

Elevated intestinal permeability has long been appreciated as a pathophysiological marker of gut inflammatory disease, in particular in inflammatory bowel disease (IBD). In addition to the association of IBD risk genetic variants with multiple aspects of intestinal permeability and barrier dysfunction, increased permeability has been identified as a predictor of IBD onset in healthy first-degree relatives of Crohn’s disease patients, and of relapse in Crohn’s patients.1–4 However, despite extensive investigation of the role of inflammation in disease-associated permeability, development of barrier-restoring agents beyond broad-acting anti-inflammatory agents has proven extremely challenging.
In this issue of GUT, Zuo et al. present an innovative study identifying that the tacrolimus-binding protein FKBP8 is a specific binding partner for the tight junction regulatory mediator, myosin light-chain kinase 1 (MLCK1).5 MLCK1 is one of two splice variants of the canonical tight junction regulatory protein, MLCK. MLCK phosphorylates myosin II regulatory light chain (MLC) within the perijunctional actomyosin ring to increase tight junction permeability to molecules with diameters up to ~100 Å.6 This route of paracellular permeability is often referred to as the ‘leak’ pathway.7 In studies using genetically-modified mice and enzymatic inhibitors, MLCK has been identified as a driver of acute cytokine-induced diarrhea as well as chronic immune-mediated experimental IBD.6 Moreover, MLCK acts as a critical downstream mediator of the inflammatory cytokines TNF-α and IFN-γ, culminating in alterations of epithelial tight junctions the key protein aggregate structures that regulate epithelial permeability.6, 8, 9 However, targeting of MLCK for therapeutic intervention to restore barrier function has been greatly hampered by the difficulty in avoiding unwanted side effects on MLCK expressed in cardiac, smooth and skeletal muscle.
This paper addresses a significant problem by determining a route to circumvent the non-specific effects of broad MLCK inhibition, by identifying FKBP8 as an additional factor that can be targeted to reduce accumulation of MLCK1 at tight junctions. In their prior work, the authors elucidated that TNF-induced recruitment of MLCK1 – but not the MLCK2 splice variant – to the perijunctional actomyosin ring was sensitive to small molecule blockade of an amino-terminal immunoglobulin-like domain, IgCAM3, that is unique to MLCK1.8 They hypothesized that the small molecule that blocked MLCK1 recruitment acted by interfering with IgCAM3 binding to another – as yet unidentified - protein. To identify this putative mediator, they used a yeast 2-hybrid screen to probe a human intestinal epithelial cDNA library for MLCK1-specific binding proteins. They identified FK506-binding protein FKBP8, also known as FKBP38, a protein that has been linked to autophagy, mitophagy, and the unfolded protein response but had not previously been functionally characterized in intestinal epithelial cells.10 Using protein binding and proximity ligation approaches, they showed that MLCK1 binds directly to FKBP8 and that these interactions are essential for MLCK1 recruitment, MLC phosphorylation, and TNF-induced barrier loss in intestinal epithelial cell lines and human intestinal organoid cultures (see Figure 1). Notably, while TNF increases MLCK1 expression, the effect of TNF on FKBP8 was specific in promoting MLCK-FKBP8 interactions as no change in FKBP8 expression or distribution levels was observed in response to TNF treatment. The translational importance of MLCK1-FKBP8 interactions was indicated by examination of biopsies from Crohn’s disease patients that also showed increased MLCK1- FKBP8 interactions relative to control subjects. In intervention studies, MLCK1-FKBP8 binding was inhibited by the immunosuppressive agent, tacrolimus, which displaced MLCK1 from the perijunctional actomyosin ring. This reversed TNF-induced MLCK1-FKBP8 interactions, MLCK1 recruitment, and barrier loss both in vitro and in an in vivo mouse model of T-cell-driven colitis. It is important to note that the effect of tacrolimus was specific to FKBP8 as MLCK1 enzymatic activity was not affected, while the inhibitory effect of tacrolimus was replicated in vitro by epithelial deletion of FKBP8.
Figure 1.
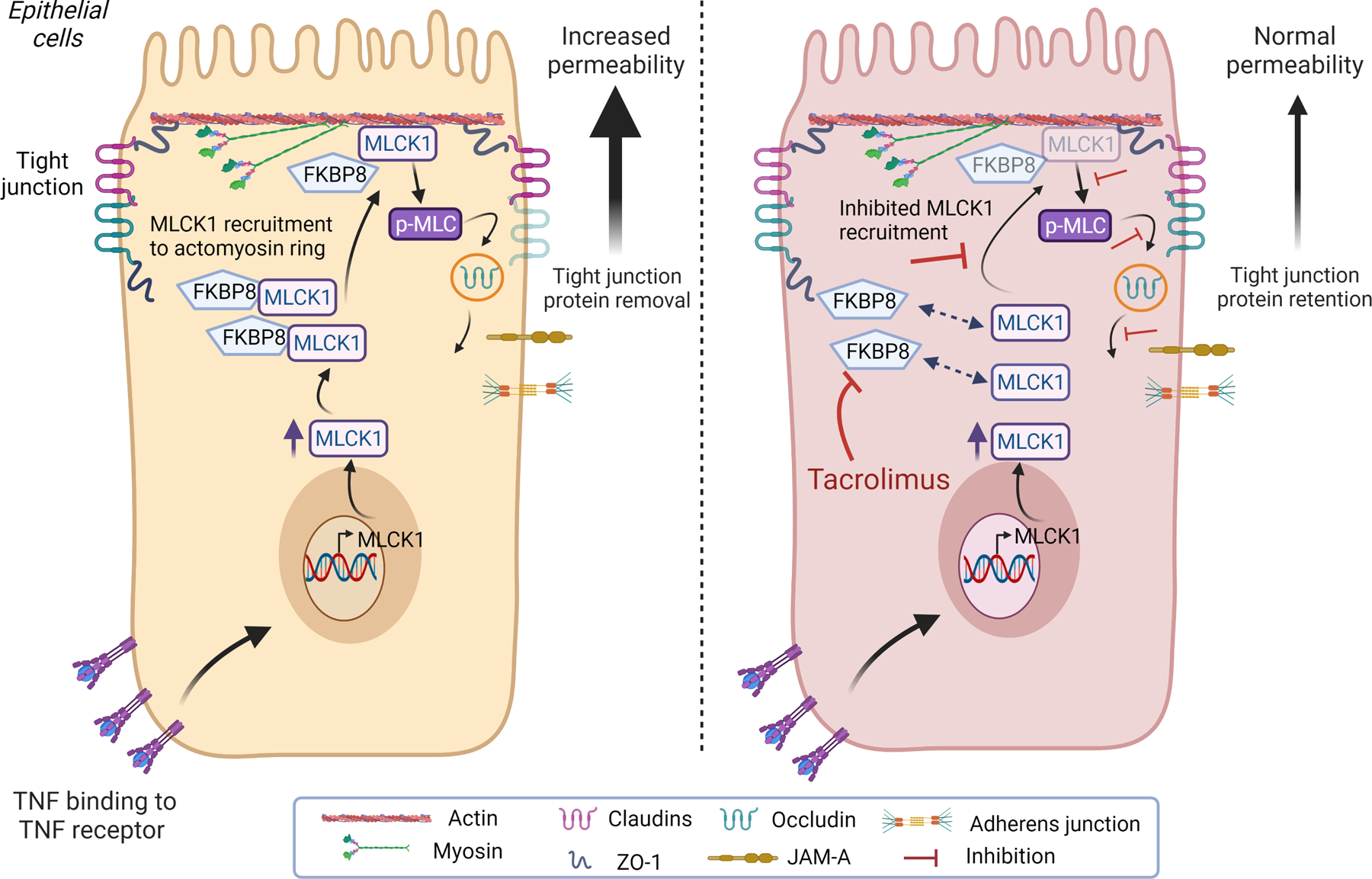
FKBP8 binding to MLCK1 mediates TNF-induced tight junction protein internalisation and increased permeability. TNF-α induces MLCK1 expression in intestinal epithelial cells. MLCK1 requires binding to the tacrolimus-sensitive protein FKBP8 in order to be recruited to the perijunctional actomyosin ring. MLCK1 can then induce phosphorylation of myosin light chain (pMLC), which drives internalisation of the transmembrane tight junction protein occludin. Removal of occludin from the tight junction increases paracellular permeability. Treatment of intestinal epithelial cells with tacrolimus has no effect on TNF induction of MLCK1 expression, but it causes dissociation of FKBP8 from MLCK1 thereby inhibiting MLCK1 recruitment to the perijunctional actomyosin ring. This inhibits MLCK1-mediated p-MLC, occludin internalisation and TNF-induced permeability increases. (figure created with BioRender.com). MLCK1, myosin light-chain kinase 1; TNF, tumour necrosis factor.
In summary, the authors have uncovered a molecular mechanism of MLCK1 recruitment and identified FKBP8 as a therapeutic target for intestinal barrier restoration in inflammatory disorders. While there are some remaining questions to be answered, such as the exact nature and exclusivity of the interaction between MLCK1 and FKBP8, given the capacity of FKBP8 to act as a scaffolding protein that may recruit additional binding partners, this study does represent an exciting advance that might lay the foundation for future therapeutic interventions targeting FKBP8-MLCK1 interactions. From a conceptual perspective, the authors have removed a significant roadblock in the potential clinical applicability of MLCK-mediated permeability defects that may invigorate exploration of barrier-restoring agents to treat disease.
Acknowledgments:
Studies in the author’s laboratory are supported by NIH grants 2R01DK091281, 1R01AI153314, 1R01DK130373, R21AI152017 (to DFM); a Pfizer Global Medical Research Award 59444033_ISR (to DFM).
Abbreviations:
- CD
Crohn’s disease
- FKBP8
FK506-binding protein 8
- IgCAM3
immunoglobulin cell adhesion molecule domain 3
- IFN-γ
interferon-gamma
- MLC
myosin II regulatory light chain
- MLCK
myosin-light chain kinase
- TNF-α
tumor necrosis factor-alpha
Footnotes
Conflict of Interest: The author declares that no conflict of interest exists.
REFERENCES
- 1.McCole DF. IBD candidate genes and intestinal barrier regulation. Inflamm Bowel Dis 2014;20:1829–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchelletta RR, Krishnan M, Spalinger MR, et al. T cell protein tyrosine phosphatase protects intestinal barrier function by restricting epithelial tight junction remodeling. J Clin Invest 2021;131. doi: 10.1172/JCI138230. [Epub ahead of print: 01 Sep 2021]. [DOI] [PMC free article] [PubMed]
- 3.Turpin W, Lee S-H, Raygoza Garay JA, et al. Increased intestinal permeability is associated with later development of Crohn’s disease. Gastroenterology 2020;159:2092–100. [DOI] [PubMed] [Google Scholar]
- 4.Wyatt J, Vogelsang H, Hübl W, et al. Intestinal permeability and the prediction of relapse in Crohri’s disease. The Lancet 1993;341:1437–9. [DOI] [PubMed] [Google Scholar]
- 5.Zuo L, Kuo W-T, Cao F, et al. Tacrolimus-binding protein FKBP8 directs myosin light chain kinasedependent barrier regulation and is a potential therapeutic target in Crohn’s disease. Gut 2022;10:gutjnl-2021–326534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest 2005;115:2702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai P-Y, Zhang B, He W-Q, et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017;21:671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham WV, He W, Marchiando AM, et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat Med 2019;25:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Utech M, Ivanov AI, Samarin SN, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell 2005;16:5040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhujabal Z, Birgisdottir Åsa B, Sjøttem E, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 2017;18:947–61. [DOI] [PMC free article] [PubMed] [Google Scholar]