

Abstract
For improved control of biomaterial property design, a better understanding of complex coacervation involving anionic polysaccharides and proteins is needed. Here, we address the initial steps in condensate formation of β‐lactoglobulin A (β‐LgA) with nine defined alginate oligosaccharides (AOSs) and describe their multivalent interactions in structural detail. Binding of AOSs containing four, five, or six uronic acid residues (UARs), either all mannuronate (M), all guluronate (G), or alternating M and G embodying the block structural components of alginates, was characterized by isothermal titration calorimetry, nuclear magnetic resonance spectroscopy (NMR), and molecular docking. β‐LgA was highly multivalent exhibiting binding stoichiometries decreasing from five to two AOSs with increasing degree of polymerization (DP) and similar affinities in the mid micromolar range. The different AOS binding sites on β‐LgA were identified by NMR chemical shift perturbation analyses and showed diverse compositions of charged, polar and hydrophobic residues. Distinct sites for the shorter AOSs merged to accommodate longer AOSs. The AOSs bound dynamically to β‐LgA, as concluded from saturation transfer difference and 1H‐ligand‐targeted NMR analyses. Molecular docking using Glide within the Schrödinger suite 2016‐1 revealed the orientation of AOSs to only vary slightly at the preferred β‐LgA binding site resulting in similar XP glide scores. The multivalency coupled with highly dynamic AOS binding with lack of confined conformations in the β‐LgA complexes may help explain the first steps toward disordered β‐LgA alginate coacervate structures.
Keywords: complex coacervation, nuclear magnetic resonance, percolation, phase separation, protein carbohydrate interaction, small molecule binding
1. INTRODUCTION
Liquid–liquid‐phase separation (LLPS) describes a reversible emulsoid stage existing between a solution and condensed structures, where coacervates are formed, leading to a thermodynamic co‐existence of a dense and a dilute phase (Nakashima et al., 2019; Wang et al., 2021). Recently, coacervations and LLPS have been described in detail for multivalent proteins interacting with other proteins, RNA, or DNA (Mimura et al., 2021; Sottini et al., 2020; Wang et al., 2021). Related to this, interactions between anionic polysaccharides and positively charged proteins, coined complex coacervation (Timilsena et al., 2019), result in liquid–solid phase separation. Both the initial step of complex coacervation by formation of molecular condensates, the coacervation process and the resulting particles are poorly understood at the structural level. For the commonly used model system of β‐lactoglobulin (β‐Lg) and alginate, the polymer complexes are vaguely described to be without systematic order as based on microscopy, rheology, turbidity and ITC analyses (Gorji et al., 2018; Qomarudin et al., 2015). Bovine β‐Lg constitutes more than 50% of all protein in whey (Creamer et al., 2011) and exist in three isoforms, β‐LgA, β‐LgB, and β‐LgC of which β‐LgA is the most studied (Farrell et al., 2004). β‐Lg adopts a lipocalin fold composed of nine β‐strands (A‐I) and two α‐helices (Uhrínová et al., 2000). β‐strands A‐H jointly form an inner cavity, called the calyx, characteristic for this protein family. β‐Lg is monomeric at pH below three but forms dimers at pH 3–8 (Khan et al., 2018) with β‐strand I and the flexible AB loop constituting the dimer interface (Khan et al., 2018; Sakurai & Goto, 2002). The hydrophobic calyx is well‐known to host a variety of guest molecules (Gutiérrez‐Magdaleno et al., 2013; Loch et al., 2012). Notably, β‐LgA was recently found to bind oligosaccharides on the surface in the acidic pH range where β‐Lg is monomeric (Birch et al., 2021; Stender et al., 2019). β‐Lg has been shown to either self‐coacervate induced by ethylene glycol, monosaccharides or amino acids (Zhang et al., 2021), or to coacervate with other proteins such as lactoferrin (Yan et al., 2013). Further, complex coacervation has been achieved with a number of carbohydrate polymers, for example, acacia gum, κ‐carrageenan, and alginates (Hosseini et al., 2013; Madsen et al., 2021; Sanchez et al., 2006). In the current understanding, electrostatic interactions govern alginate and β‐LgA complex coacervation (Gorji et al., 2018; Qomarudin et al., 2015; Stender et al., 2018).
Alginates are linear 1,4‐linked anionic polysaccharides consisting of two uronic acid residues (UARs), β‐D‐mannuronate (M) and α‐L‐guluronate (G) (Craigie et al., 1984). It is found in cell walls of brown macroalgae and secreted by certain bacteria (Gacesa, 1998; Stokke et al., 2000). α‐L‐guluronate residues, the C‐5 epimer of β‐D‐mannuronate, are generated at the polymer level by the action of mannuronan C‐5 epimerases (Valla et al., 2001), resulting in linear anionic polysaccharides of varying degree of polymerization (DP) composed of M‐, MG‐, and G‐blocks (Figure 1a) (Gawin et al., 2020). Due to their physicochemical characteristics, alginates are widely applied as gelation and thickening agents and in formation of protein complexes in the food and pharma sectors (Guo et al., 2020; Wu et al., 2013). β‐LgA was recently shown to bind alginate stretches of 8 to 22 UARs, depending on the M/G content (Madsen et al., 2021; Stender et al., 2018). The two trisaccharides, ΔGG and ΔMG, with the alginate lyase produced unsaturated 4‐deoxy‐α‐L‐erythro‐hex‐4‐enopyranuronic acid (Δ) residue at the non‐reducing end, have by NMR been shown to form contacts with lysines at two sites on β‐LgA. With only few residue contacts determined, the understanding of early β‐LgA alginate condensate states was not fully achieved (Stender et al., 2019).
FIGURE 1.
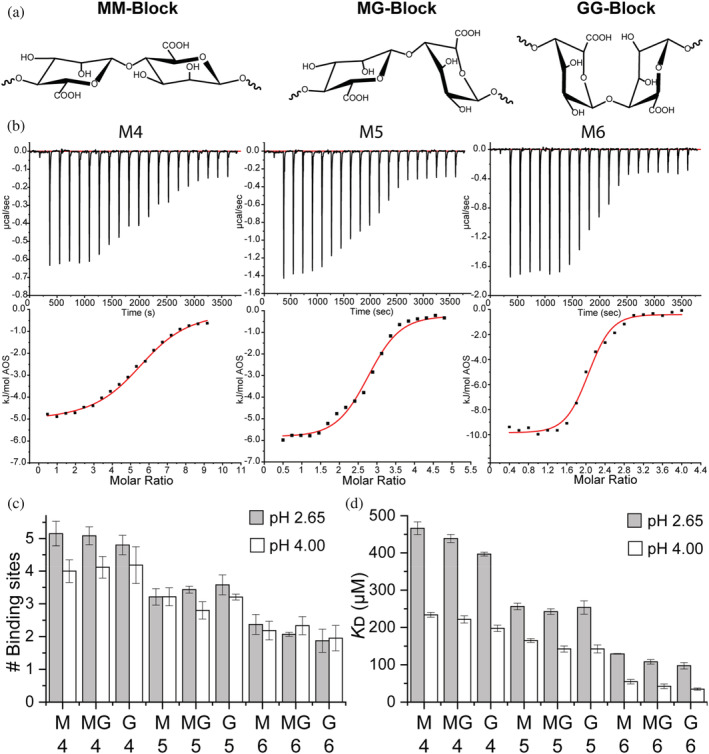
Chemical structures of alginate oligosaccharides (AOSs) and ITC titration of 500 μM β‐LgA with different AOSs. (a) M‐block: 1,4‐linked β‐D‐mannuronic acids, MG‐block: Alternating 1,4‐linked β‐D‐mannuronic and α‐L‐guluronic acids. G‐block: 1,4‐linked α‐L‐guluronic acids. (b) Raw baseline‐corrected ITC data (top) with enthalpograms (bottom) obtained for M4–M6 at pH 2.65. Fitted curves are in red. (c) Number of AOS binding sites on monomeric β‐LgA, calculated from the fit of the corresponding ITC curve fits. (d) K D calculated from the fit in (a). All titrations were performed in triplicate. Errors are SEM
To understand the amorphicity of complex coacervate structures, the pathway of coacervation, and to improve the use and design of alginate―β‐Lg coacervates for microencapsulation, we investigated complex structures of β‐LgA with longer defined AOSs of pure M, G, and MG blocks (Figure 1a). These complex structures may represent the early steps in the multivalent interaction of β‐LgA with alginate that leads to coacervation. By integration of data from ITC, protein‐ and ligand‐based NMR, and MD docking, we could identify new extended binding sites on the β‐LgA surface. These data provided insights into the contact freedom of AOSs on the surface of β‐LgA, which may allow for many different early condensate states driving the formation of amorphous over ordered coacervate structures.
2. RESULTS
2.1. β‐LgA complexes depend on AOS DP and pH
Complex coacervation between proteins and anionic polysaccharides relies on multivalent interactions. As pKa values of M and G are 3.4 and 3.7 (Draget et al., 1994), respectively, and β‐LgA becomes monomeric when pH approaches 2 (Taulier & Chalikian, 2001), affinity and stoichiometry of the AOS's binding to β‐LgA were determined at pH 2.65 and 4.00 by ITC (Figure 1b) using the same conditions (50 mM KH2PO4) as for NMR assignment of β‐LgA (Stender et al., 2019; Uhrínová et al., 1998). At pH 2.65 and 4.00, AOS /β‐LgA stoichiometry varied from 5.2 ± 0.4 to 1.9 ± 0.4, and from 4.2 ± 0.3 to 2.0 ± 0.4, respectively, decreasing significantly with increasing size of the AOSs (DP4–DP6) (p < 0.05) (Figure 1c, Table S1). This corresponds to a total of 12–20 bound UARs, represented by the different AOSs at the respective pH values where the ligands essentially carry a low and a high negative charge. The stoichiometry only varied for DP4, with β‐LgA binding 5.0 ± 0.2 AOSs at pH 2.65 and 4.1 ± 0.1 at pH 4.00 (Figure 1c). Docking experiments, discussed in more detail in Section 2.2 and Data S3, were performed on β‐LgA structures prepared either at pH 4.00 or pH 2.65 (Shelley et al., 2007). Docking yielded XP glide scores (XP Gscores) of the AOSs, which were either lower or larger at pH 2.65 than at pH 4.00, thus indicating that not all β‐LgA surfaces, which are favorable for binding uncharged AOSs (pH 2.65), are favorable for binding negatively charged AOSs (pH 4.00) (Table 1).
TABLE 1.
Results from molecular docking of the different AOSs at all sites at either pH 2.65 or pH 4.00. The XP glide scores are given in kcal/mol
| pH 2.65 | pH 4.00 | pH 2.65 | pH 4.00 | pH 2.65 | pH 4.00 | pH 2.65 | pH 4.00 | |
|---|---|---|---|---|---|---|---|---|
| Site 1 | Site 1 | Site 2 | Site 2 | Site 3 | Site 3 | Site 4 | Site 4 | |
| M4 | −6.4 | −12.2 | −10.1 | −6.1 | −6.3 | −9.0 | −8.3 | −12.1 |
| M5 | −9.8 | −12.1 | −11.8 | −7.9 | −9.8 | −12.7 | −8.4 | −10.6 |
| M6 | −8.3 | −14.1 | −13.1 | −10.6 | −8.0 | −12.4 | −9.2 | −13.2 |
| MG4 | −7.6 | −12.7 | −8.1 | −8.5 | −7.0 | −12.1 | −7.6 | −10.0 |
| MG5 | −9.4 | −10.8 | −8.9 | −9.5 | −8.6 | −12.9 | −10.3 | −11.3 |
| MG6 | −9.0 | −16.5 | −12.0 | −8.7 | −10.6 | −13.5 | −10.8 | −15.0 |
| G4 | −8.6 | −12.1 | −8.8 | −7.8 | −6.6 | −10.9 | −7.5 | −10.9 |
| G5 | −11.5 | −12.1 | −11.1 | −11.9 | −9.4 | −9.7 | −10.4 | −9.6 |
| G6 | −9.3 | −15.1 | −12.1 | −7.8 | −9.7 | −14.3 | −10.3 | −12.5 |
K D values decreased three–fourfold for AOSs of DP4 through DP6, regardless of the M, MG, or G content, and were in most cases around twofold higher at pH 2.65 than at pH 4.00 (p < 0.0001) (Figure 1d, Table S1). At pH 4.0, K D decreased from 234 ± 7 to 55 ± 6 μM for M, 222 ± 10 to 42 ± 6 μM for MG, and 198 ± 9 to 35 ± 3 μM for G AOSs, with the same effect being observed at pH 2.65 (Figure 1d; Table S1). At pH 2.65 the enthalpy change (ΔH) decreased from −5 (6) ± 0.5 to −10 ± 0.6 kJ/(mol AOS) for binding M4 (MG,G) through M6 (MG,G) but at pH 4.00, ΔH for M4–M6 became more exothermic from −18.0 ± 0.7 to −29.0 ± 0.1 kJ/mol and for both MG and G types from −23.0 ± 0.2 to −31.0 ± 0.3 kJ/mol (Figure S1). Plotting enthalpy changes against the theoretical number of charges per AOS (calculated from pK a values of M and G monomers, 3.38 and 3.65, respectively) at both pH 2.65 and pH 4.00 revealed a linear correlation (R 2 = 0.99) (Figure 2a). Thus, the enthalpic contribution comes from the mutual interaction of opposite charges on the two molecules, with the negative charges on AOS being the limiting factor. The persistence of the interaction at pH 2.65, driven by entropy, stems from, for example, hydrophobicity, release of counter ions or water molecules, and polar interactions. Docking XP Gscores of AOSs on β‐LgA at pH 2.65 and pH 4.00 agreed with ITC, showing lower energies for certain poses at pH 4.00, being dependent on AOS length, but not its composition (Table 1).
FIGURE 2.
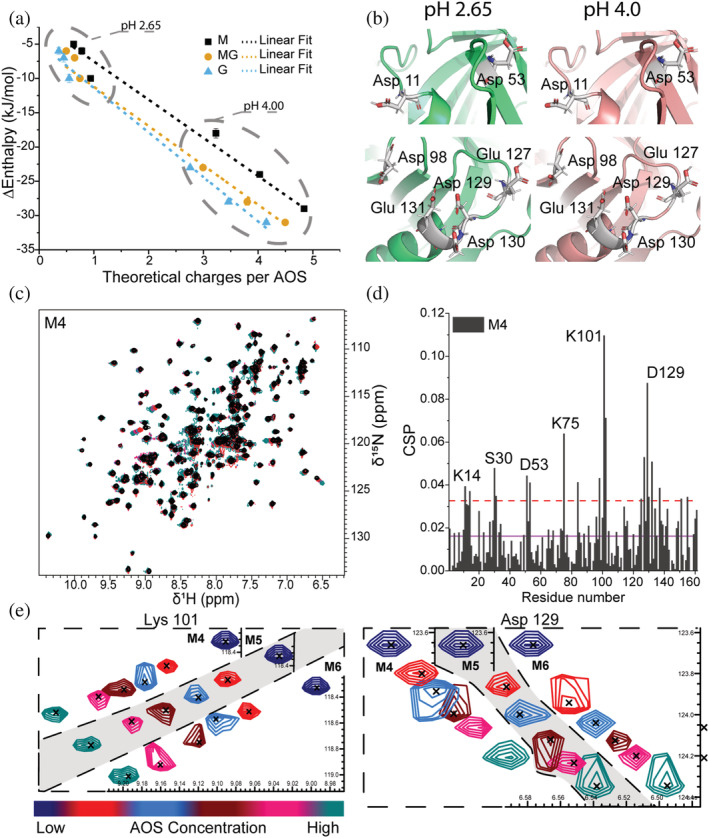
Details of AOSs binding to β‐LgA. (a) Changes in enthalpy determined by ITC plotted against the theoretical number of charges on the AOSs at pH 2.65 and pH 4.00. The number of charges was calculated using pKa values of monomeric mannuronate or guluronate. Linear curves (dashed lines) were fitted to the data to visualize correlation (R 2 = 0.99). (b) Zoom on acidic residues of sites 1 (top) and 4 (bottom) changing protonation state depending on pH. β‐LgA backbone is shown in green (pH 2.65) and pink (pH 4.00); side chains are gray. (c) Overlay of 1H,15N‐HSQC spectra of β‐LgA titrated by 0–10 mM M4 at pH 2.65 and 37°C. (d) Chemical shift perturbations of β‐LgA per residue induced by addition of 10 mM M4. The purple line represents the average CSP, the dashed red line CSP_AVG + 1SD. (e) Changes in peak positions of Lys101 and Asp129 of site 4 upon titration with M4, M5 or M6
2.2. Mapping the β‐LgA AOS binding surface and analysis of bound AOS conformation
1H,15 N‐heteronuclear single quantum coherence (HSQC) NMR spectra and resonance assignments have previously been reported of monomeric β‐LgA in 50 mM KH2PO4 at pH 2.65 (Birch et al., 2021; Stender et al., 2019; Uhrínová et al., 1998). Here, well matched β‐LgA 1H,15 N‐HSQCs were obtained under the same conditions, and the original 151 identified peaks were assigned (Figure S2). Titrating 15 N‐β‐LgA with the different AOSs and recording the corresponding 1H,15 N‐HSQC spectra showed changes in chemical shifts (Figure 2), and chemical shift perturbation (CSP) analyses revealed the binding surfaces of the AOSs on β‐LgA (Figures 2d and S3).
Titrations at pH 2.65 with each of the three AOSs of DP4, DP5, and DP6 were done in 5 steps with 10, 3.75, and 1.6 mM as highest concentration, respectively (Figures 2d, S3 and S4). Highly perturbed residues were identified from CSPs above the average CSP + 1SD (AVG + 1SD). The affected chemical shifts of β‐LgA were annotated to represent 4 of the 5 differing sites determined by ITC. Site 1 involved Asp11‐Lys14 (Figure S4), site 2 Ile29‐Leu31, site 3 Asp53, Lys75 and Ile84 and site 4 Lys101, Tyr102, Glu127, and Asp129 (Figure 2d). While sites 1, 2, and 4 each spanned approximately 10 Å, site 3 was about 20 Å in length from edge to edge. As site 3 is the largest, this may harbor the fifth binding site from ITC, but our data do not provide such detail. The calculated hydrophobicity differed from site to site, with sites 1 and 2 being most hydrophobic, while site 3 was slightly hydrophilic, and site 4 was mostly hydrophilic (Cowan & Whittaker, 1990). The effect of increasing DP was most prominent for MG. Thus, MG4 interacted with residues Asp11‐Lys14, Ile29, Ser30, Asp53, Lys75, Ile84, Lys101, Tyr102, Glu127 and Asp129, while MG6 additionally affected the neighboring residues Lys8, Ile56, Ile71, Thr76, Glu115, Val128, Asp130 and Ala132 (Figures 3a,b and S4). To determine residue‐specific K D values, CSPs larger than the AVG + 1SD were plotted against AOS concentration (Figure 4) (see Materials and Methods 4.6 and 2.3 in Data S1). These K D values overall matched the K D values determined by ITC for the different AOSs (Figures 1 and 4). The β‐LgA surface charge changes from pH 2.65 to pH 4.00 where β‐LgA also becomes dimeric (Figures 2b and S5). Docking of G4 to site 4 at either pH 2.65 or pH 4.00, for instance, showed two different poses. At pH 2.65 G4 interacts with protonated acidic side chains (127–131) and at pH 4.00 the orientation shifts, such that it instead interacts with positively charged side chains (Lys100 and Lys135) (Figures S5 and S6). The pose at pH 4.00 is energetically more favorable than the pose at pH 2.65, Table 1 should be here reflecting the pH‐dependent changes in the binding surfaces. This pattern was generally also observed for the other AOSs (Table 1). XP glidescore of site 2 was found to increase by 1–4 kcal/mol, reflecting that the binding surface at higher pH can become unfavorable, as also found in ITC data with stoichiometry changing from 5 at pH 2.65 to 4 at pH 4.00. Interaction populations for each docked site at either pH 2.65 or pH 4.00 showed broad interaction profiles, but with matching residues for the two different pH values (Figures 3d and S7–S14). Interaction maps revealed that charged residues interacted with AOSs through either salt bridging (pH 4.00) or hydrogen bonding (pH 2.65) (Figure S6), thus supporting the observation of differences in binding from ITC experiments.
FIGURE 3.
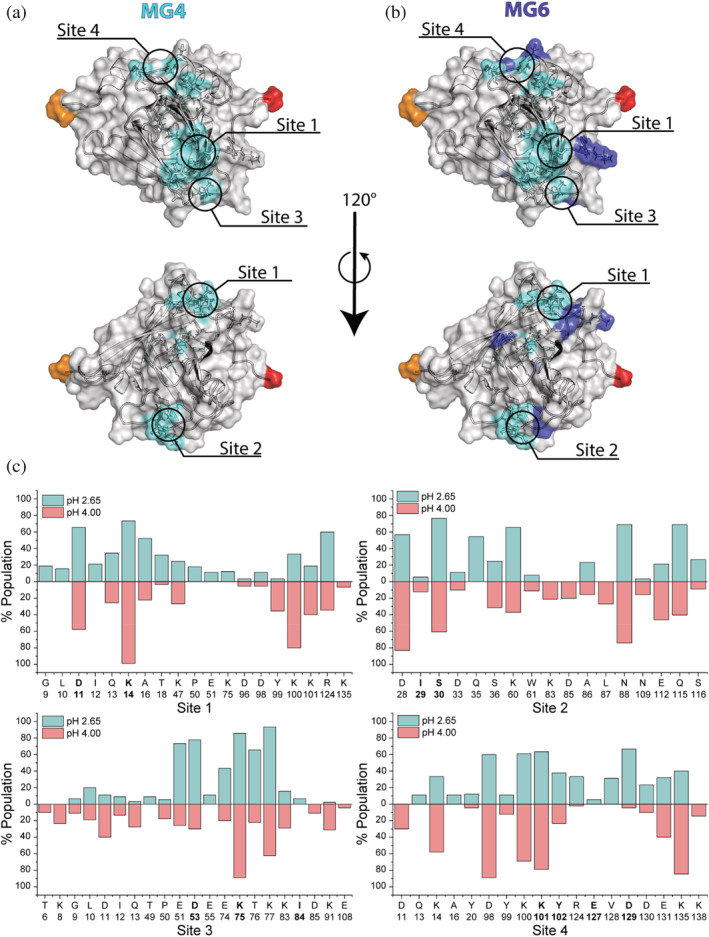
Binding sites on β‐LgA. Residues perturbed by MG AOSs (CSP > AVG + 1SD) identifying the binding sites on β‐LgA (RCSB: 1DV9 1) shown in transparent surface representation. (a) Residues perturbed by MG4 (nITC = 5) are colored cyan. (b) Residues perturbed by MG6 (nITC = 2) are colored dark blue. N‐ and C‐terminal residues are colored in red and orange, respectively, unaffected surface is gray. (c) Population percent of β‐LgA residues found to interact with AOSs by molecular docking simulation at either pH 2.65 (light blue) or pH 4.00 (light red) for each of the NMR determined sites (bold letters)
FIGURE 4.
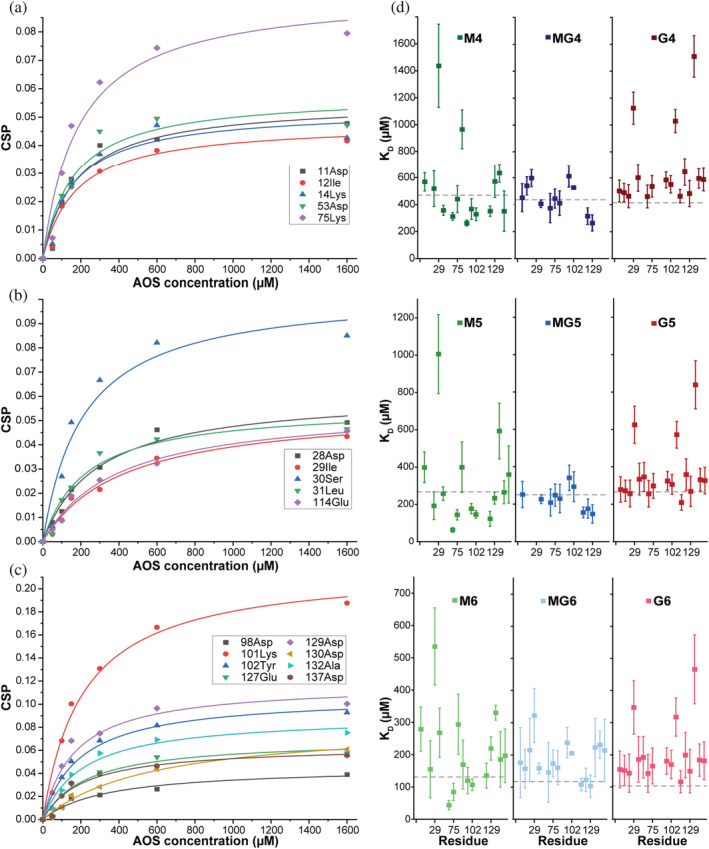
Affinity determination of individual sites from CSPs. NMR CSPs of 15 N‐β‐LgA (200 μM) titrated with AOS (1.6, 3.75, and 10 mM for DP 6, 5, and 4, respectively) as a function of ligand concentration. CPS induced by M6 are fitted to a hyperbolic binding curve to obtain KD for residues with CSP > AVG + 1SD. (a) Sites 1 + 3, (b) site 2, (c) site 4. (d) KD values of individual residues with CSP > AVG + 1SD extracted from NMR (Figure S3). Gray dotted lines correspond to global KD values determined by ITC
2.3. AOSs exhibit highly dynamic binding to β‐LgA
1H‐ligand‐targeted NMR allows determination of specific close distance interactions between protein and ligands and is commonly used in discovery, design, and development of drugs (Fielding, 2007; Mayer & James, 2004). Saturation transfer difference (STD), T1ρ and WaterLOGSY were conducted to examine orientation preferences of the AOSs on β‐LgA (Raingeval et al., 2019; Viegas et al., 2011). Proton peaks were assigned by recording of 1D 1H spectra and a series of 2D 1H ‐1H and 1H ‐13C correlated spectra at pH 5.00 and pH 2.65 (Tables S2‐S7, Figures S15‐S18). Proton peaks for the AOSs are found in the region 3.8–5.3 ppm (Figures S15‐S18), but individual peaks cannot be resolved in the proton dimension due to poor chemical shift dispersion (Figure 5c, Tables S2–S7).
FIGURE 5.
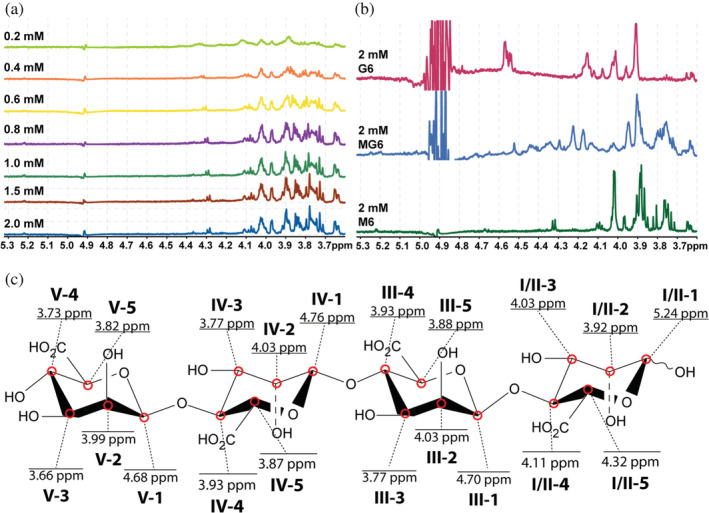
Saturation transfer difference 1H‐spectra. (a) Seven‐step titration of 20 μM β‐LgA by 0.2–2 mM M4 at pH 2.65. (b) 1H‐STD spectra of 20 μM β‐LgA with 2 mM G6 (cyan), MG6 (blue) or M6 (green) at pH 2.65. (c) Structural illustration of mannuronic acid DP4 (M4) with proton chemical shift annotations, roman numerals (I–V) represent the in‐chain position of the uronic acid and numbers (1–5) represent the in‐ring carbon number the proton is associated to
STD titration experiments were initiated by establishing a suitable saturation transfer time to 2 s, which yielded 50% of the maximal signal (Figure S19) (Viegas et al., 2011). Multiple peaks, even at low AOS concentration, showed a response in 1H‐STDs when titrating β‐LgA with M4 (Figure 5a) and the same was observed for M5 and M6 (Figures S20 and S21) as well as MG6 and G6 (Figure 5b). Thus, these AOSs seemed not to adopt a preferred conformation in complexes with β‐LgA, but rather exist in an ensemble of structural heterogenic states, with several conformations interconverting on a fast timescale, which all contribute to complex formation (Olsen et al., 2017; Warfield et al., 2014). WaterLOGSY and T1ρ experiments supported the STD results by showing that all peaks for M6, MG6, and G6 gave signals in the presence of β‐LgA (Figures S22–S24). NMR peak Intensities of identified proton signals resolved by the STD titration were plotted against AOS concentrations to estimate K D globally and individually for M4 (Figure 6), M5 and M6 (Figures S16 and S17) (see Materials and Methods 4.7.2). The global K D values for M6, M5, and M4 of 147 ± 40 μM, 228 ± 30 μM, and 520 ± 33 μM, respectively, agree with K D values determined by both 1H,15N‐HSQC titration (Figure 4d) and ITC (Figure 1c and Table S1). Individually determined K D values did not significantly differ from the globally determined K D, suggesting that all protons are equally specific in the interaction (Figures 6, S25, and S26). Molecular docking resulted in absolute binding orientations, with the top 5 best scoring poses within 1.5 kJ/mol XP Gscore and showed 5.3 to 15.9 Å difference in orientation (Figures S18 and S19) with many interacting residues (Figure 3c), which also supports the highly dynamic interaction indicated by STD NMR (Figure 7).
FIGURE 6.
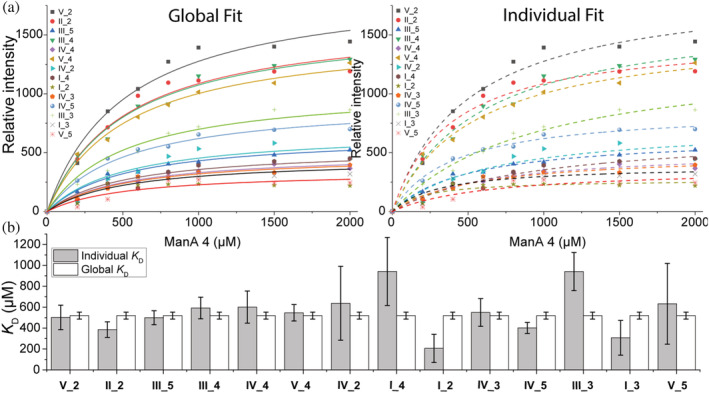
KD determination by following peak intensities from STD 1H‐spectra. (a) Integrated peak volumes from M4 STD and resulting global and individual fit to determine K D. (b) Resulting K D values from M4 STD, numbers 1–6 represent ring carbon numbers, roman numbers (I–V) represent the chain position of the ring carbon (see Figure 5c)
FIGURE 7.
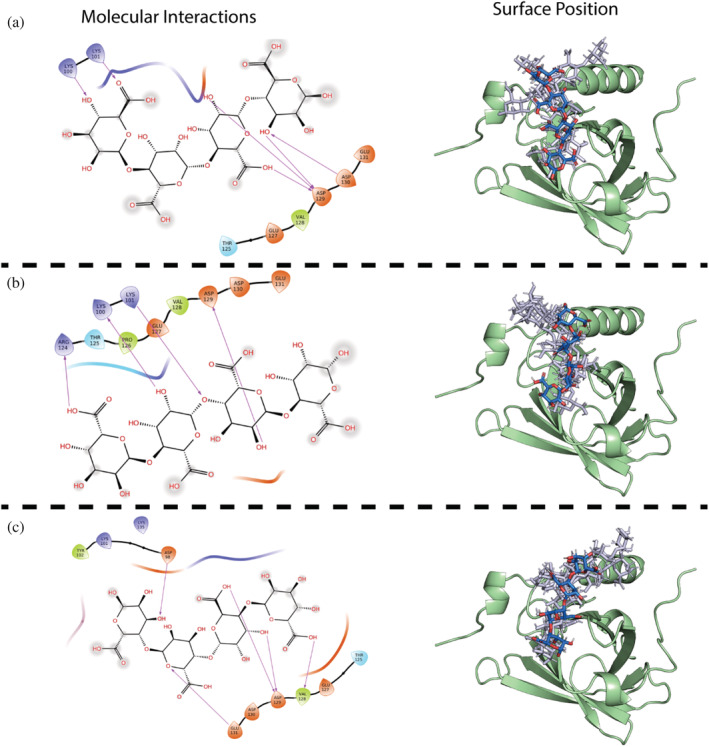
Orientation of AOSs docked onto β‐LgA (green) binding site 4 (Lys101). Right hand side shows surface position of (a) M4 best XP GScore (blue), top 2–5 XP GScore (gray), (b) MG4 best XP GScore (blue), top 2–5 XP GScore (gray), (c) G4 best XP GScore (blue), top 2–5 XP GScore (gray). To the left are shown interaction schemes for the best XP GScore of OH, carboxyl and glycosidic oxygen groups in individual AOSs to residues at binding site 4. Acidic side chains are colored orange, basic side chains blue, hydrophobic side chains green and polar side chains cyan. Arrows mark side chain AOSs interactions
3. DISCUSSION
Mixtures of full length alginate (DP > 600) and β‐LgA at a pH below 4.5 have been reported to complex coacervate and form predominantly insoluble complexes (Gorji et al., 2018; Madsen et al., 2021; Qomarudin et al., 2015), attributed to charge‐neutralization of the polysaccharide by engaging in multivalent interactions (Harnsilawat et al., 2006; Madsen et al., 2021). However, equal amounts (wt/wt) of alginate oligomers of DP6 and β‐LgA can form soluble complexes (Stender et al., 2018). This motivated the use of AOSs as ligands to gain insights into the intermolecular forces and impact of M and G composition on the interaction with β‐LgA and evaluate the system as a model of the early multivalent stages leading to coacervation.
The reported small number of UARs bound to β‐Lg for soluble complexes (6 UARs (Stender et al., 2018)) does not agree with polymer coacervation measured by ITC, in which a stretch of 8–22 UARs (calculated based on literature data of stoichiometry and alginate size as in Madsen et al. (Madsen et al., 2021)) bound to one β‐LgA molecule. Higher ionic strength narrowed the range of UARs that one β‐LgA molecule can bind to 16–20 (Madsen et al., 2021), equivalent to 4–5 DP4, 3–4 DP5, and 3 DP6 AOSs. These polymer level stoichiometries (UARs/β‐LgA) matched very well with the presented ITC data for the AOSs. Here 20–12 UARs bound per β‐LgA for shorter or longer AOSs, respectively. Thus, compared to previous stoichiometries of AOS binding to β‐LgA (Stender et al., 2018), a larger molecular coverage was achieved. Affinities and enthalpies clearly increased for all AOSs from pH 2.65 to pH 4.00, in addition to apparent changes in stoichiometry (from 5 to 4) for M4, MG4, and G4, albeit not for AOSs of DP5 and DP6. Both AOSs and β‐LgA underwent changes in charge states from pH 2.65 to 4.00 reflected in the different binding energy contributions, indicating that complex coacervation is driven by entropy at pH 2.65 and by enthalpy at pH 4.00. This suggests a difference in residue–AOS contacts when AOSs bind at pH 2.65 as compared to pH 4.00, as demonstrated by the difference in docking poses at pH 2.65 or pH 4.00 (Figures 3c, S5 and S27). β‐Lg changes from monomeric at pH 2.65 to dimeric at pH 4.00, however the dimer interphase does not coincide with the AOS binding sites as determined by NMR.
Stoichiometries obtained from ITC were used to guide the annotation of individual binding sites on the β‐Lg surface based on 1H,15N‐HSQC data. β‐LgA residues perturbed by AOSs of DP4‐6 at pH 2.65 coincided in part with two sites previously identified by NMR to bind enzymatically produced trisaccharides from alginate (Stender et al., 2019). These correlated with sites 1 and 3 in the present study (Figure 4) (Stender et al., 2019), and merged for AOSs > DP4 with site 4 to form a continuous binding surface composed of sites 1, 3, and 4 (Figure 4). Docking showed that site 1 was slightly favored over site 4, but with shared residues in the interaction population (Figure 3c). Site 2 (around Ser30), does not bind AOSs of DP3 (Stender et al., 2019), but has been suggested to be involved in hydrophobic binding of buformin (Sahihi & Ghayeb, 2014). CSP NMR showed a few individual residues to have lower affinity than found by ITC. Especially, Ile29 showed a three–fivefold higher K D for M4‐M6, indicating that this residue is only affected at high AOS concentration. Based on ITC measurements, M‐ and G‐dependent effects on K D and stoichiometry were found for polymeric alginate (DP > 600) interaction with β‐LgA (Madsen et al., 2021), which correlated with differences in structural flexibility (Draget et al., 2001). By using AOSs of DP 4–6, ligand flexibility seemed not to discriminate β‐LgA surface coverage, as opposed to polymer interaction (Madsen et al., 2021; Stender et al., 2018).
Neither the spatial orientation nor the conformation of AOSs bound to protein have been reported earlier. In our work, STD, WaterLOGSY and T1ρ showed that no specific conformation existed, but that the AOSs instead adopted several orientations and conformations on the surface of β‐LgA, similarly to the highly dynamic complexes involving intrinsically disordered proteins (Olsen et al., 2017; Sottini et al., 2020). Modeling of the AOSs onto the different identified binding regions on β‐LgA, indicated that all the AOSs could adopt a number of different orientations of similar XP Gscores, in agreement with the perception gained from 1H‐ligand targeted NMR (STD, WaterLOGSY and T1ρ).
Even though the interaction between AOSs and β‐LgA is best represented by an ensemble of dynamically interconverting states, as shown by 1H‐ligand targeted NMR, docking of the AOSs to the β‐LgA surface indicated small, but distinct differences in the AOSs orientation (RMSDs of 5.3 to 15.9 Å, Figure S28), governed by their composition. Strikingly, despite these differences in orientations, XP Gscores were very similar for AOSs of the same DP. Of special note, site 4 harbored many acidic residues causing electrostatic repulsion of AOSs at pH 4.00, but little to none at pH 2.65. This agrees well with previous suggestions that binding patterns change according to the charge state of AOSs and surface charge of β‐LgA (Stender et al., 2018; Stender et al., 2019).
The multivalency of both the long alginate polymer, which can accommodate several β‐Lg proteins along its chain, and β‐Lg itself, where the distribution of several energetically similar, but chemically diverse surface binding sites exist, is a prime prerequisite for the formation of condensates (Li et al., 2012) and for percolation (Harmon et al., 2017). The highly dynamic states we have uncovered here using shorter alginate building blocks may represent some of the early states in condensate formation. These low affinities, highly dynamic contacts and charge dependent states allow for fast un‐ and rebinding and optimization of the network driving phase separation. These results support a current hypothesis for phase separation coupled to percolation, by showing how highly dynamic multivalent contacts give rise to dense phase separated networks (Mittag & Pappu, 2022). Whether the low affinity interactions and the highly dynamic ensemble with interconverting binding sites are maintained in the final condensates cannot be inferred from the current study. However, a study on β‐LgA interaction with acacia gum suggests a nucleation growth dependent coacervation such that the initially formed condensate connections remain in the final coacervate structures (Sanchez et al., 2006).
4. CONCLUSION
Distinct AOSs of M, MG, and G types engage in highly dynamic complexes with β‐LgA at pH 2.65 as shown by ligand targeted NMR. The AOSs exhibit no preferred conformational pose on β‐Lg, but rather, depending on the surface of β‐LgA, form an ensemble of interconverting states. The interaction is limited by the AOS charge state where there is a small but relevant electrostatic contribution at low pH, which becomes more distinct at pH 4.0 as well as increasingly dominated by enthalpic forces as supported by stoichiometry, enthalpy, and K D. It is thus unlikely that all β‐LgA residues involved in AOS binding at pH 2.65, where β‐LgA is monomeric, are engaged at pH 4.00 where β‐LgA is dimeric and vice versa. The contact freedom observed for AOSs in binding to β‐LgA, translates to the understanding of the coacervation between β‐LgA and full‐length alginate, as it gives the first notion of the promiscuity that alginates have on the surface of β‐LgA, leading to the formation of disordered coacervate structures.
5. MATERIALS AND METHODS
AOSs are described by M, G, or MG with a DP of 4–6. For example, M4, G4 and MG4 are tetrasaccharides of mannuronic, guluronic, and alternating mannuronic‐ and guluronic acid residues, respectively.
5.1. AOS preparation
Poly‐M (intrinsic viscosity [η] = 940 ml/g; FG = 0.0) was produced by an epimerase (AlgG) negative strain of Pseudomonas fluorescens and poly‐G (FG = 0.97) was prepared by in vitro epimerization of poly‐M using AlgE1 (Aarstad et al., 2019). Poly‐MG of alternating structure (FG = 0.46, FGG = 0.0) was made by in vitro epimerization of poly‐M using AlgE4 (Donati et al., 2005). The M, G, and MG block AOSs were subsequently produced from these polymers by mild acid hydrolysis and purified as previously described (Ballance et al., 2005; Campa et al., 2004).
5.2. Production and purification of recombinant β‐LgA and 15N‐β‐LgA
The plasmid pPICZαA–β‐LgA encoding β‐LgA used for previous NMR studies (Birch et al., 2021; Stender et al., 2019), was transformed into E. coli DH5α, selected for zeocin resistance and verified by sequencing. pPICZαA–β‐LgA (20 μg) was linearized by PmeI (37°C, 1 h), purified with GeneJET PCR Purification Kit (ThermoFischer Scientific) and transfected into P. pastoris X‐33 by electroporation of 5 μg linear DNA to 100 μl competent cells, which were plated on zeocin containing yeast extract peptone dextrose agar, incubated (3 days, 30°C) to select for resistance and screened for β‐LgA production level. Production and purification of β‐LgA and 15N‐β‐LgA was performed as previously described (see Data S1) (Stender et al., 2019).
5.3. Sample preparation for 1H, 15N‐HSQC NMR, and ITC titration studies
β‐LgA in 50 mM KH2PO4 pH 4.00 or 2.65 and 15N‐β‐LgA in 55 mM KH2PO4 pH 2.65 were dialyzed (3× 100‐fold dilution, 3000 MWCO Spectra/Por membrane, Spectrumlabs) against the respective buffers. Protein concentration was measured spectrophotometrically (Nanodrop, Thermo Scientific) at 280 nm using a εM = 18,700 M−1 cm−1 as predicted by ProtParam (Gasteiger et al., 2005). Concentrations of AOSs (in dH2O) were determined by the phenol sulfuric acid method (Dubois et al., 1956), and the AOSs were aliquoted as 55 and 255 μl portions of 50 and 20 mM, respectively, lyophilized and stored in a vacuum desiccator until use.
5.4. Isothermal titration calorimetry
β‐LgA (1.0 mM) was titrated by AOSs (20 or 50 mM) dissolved in β‐LgA dialysates of pH 4.00 or 2.65. Degassed (2 min) β‐LgA (200 μl) and AOS (40 μl) were loaded into the cell and syringe, respectively. Titration at 37°C with stirring at 750 rpm involved an initial 0.4 μl injection followed by 19 injections of 2 μl (ITC200, Thermo Scientific). Raw data were corrected for heat of dilution by subtracting a blank titration. Normalized, integrated raw data were plotted as enthalpograms and fitted with an independent one‐site binding model, to determine dissociation constant (K D), stoichiometry (n) and enthalpy (ΔH) using the instrument Origin‐based software (Microcal Analysis).
5.5. 1H, 15N‐HSQC NMR, and chemical shift perturbation analysis
15N‐β‐LgA and AOS (1 mM and 5–20 mM, respectively, in 0.1 mM DSS, 10% D2O, 50 mM KH2PO4 pH 2.65) were centrifuged (20,000g, 20 min, 4°C), transferred to new tubes and stored on ice. Concentrations of AOS and 15N‐β‐LgA were determined by the phenol‐sulfuric acid assay (Dubois et al., 1956) and UV‐absorbance, respectively (Birch et al., 2021; Stender et al., 2019). The 50 mM KH2PO4 pH 2.65 was identical to previous studies, allowing transfer of peak assignments (Birch et al., 2021; Stender et al., 2019; Uhrínová et al., 1998) and pH was verified throughout experiments to ensure consistency. Titrations were performed in five steps in the range 0–10, 0–3.75, and 0–1.6 mM AOSs DP4, DP5 and DP6, respectively, and 200 μM 15N‐β‐LgA in 5 mm NMR tubes. All 1H, 15N‐HSQC spectra were recorded on a 600 MHz Bruker Ascend with AVIIIHD console equipped with a 5 mm QCI cryoprobe (Bruker BioSpin AG). Free induction decays were processed with nmrPipe (Delaglio et al., 1995) and proton chemical shifts were referenced to DSS at 0.00 ppm. Chemical shifts in fast exchange were monitored during the titrations (Teilum et al., 2017), by the distance at which peaks move. The chemical shift perturbation were calculated as previously described (Mulder et al., 1999; Teilum et al., 2017).
5.6. Label free ligand mapping by NMR
5.6.1. Sample and equipment set‐up
β‐LgA was dissolved and dialyzed (as above) against 50 mM KH2PO4 pH 2.65 or 4.00. β‐LgA (20 μM) was titrated with AOSs (0.2–2.0 mM) in seven steps, corresponding to ligand molar excess of 10–100× as suggested by Viegas et al. (2008) Spectra were acquired on 800 MHz Bruker Ascend with AVIIIHD console equipped with a 5 mm TCI cryoprobe (Bruker BioSpin AG) at 37°C.
5.6.2. Saturation transfer difference
Testing for saturation frequencies and time by a build‐up experiment following Viegas et al. (2008) used a sample composed of 20 μM β‐LgA with 2 mM M6 at pH 2.65 50 mM KH2PO4. Saturation times were set from 0.5 to 5 s in 10 steps. From the build‐up experiment, the amplification factor was plotted against saturation time at saturation frequency of 0.303 ppm and a saturation time of 2 s was chosen for the saturation transfer difference (STD) titration experiment (Figure S10). The amplification factor was measured as the height of the peaks. By plotting amplification factor against ligand concentration, K D can be determined by fitting , where αSTD is the maximum amplification factor determined by the fitting and [L] is the varying ligand concentration.
AUTHOR CONTRIBUTIONS
Mikkel Madsen: Conceptualization (equal); data curation (lead); formal analysis (lead); investigation (lead); writing – original draft (lead); writing – review and editing (lead). Andreas Prestel: Validation (equal); writing – review and editing (equal). Eva Madland: Validation (supporting). Peter Westh: Validation (supporting); writing – review and editing (supporting). Anne Tøndervik: Formal analysis (supporting); investigation (supporting). Håvard Sletta: Formal analysis (supporting); investigation (supporting). Günther H. J. Peters: Formal analysis (equal); investigation (supporting); writing – review and editing (supporting). Finn L. Aachmann: Conceptualization (equal); data curation (supporting); formal analysis (supporting); funding acquisition (equal); resources (equal); validation (equal); writing – review and editing (supporting). Birthe B. Kragelund: Conceptualization (equal); funding acquisition (equal); methodology (equal); resources (equal); validation (equal); writing – review and editing (equal). Birte Svensson: Conceptualization (equal); data curation (supporting); funding acquisition (lead); resources (equal); supervision (equal); writing – review and editing (equal).
FUNDING INFORMATION
This work was supported by The Novo Nordisk Foundation (NNF) Biotechnology Synthesis and Production (#NNFOC0027616, to Birte Svensson), The Novo Nordisk Foundation Challenge grant REPIN (#NNF18OC0032996; to Birthe B. Kragelund) and The Norwegian Research Council (#226244; to Finn L. Aachmann). Villum Fonden and Novo Nordisk Foundation (#NNF18OC0032996; cOpenNMR) are thanked for generous NMR infrastructure support.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the content of this article.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
Karina Jansen (DTU Bioengineering) is gratefully acknowledged for technical support. We thank Maher Abou Hachem for the use of ITC200 (instrument grant: Carlsberg Foundation 2011‐01‐0598), with associated software.
Madsen M, Prestel A, Madland E, Westh P, Tøndervik A, Sletta H, et al. Molecular insights into alginate β‐lactoglobulin A multivalencies—The foundation for their amorphous aggregates and coacervation. Protein Science. 2023;32(2):e4556. 10.1002/pro.4556
Review Editor: Aitziber L. Cortajarena.
Funding information Norges Forskningsråd, Grant/Award Number: 226244; Novo Nordisk Fonden, Grant/Award Numbers: NNF18OC0032996, NNFOC0027616; Villum Fonden, Grant/Award Number: cOpenNMR; Carlsberg Foundation, Grant/Award Number: 2011‐01‐0598
Contributor Information
Finn L. Aachmann, Email: finn.l.aachmann@ntnu.no.
Birthe B. Kragelund, Email: bbk@bio.ku.dk.
Birte Svensson, Email: bis@bio.dtu.dk.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Aarstad OA, Stanisci A, Sætrom GI, Tøndervik A, Sletta H, Aachmann FL, et al. Biosynthesis and function of long guluronic acid‐blocks in alginate produced by Azotobacter vinelandii . Biomacromolecules. 2019;20:1613–22. [DOI] [PubMed] [Google Scholar]
- Ballance S, Holtan S, Aarstad OA, Sikorski P, Skjåk‐Bræk G, Christensen BE. Application of high‐performance anion‐exchange chromatography with pulsed amperometric detection and statistical analysis to study oligosaccharide distributions ‐ a complementary method to investigate the structure and some properties of alginates. J Chromatogr A. 2005;1093:59–68. [DOI] [PubMed] [Google Scholar]
- Birch J, Khan S, Madsen M, Kjeldsen C, Møller MS, Stender EGP, et al. Binding sites for oligosaccharide repeats from lactic acid bacteria exopolysaccharides on bovine β‐lactoglobulin identified by NMR spectroscopy. ACS Omega. 2021;6:9039–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campa C, Oust A, Skjåk‐Bræk G, Paulsen BS, Paoletti S, Christensen BE, et al. Determination of average degree of polymerisation and distribution of oligosaccharides in a partially acid‐hydrolysed homopolysaccharide: a comparison of four experimental methods applied to mannuronan. J Chromatogr A. 2004;1026:271–81. [DOI] [PubMed] [Google Scholar]
- Cowan R, Whittaker RG. Hydrophobicity indices for amino acid residues as determined by high‐performance liquid chromatography. Pept Res. 1990;3:75–80. [PubMed] [Google Scholar]
- Craigie JS, Morris ER, Rees DA, Thom D. Alginate block structure in phaeophyceae from Nova Scotia: variation with species, environment and tissue‐type. Carbohydr Polym. 1984;4:237–52. [Google Scholar]
- Creamer LK, Loveday SM, Sawyer L. Milk Proteins: β‐Lactoglobulin. Encyclopedia of dairy sciences. Second ed. Amsterdam, Holland: Elsevier Inc.; 2011. p. 787–94. [Google Scholar]
- Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- Donati I, Holtan S, Mørch YA, Borgogna M, Dentini M, Skjåk‐Bræk G. New hypothesis on the role of alternating sequences in calcium‐alginate gels. Biomacromolecules. 2005;6:1031–40. [DOI] [PubMed] [Google Scholar]
- Draget KI, Gåserød O, Aunea I, Andersen PO, Storbakken B, Stokke BT, et al. Effects of molecular weight and elastic segment flexibility on syneresis in Ca‐alginate gels. Food Hydrocoll. 2001;15:485–90. [Google Scholar]
- Draget KI, Skjåk Bræk G, Smidsrød O. Alginic acid gels: the effect of alginate chemical composition and molecular weight. Carbohydr Polym. 1994;25:31–8. [Google Scholar]
- Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem. 1956;28:350–6. [Google Scholar]
- Farrell HM, Jimenez‐Flores R, Bleck GT, Brown EM, Butler JE, Creamer LK, et al. Nomenclature of the proteins of cows' milk. J Dairy Sci. 2004;87:1641–74. [DOI] [PubMed] [Google Scholar]
- Fielding L. NMR methods for the determination of protein‐ligand dissociation constants. Prog Nucl Magn Reson Spectrosc. 2007;51:219–42. [Google Scholar]
- Gacesa P. Bacterial alginate biosynthesis ‐ recent progress and future prospects. Microbiology. 1998;144:1133–43. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook. New Jersey, USA: Humana Press; 2005. p. 571–607. [Google Scholar]
- Gawin A, Tietze L, Aarstad OA, Aachmann FL, Brautaset T, Ertesvåg H. Functional characterization of three Azotobacter chroococcum alginate‐modifying enzymes related to the Azotobacter vinelandii AlgE mannuronan C‐5‐epimerase family. Sci Rep. 2020;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorji EG, Waheed A, Ludwig R, Toca‐Herrera JL, Schleining G, Gorji SG. Complex coacervation of milk proteins with sodium alginate. J Agric Food Chem. 2018;66:3210–20. [DOI] [PubMed] [Google Scholar]
- Guo X, Wang Y, Qin Y, Shen P, Peng Q. Structures, properties and application of alginic acid: a review. Int J Biol Macromol. 2020;162:618–28. [DOI] [PubMed] [Google Scholar]
- Gutiérrez‐Magdaleno G, Bello M, Carmen Portillo‐Téllez M, Rodríguez‐Romero A, García‐Hernández E. Ligand binding and self‐association cooperativity of β‐lactoglobulin. J Mol Recognit. 2013;26:67–75. [DOI] [PubMed] [Google Scholar]
- Harmon TS, Holehouse AS, Rosen MK, Pappu RV. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. elife. 2017;6:1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnsilawat T, Pongsawatmanit R, McClements DJ. Characterization of β‐lactoglobulin‐sodium alginate interactions in aqueous solutions: a calorimetry, light scattering, electrophoretic mobility and solubility study. Food Hydrocoll. 2006;20:577–85. [Google Scholar]
- Hosseini SMH, Emam‐Djomeh Z, Razavi SH, Moosavi‐Movahedi AA, Saboury AA, Mohammadifar MA, et al. Complex coacervation of β‐lactoglobulin ‐ κ‐carrageenan aqueous mixtures as affected by polysaccharide sonication. Food Chem. 2013;141:215–22. [DOI] [PubMed] [Google Scholar]
- Khan S, Ipsen R, Almdal K, Svensson B, Harris P. Revealing the dimeric crystal and solution structure of β‐lactoglobulin at pH 4 and its pH and salt dependent monomer − dimer equilibrium. Biomacromolecules. 2018;19:4–11. [DOI] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loch JI, Polit A, Bonarek P, Olszewska D, Kurpiewska K, Dziedzicka‐Wasylewska M, et al. Structural and thermodynamic studies of binding saturated fatty acids to bovine β‐lactoglobulin. Int J Biol Macromol. 2012;50:1095–102. [DOI] [PubMed] [Google Scholar]
- Madsen M, Westh P, Khan S, Ipsen R, Almdal K, Aachmann FL, et al. Impact of alginate mannuronic‐guluronic acid contents and pH on protein binding capacity and complex size. Biomacromolecules. 2021;22:649–60. [DOI] [PubMed] [Google Scholar]
- Mayer M, James TL. NMR‐based characterization of phenothiazines as a RNA binding scaffold. J Am Chem Soc. 2004;126:4453–60. [DOI] [PubMed] [Google Scholar]
- Mimura M, Tomita S, Sugai H, Shinkai Y, Ishihara S, Kurita R. Uncharged components of single‐stranded DNA modulate liquid–liquid phase separation with cationic linker histone H1. Front Cell Dev Biol. 2021;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T, Pappu RV. A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol Cell. 2022;82:2201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder FAA, Schipper D, Bott R, Boelens R. Altered flexibility in the substrate‐binding site of related native and engineered high‐alkaline bacillus subtilisins. J Mol Biol. 1999;292:111–23. [DOI] [PubMed] [Google Scholar]
- Nakashima KK, Vibhute MA, Spruijt E. Biomolecular chemistry in liquid phase separated compartments. Front Mol Biosci. 2019;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JG, Teilum K, Kragelund BB. Behaviour of intrinsically disordered proteins in protein–protein complexes with an emphasis on fuzziness. Cell Mol Life Sci. 2017;74:3175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qomarudin Q, Orbell JD, Ramchandran L, Gray SR, Stewart MB, Vasiljevic T. Properties of β‐lactoglobulin/alginate mixtures as a function of component ratio, pH and applied shear. Food Res Int. 2015;71:23–31. [Google Scholar]
- Raingeval C, Cala O, Brion B, Le Borgne M, Hubbard RE, Krimm I. 1D NMR WaterLOGSY as an efficient method for fragment‐based lead discovery. J Enzyme Inhib Med Chem. 2019;34:1218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahihi M, Ghayeb Y. Binding of biguanides to β‐lactoglobulin: molecular‐docking and molecular dynamics simulation studies. Chem Pap. 2014;68:1601–7. [Google Scholar]
- Sakurai K, Goto Y. Manipulating monomer‐dimer equilibrium of bovine β‐lactoglobulin by amino acid substitution. J Biol Chem. 2002;277:25735–40. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Mekhloufi G, Renard D. Complex coacervation between β‐lactoglobulin and acacia gum: a nucleation and growth mechanism. J Colloid Interface Sci. 2006;299:867–73. [DOI] [PubMed] [Google Scholar]
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK a prediction and protonation state generation for drug‐like molecules. J Comput Aided Mol Des. 2007;21:681–91. [DOI] [PubMed] [Google Scholar]
- Sottini A, Borgia A, Borgia MB, Bugge K, Nettels D, Chowdhury A, et al. Polyelectrolyte interactions enable rapid association and dissociation in high‐affinity disordered protein complexes. Nat Commun. 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender EGP, Birch J, Kjeldsen C, Nielsen LD, Duus JØ, Kragelund BB, et al. Alginate trisaccharide binding sites on the surface of β‐lactoglobulin identified by NMR spectroscopy: implications for molecular network formation. ACS Omega. 2019;4:6165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender EGP, Khan S, Ipsen R, Madsen F, Hägglund P, Abou Hachem M, et al. Effect of alginate size, mannuronic/guluronic acid content and pH on particle size, thermodynamics and composition of complexes with β‐lactoglobulin. Food Hydrocoll. 2018;75:157–63. [Google Scholar]
- Stokke BT, Draget KI, Smidsrød O, Yuguchi Y, Urakawa H, Kajiwara K. Small‐angle x‐ray scattering and rheological characterization of alginate gels. 1. Ca−alginate gels. Macromolecules. 2000;33:1853–63. [DOI] [PubMed] [Google Scholar]
- Taulier N, Chalikian TV. Characterization of pH‐induced transitions of β‐lactoglobulin: ultrasonic, densimetric, and spectroscopic studies. J Mol Biol. 2001;314:873–89. [DOI] [PubMed] [Google Scholar]
- Teilum K, Kunze MBA, Erlendsson S, Kragelund BB. (S)pinning down protein interactions by NMR. Protein Sci. 2017;26:436–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timilsena YP, Akanbi TO, Khalid N, Adhikari B, Barrow CJ. Complex coacervation: principles, mechanisms and applications in microencapsulation. Int J Biol Macromol. 2019;121:1276–86. [DOI] [PubMed] [Google Scholar]
- Uhrínová S, Smith MH, Jameson GB, Uhrín D, Sawyer L, Barlow PN. Structural changes accompanying pH‐induced dissociation of the β‐lactoglobulin dimer. Biochemistry. 2000;39:3565–74. [DOI] [PubMed] [Google Scholar]
- Uhrínová S, Uhrín D, Denton H, Smith M, Sawyer L, Barlow PN. Complete assignment of 1H, 13C and 15 N chemical shifts for bovine β‐lactoglobulin: secondary structure and topology of the native state is retained in a partially unfolded form. J Biomol NMR. 1998;12:89–107. [DOI] [PubMed] [Google Scholar]
- Valla S, Li JP, Ertesvåg H, Barbeyron T, Lindahl U. Hexuronyl C5‐epimerases in alginate and glycosaminoglycan biosynthesis. Biochimie. 2001;83:819–30. [DOI] [PubMed] [Google Scholar]
- Viegas A, Brás NF, Cerqueira NMFSA, Fernandes PA, Prates JAM, Fontes CMGA, et al. Molecular determinants of ligand specificity in family 11 carbohydrate binding modules ‐ an NMR, X‐ray crystallography and computational chemistry approach. FEBS J. 2008;275:2524–35. [DOI] [PubMed] [Google Scholar]
- Viegas A, Manso J, Nobrega FL, Cabrita EJ. Saturation‐transfer difference (STD) NMR: a simple and fast method for ligand screening and characterization of protein binding. J Chem Educ. 2011;88:990–4. [Google Scholar]
- Wang B, Zhang L, Dai T, Qin Z, Lu H, Zhang L, et al. Liquid–liquid phase separation in human health and diseases. Signal Transduct Target Ther. 2021;6:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield L, Tuttle LM, Pacheco D, Klevit RE, Hahn S. A sequence‐specific transcription activator motif and powerful synthetic variants that bind mediator using a fuzzy protein interface. Proc Natl Acad Sci U S A. 2014;111:E3506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Ni C, Yao B, Zhu C, Huang B, Zhang L. Covalently cross‐linked and hydrophobically modified alginic acid hydrogels and their application as drug carriers. Polym Eng Sci. 2013;53:1583–9. [Google Scholar]
- Yan Y, Kizilay E, Seeman D, Flanagan S, Dubin PL, Bovetto L, et al. Heteroprotein complex coacervation: bovine β‐lactoglobulin and lactoferrin. Langmuir. 2013;29:15614–23. [DOI] [PubMed] [Google Scholar]
- Zhang T‐D, Deng X, Wang MY, Chen LL, Wang XT, Li CY, et al. Formation of β‐lactoglobulin self‐assemblies via liquid‐liquid phase separation for applications beyond the biological functions. ACS Appl Mater Interfaces. 2021;13:46391–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.