in male versus female mice. However, the link between stimulation of MOR on microglial cells and the development of tolerance is in dispute (29, 74-76), as MOR mRNA appears absent in microglia (29). Recently, the critical role of MOR’s on peripheral sensory neurons in driving pro-nociceptive plasticity thought to underlie the development of tolerance to opiates has been demonstrated (29). The results of the current study support these findings, as the tolerance sparing and reversing effects of the CB2 agonist LY2828360 were completely absent in male and female mice lacking CB2 on peripheral sensory neurons. Future studies should examine the particular subtypes of sensory neurons (e.g., nociceptors vs. mechanoreceptors) which underlie the tolerance sparing effects of CB2 agonists on morphine anti-nociception, as well as the signaling mechanisms underlying these effects. Additionally, whether CB2 receptor stimulation decreases pro-nociceptive plasticity induced by morphine in peripheral sensory neurons via direct (e.g., CB2 stimulation directly decreasing firing rates of peripheral sensory neurons) or indirect mechanisms (e.g., CB2 stimulation on microglia decreasing the release of proinflammatory factors driving tolerance) is poorly understood. These factors may be influenced by pathological states differentially in males and females. Finally, although we did not observe potency differences in CB2 agonist-mediated anti-allodynic efficacy between sexes, our studies do not preclude the possibility that the potency (or efficacy) of LY2828360 in preventing or reversing morphine tolerance may be lower in female versus male mice. Future studies may examine whether higher doses of CB2 agonist would more effectively prevent or reverse morphine tolerance in female mice.
In conclusion, the present studies support the use of CB2 agonists in alleviating HIV-associated antiretroviral neuropathy and demonstrate, for the first time, that CB2 expression on peripheral sensory neurons are necessary for the antinociceptive efficacy of CB2 agonists. Our studies also demonstrate that the CB2 agonist LY2828360 prevents the development of antinociceptive tolerance to morphine and provides the first evidence that a CB2 receptor agonist reverses established tolerance to morphine antinociceptive efficacy in a neuropathic pain model. Furthermore, our studies have provided the first evidence that CB2 receptor expression on peripheral sensory neurons is necessary for the anti-allodynic efficacy of CB2 agonists as well as the ability of a CB2 agonist to prevent or reverse morphine tolerance.
Supplementary Material
Acknowledgements
This work was supported by National Institute on Drug Abuse Grants DA047858, DA041229 (A.G.H. and K.M.), DA042584 (A.G.H.), DA009158 (A.G.H., K.M., and A.M.), the National Cancer Institute Grant CA200417 (A.G.H.), an Indiana Addiction Grand Challenge Grant (A.G.H.), the Research and Education Component of the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin (C.J.H) and the Ministerio de Economía y Competitividad (SAF 2016-75959-R and SAF PID2019-108992RB-I00 to JR). L.M.C. was supported by National Institute on Drug Abuse T32 training grant DA024628 and the Harlan Scholars Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Lawrence Carey: conceptualization, formal analysis, investigation, writing-original draft, writing review and editing; Zhili Xu: investigation, formal analysis, writing-original draft; Gabriela Rajic: investigation; Alexandros Makriyannis: resources; Julian Romero: resources; Cecilia Hillard: resources, Ken Mackie: resources; Andrea Hohmann: conceptualization, writing-original draft, writing-review and editing, supervision, funding acquisition.
Declarations of interest: none.
Conflict of interest statement
The authors have no conflicts of interest, financial or otherwise, to declare.
References
- 1.Kamerman PR, Wadley AL, Cherry CL. HIV-associated sensory neuropathy: risk factors and genetics. Curr Pain Headache Rep. 2012;16(3):226–36. [DOI] [PubMed] [Google Scholar]
- 2.Schutz SG, Robinson-Papp J. HIV-related neuropathy: current perspectives. HIV AIDS (Auckl). 2013;5:243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace VC, Blackbeard J, Pheby T, Segerdahl AR, Davies M, Hasnie F, et al. Pharmacological, behavioural and mechanistic analysis of HIV-1 gp120 induced painful neuropathy. Pain. 2007;133(1–3):47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Monte SM, Gabuzda DH, Ho DD, Brown RH Jr., Hedley-Whyte ET, Schooley RT, et al. Peripheral neuropathy in the acquired immunodeficiency syndrome. Ann Neurol. 1988;23(5):485–92. [DOI] [PubMed] [Google Scholar]
- 5.Chaunu MP, Ratinahirana H, Raphael M, Henin D, Leport C, Brun-Vezinet F, et al. The spectrum of changes on 20 nerve biopsies in patients with HIV infection. Muscle Nerve. 1989;12(6):452–9. [DOI] [PubMed] [Google Scholar]
- 6.Rizzuto N, Cavallaro T, Monaco S, Morbin M, Bonetti B, Ferrari S, et al. Role of HIV in the pathogenesis of distal symmetrical peripheral neuropathy. Acta Neuropathol. 1995;90(3):244–50. [DOI] [PubMed] [Google Scholar]
- 7.Wallace VC, Blackbeard J, Segerdahl AR, Hasnie F, Pheby T, McMahon SB, et al. Characterization of rodent models of HIV-gp120 and anti-retroviral-associated neuropathic pain. Brain. 2007;130(Pt 10):2688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Husstedt IW, Bockenholt S, Kammer-Suhr B, Evers S. [Pain therapy in HIV-associated polyneuropathy]. Schmerz. 2001;15(2):138–46. [DOI] [PubMed] [Google Scholar]
- 9.Phillips TJ, Cherry CL, Cox S, Marshall SJ, Rice AS. Pharmacological treatment of painful HIV-associated sensory neuropathy: a systematic review and meta-analysis of randomised controlled trials. PLoS One. 2010;5(12):e14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrams DI, Jay CA, Shade SB, Vizoso H, Reda H, Press S, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68(7):515–21. [DOI] [PubMed] [Google Scholar]
- 11.Ellis RJ, Toperoff W, Vaida F, van den Brande G, Gonzales J, Gouaux B, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34(3):672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woolridge E, Barton S, Samuel J, Osorio J, Dougherty A, Holdcroft A. Cannabis use in HIV for pain and other medical symptoms. J Pain Symptom Manage. 2005;29(4):358–67. [DOI] [PubMed] [Google Scholar]
- 13.Rahn EJ, Hohmann AG. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 2009;6(4):713–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153(2):319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatry. 2015;77(5):475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aly E, Khajah MA, Masocha W. beta-Caryophyllene, a CB2-Receptor-Selective Phytocannabinoid, Suppresses Mechanical Allodynia in a Mouse Model of Antiretroviral-Induced Neuropathic Pain. Molecules. 2019;25(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchalant Y, Brownjohn PW, Bonnet A, Kleffmann T, Ashton JC. Validating Antibodies to the Cannabinoid CB2 Receptor: Antibody Sensitivity Is Not Evidence of Antibody Specificity. J Histochem Cytochem. 2014;62(6):395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang HY, Shen H, Jordan CJ, Liu QR, Gardner EL, Bonci A, et al. CB2 receptor antibody signal specificity: correlations with the use of partial CB2-knockout mice and anti-rat CB2 receptor antibodies. Acta Pharmacol Sin. 2019;40(3):398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160(3):467–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin X, Dhopeshwarkar AS, Huibregtse M, Mackie K, Hohmann AG. Slowly Signaling G Protein-Biased CB2 Cannabinoid Receptor Agonist LY2828360 Suppresses Neuropathic Pain with Sustained Efficacy and Attenuates Morphine Tolerance and Dependence. Mol Pharmacol. 2018;93(2):49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin X, Xu Z, Carey L, Romero J, Makriyannis A, Hillard CJ, et al. A peripheral CB2 cannabinoid receptor mechanism suppresses chemotherapy-induced peripheral neuropathy: evidence from a CB2 reporter mouse. Pain. 2022;163(5):834–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahn EJ, Thakur GA, Wood JA, Zvonok AM, Makriyannis A, Hohmann AG. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: antinociception without central nervous system side-effects. Pharmacol Biochem Behav. 2011;98(4):493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atwood BK, Wager-Miller J, Haskins C, Straiker A, Mackie K. Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol Pharmacol. 2012;81(2):250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhopeshwarkar A, Mackie K. Functional Selectivity of CB2 Cannabinoid Receptor Ligands at a Canonical and Noncanonical Pathway. J Pharmacol Exp Ther. 2016;358(2):342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li AL, Lin X, Dhopeshwarkar AS, Thomaz AC, Carey LM, Liu Y, et al. Cannabinoid CB2 Agonist AM1710 Differentially Suppresses Distinct Pathological Pain States and Attenuates Morphine Tolerance and Withdrawal. Mol Pharmacol. 2019;95(2):155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez A, Aparicio N, Pazos MR, Grande MT, Barreda-Manso MA, Benito-Cuesta I, et al. Cannabinoid CB2 receptors in the mouse brain: relevance for Alzheimer's disease. J Neuroinflammation. 2018;15(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin X, Xu Z, Carey L, Romero J, Makriyannis A, Hillard CJ, et al. A peripheral CB2 cannabinoid receptor mechanism suppresses chemotherapy-induced peripheral neuropathy. Pain. 2021;Publish Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zurborg S, Piszczek A, Martinez C, Hublitz P, Al Banchaabouchi M, Moreira P, et al. Generation and characterization of an Advillin-Cre driver mouse line. Mol Pain. 2011;7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, et al. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med. 2017;23(2):164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carey LM, Slivicki RA, Leishman E, Cornett B, Mackie K, Bradshaw H, et al. A pro-nociceptive phenotype unmasked in mice lacking fatty-acid amide hydrolase. Mol Pain. 2016;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carey LM, Lee WH, Gutierrez T, Kulkarni PM, Thakur GA, Lai YY, et al. Small molecule inhibitors of PSD95-nNOS protein-protein interactions suppress formalin-evoked Fos protein expression and nociceptive behavior in rats. Neuroscience. 2017;349:303–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carey LM, Gutierrez T, Deng L, Lee WH, Mackie K, Hohmann AG. Inflammatory and Neuropathic Nociception is Preserved in GPR55 Knockout Mice. Sci Rep. 2017;7(1):944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abrams DI, Hilton JF, Leiser RJ, Shade SB, Elbeik TA, Aweeka FT, et al. Short-term effects of cannabinoids in patients with HIV-1 infection: a randomized, placebo-controlled clinical trial. Ann Intern Med. 2003;139(4):258–66. [DOI] [PubMed] [Google Scholar]
- 34.Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, et al. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. 2012;153(5):1091–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munawar N, Oriowo MA, Masocha W. Antihyperalgesic Activities of Endocannabinoids in a Mouse Model of Antiretroviral-Induced Neuropathic Pain. Front Pharmacol. 2017;8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy A, Barbaro J, Martinez-Aguado P, Chilunda V, Jaureguiberry-Bravo M, Berman JW. The Effects of Opioids on HIV Neuropathogenesis. Front Immunol. 2019;10:2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo CJ, Li Y, Tian S, Wang X, Douglas SD, Ho WZ. Morphine enhances HIV infection of human blood mononuclear phagocytes through modulation of beta-chemokines and CCR5 receptor. J Investig Med. 2002;50(6):435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyagi T, Chuang LF, Doi RH, Carlos MP, Torres JV, Chuang RY. Morphine induces gene expression of CCR5 in human CEMx174 lymphocytes. J Biol Chem. 2000;275(40):31305–10. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Merrill JD, Mooney K, Song L, Wang X, Guo CJ, et al. Morphine enhances HIV infection of neonatal macrophages. Pediatr Res. 2003;54(2):282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson NM, Jung H, Ripsch MS, Miller RJ, White FA. CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav Immun. 2011;25(3):565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhangoo SK, Ren D, Miller RJ, Chan DM, Ripsch MS, Weiss C, et al. CXCR4 chemokine receptor signaling mediates pain hypersensitivity in association with antiretroviral toxic neuropathy. Brain Behav Immun. 2007;21(5):581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhangoo SK, Ripsch MS, Buchanan DJ, Miller RJ, White FA. Increased chemokine signaling in a model of HIV1-associated peripheral neuropathy. Mol Pain. 2009;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu S, Sheng WS, Rock RB. CB2 receptor agonists protect human dopaminergic neurons against damage from HIV-1 gp120. PLoS One. 2013;8(10):e77577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purohit V, Rapaka RS, Rutter J. Cannabinoid receptor-2 and HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2014;9(4):447–53. [DOI] [PubMed] [Google Scholar]
- 45.Hollinshead SP, Tidwell MW, Palmer J, Guidetti R, Sanderson A, Johnson MP, et al. Selective cannabinoid receptor type 2 (CB2) agonists: optimization of a series of purines leading to the identification of a clinical candidate for the treatment of osteoarthritic pain. J Med Chem. 2013;56(14):5722–33. [DOI] [PubMed] [Google Scholar]
- 46.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310(5746):329–32. [DOI] [PubMed] [Google Scholar]
- 47.Onaivi ES. Neuropsychobiological evidence for the functional presence and expression of cannabinoid CB2 receptors in the brain. Neuropsychobiology. 2006;54(4):231–46. [DOI] [PubMed] [Google Scholar]
- 48.Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071(1):10–23. [DOI] [PubMed] [Google Scholar]
- 49.Stempel AV, Stumpf A, Zhang HY, Ozdogan T, Pannasch U, Theis AK, et al. Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron. 2016;90(4):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ, et al. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A. 2014;111(46):E5007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li MH, Suchland KL, Ingram SL. Compensatory Activation of Cannabinoid CB2 Receptor Inhibition of GABA Release in the Rostral Ventromedial Medulla in Inflammatory Pain. J Neurosci. 2017;37(3):626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, et al. Brain cannabinoid CB(2) receptors modulate cocaine's actions in mice. Nat Neurosci. 2011;14(9):1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia C, Palomo-Garo C, Garcia-Arencibia M, Ramos J, Pertwee R, Fernandez-Ruiz J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Delta(9)-THCV in animal models of Parkinson's disease. Br J Pharmacol. 2011;163(7):1495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma Z, Gao F, Larsen B, Gao M, Luo Z, Chen D, et al. Mechanisms of cannabinoid CB2 receptor-mediated reduction of dopamine neuronal excitability in mouse ventral tegmental area. EBioMedicine. 2019;42:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ignatowska-Jankowska BM, Muldoon PP, Lichtman AH, Damaj MI. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology (Berl). 2013;229(4):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ortega-Alvaro A, Aracil-Fernandez A, Garcia-Gutierrez MS, Navarrete F, Manzanares J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garcia-Gutierrez MS, Manzanares J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J Psychopharmacol. 2011;25(1):111–20. [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Gutierrez MS, Perez-Ortiz JM, Gutierrez-Adan A, Manzanares J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB(2) receptors. Br J Pharmacol. 2010;160(7):1773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmole AC, Lundt R, Gennequin B, Schrage H, Beins E, Kramer A, et al. Expression Analysis of CB2-GFP BAC Transgenic Mice. PLoS One. 2015;10(9):e0138986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim J, Li Y. Chronic activation of CB2 cannabinoid receptors in the hippocampus increases excitatory synaptic transmission. J Physiol. 2015;593(4):871–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Kim J. Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience. 2015;311:253–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, Kim J. CB2 Cannabinoid Receptor Knockout in Mice Impairs Contextual Long-Term Memory and Enhances Spatial Working Memory. Neural Plast. 2016;2016:9817089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Svizenska IH, Brazda V, Klusakova I, Dubovy P. Bilateral changes of cannabinoid receptor type 2 protein and mRNA in the dorsal root ganglia of a rat neuropathic pain model. J Histochem Cytochem. 2013;61(7):529–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006;23(6):1530–8. [DOI] [PubMed] [Google Scholar]
- 65.Wotherspoon G, Fox A, McIntyre P, Colley S, Bevan S, Winter J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience. 2005;135(1):235–45. [DOI] [PubMed] [Google Scholar]
- 66.Nent E, Nozaki C, Schmole AC, Otte D, Zimmer A. CB2 receptor deletion on myeloid cells enhanced mechanical allodynia in a mouse model of neuropathic pain. Sci Rep. 2019;9(1):7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cabanero D, Ramirez-Lopez A, Drews E, Schmole A, Otte DM, Wawrzczak-Bargiela A, et al. Protective role of neuronal and lymphoid cannabinoid CB2 receptors in neuropathic pain. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belvisi MG, Patel HJ, Freund-Michel V, Hele DJ, Crispino N, Birrell MA. Inhibitory activity of the novel CB2 receptor agonist, GW833972A, on guinea-pig and human sensory nerve function in the airways. Br J Pharmacol. 2008;155(4):547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anand U, Otto WR, Sanchez-Herrera D, Facer P, Yiangou Y, Korchev Y, et al. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain. 2008;138(3):667–80. [DOI] [PubMed] [Google Scholar]
- 70.Dumas EO, Pollack GM. Opioid tolerance development: a pharmacokinetic/pharmacodynamic perspective. AAPS J. 2008;10(4):537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Craft RM. Sex differences in opioid analgesia: "from mouse to man". Clin J Pain. 2003;19(3):175–86. [DOI] [PubMed] [Google Scholar]
- 72.Zhang M, Wang K, Ma M, Tian S, Wei N, Wang G. Low-Dose Cannabinoid Type 2 Receptor Agonist Attenuates Tolerance to Repeated Morphine Administration via Regulating mu-Opioid Receptor Expression in Walker 256 Tumor-Bearing Rats. Anesth Analg. 2016;122(4):1031–7. [DOI] [PubMed] [Google Scholar]
- 73.Zhang M, Dong L, Zou H, Li J, Li Q, Wang G, et al. Effects of Cannabinoid Type 2 Receptor Agonist AM1241 on Morphine-Induced Antinociception, Acute and Chronic Tolerance, and Dependence in Mice. J Pain. 2018;19(10):1113–29. [DOI] [PubMed] [Google Scholar]
- 74.Mattioli TA, Leduc-Pessah H, Skelhorne-Gross G, Nicol CJ, Milne B, Trang T, et al. Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One. 2014;9(5):e97361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fukagawa H, Koyama T, Kakuyama M, Fukuda K. Microglial activation involved in morphine tolerance is not mediated by toll-like receptor 4. J Anesth. 2013;27(1):93–7. [DOI] [PubMed] [Google Scholar]
- 76.Kao SC, Zhao X, Lee CY, Atianjoh FE, Gauda EB, Yaster M, et al. Absence of mu opioid receptor mRNA expression in astrocytes and microglia of rat spinal cord. Neuroreport. 2012;23(6):378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.