

Abstract
Type 1 diabetes (T1D) is classified as an autoimmune disease where pancreatic β-cells are specifically targeted by cells of the immune system. The molecular mechanisms underlying this process are not completely understood. Herein, we identified that the Icam1 gene and ICAM-1 protein were selectively elevated in female NOD mice relative to male mice, fitting with the sexual dimorphism of diabetes onset in this key mouse model of T1D. In addition, ICAM-1 abundance was greater in hyperglycemic female NOD mice compared with age-matched normoglycemic female NOD mice. Moreover, we discovered that the Icam1 gene was rapidly upregulated in response to IL-1β in mouse, rat, and human islets and in 832/13 rat insulinoma cells. This early temporal genetic regulation requires key components of the NF-κB pathway and was associated with rapid recruitment of the p65 transcriptional subunit of NF-κB to corresponding κB elements within the Icam1 gene promoter. In addition, RNA polymerase II recruitment to the Icam1 gene promoter in response to IL-1β was consistent with p65 occupancy at κB elements, histone chemical modifications, and increased mRNA abundance. Thus, we conclude that β-cells undergo rapid genetic reprogramming by IL-1β to enhance expression of the Icam1 gene and that elevations in ICAM-1 are associated with hyperglycemia in NOD mice. These findings are highly relevant to, and highlight the importance of, pancreatic β-cell communication with the immune system. Collectively, these observations reveal a portion of the complex molecular events associated with onset and progression of T1D.
Keywords: autoimmunity, cytokine, diabetes, transcription
Introduction
Type 1 diabetes (T1D) is classified as an organ-specific autoimmune disease that arises when blood glucose control can no longer be adequately maintained. While the initial triggering event(s) is currently unknown, major outcomes of this disease are the onset of hyperglycemia secondary to decreases in circulating insulin due to loss of β-cell mass, insulin secretion, or both (1). There is a longstanding view that immune cell targeting of the pancreatic β-cells within the islets of Langerhans is a critical component of the disease process (2–4). Pro-inflammatory cytokines, such as IL-1β and IFN-γ, have been viewed as part of this process for decades (5–7). The ability of pro-inflammatory cytokines to promote changes in β-cell gene transcription is likely to be a critical part of the disease component at least in part by regulating the production and secretion of chemokines (8–10). Once secreted, chemokines influence immune cell trafficking to sites of inflammation (11, 12).
Pancreatic β-cells exhibit rapid alterations in gene expression patterns upon exposure to IL-1β, IFN-γ, or both cytokines in combination (13–17). This process involves the NF-κB pathway, with rapid nuclear entry by RelA/p65, occupancy of κB elements within promoter regions of the chemokine genes, and histone chemical modifications (13–19). The robust transcriptional response to cytokines promotes β-cells to produce and secrete large amounts of chemokine proteins concomitantly with reductions in insulin secretion (14). The actions of the NF-κB pathway are often augmented by signaling through IFN-γ receptor and activation of STAT1 (8, 9).
Once immune cells arrive within a site of inflammation, often having been recruited by chemokine gradients and primed by exposure to antigen(s), there is interaction between the discrete types of immune cells as well as communication with the inflamed tissue (20, 21). Part of this interactive process includes the increased abundance of cell surface proteins that allow for adhesion. One such example is the intracellular adhesion molecule-1 (ICAM-1), a transmembrane domain containing protein that interacts with specialized integrin proteins typically present on immune cells (22, 23). One such specialized set of proteins makes up the lymphocyte function-associated antigen-1 (LFA-1, aka αLβ2), a heterodimer composed of CD11a and CD18 (24). Upon activation, LFA-1 interacts primarily with ICAM-1, but can also interact with other members of the ICAM family (25). Thus, fine tuning an immune response is balanced by a number of important events, including cytokine and chemokine production, ICAM-1 upregulation, and the ICAM-1/LFA-1 interaction. The direct involvement of ICAM-1 in organ-specific autoimmunity and rejection of grafted tissue has been demonstrated in various studies (26–30).
In this study, we have investigated the signal-specific induction of ICAM-1 in pancreatic β-cells. Using bulk RNA-sequencing (RNA-Seq), we found that the 832/13 rat β-cell line and human islets exposed to IL-1β each displayed strikingly enhanced Icam1 transcript levels over untreated cells. In addition, we observed that the Icam1 gene is rapidly induced in β-cell lines as well as isolated mouse, rat, and human islets in response to interleukin-1β. Upon further investigation of the molecular determinants underlying these responses, we found that RNA polymerase II and the RelA/p65 subunit of NF-κB occupied genomic regions controlling Icam1 in a temporal manner consistent with the first appearance of transcript over baseline levels. These transcriptional data are supported by two key in vivo approaches: 1) ICAM-1 protein is enhanced in pancreatic islets four hours after systemic injection of IL-1β, 2) Female NOD mice display more Icam1 expression in islet β-cells when compared with male NOD mice, and 3) female NOD mice display elevations in ICAM-1 protein days after becoming hyperglycemic when compared with age-matched normoglycemic controls. Collectively, these genetic and molecular approaches offer novel insights into a key gene regulating immune system function and thus add to the existing molecular framework explaining events critical for onset and progression of T1D.
Materials and Methods
Cell Culture, Adenoviruses, and Reagents
Culture and passage of the 832/13 rat insulinoma cell line has been described (31). Cell lines were confirmed to be free of mycoplasma contamination using the Lonza MycoAlert Mycoplasma Detection Kit. Recombinant adenoviruses expressing GFP, p65, p65S276A, β-galactosidase (βGAL), CA (constitutively active) IKKβ (S177E/S181E) and IκBα super-repressor (S32A/S36A) have been described (15, 32). TPCA was from Bio-Techne (Minneapolis, MN, USA). Recombinant IL-1β and IFN-γ were from Peprotech (Cranbury, NJ, USA).
Experimental Animals, Islet Isolation, Pancreas Histology, and Human Islets
Seven week old male and female NOD (stock # 001976), and male C57BL/6J (stock # 000664) mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). Seven-week-old male Wistar rats (Strain #003) were purchased from Charles River (Wilmington, MA). Various ages of mice with the following conditional alleles were all from the Jackson Laboratory: IL-1R floxed (Jax #: 028398), RelA (p65) floxed (Jax #: 024342), and Pdx1-cre (Jax #: 014647). All animals were multi-housed with a 12-h light/dark cycle, and were allowed to acclimate to the facility for a minimum of one week prior to beginning experimentation. All animals had ad libitum access to food and drink prior to isolation of islets or pancreata for fixation. Female NOD mice were checked two-three times weekly for the presence of hyperglycemia (≥ 250 mg/dL) and were collected within two days of two consecutive hyperglycemic values. An age-matched normoglycemic control was also collected at the same time. C57BL/6J mice were injected with either saline or IL-1β (1 μg/kg body weight) and four hours later pancreatic tissue was collected for fixation in neutral buffered formalin. Procedures for sectioning and staining of pancreatic tissue have been reported (33, 34). The primary antibodies used were: STAT-1, Cell Signaling #14994, (1:400); ICAM-1, LSBio, LS-313412, (1:200); glucagon, eBioscience #14–9743-82, (1:1500); insulin, BioRad #5330–0104G, (1:1000). The secondary antibodies used for fluorescence imaging were from Jackson ImmunoResearch: Donkey anti-Guinea Pig Alexa Fluor 488, #706–545-148, (1:300) and Donkey anti-Rabbit Alexa Fluor 594, #711–586-152, (1:300). For the chromogenic stains, we used the Leica Biosystems ChromoPlex 1 Dual Detection kit, DS9665 which provided red plus brown detection. For comparisons of tissues from 8 and 12 week old C57BL/6J and NOD female mice, we used a Nanozoomer HT slide scanner equipped with a triple bandpass filter set for DAPI/FITC/TxRed fluorescence detection and a 20x /0.8NA objective. For the comparison of the normoglycemic versus hyperglycemic tissues from 18 week old female NOD mice, we used a Leica DM6000 microscope with 40x /0.95 NA objective. Our techniques for isolation of both mouse and rat islets have been described in detail previously (14, 18, 33). Human islets were obtained from Lonza (Clonetics™ Fresh Human Pancreatic Islets) with donor information described previously (33). All animal procedures were approved by the respective Pennington Biomedical Research Center or University of Tennessee Medical Center Institutional Care and Use Committees.
Identification of Predicted κB Genomic Regions and Construction of Luciferase Plasmids
Six putative NF-κB elements were identified in the −3kb region upstream of the rat Icam1 gene using the JASPAR web-based promoter analysis tool (35) and confirmed using PROMO, a separate web-based program (36). Of the six sites identified by each of these computer based approaches, four Icam1 promoter sequences were found to be conserved between rat, mouse, and human. These four sites were given the identifiers κB 1–4 relative to the transcriptional start site and were carried forward for further analysis. Using genomic DNA from rat 832/13 cells, 2.9 kb of the rat Icam1 promoter sequence was amplified with AccuPrime Pfx SuperMix (Invitrogen) and Icam1 3kb cloning primers (Supplementary Table 1). This amplicon was digested with SacI and HindIII and inserted into pNL1.2 (Promega) at the multiple cloning site. The same reverse primer was used with the Icam1 1kb cloning primer to amplify 1.1 kb of the rat Icam1 promoter. This amplicon was digested with HindIII and inserted into pNL1.2 at the multiple cloning site. Mutations were generated in the 2.9 kb pNL1.2 construct by site-directed mutagenesis using QuikChange II Site-Directed Mutagenesis Kit (Agilent). Primers for mutant sequences can be found in Supplemental Table 1. Each construct and successful site-directed mutagenesis event were confirmed by sequencing at the Pennington Biomedical Research Center Genomics Core Facility.
Transient Transfections and Luciferase Assays
For luciferase assays, 832/13 cells were grown to 75% confluence in 24 well plates. Luciferase reporter plasmids and siRNA duplexes were transfected into cells using TransFectin Lipid Reagent (Bio-Rad) according to the manufacturer’s instructions. 24 hours (h) post-transfection, cells were treated as indicated in the respective figure legends. Cells were lysed in 50 μL Nano-Glo Luciferase Assay Reagent (Promega) for 10 minutes (min) with rocking at room temperature. Luminescence was measured with a Glo-Max Multi+ Luminometer (Promega). Silencer Select siRNA duplexes (Invitrogen, Waltham, MA) used in this study are as follows: negative control siScramble (AM4611), sip65 (siRNA ID no. s159516) and sip50 (siRNA ID no. s135617).
RNA extraction, cDNA synthesis, and Gene Expression Analysis
Our procedures and reagents for isolation of RNA from cell lines and islets, cDNA synthesis, and transcript analysis by real-time PCR have all been reported (33, 37). PCR analysis was conducted using a Bio-Rad CFX 1000 thermal cycler. Primer sequences are listed in Supplementary Table 1.
Serial Analysis of Gene Expression (SAGE), Gene Expression Data Analysis, and Pathway Enrichment Analysis
SAGE:
RNA content and quality (260/280 ratio range 1.9 – 2.1) were assessed using a Nanodrop 1000 and then used to perform the SAGE analysis (38, 39). Briefly, gene expression profiling was performed by expression tag sequencing (SAGE) on an AB SOLiD 5500XL next-generation sequencing instrument using reagent kits from the manufacturer (Applied Biosystems, Foster City, CA). Sequence reads were aligned to human (hg38) or rat (rn6) reference RefSeq transcripts, respectively, via SOLiDSAGE (Applied Biosystems). Only uniquely mapped sequence reads were counted to generate the expression count level for each respective RefSeq gene. Expression levels of genes of interest were confirmed by RT-PCR using methods described above. The SAGE dataset has been uploaded to the Gene Expression Omnibus (GEO) website (https://www.ncbi.nlm.nih.gov/geo/) using Accession # GSE124166.
Gene expression data analysis:
Differential analysis of RNA read count data was performed using DESeq2 v1.4.5 software (40), which models read counts as a negative binomial distribution and uses an empirical Bayes shrinkage-based method to estimate priors for signal dispersion and fold-changes, and to calculate posterior estimates of these parameters. Gene expression signals were logarithmically transformed (to base 2) for all downstream analyses (the lowest expression value being set to 1 for this purpose). Gene expression based principal components analysis using JMP Genomics v6.0 was carried out as a quality control measure in the human and rat studies with two outlier samples detected in the human study, which were removed from further analysis (PCA shown in Supplementary Figure 1).
Pathway enrichment analysis:
Pathway enrichment was conducted via the competitive gene-scoring based gene set enrichment analysis tool (GSEA) (41). GSEA was performed by first ranking the expression of all genes in the untreated and IL-1β treated islets and beta cells via the signal-to-noise ratio (SNR) metric, and then employing a weighted Kolmogorov-Smirnov test to determine if the gene SNRs deviate significantly from a uniform distribution in a priori defined gene-sets (pathways) obtained from Wikipathways (www.wikipathways.org). Human and rat gene expression data was analyzed against human and rat specific pathway lists, respectively. Statistical significance for the observed enrichment was ascertained by permutation testing over size-matched gene-sets. Significant gene-sets were selected by control of the false discovery rate, FDR at ≤5% (42). The per-sample expression profiles of genes contributing to core enrichment of the significant pathways were visualized via row-normalized blue-red heatmaps with blue representing lower, and red representing higher gene expression levels.
Preparation of Whole Cell Extracts and Immunoblotting
832/13 cells were seeded in 6 well plates and treated as indicated in figure legends. Cells were then lysed in 100 μl M-PER lysis reagent (ThermoFisher Scientific) supplemented with Halt Protease Inhibitor Cocktail (ThermoFisher Scientific). Whole cell lysates quantified using a BCA assay (ThermoFisher Scientific). Denaturation of samples and immunoblotting conditions have been described in detail (34, 43). Antibodies used are listed in Supplementary Table 2.
Chromatin Immunoprecipitation
Preparation:
Cells were grown to 75% confluence in 10 cm dishes. Cells were serum starved in RPMI for 1 h before treatment with cytokine. After treatment, the cells were washed twice with 5 mL PBS and then crosslinked with ChIP Crosslink Gold (Diagenode) in PBS for 30 min at room temperature according to the manufacturer’s instructions. The plates were washed twice with 5 mL PBS again and then crosslinked in 1% methanol-free formaldehyde in 4.5 mL PBS for 10 min at room temperature. The crosslinking reaction was quenched with glycine at a final concentration of 125 mM for 5 min. The plates were washed twice with ice-cold PBS and cells scraped into 1 mL PBS with Halt Protease Inhibitor Cocktail (Thermo Scientific). Cells were pelleted by spinning 2 min 4°C at 5000 × g. Pellets were resuspended in 950 μL lysis buffer (1% SDS, 0.5% Triton X-100, 50 mM Tris, 10 mM EDTA, 0.5 mM DTT, pH = 8.0) and incubated 30 min at 4°C with rotation. Lysates were divided into 3 × 300 μL aliquots and dispensed into polystyrene sonication tubes (Evergreen Scientific). Chromatin was sheared to an average fragment size of 200–250 bp by sonicating 12 cycles (30 sec ON/30 sec OFF) in a Bioruptor Pico (Diagenode). Chromatin was cleared by centrifugation at 12,000 × g for 10 mins at 4°C. Fragment size was confirmed by addition of 1 μL of sheared chromatin to 19 μL of TE, incubation for 30 mins with RNase A (Thermo Scientific) at 37°C, addition of proteinase K, incubation for 1 h at 65°C, and running for 30 min on a 1.5% agarose gel. Samples were diluted by adding 100 μL of lysate to 900 μL of Dilution Buffer (1.1% Triton X-100, 0.01% SDS, 17 mM Tris, 1.2 mM EDTA, 167 mM NaCl, pH = 8) and 2 μL of protease inhibitors. Diluted material was precleared overnight with 5 μL Protein G Dynabeads (Thermo Scientific) by incubation at 4°C with rotation. Input samples were prepared by adding 2 μL lysate to 150 μL ChIP Elution Buffer (1% SDS, 50 mM Tris, 10 mM EDTA). These 2% input samples were set aside at 4°C overnight.
Immunoprecipitation:
5 μL of Dynabeads were prepared for each IP in the following manner. Beads were incubated in PBS-TWEEN with 1 μg antibody for 30 min with rotation at room temperature. Beads were then blocked by incubation in 1% BSA in PBS for 30 minutes at room temperature. After preclearing beads were removed from IP reactions, 5 μL of antibody-bound immunoprecipitation beads were added to each reaction and rotated at 4°C for 1 h. IP reactions were then transferred to clean microfuge tubes and washed with the following buffers: twice with Low Salt Wash (0.1% SDS, 1% Triton X-100, 0.05% TWEEN, 20 mM Tris, 2 mM EDTA, 150 mM NaCl), twice with High Salt Wash (0.1% SDS, 1% Triton X-100, 20 mM Tris, 2 mM EDTA, 500 mM NaCl), twice with LiCl Wash (1% NP-40, 1% sodium deoxycholate, 0.25 mM LiCl, 10 mM Tris, 1 mM EDTA), and once with TE (10 mM Tris, 1 mM EDTA). All washes were 5 min with rotation at 4°C except the TE wash, which was performed at room temperature. Beads were then resuspended in 150 μL ChIP Elution Buffer and, along with input samples, were incubated at 65°C for 30 min with shaking in a thermomixer. After elution, supernatants were transferred to clean microcentrifuge tubes for overnight decrosslinking with Proteinase K and 200 mM NaCl at 65°C. DNA was extracted from ChIP samples and inputs using GenCatch Advanced PCR Extraction Kit (Epoch Life Science). Primer sequences are available in Supplementary Table 1. Antibodies used are listed in Supplementary Table 2.
Statistical analysis
Statistical analysis was conducted using Prism version 9.3 (GraphPad). One or two-way ANOVA analyses were used to calculate the p-values indicated in the figure legends.
Results
RNA-Seq reveals the gene encoding Icam1 was rapidly upregulated in response to IL-1β in human islets and the 832/13 rat β-cell line.
Human islets and the 832/13 rat β-cell line were exposed to the cytokine IL-1β for 3 h or left untreated. Using an adjusted p-value of p < 0.1 and absolute fold-change of ≥ 1.5 for differential gene expression in both human and rat-derived cells, the top 50 responsive genes under these conditions were analyzed and shown in heatmap format for human islets (Figure 1A) and rat 832/13 cells (Figure 1B). Among the highest responding genes in human islets were various chemokines (e.g., CXCL1, CXCL2, CCL20, etc.) and cytokines (e.g., TNF-α). Similar upregulation of chemokine and inflammatory genes were seen in 832/13 cells (e.g. Cxcl1, Cxcl3, and Tlr2). This finding using unbiased bulk RNA-Sequencing is consistent with gene expression observations from our group (8, 14, 15, 18, 19) and others (10). From this analysis, we identified Icam1 as one of the most highly responsive genes (Figure 1A and 1B). Pathway enrichment analysis GSEA was consistent with IL-1β signaling (Figure 1C). Further analysis of the IL-1β signaling pathway showed 61 genes upregulated in human islets, 21 in 832/13 cells, and 11 of those genes are upregulated in both species (Figure 1C Venn diagram). There was also a high significant overlap (overlap p-value<2.2e-16 by Fisher’s exact test) between significantly differentially expressed genes from both human islet and rat cell lines (283 human genes and 554 rat genes at an absolute fold-change ≥2, adjusted p-value ≤ 0.1; not shown). The PCA analyses for these samples are provided in Supplementary Figure 1.
Figure 1: RNA-Seq reveals the gene encoding Icam1 was rapidly upregulated in response to IL-1β in human islets and the 832/13 rat β-cell line.
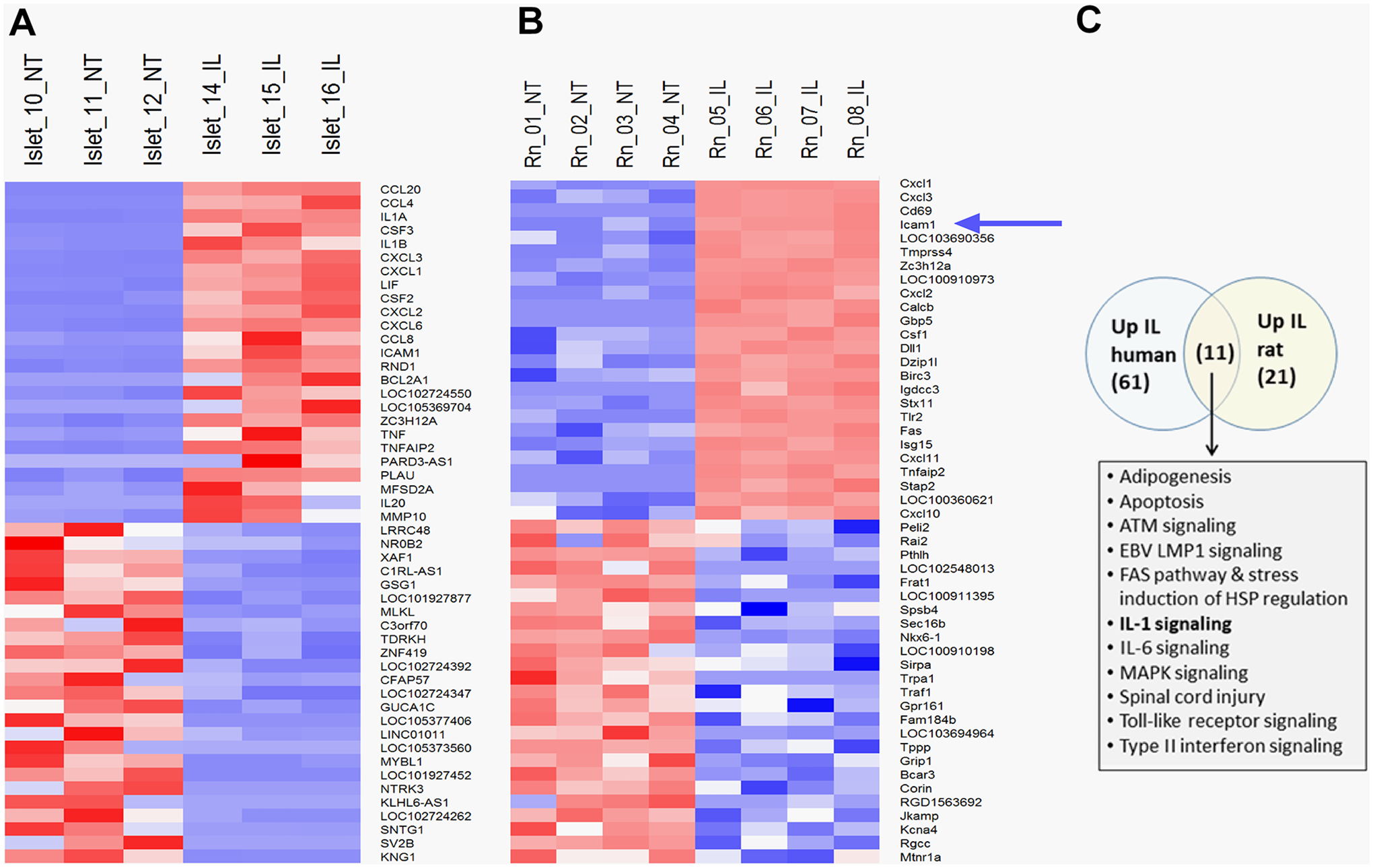
Clustered heatmap of the top 50 most up-regulated and down-regulated genes in human islets (A) and 832/13 rat insulinoma cells (B) exposed to IL-1β for 3 h (selected by magnitude of fold-change). Treatment conditions across replicates are indicated at the top of the heatmap and genes are indicated in rows. Gene expression signals were row-normalized with higher gene expression is expressed in shades of red and lower expression in shades of blue. The blue arrow indicates the Icam1 gene. C. Pathway enrichment analysis of human islets and 832/13 rat β-cells as determined via GSEA. The top enriched pathways from each study (FDR≤5%) were compared via a Venn analysis. Numbers in parentheses refer to the number of significantly enriched pathways in each set. Pathways identified in common in both species are listed in bulleted format. NT = no treatment; IL = IL-1β- treated.
Expression of Icam1 is inducible ex vivo in mouse, rat, and human islets and in vivo in both C57BL/6J and NOD mice.
Based on the RNA-Seq results shown in Figure 1, we next collected RNA from human islets after being left untreated or exposed to 10 ng/mL IL-1β for 1, 2, or 3 h and found that Icam1 was markedly elevated by 3 h (Figure 2A). This finding was also reproduced in cultured rat islets (Figure 2B). We next cultured mouse islets in 10 ng/mL IL-1β for 3 h and found that Icam1 was also induced by exposure to this cytokine (Figure 2C). After these observations across multiple species, we next tested the hypothesis that islet ICAM-1 protein would be inducible in response to a systemic cytokine signal; this was accomplished with intraperitoneal injection of recombinant mouse IL-1β (1 μg/kg body weight), which is a dose used previously for physiological outcomes (44). Male C57BL/6J mice injected i.p. with IL-1β displayed robust staining for ICAM-1 (Figure 2D; shown in pink in the right hand panel). We interpret this data as clear evidence that the ICAM-1 protein was markedly and rapidly upregulated in pancreatic islets in vivo after systemic delivery of IL-1β, which is consistent with the ex vivo results using cultured mouse, rat, and human islets. Moreover, using an established mouse model of autoimmunity and T1D, we found that female NOD mice display more expression of Icam1 relative to male NOD mice (Figure 2E). In addition, when female NOD mice convert to hyperglycemia (>250 mg/dL), Icam1 expression is elevated when compared with age-matched normoglycemic female NOD mice (Figure 2F). This phenotype is also observed at the protein level in pancreatic tissue isolated from normoglycemic relative to hyperglycemic female NOD mice (Figure 3) Importantly, we note that a portion of the immune cells surrounding the islets and also infiltrating the islets are ICAM-1 positive. In addition, the β-cells also display increased ICAM-1 protein in hyperglycemic NOD mice (Figure 3). At NOD mice aged eight and twelve weeks, only a subpopulation of the immune cells are positive for ICAM-1 (Supplementary Figure 2). Using C57BL/6J mice as a model that does not display autoimmunity or hyperglycemia, we note the tissue-resident immune cells are not likely to express ICAM-1 at levels observed in the NOD mice (Supplementary Figure 2).
Figure 2. Expression of Icam1 is inducible ex vivo in mouse, rat, and human islets and in vivo in both C57BL/6J and NOD mice.
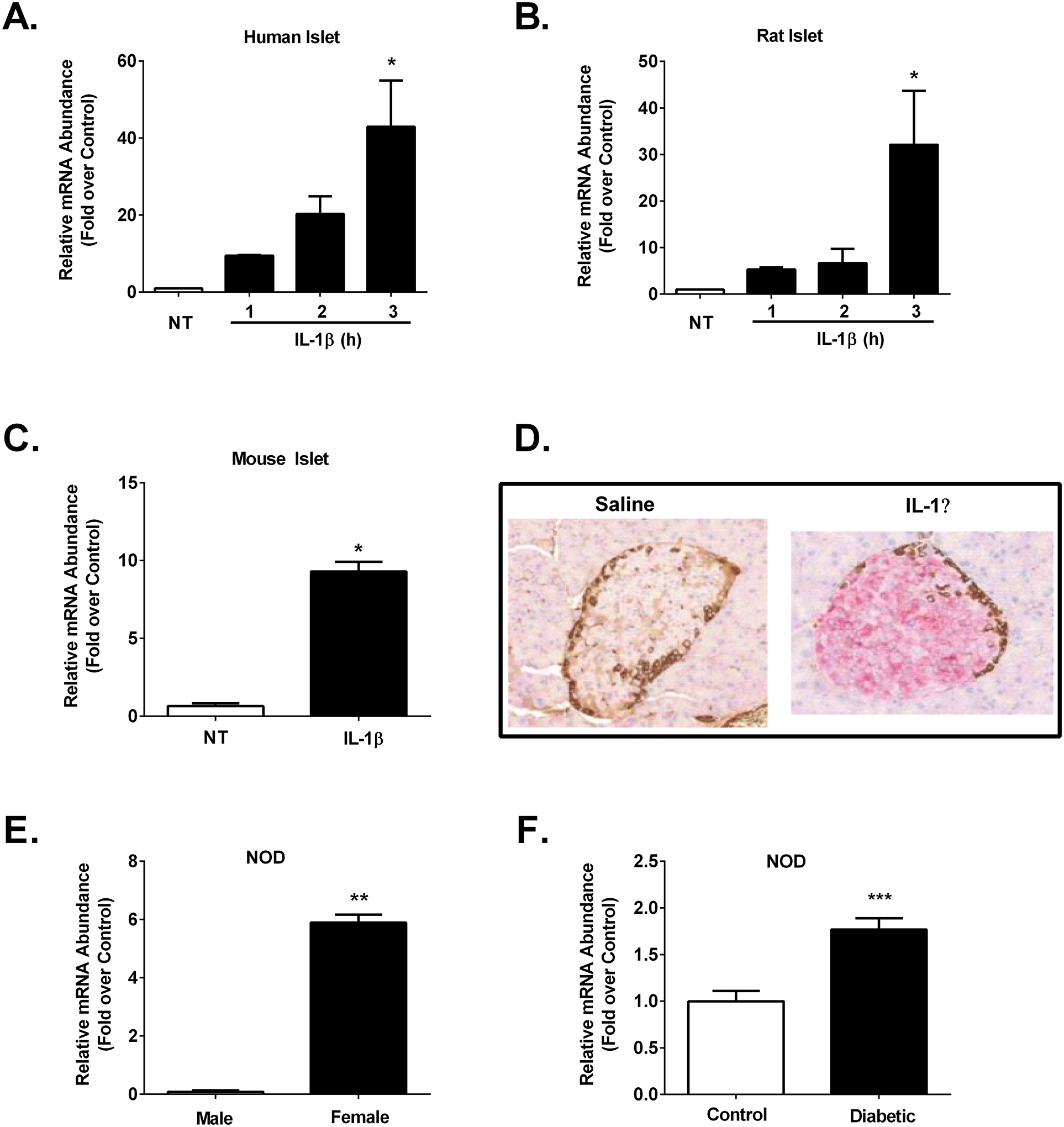
A. Human islets (n=3) were untreated (NT) or stimulated with 10 ng/mL IL-1β for the indicated times. B. Rat islets (n=3) were NT or stimulated with 10 ng/mL IL-1β for the indicated times. C. Mouse islets (n=3) were NT or stimulated for 3 h with 10 ng/mL IL-1β. D. Formalin-fixed paraffin-embedded (FFPE) pancreatic tissue was sectioned and stained for ICAM-1 (pink) and glucagon (brown) in 12 week old male C57BL/6J mice injected with either saline control or IL-1β (1 μg/kg body weight). E. Icam1 mRNA abundance in islets isolated from 12 week old normoglycemic male (n=3) and female (n=7) NOD mice. F. Icam1 expression levels in islets isolated from 16 week old female control (n=10) or hyperglycemic (n=9) NOD mice. The transcript data (A-C, E-F) are shown as means ± SEM. ***, p < 0.001; **, p < 0.01; *, p < 0.05. p values are vs. respective NT controls.
Figure 3. Hyperglyemic female NOD mice display ICAM-1 expression in pancreatic β-cells.
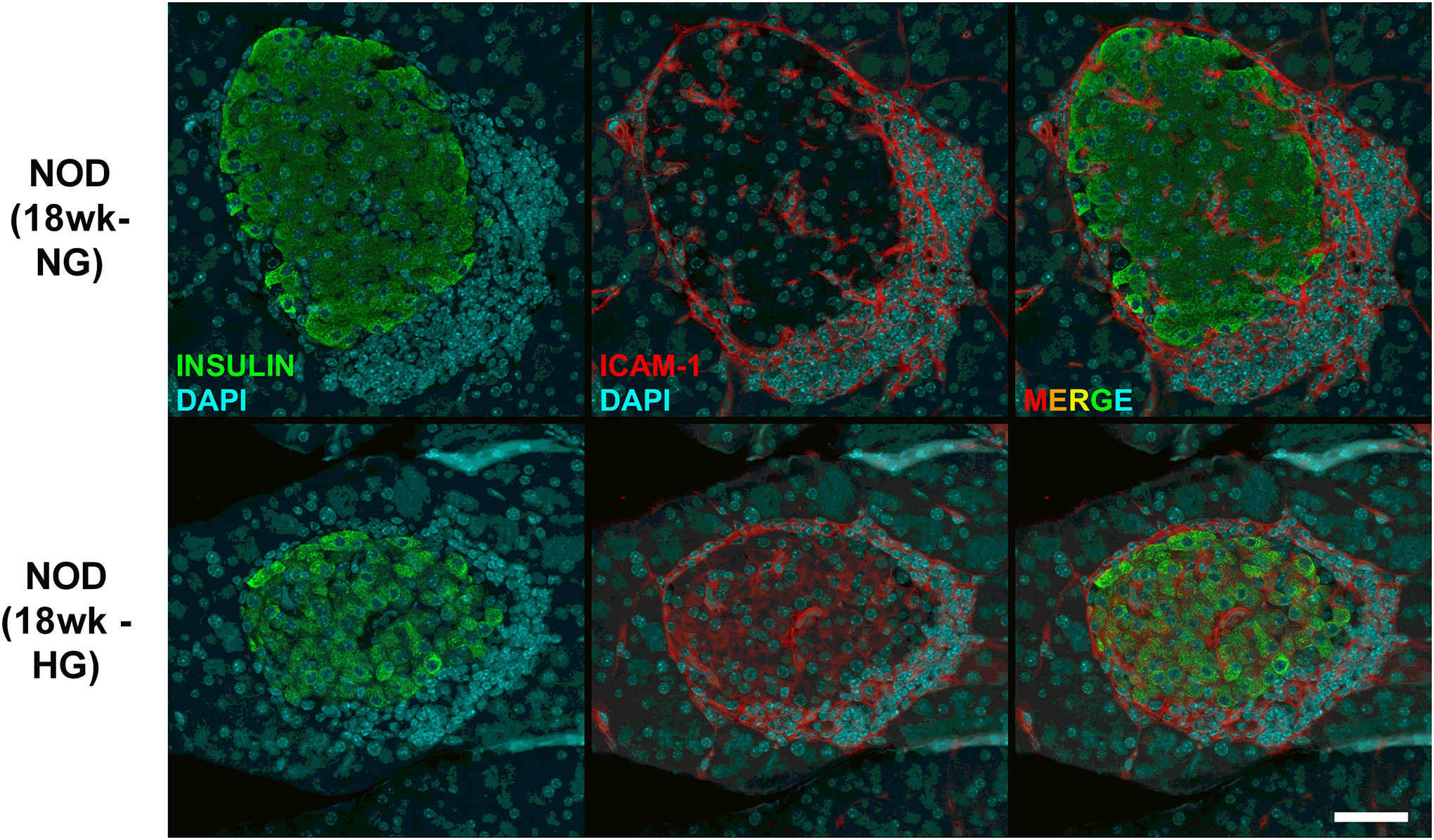
Pancreatic tissue from female NOD mice were collected at 18 weeks of age after two consecutive measurements of fed blood glucose values > 250 mg/dL (HG). Age matched normoglycemic controls (NG) were also collected at the same necropsy. Formalin-fixed paraffin-embedded pancreatic tissue was sectioned and stained using antibodies against insulin (shown in green) and ICAM-1 (shown in red) and imaged using a 40x objective. The scale bar represents 50 microns.
IL-1β increases Icam1 gene promoter activity, mRNA content, and protein abundance in 832/13 cultured rat β-cells.
832/13 cells were exposed to IL-1β for time points ranging from 5 min to 24 h, with maximal expression occurring around 2–4 h (Figure 4A), consistent with elevations at 3 h observed in the RNA-Seq presented in Figure 1. A concentration response demonstrated that the Icam1 gene responds to very small amounts of IL-1β, with as low as 0.01 ng/mL providing 13.6-fold increase in mRNA abundance (Figure 4B). We next investigated the role of interferon-gamma (IFN-γ) to potentiate the response of the Icam1 gene to IL-1β. We found that the addition of IFN-γ increased the expression of Icam1 by 1.63-fold over the IL-1β response (Figure 4C), although this observation did not reach the threshold for statistical significance used in this study. Using luciferase reporters with either −3kb or −1kb regions of the proximal gene promoter, we observed analogous responsiveness to cytokines as the endogenous gene, with the −3kb region, containing greater quantities of key NF-κB and GAS genomic regions, required for the greater response to cytokines. In addition, ICAM-1 protein abundance was also elevated in response to IL-1β and IL-1β plus IFN-γ (Figure 4E). Secretion of ICAM-1 was significantly increased in 832/13 cells following a 6 h exposure to IL-1β (data not shown). The iNOS protein is shown as a known control for cytokine responsiveness (16, 45), while β-actin serves as a control for protein loading. Taken together, these results are consistent with the Icam1 gene responding to β-cell exposure to IL-1β.
Figure 4. IL-1β increases Icam1 gene promoter activity, mRNA content, and protein abundance in 832/13 cultured rat β-cells.
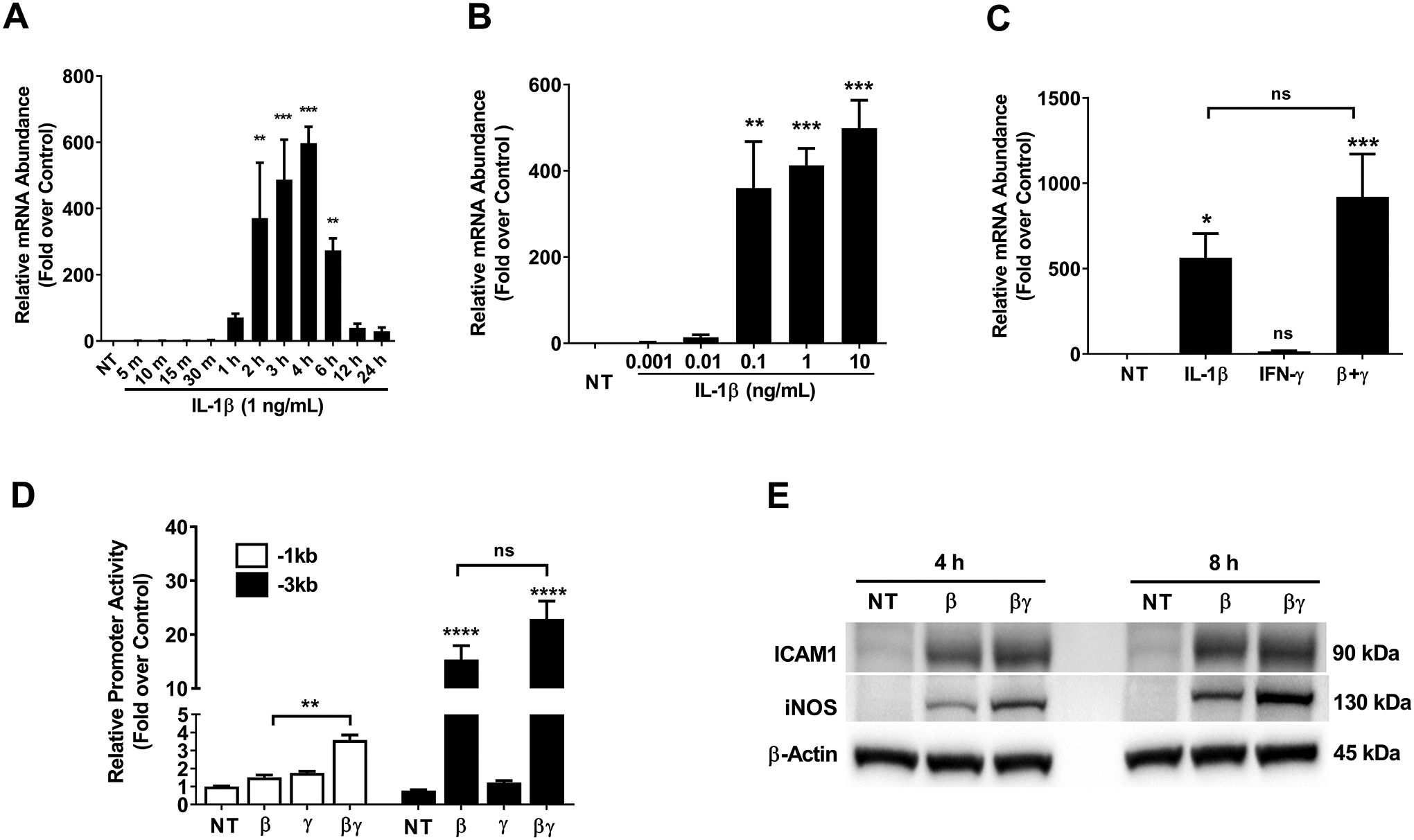
A. 832/13 insulinoma cells were not treated (NT) or stimulated with 1 ng/mL IL-1β for the indicated times. B. 832/13 cells were either untreated (NT) or treated with the indicated concentrations of IL-1β for 3 h. C. 832/13 cells were treated with 1 ng/mL IL-1β, 100 U/mL IFN-γ, or a combination of both cytokines for 3 h. D. 832/13 cells were transfected with Icam1 promoter luciferase constructs containing either −1kb or −3kb of the Icam1 gene promoter. 24 h post-transfection, cells were either untreated or stimulated with 1 ng/mL IL-1β, 100 U/mL IFN-γ, or a combination of both cytokines for 4 h. Data are normalized to the respective NT control for each construct (−1kb and −3kb). E. Cells were untreated or treated with 1 ng/mL IL-1β or a combination of both IL-1β and 100 U/mL IFN-γ for either 4 h or 8 h. β-actin is shown as a control for protein loading. An image representative of two independent experiments is shown. ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05. p values are vs. respective NT controls unless indicated otherwise in figures. ns = not significant.
Interleukin-1 receptor activation uses the NF-κB pathway to support enhanced Icam1 mRNA synthesis.
Islet β-cells display highly enriched expression of the interleukin-1 receptor (IL-1R) relative to other tissues (46, 47). Using islets isolated from mice with pancreatic islet deletion of the IL-1R, we observed a 49% reduction in Icam1 gene expression in response to IL-1β relative to control islets (Figure 5A). We next used TPCA, an inhibitor of the IKK complex downstream of the IL-1R, which reduced the ability of IL-1β to support Icam1 gene expression (Figure 5B). As a complementary approach, we overexpressed a constitutively-active form of IKKβ (CA IKKβ), known to drive NF-κB response genes in β-cells and other tissues (32, 48), coupled with inhibition of NF-κB by a mutated form of the IκBα protein [termed the super-repressor; refs. (32, 49)]. We found that IKKβ alone drove a near 100-fold increase in expression of the Icam1 gene and that the IκBαSR largely suppressed this response (Figure 5C; white bars). In response to IL-1β, CA IKKβ overexpression was redundant, which we interpret to indicate that IL-1R activation is maximal (Figure 5C; black bars). The IκBαSR dose-dependently reduced the expression of the Icam1 gene in response to IL-1β even in the presence of CA IKKβ (Figure 5C; black bars).
Figure 5. Interleukin-1 receptor activation uses the NF-κB pathway to support enhanced Icam1 mRNA synthesis.
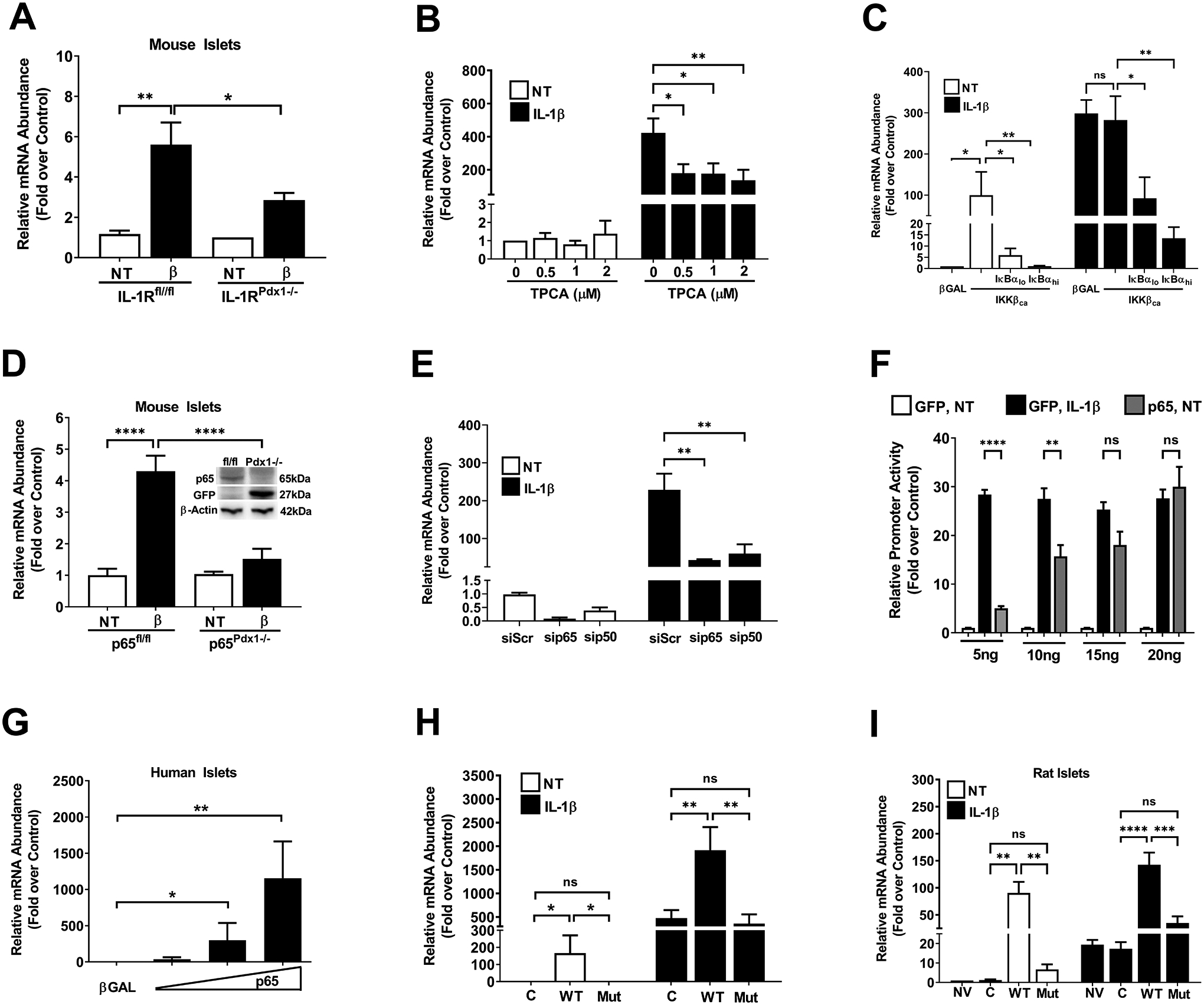
A. Islets isolated from either control mice (IL-1Rfl/fl), or mice with a pancreas-specific deletion of the IL-1R (IL-1RPdx1−/−) were either untreated (NT) or stimulated with 10 ng/mL IL-1β for 3 h (n=5–8 per group). B. 832/13 cells were pre-treated with the indicated concentrations of TPCA for 1 h, followed by 3 h treatment with 1 ng/mL IL-1β. C. 832/13 cells were transduced with either a βGal control adenovirus or adenoviruses overexpressing either caIKKβ alone or a combination of the caIKKβ and IκBαSR. At 18 h post-transduction, cell were either left untreated (white bars) or exposed to 1 ng/mL IL-1β for 3 h (black bars). D. Islets isolated from either control mice (p65fl/fl), or mice with a pancreas-specific deletion of p65 (p65Pdx1−/−), were either NT or exposed to 10 ng/mL IL-1β for 3 h (n=6 per group). Inset shows an immunoblot for p65 and GFP with beta-actin as a loading control. E. 832/13 cells were transfected with siRNA duplexes targeting either the p65 or p50 subunits of NF-κB. At 18 h after transfection, the cells were either left untreated (white bars) or exposed to 1 ng/mL IL-1β for 3 h (black bars). F. 832/13 cells were transfected with 5 ng per well of the −3kB Icam1 promoter-luciferase vector in combination with GFP (control) or the indicated concentrations of p65 plasmid. At 24 h post-transfection, cells were either NT or exposed to 1 ng/mL for 4 h. G. Human islets were transduced with adenovirus expressing either βGal or p65. At 24 h post-transduction, islets were harvested for RNA extraction. H. 832/13 cells were transduced with adenoviruses expressing either βGal, p65, or p65S276A. At 18 h after viral administration, cells were either NT (white bars) or exposed to 1 ng/mL IL-1β (black bars). I. Isolated rat islets were untreated or transduced with adenoviruses expressing either βGal, p65, or p65S276A. At 18 h after viral administration, islets were either NT (white bars) or exposed to 10 ng/mL IL-1β (black bars).
We next used islets isolated from mice with pancreatic islet deletion of the p65 subunit of NF-κB, which is the major transcriptional subunit associated with activation of this pathway. The conditional alleles for p65 also include a mechanism for activating GFP expression using cre-mediated recombination (50). Indeed, we saw that as p65 abundance decreased, GFP protein increased (Figure 5D; inset). In addition, IL-1β was unable to induce expression of the Icam1 gene in islets from the p65pdx1−/− mice (Figure 5D). In 832/13 cells, siRNA-mediated silencing of p65 and p50 subunits of NF-κB also reduced the expression of the Icam1 gene in response to IL-1β (Figure 5E). Because of the clear dependence of the Icam1 gene on NF-κB, we next investigated whether increasing the abundance of p65 activated Icam1 promoter activity to the level induced by exposure to IL-1β. Using a plasmid to deliver increasing expression of p65, we found that promoter luciferase activity was enhanced in a concentration-dependent manner (Figure 5F). We note that a plasmid expressing GFP was used to ensure that all cells received the same concentration of transfected DNA.
Similar to what we observed with cultured 832/13 β-cells, we found that human islets transduced with a recombinant adenovirus expressing p65 also demonstrated a dose-dependent increase in the expression of the Icam1 gene (Figure 5G). We next expressed either wild-type p65 or p65 with a Ser276Ala mutation, a site which regulates transcriptional activity of this NF-κB subunit (51, 52). We found that in 832/13 cells, similar to human islets, p65 drove increased expression of the Icam1 gene in the absence of IL-1β while the p65S276A did not have this ability (Figure 5H; white bars). Moreover, overexpression of p65 augmented expression of the Icam1 gene in response to IL-1β while expressing the p65S276A construct did not (Figure 5H; black bars). Similar results were obtained using isolated rat islets (Figure 5I). Taken together, it is clear that p65 is necessary for the IL-1β induction of the Icam1 gene and that p65 overexpression alone (in the absence of IL-1β) was sufficient to drive increased Icam1 transcript abundance.
Site-directed mutation analyses within the Icam1 gene promoter reveal key genomic elements controlling transcriptional responses.
In silico analyses revealed the presence of several putative response elements, which included four κB elements and one gamma activated sequence (GAS; Figure 6A). These promoter sequences are conserved across mouse, rat, and human genomic DNA. Using a promoter construct with only the GAS element mutated (to prevent STAT1 binding), we found that there was a 25.2% decrease in IL-1β driven promoter activity and a 56.1% decrease in the IL-1β plus IFN-γ response (Figure 6B). This is consistent with other IL-1β responsive genes in β-cells that use STAT1 as an accessory factor to support the IL-1β response as well as signaling input from IFN-γ (15, 16). Because of the clear dependence on the IKK/NF-κB pathway to support Icam1 expression in response to IL-1β, we made mutations in each of the predicted κB elements within the Icam1 gene promoter (noted κB1 – κB4 in Figure 6A). Mutations in the κB1 element reduced IL-1β-mediated activation of the Icam1 promoter by 85.9% (Figure 6C), while mutations to the κB2 element were less severe, producing only a 34.2% decrease (Figure 6D). Moving further distal from the transcriptional start site, mutations in the κB3 element also strongly reduced promoter activity in response to IL-1β (Figure 6E) while κB4 mutations were more modest (Figure 6F). Collectively, we interpret this data to indicate that multiple κB elements within the Icam1 gene promoter support transcriptional responses after exposure to IL-1β.
Figure 6. Site-directed mutation analyses within the Icam1 gene promoter reveal key genomic elements controlling transcriptional responses.
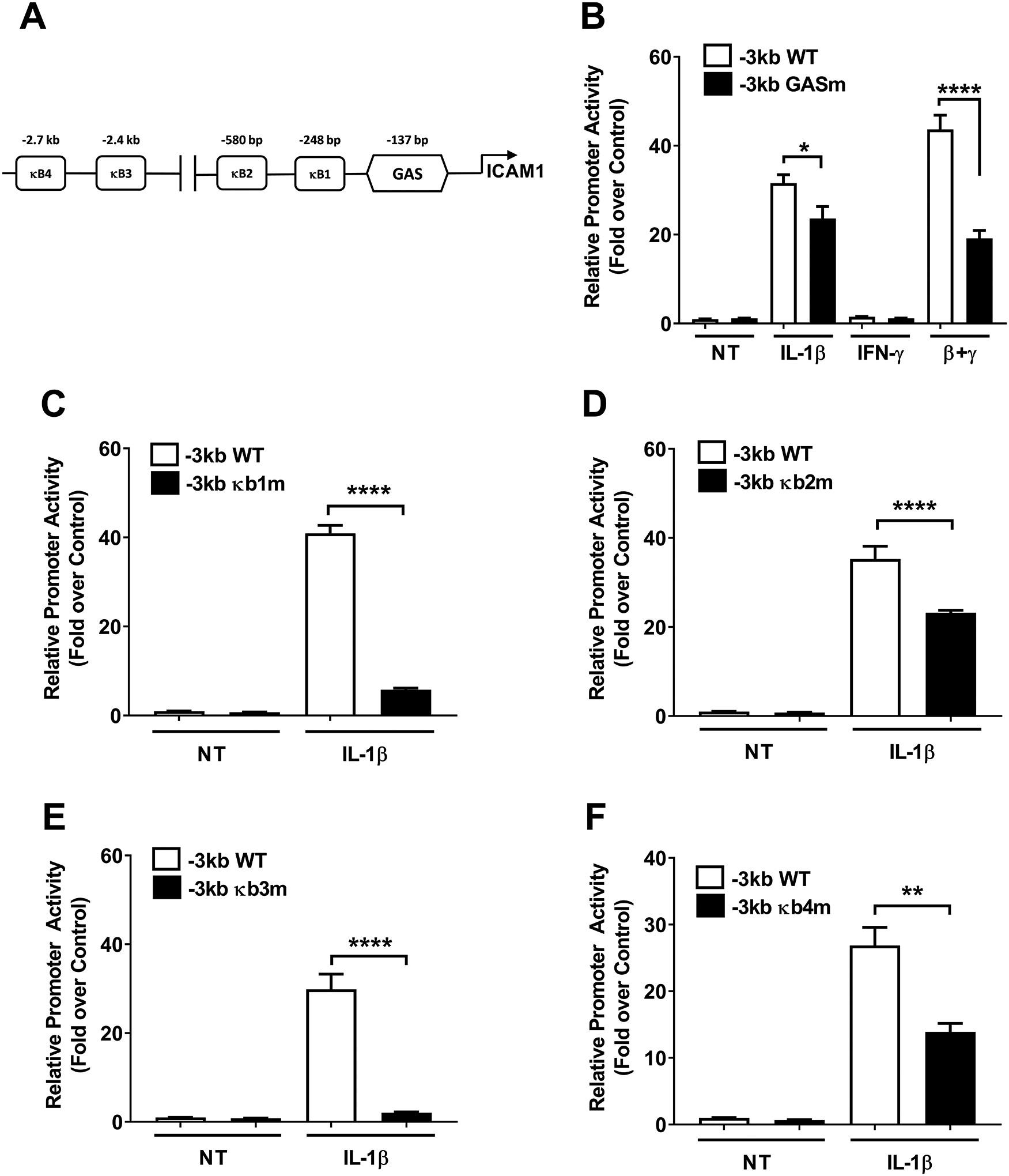
A. Schematic indicating the four computer software predicted NF-κB sites and one GAS element within the −3kb promoter region of the Icam1 gene. B-F. Promoter luciferase data obtained using either wild-type (WT) or promoter constructs with each individual predicted genomic element mutated. 832/13 cells were transfected with promoter luciferase plasmids. At 24 h post-transfection, the cells were either untreated (NT) or exposed to 1 ng/mL IL-1β, 100 U/mL IFN-γ, or both cytokines for 4 h. Data are shown as means of the luciferase signal normalized to the NT control for each group. Error bars represent standard error of the means. * p < 0.05, ** p < 0.01, **** p < 0.0001.
IL-1β induces recruitment of the NF-κB p65 subunit to the Icam1 gene promoter regions containing functional κB elements.
Mutations within the κB1 region of the Icam1 gene promoter strongly reduced transcriptional activity in response to IL-1β (Figure 6C and 6E). We next used chromatin immunoprecipitation (ChIP) assays to determine occupancy of p65 at each of the kB elements within the Icam1 gene; the regions analyzed by ChIP are shown schematically in Figure 7A. Congruent with this finding, we observed occupancy of the κB1 site by p65 after exposure to IL-1β (Figure 7B) with a detectable trend for increased binding within 15 min that increased to 9.2- and 13.2-fold over IgG controls at 30 and 60 min after IL-1β exposure (Figure 7B). The binding of p65 at the κB2 site did not reach statistical significance (Figure 7C). We found that p65 occupancy at the κB3 site was also robust at 30 and 60 min after IL-1β exposure (Figure 7D), while binding at the κB4 site was considerably less proficient (Figure 7E).
Figure 7. IL-1β induces recruitment of the NF-κB p65 subunit to the Icam1 gene promoter regions containing functional κB elements.
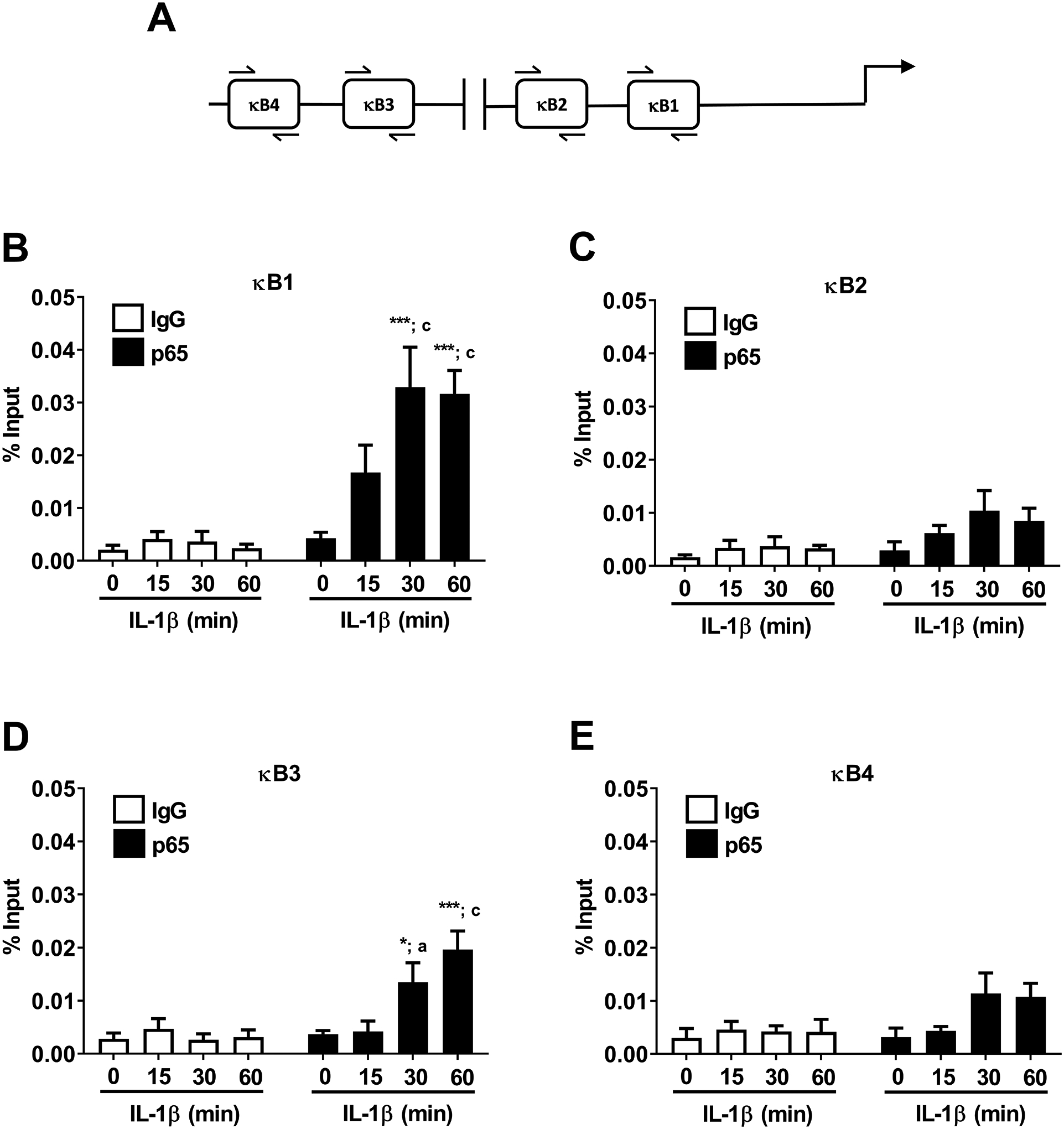
A. Schematic indicating the four NF-κB sites investigated using ChIP assays. B-E. 832/13 cells were untreated (0 min) or stimulated with 1 ng/mL IL-1β for the indicated times. ChIP assays were performed to determine p65 promoter occupancy at the indicated sites. Rabbit IgG was used as a negative control for the immunoprecipitation. Data is represented as percent of input, with 4–5 replicates of each condition. * p < 0.05 vs. no IL-1β treatment for p65 antibody (black bar), *** p < 0.001 vs. no IL-1β treatment for p65 antibody (black bar), a p < 0.05 vs. respective IL-1β treatment for IgG antibody (white bar), c p < 0.001 vs. respective IL-1β treatment for IgG antibody (white bar).
IL-1β promotes phosphorylation of carboxy-terminal domain regions of RNA polymerase II associated with the Icam1 gene promoter and coding regions.
RNA polymerase II displays a large tail region within the C-terminus of the protein that is subjected to phosphorylation during the initiation and elongation stages of transcription (53). The genomic regions analyzed by ChIP are shown schematically in Figure 8A. We found that phosphorylation of Ser5, a marker of transcription initiation, was enhanced at the transcriptional start site of the Icam1 gene promoter after exposure to IL-1β (Figure 8B; green bars). Phosphorylation of Ser2, a marker associated with transcriptional elongation (i.e., moving of the RNA Pol II through the coding region), was not enriched at the transcriptional start site, as would be expected (Figure 8B; red bars). Indeed, after 60 min of IL-1β exposure, there was an 85.5-fold increase in Ser2 phosphorylation over IgG control at the +5kB region (downstream of the transcriptional start site; Figure 8C; red bars), which was maintained out to the +10kB region (Figure 8D; red bars). The overall signal strength for Pol II Ser5, while still present over baseline, was reduced in magnitude at the +5kB and +10kB genomic regions (Figures 8C and 8D; green bars). This is consistent with observations at other cytokine responsive genes in pancreatic β-cells (15) and signal-specific gene regulation in other model systems (54).
Figure 8. IL-1β promotes phosphorylation of carboxy-terminal domain regions of RNA polymerase II associated with the Icam1 gene promoter and coding regions.
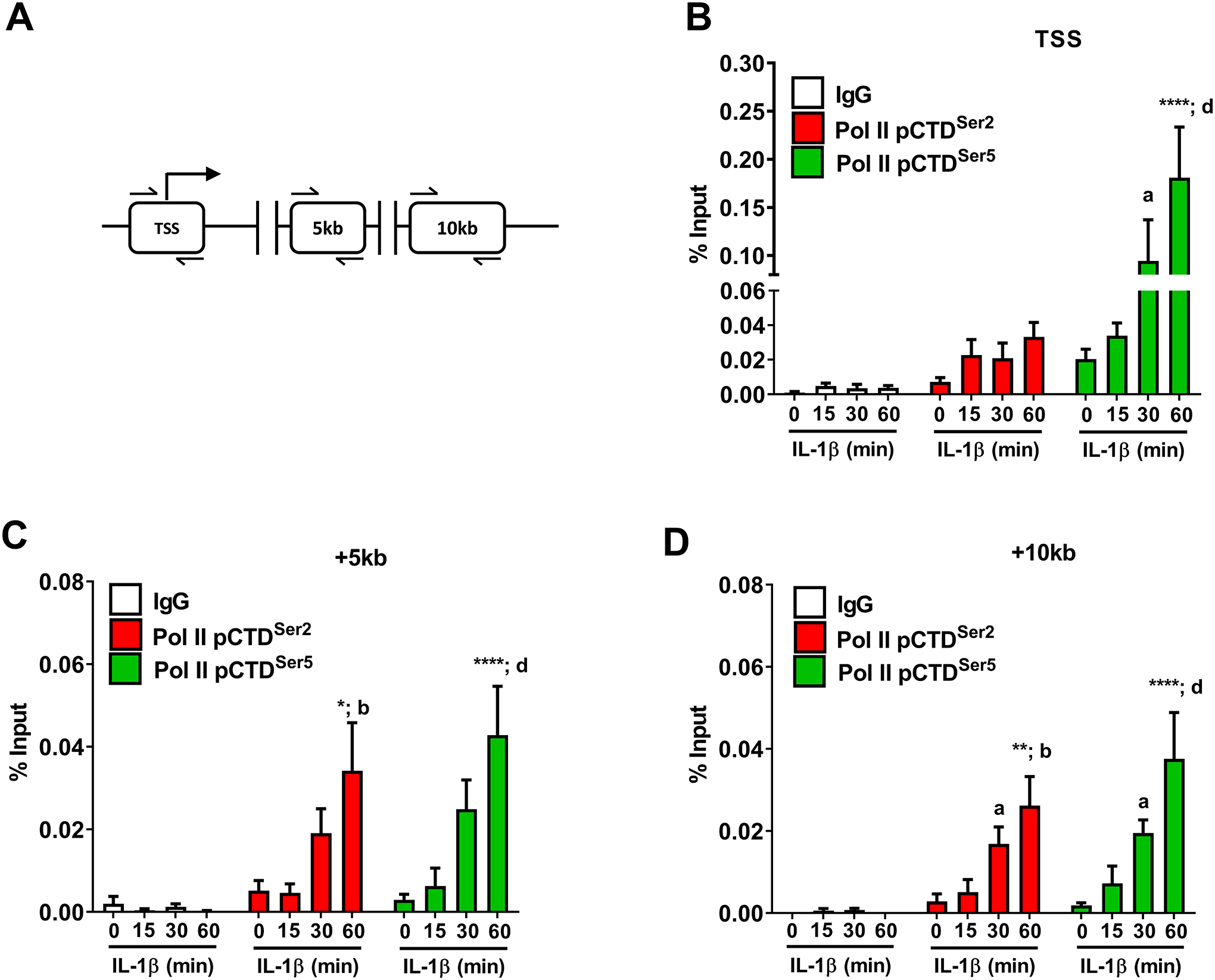
A. Schematic indicating the transcriptional start site (TSS) and two coding regions investigated using ChIP assays. B-D. 832/13 cells were untreated (0 min) or stimulated with 1 ng/mL IL-1β for the indicated times. ChIP assays were conducted to investigate RNA Polymerase II (phospho-S5 or phospho S-2) occupancy for the indicated sites. Rabbit IgG was used as a negative control for the immunoprecipitation. Data is represented as percent of input, with 4 replicates of each condition. *, p < 0.05; **, p < 0.01; ****, p < 0.0001, a p < 0.05, b p < 0.01, d p < 0.0001. Letters compare to same IL-1β treatment time for IgG antibody; asterisks compare to 0 min time-point of respective antibody.
Histone chemical modifications associated with the Icam1 gene are regulated in response to IL-1β.
Signal-specific gene transcription is often associated with alterations in chemical modifications to histones. Modifications of histone H3 at lysine 4 (H3K4) are often found within gene promoters and are typically well conserved across species (55). Here we investigated changes in both mono- (H3K4me1) and tri-methylation (H3K4me3) of H3K4 as well as acetylation of H3K4 (H3K4ac) in the Icam1 gene in response to IL-1β (regions shown schematically in Figure 9A). We found that the H3K3me1 and H3K4me3 signals were not above what was observed for the IgG controls in the genomic region containing the κB3 element while the H3K4ac modification was responsive to IL-1β in this region (Figure 9B; blue bars). The κB1 element displayed robust H3K4me3 signal, which decreased in response to IL-1β (Figure 9C; green bars) while the H3K4ac signal in the region containing κB1 remained similar to that seen in κB3 containing portion of the Icam1 gene promoter (Figure 9C; blue bars). The genomic regions containing the Icam1 transcriptional start site displayed strong enrichment for H3K4me3 and this chemical signature was not affected by IL-1β exposure (Figure 9D; green bars). H3K4 acetylation was enriched over baseline and increased significantly in response to IL-1β (Figure 9D; blue bars). When examining the region of the Icam1 gene 5kb downstream of the transcriptional start site (+5kb), we note that H3K4 acetylation was present in the baseline state (no IL-1β) and stayed enriched during IL-1β exposure (Figure 9E; blue bars). A schematic representation showing p65 occupancy at the genomic regions of greatest occupancy (κB1 and κB3) with associated histone markings associated with the Icam1 gene is given in Figure 10.
Figure 9. Histone chemical modifications associated with the Icam1 gene are regulated in response to IL-1β.
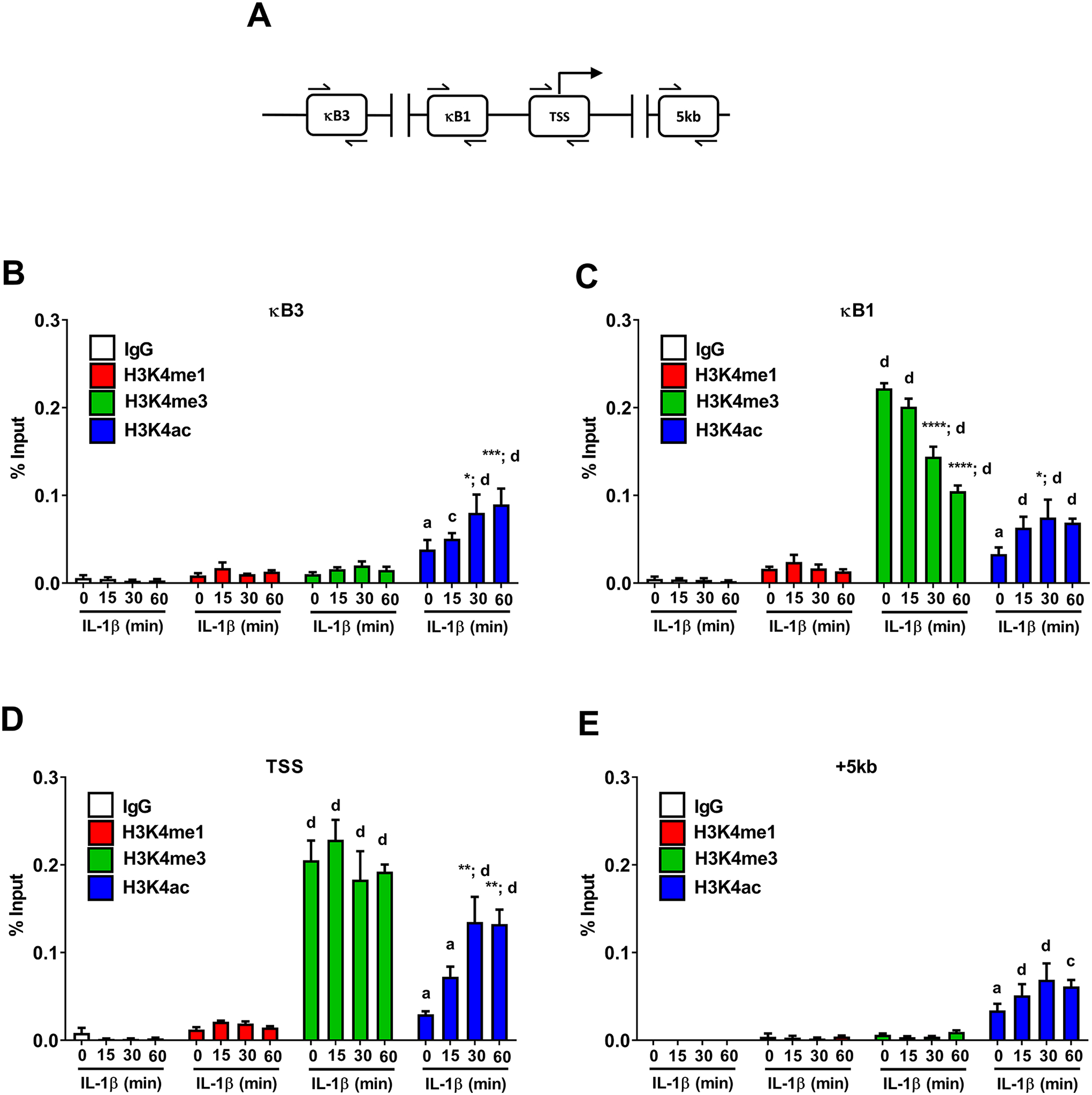
A. Schematic indicating the sites investigated for binding events using ChIP assays. B-E. 832/13 cells were untreated (0 min) or stimulated with 1 ng/mL IL-1β for the indicated times. ChIP assays were used to determine histone chemical modification (H3K4me1, H3K4me3, or H3K4ac) at the indicated sites. Rabbit IgG was used as a negative control. Data is represented as percent of input, with 3–4 replicates of each condition. Letters compare same IL-1β treatment time for IgG antibody; asterisks compare to 0 min time-point of respective antibody. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. a p < 0.05, c p < 0.001, d p < 0.0001.
Figure 10. Schematic representation of the Icam1 transcriptional response to IL-1β.
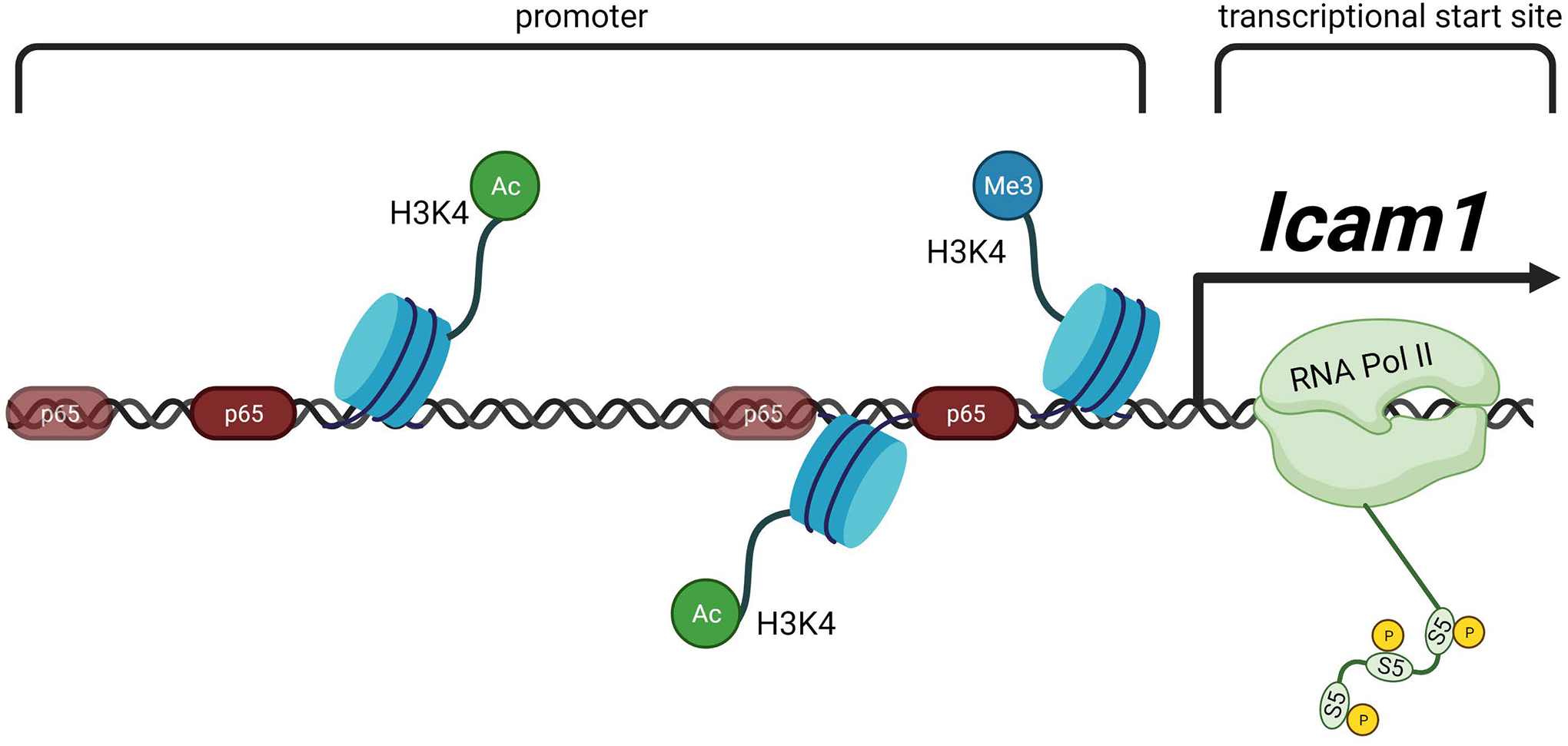
In response to IL-1R activation by IL-1β, β-cells increase the abundance of Icam1 mRNA. This transcriptional activity requires recruitment of the NF-κB p65 transcriptional subunit, histone chemical modifications, and occupancy of RNA polymerase II at sites within the proximal Icam1 gene promoter. Figure created using BioRender.com.
Discussion
The Icam1 gene encodes a membrane-associated protein that contributes to various immune system functions, including trafficking, docking, and signaling-based alterations in leukocyte activity (25, 56). For example, T-cells express the integrin LFA-1, which is composed of CD11a and CD18, and interacts with ICAM proteins, particularly ICAM-1 (22). The activities of both CD4 and CD8 T-cells are influenced through this LFA-1/ICAM-1 interaction (57, 58). Consequently, ICAM-1 is a critical contributor to a multitude of immune cell activities and is thus important for physiological responses. Dysregulation of ICAM-1 expression or the ICAM-1 / LFA-1 interactions also can become pathological and contribute to autoimmune and other diseases. One such autoimmune disease clearly influenced by ICAM-1 is Type 1 diabetes (26).
Many cell types, including pancreatic β-cells, express Icam1; the Icam1 gene is highly responsive to cytokines, with positive regulation by interleukin-1β and interferon-gamma (23, 59). However, many studies investigate the regulation of the Icam1 gene after 18–24 hours of cellular exposure to an inflammatory stimulus, such as a pro-inflammatory cytokine. Because we are interested in early genetic reprogramming events that are likely to change the status of pancreatic β-cells and their interaction with the immune system, we investigated β-cell exposure to IL-1β at much earlier time points. We started with a bulk RNA-seq approach using human islets and 832/13 rat insulinoma cells (Figure 1) and found that Icam1 was an early response gene in both human islets and rat insulinoma cells. Consequently, we report several key discoveries about the early regulation of the Icam1 gene in pancreatic β-cells and also show that the abundance of the ICAM-1 protein is enhanced by both systemic inflammatory signals and in situations of organ-specific autoimmunity.
We first identified Icam1 as highly and rapidly responsive (within 3 hours) to the cytokine interleukin-1β in both rat β-cell line 832/13 and in cultured human islets (Figure 1). Further studies using mouse, rat, and human islets indeed showed that the Icam1 gene was also an early response gene across species after β-cell exposure to IL-1β (Figure 2A, 2B, and 2C). These increases in gene activity in culture can also be recapitulated in vivo using C57BL/6J mice injected with IL-1β, where ICAM-1 protein abundance was greater in pancreatic tissue four hours after cytokine injection (Figure 2D). Further supporting a role for ICAM-1 in situations of autoimmunity, we also found that islets isolated from female NOD mice have increased Icam1 gene expression relative to male mice (Figure 2E). Moreover, hyperglycemic female NOD mice show elevated Icam1 transcripts (Figure 2F) and protein abundance (Figure 3) relative to their age-matched normoglycemic counterparts. Collectively, this data helps to partially explain the sexual dimorphism in NOD mice (females have much higher incidence of disease) and supports the notion that ICAM-1 is a critical contributor to the autoimmune responses that lead to Type 1 diabetes (26). We note that both immune cells and islet β-cells are capable of expressing ICAM-1 in the NOD mouse (Figure 3 and Supplementary Figure 2). This is likely to be an important component of the disease phenotype that will be explored in further detail in future studies.
While it is clear that ICAM-1 participates in the severity of insulitis and eventual onset of diabetes in NOD mice (26), the genetic control of this gene in pancreatic β-cells is incompletely understood. Using a multitude of genetic and molecular approaches, our novel data now reveals that expression of the Icam1 gene is clearly dependent on the NF-κB pathway. Tracing the NF-κB pathway from cell membrane to nucleus demonstrates a reliance on IL-1R signaling to support IL-1β induced-expression (Figure 5A). Pharmacological inhibition of IKK activity reduces IL-1β-mediated Icam1 expression (Figure 5B), while overexpression of constitutively-active IKKβ enhances expression of the Icam1 gene (Figure 5C). In addition, genetic reduction of the p65 subunit of NF-κB reduces expression of the Icam1 gene during exposure to IL-1β (Figure 5D). Furthermore, siRNA-mediated silencing of p65 in 832/13 cells also reduced the ability of IL-1β to induce expression of the Icam1 gene (Figure 5E). We further found that the Icam1 gene promoter (Figure 5F) and human Icam1 gene are sensitive to increases in p65 abundance (Figure 5G). Moreover, eliminating the phosphoacceptor site at position 276 within the p65 subunit also reduced the ability of the Icam1 gene to respond to increases in p65 abundance (Figure 5H and 5I; white bars) as well as to IL-1β (Figure 5H and 5I; black bars). These observations are also consistent with elevated Icam1 expression in NOD mice, a model of autoimmunity, and in a separate model of systemic cytokine signaling (using i.p. injection of IL-1β) which each revealed increases in Icam1 expression in the pancreatic islets (Figure 2D, Figure 2E, Figure 2F, and Figure 3).
We next sought to understand the molecular details underlying genomic control of the Icam1 gene in response to IL-1β. Computer-based prediction software identified several possible κB elements and one GAS element in the −3kb region of the gene promoter (shown schematically in Figure 6A). Site-directed mutagenesis revealed that the κB elements designated κB1 and κB3 contributed strongly to the ability of IL-1β to activate this gene promoter (Figures 6C and 6E). These data are consistent with the observed occupancy of p65 at genomic regions containing these sites (Figures 7B and 7D). Moreover, total RNA polymerase II is recruited to the gene promoter in response to IL-1β prior to the first appearance of transcript (data not shown). In addition, RNA polymerase II phosphorylated at Ser5 within the carboxy terminal domain was present at the transcriptional start site within 30 and 60 minutes after cellular exposure to IL-1β (Figure 8B). This observation is consistent with initiation of gene transcription (54) and with specific appearance of Icam1 mRNA at 2–4 hours after exposure to IL-1β (Figure 4A). Phosphorylation of RNA Pol II at Ser5 increased within the coding region of Icam1 consistent with elongation of transcription (Figures 8C and 8D). We interpret this data to indicate that Icam1 is an important early responsive gene that is transcriptionally activated in β-cells after exposure to IL-1β.
The increase in transcription of the Icam1 gene also correlated with changes in specific histone chemical modifications. Acetylation of histone H3 at lysine 4 (H3K4) increased in the genomic region containing the κB3 element in response to IL-1β (Figure 9B). This change in chemical modification could be to facilitate p65 binding, or alternatively, may occur in response to p65 occupancy and associated assembly of a multi-regulatory transcriptional complex. We further found that the region containing the κB1 element demonstrated greater H3K4 trimethylation (H3K3me3) at baseline, which was reduced in response to IL-1β (Figure 9C). The genomic region containing the transcriptional start site retained H3K4me3 markings during all times IL-1β was present (Figure 9D). We suspect these markings indicate an active gene promoter and that perhaps the biological significance of the reduction in H3K4me3 at the κB1 site indicates RNA Pol II clearance. This could be consistent with H3K4me4 markings retained at the transcriptional start site for anchoring another round of TFIID for re-initiation of successive rounds of transcription (60). Collectively, p65 occupancy, histone chemical modifications, and recruitment of RNA Pol II occur in response to IL-1β to enhance Icam1 transcription in β-cells (Figure 10).
Taken together with existing knowledge that genes encoding chemokines, such as CCL2, CXCL1, CXCL2, CXCL10, and CCL20, are also highly responsive to IL-1β in pancreatic β-cells (10, 15, 17, 18, 32), it is likely that this coordinated program of gene expression (i.e., chemokines plus Icam1) will both recruit and retain immune cells in close proximity to islet β-cells. The dysregulation of this genetic control may lead to overactive chemokine production, enhanced ICAM-1 abundance, and the insulitis observed in T1D. Even if insulitis is mild, as may be true for many cases of T1D in humans (61), the combination of elevated chemokines and ICAM-1 could influence severe targeting of β-cells by antigen-primed leukocytes that leads to reduced insulin secretion and eventual decreases in total numbers of β-cells. Thus, strategies aimed at a greater understanding of the molecular events that lead to dysregulated crosstalk between β-cells and immune cells will provide greater understanding of the T1D disease process.
Supplementary Material
Key Points:
Icam1 mRNA is elevated in islets from female versus male NOD mice
ICAM-1 protein abundance is greater in hyperglycemic female NOD mice
IL-1β rapidly upregulates expression of the Icam1 gene in β-cells
Acknowledgments
We thank Dr. Danhong Lu (Duke University) and Kristin Rohli (PBRC) for technical assistance.
Funding
This work was supported by grants R01 DK123183, R03 AI151920, P20 GM135002, P30 GM118430, P30 DK072476, and U54 GM104940 obtained from the National Institutes of Health. This project also used the Cell Biology and Bioimaging Core that is supported in part by COBRE (P20 GM135002 & P20 GM103528) and NORC (P30 DK072476) center grants from the National Institutes of Health.
Footnotes
Competing Interests
The authors have no conflicts of interest to declare.
References
- 1.Atkinson MA, Eisenbarth GS, and Michels AW. 2014. Type 1 diabetes. Lancet 383: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castano L, and Eisenbarth GS. 1990. Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol 8: 647–679. [DOI] [PubMed] [Google Scholar]
- 3.Eisenbarth GS 1986. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 314: 1360–1368. [DOI] [PubMed] [Google Scholar]
- 4.Ize-Ludlow D, Lightfoot YL, Parker M, Xue S, Wasserfall C, Haller MJ, Schatz D, Becker DJ, Atkinson MA, and Mathews CE. 2011. Progressive erosion of beta-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes 60: 2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mandrup-Poulsen T 1996. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 39: 1005–1029. [DOI] [PubMed] [Google Scholar]
- 6.Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, and Svenson M. 1986. Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science 232: 1545–1547. [DOI] [PubMed] [Google Scholar]
- 7.Campbell IL, Wong GH, Schrader JW, and Harrison LC. 1985. Interferon-gamma enhances the expression of the major histocompatibility class I antigens on mouse pancreatic beta cells. Diabetes 34: 1205–1209. [DOI] [PubMed] [Google Scholar]
- 8.Collier JJ, Sparer TE, Karlstad MD, and Burke SJ. 2017. Pancreatic islet inflammation: an emerging role for chemokines. J Mol Endocrinol 59: R33–R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke SJ, and Collier JJ. 2015. Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation? Biomolecules 5: 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarkar SA, Lee CE, Victorino F, Nguyen TT, Walters JA, Burrack A, Eberlein J, Hildemann SK, and Homann D. 2012. Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes 61: 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baggiolini M 1998. Chemokines and leukocyte traffic. Nature 392: 565–568. [DOI] [PubMed] [Google Scholar]
- 12.Godessart N, and Kunkel SL. 2001. Chemokines in autoimmune disease. Curr Opin Immunol 13: 670–675. [DOI] [PubMed] [Google Scholar]
- 13.Burke SJ, Karlstad MD, Eder AE, Regal KM, Lu D, Burk DH, and Collier JJ. 2016. Pancreatic beta-Cell production of CXCR3 ligands precedes diabetes onset. BioFactors 42: 703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke SJ, Stadler K, Lu D, Gleason E, Han A, Donohoe DR, Rogers RC, Hermann GE, Karlstad MD, and Collier JJ. 2015. IL-1beta reciprocally regulates chemokine and insulin secretion in pancreatic beta-cells via NF-kappaB. Am J Physiol Endocrinol Metab 309: E715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burke SJ, Lu D, Sparer TE, Masi T, Goff MR, Karlstad MD, and Collier JJ. 2014. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab 306: E131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke SJ, Updegraff BL, Bellich RM, Goff MR, Lu D, Minkin SC Jr., Karlstad MD, and Collier JJ. 2013. Regulation of iNOS Gene Transcription by IL-1beta and IFN-gamma Requires a Coactivator Exchange Mechanism. Mol Endocrinol 27: 1724–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke SJ, Goff MR, Lu D, Proud D, Karlstad MD, and Collier JJ. 2013. Synergistic Expression of the CXCL10 Gene in Response to IL-1beta and IFN-gamma Involves NF-kappaB, Phosphorylation of STAT1 at Tyr701, and Acetylation of Histones H3 and H4. J Immunol 191: 323–336. [DOI] [PubMed] [Google Scholar]
- 18.Burke SJ, Karlstad MD, Regal KM, Sparer TE, Lu D, Elks CM, Grant RW, Stephens JM, Burk DH, and Collier JJ. 2015. CCL20 is elevated during obesity and differentially regulated by NF-kappaB subunits in pancreatic beta-cells. Biochim Biophys Acta 1849: 637–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke SJ, Lu D, Sparer TE, Karlstad MD, and Collier JJ. 2014. Transcription of the gene encoding TNF-alpha is increased by IL-1beta in rat and human islets and beta-cell lines. Mol Immunol 62: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallberg M, and Cooke A. 2013. Immune mechanisms in type 1 diabetes. Trends Immunol 34: 583–591. [DOI] [PubMed] [Google Scholar]
- 21.Lehuen A, Diana J, Zaccone P, and Cooke A. 2010. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 10: 501–513. [DOI] [PubMed] [Google Scholar]
- 22.Makgoba MW, Sanders ME, Ginther Luce GE, Dustin ML, Springer TA, Clark EA, Mannoni P, and Shaw S. 1988. ICAM-1 a ligand for LFA-1-dependent adhesion of B, T and myeloid cells. Nature 331: 86–88. [DOI] [PubMed] [Google Scholar]
- 23.Dustin ML, Rothlein R, Bhan AK, Dinarello CA, and Springer TA. 1986. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J Immunol 137: 245–254. [PubMed] [Google Scholar]
- 24.Springer TA 1990. Adhesion receptors of the immune system. Nature 346: 425–434. [DOI] [PubMed] [Google Scholar]
- 25.Long EO 2011. ICAM-1: getting a grip on leukocyte adhesion. J Immunol 186: 5021–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin S, van den Engel NK, Vinke A, Heidenthal E, Schulte B, and Kolb H. 2001. Dominant role of intercellular adhesion molecule-1 in the pathogenesis of autoimmune diabetes in non-obese diabetic mice. J Autoimmun 17: 109–117. [DOI] [PubMed] [Google Scholar]
- 27.Anderson ME, and Siahaan TJ. 2003. Targeting ICAM-1/LFA-1 interaction for controlling autoimmune diseases: designing peptide and small molecule inhibitors. Peptides 24: 487–501. [DOI] [PubMed] [Google Scholar]
- 28.Katz SM, Bennett F, Stecker K, Clark JH, Pham T, Wang ME, Kahan BD, and Stepkowski SM. 2000. ICAM-1 antisense oligodeoxynucleotide improves islet allograft survival and function. Cell Transplant 9: 817–828. [DOI] [PubMed] [Google Scholar]
- 29.Balasa B, La Cava A, Van Gunst K, Mocnik L, Balakrishna D, Nguyen N, Tucker L, and Sarvetnick N. 2000. A mechanism for IL-10-mediated diabetes in the nonobese diabetic (NOD) mouse: ICAM-1 deficiency blocks accelerated diabetes. J Immunol 165: 7330–7337. [DOI] [PubMed] [Google Scholar]
- 30.Hasegawa Y, Yokono K, Taki T, Amano K, Tominaga Y, Yoneda R, Yagi N, Maeda S, Yagita H, Okumura K, and et al. 1994. Prevention of autoimmune insulin-dependent diabetes in non-obese diabetic mice by anti-LFA-1 and anti-ICAM-1 mAb. Int Immunol 6: 831–838. [DOI] [PubMed] [Google Scholar]
- 31.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, and Newgard CB. 2000. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49: 424–430. [DOI] [PubMed] [Google Scholar]
- 32.Burke SJ, Goff MR, Updegraff BL, Lu D, Brown PL, Minkin SC Jr., Biggerstaff JP, Zhao L, Karlstad MD, and Collier JJ. 2012. Regulation of the CCL2 Gene in Pancreatic beta-Cells by IL-1beta and Glucocorticoids: Role of MKP-1. PLoS One 7: e46986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collier JJ, Batdorf HM, Martin TM, Rohli KE, Burk DH, Lu D, Cooley CR, Karlstad MD, Jackson JW, Sparer TE, Zhang J, Mynatt RL, and Burke SJ. 2021. Pancreatic, but not myeloid-cell, expression of interleukin-1alpha is required for maintenance of insulin secretion and whole body glucose homeostasis. Molecular metabolism 44: 101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burke SJ, Batdorf HM, Eder AE, Karlstad MD, Burk DH, Noland RC, Floyd ZE, and Collier JJ. 2017. Oral Corticosterone Administration Reduces Insulitis but Promotes Insulin Resistance and Hyperglycemia in Male Nonobese Diabetic Mice. Am J Pathol 187: 614–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, Baranasic D, Arenillas DJ, Sandelin A, Vandepoele K, Lenhard B, Ballester B, Wasserman WW, Parcy F, and Mathelier A. 2018. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46: D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, and Alba MM. 2002. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18: 333–334. [DOI] [PubMed] [Google Scholar]
- 37.Burke SJ, Batdorf HM, Burk DH, Noland RC, Eder AE, Boulos MS, Karlstad MD, and Collier JJ. 2017. db/db Mice Exhibit Features of Human Type 2 Diabetes That Are Not Present in Weight-Matched C57BL/6J Mice Fed a Western Diet. Journal of diabetes research 2017: 8503754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salbaum JM, Kruger C, MacGowan J, Herion NJ, Burk D, and Kappen C. 2015. Novel Mode of Defective Neural Tube Closure in the Non-Obese Diabetic (NOD) Mouse Strain. Scientific reports 5: 16917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumura H, Kruger DH, Kahl G, and Terauchi R. 2015. SuperSAGE as an analytical tool for host and viral gene expression. Methods Mol Biol 1236: 181–195. [DOI] [PubMed] [Google Scholar]
- 40.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiner A, Yekutieli D, and Benjamini Y. 2003. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19: 368–375. [DOI] [PubMed] [Google Scholar]
- 43.Collier JJ, Fueger PT, Hohmeier HE, and Newgard CB. 2006. Pro- and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and beta-cell lines. Diabetes 55: 1398–1406. [DOI] [PubMed] [Google Scholar]
- 44.Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, Item F, Vallois D, Pattou F, Kerr-Conte J, Lavallard V, Berney T, Thorens B, Konrad D, Boni-Schnetzler M, and Donath MY. 2017. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol 18: 283–292. [DOI] [PubMed] [Google Scholar]
- 45.Heitmeier MR, Scarim AL, and Corbett JA. 1997. Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J Biol Chem 272: 13697–13704. [DOI] [PubMed] [Google Scholar]
- 46.Scarim AL, Arnush M, Hill JR, Marshall CA, Baldwin A, McDaniel ML, and Corbett JA. 1997. Evidence for the presence of type I IL-1 receptors on beta-cells of islets of Langerhans. Biochim Biophys Acta 1361: 313–320. [DOI] [PubMed] [Google Scholar]
- 47.Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, Kerr-Conte J, Pattou F, Ehses JA, Schuit FC, and Donath MY. 2009. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150: 5218–5229. [DOI] [PubMed] [Google Scholar]
- 48.Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, and Xu H. 2012. Constitutive activation of IKKbeta in adipose tissue prevents diet-induced obesity in mice. Endocrinology 153: 154–165. [DOI] [PubMed] [Google Scholar]
- 49.Jobin C, Panja A, Hellerbrand C, Iimuro Y, Didonato J, Brenner DA, and Sartor RB. 1998. Inhibition of proinflammatory molecule production by adenovirus-mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J Immunol 160: 410–418. [PubMed] [Google Scholar]
- 50.Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, and Klein U. 2014. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med 211: 2103–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong H, Voll RE, and Ghosh S. 1998. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1: 661–671. [DOI] [PubMed] [Google Scholar]
- 52.Prasad RC, Wang XL, Law BK, Davis B, Green G, Boone B, Sims L, and Law M. 2009. Identification of genes, including the gene encoding p27Kip1, regulated by serine 276 phosphorylation of the p65 subunit of NF-kappaB. Cancer Lett 275: 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phatnani HP, and Greenleaf AL. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 20: 2922–2936. [DOI] [PubMed] [Google Scholar]
- 54.Hsin JP, and Manley JL. 2012. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26: 2119–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ 3rd, Gingeras TR, Schreiber SL, and Lander ES. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181. [DOI] [PubMed] [Google Scholar]
- 56.Marlin SD, and Springer TA. 1987. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 51: 813–819. [DOI] [PubMed] [Google Scholar]
- 57.Dang LH, Michalek MT, Takei F, Benaceraff B, and Rock KL. 1990. Role of ICAM-1 in antigen presentation demonstrated by ICAM-1 defective mutants. J Immunol 144: 4082–4091. [PubMed] [Google Scholar]
- 58.Salomon B, and Bluestone JA. 1998. LFA-1 interaction with ICAM-1 and ICAM-2 regulates Th2 cytokine production. J Immunol 161: 5138–5142. [PubMed] [Google Scholar]
- 59.Vives M, Soldevila G, Alcalde L, Lorenzo C, Somoza N, and Pujol-Borrell R. 1991. Adhesion molecules in human islet beta-cells. De novo induction of ICAM-1 but not LFA-3. Diabetes 40: 1382–1390. [DOI] [PubMed] [Google Scholar]
- 60.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, and Timmers HT. 2007. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131: 58–69. [DOI] [PubMed] [Google Scholar]
- 61.Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, and Atkinson MA. 2016. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 65: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.