

SUMMARY
Despite the growing number of GPCR structures, only 39 structures have been co-crystallized with allosteric inhibitors. These structures have been studied by protein mapping using the FTMap server, which determines the clustering of small organic probe molecules distributed on the protein surface. The method has found druggable sites overlapping with the co-crystallized allosteric ligands in 21 GPCR structures. Mapping of Alphafold2 generated models of these proteins confirm that same sites can be identified without the presence of bound ligands. We then mapped the 394 GPCR X-ray structures available at the time of the analysis (September 2020). Results show that for each of the 21 structures with bound ligands there exist many other GPCRs that have a strong binding hot spot at the same location, suggesting potential allosteric sites in a large variety of GPCRs. These sites cluster at nine distinct locations, each found in many different proteins. However, ligands binding at the same location generally show little or no similarity, and the amino acid residues interacting with these ligands also differ. Results confirm the possibility of specifically targeting these sites across GPCRs for allosteric modulation and help to identify the most likely binding sites among the limited number of potential locations. The FTMap server is available free for academic and governmental use at https://ftmap.bu.edu/.
Graphical Abstract
INTRODUCTION
G protein-coupled receptors (GPCRs) are one of the most populated groups of transmembrane proteins encoded by more than 1000 human genes.1, 2 GPCRs play a major role in mediating cellular response to different endogenous ligands by translating extracellular signals into the cell. Ligand binding at the extracellular side of the GPCRs results in conformational changes in the seven transmembrane (7TM) helices that rearrange the intracellular interface used by G protein and β-arrestin type signaling proteins. Endogenous ligands bind at the orthosteric binding site that serves as a potential site for therapeutic interventions including the activation (by full or partial agonists) or blocking (by inverse agonists or antagonists) the receptor function. In fact, almost 500 drugs targeting more than 100 different GPCRs are in current clinical use representing about 35% of all drugs approved by the FDA.3 Although most of these drugs target the corresponding orthosteric binding site, developing new therapies acting at these sites might be challenging due to multiple factors. First is the limited selectivity and potential side effects connected to the conserved nature of homologous receptor orthosteric sites. Second, many peptides binding to peptidergic GPCRs do not overlap spatially with the orthosteric site of small molecule ligands. Finally, targeting the same orthosteric site used by the endogenous ligands might interrupt physiological signaling patterns.
Allosteric modulation of G protein-coupled receptors represents an alternative mechanism of pharmacological intervention and has been extensively studied.4–7 By definition, allosteric modulators (AMs) bind to binding pockets different from the orthosteric site, however, they can impact the functional activity of the receptor in the presence of the endogenous ligand. Positive allosteric modulators (PAMs) potentiate while negative allosteric modulators (NAMs) suppress the functional response of the receptor to the endogenous ligand. In contrast, neutral allosteric ligands (NALs) bind to an allosteric site but have no impact on receptor signaling. Allosteric sites have less conserved amino acid sequences, which increases the chance to identify selective ligands with potentially less side effects. In addition, allosteric modulators with no inherent activity would only function in the presence of the endogenous agonist, without disrupting endogenous signaling patterns. The Allosteric Database (ASD) lists over 14,000 allosteric ligands binding to GPCRs,8 however, up to now only a few reached the market. This reflects to the challenges associated with the optimization of allosteric ligands that prompted the use of structural information in drug discovery programs. During the last couple of years the number of GPCR X-ray structures also increased, and by September 2020 reached 394.9 GPCR-AM complex structures have been reported across the four major GPCR Classes (Classes A, B, C, and F), however, the total number of X-ray structures co-crystallized with allosteric modulators in the 7 transmembrane domain is only 39, and hence information on the location of allosteric sites is far from exhaustive. The structures demonstrate that the allosteric sites are widely distributed along the protein surfaces including extracellular ligand entry sites (secondary binding pockets or extracellular vestibule), ancestral sites that are evolutionally abandoned orthosteric sites within the transmembrane domain, allosteric sites at the conformational lock to influence the conformational state of the receptor, or sites located at the intracellular signaling interface stabilizing or preventing the binding of signaling proteins.
In our previous work10 we have used the protein mapping program FTMap to investigate the binding properties of GPCRs that were co-crystallized with allosteric ligands. FTMap (http://ftmap.bu.edu/) places small organic probe molecules on a grid around the surface of the protein to be studied, finds the most favorable positions for each probe type, clusters the probes, and ranks the clusters based on their average energy. 11, 12 Regions that bind several low energy probe clusters are called consensus clusters or consensus sites (CSs) and predict binding hot spots, small regions on the protein surface that can contribute a disproportionate amount to the binding free energy, and hence are important for the binding of any ligand. It was shown that FTMap was capable of correctly identifying the known intrahelical and intercellular binding sites in the majority of the considered GPCR X-ray structures, and about half of these sites (21 of the 39) are predicted to be capable of binding ligands with micromolar or higher affinity 13. In the remaining 18 structures the site is either relatively weak or is located at the protein-membrane interface that currently FTMap is unable to identify.
For soluble proteins mapping ligand-bound structures (after removal of the ligand) is generally followed by mapping ligand-free structures of the same protein to demonstrate that FTMap also works well on such structures. However, we have only four GPCRs that have been crystallized both with and without an allosteric ligand. As an alternative approach to validation we have therefore mapped models of the proteins generated by Alphafold2,14, 15 a deep neural network-based program that was shown to predict protein structures with very high accuracy from the amino acid sequence. As will be discussed, this approach shows that the presence of bound ligands is not required for finding the binding sites.
We then asked whether the known allosteric binding sites identified in specific receptor X-ray structures are conserved between receptors. This comparative approach can be illustrated by the smallest example of two GPCR proteins that both have a strong binding hot spot at the same location, but only one protein has a known allosteric ligand binding at the hot spot. Our basic hypothesis is that the same hot spot in the other protein is also capable of binding allosteric ligands, and that ligand binding will – in most cases – have some modulatory effect. To explore this idea, we mapped the 394 GPCR structures available at September 2020, and checked whether they have strong binding hot spots at the locations observed in any of the 21 structures co-crystallized with allosteric ligands. For each of the 21 structures we identified a set of structures that have such hot spots and thus predicted ligand binding sites at the same location as the “parent” structure. The GPCRs within such clusters include proteins from the same family, but also proteins that are not closely related, with sequence identities below 60% and RMSD values greater than 5Å. In some cases, the clusters include even GPCRs from different classes. As will be described, the sites in all these structures essentially map to nine distinct consensus sites that predicted to bind a large variety of allosteric ligands in different GPCRs. The mapping also revealed that most individual GPCRs have only three or fewer sites that are predicted to be capable of binding a ligand with high affinity, and that these locations are among the nine sites we identified in the vast majority of GPCRs. However, the ligands binding at the same location in different GPCRs generally show little or no similarity, and the amino acid residues interacting with these ligands generally also differ. As will be discussed, this observation is somewhat similar to the recent finding that the cholesterol binding sites in all GPCRs are located at 12 distinct locations but lack any consensus motif.16
RESULTS
FTMap identifies allosteric sites in GPCRs with bound ligands
We considered 394 X-ray crystallographic structures representing 77 distinct GPCRs. Most of the crystallized proteins belong to Class A (360); rhodopsin, adenosine A2A and beta adrenergic receptor structures cover almost 44% of the published structures. Receptor structures from other classes (B-F) show more balanced distributions. There were 15 Class B structures from four different receptors. Class C had a total of 6 structures from 2 receptors. Of the 13 Class F structures (Frizzled) included in our set, 77% of structures were Smoothened Homolog (SMO) proteins. The set includes 39 structures co-crystallized with allosteric ligands (Table 1). To assess how well were the ligands resolved in these structures we have checked the ligand structure quality parameters that are reported in the respective PDB entries (a brief summary of these parameters is available at rcsb.org: https://www.rcsb.org/docs/general-help/ligand-structure-quality-in-pdb-structures#what-is-ligand-structure-quality). These parameters were collected for all 39 allosteric ligand-bound structures, except for 3OE0 (with a peptide ligand), where they were not reported in PDB. The average occupancy is 1.0 in all structures except for 5X7D and 6OBA (with 0.82 and 0.79, respectively), meaning that the ligands are unambiguously resolved in the overwhelming majority of the structures. The real space correlation coefficient (a local electron density goodness-of-fit indicator) is 0.911 when averaged over all structures, and slightly lower, but still acceptable (0.886) for the 13 structures with resolutions >3 Å. The average number of atomic clashes is 1.1 (1.2 for low-resolution structures), the average numbers of bond length and bond angle outliers are also reasonably low (6.6 and 6.1, or 7.0 and 8.0 for the low-resolution structures, respectively), and there are no stereochemical errors in any of the structures.
Table 1.
X-ray structures of GPCRs co-crystallized with small molecule allosteric ligands
| Target | Ligand ID | Ligand name | PDB ID | Statea | Site typeb | Site locationc |
|---|---|---|---|---|---|---|
| Class A | ||||||
| A2A | 8D1 | Cmpd-1 | 5UIG | Inactive | HC | TM-EC |
| β2 | 8VS | CMPD-15PA | 5X7D | Inactive | SI | IC |
| β2 | KBY | Compound-6FA | 6N48 | Active | CL | EH-IC |
| β2 | M3J | AS408 | 6OBA | Inactive | CL | EH |
| C5a1 | 9P2 | NDT9513727 | 5O9H | Inactive | CL | EH |
| C5a1 | 9P2 | NDT9513727 | 6C1Q | Inactive | CL | EH |
| C5a1 | EFD | Avacopan | 6C1R | Inactive | CL | EH |
| CCR2 | VT5 | CCR2-RA-[R] | 5T1A | Inactive | SI | IC |
| CCR5 | MRV | Maraviroc | 4MBS | Inactive | HC | TM-EC |
| CCR7 | JLW | Cmp2105 | 6QZH | Inactive | SI/CL | IC |
| CCR9 | 79K | Vercirnon | 5LWE | Inactive | SI | IC |
| CB1 | 9GL | ORG27569 | 6KQI | Inactive | CL | EH |
| CXCR4 | ITD | IT1t | 3ODU | Inactive | HC | TM-EC |
| CXCR4 | PRD | CVX15 | 3OE0 | Inactive | HC | TM-EC |
| FFA1 | 2YB | TAK-875 | 4PHU | Intermediate | CL | EH-EC-TM |
| FFA1 | 6XQ | Compound 1 | 5KW2 | Intermediate | CL | EH |
| FFA1 | MK6 | MK-8666 | 5TZR | Intermediate | CL | EH-EC-TM |
| FFA1 | 7OS | AP8 | 5TZY | Intermediate | CL | EH |
| GPR52 | EN6 | C17 | 6LI0 | Inactive | CL | TM-EC |
| M2 | 2CU | LY2119620 | 4MQT | Active | HC | TM-EC |
| P2Y1 | BUR | BPTU | 4XNV | Intermediate | CL | EH-EC |
| PAR2 | 8TZ | AZ8838 | 5NDD | Intermediate | HC/CL | TM |
| PAR2 | 8UN | AZ3451 | 5NDZ | Intermediate | HC/CL | EH |
| Class B | ||||||
| CRF1 | 1Q5 | CP-376395 | 4K5Y | Inactive | CL | TM (IC) |
| GLP-1 | 97Y | PF-0637222 | 5VEW | Inactive | SI | EH-IC |
| GLP-1 | 97V | NNC0640 | 5VEX | Inactive | SI | EH-IC |
| GLP-1 | 97Y | NNC0640 | 6KJV | Inactive | SI | EH-IC |
| GLP-1 | 97Y | NNC0640 | 6KK7 | Inactive | SI | EH-IC |
| GLP-1 | 97Y | NNC0640 | 6LN2 | Inactive | SI | EH-IC |
| GCGR | 5MV | MK-0893 | 5EE7 | Inactive | CL | EH-IC |
| GCGR | 97V | NNC0640 | 5XEZ | Inactive | CL | EH-IC |
| Class C | ||||||
| mGlu1 | FM9 | FITM | 4OR2 | Inactive | HC | TM |
| mGlu5 | 2U8 | Mavoglurant | 4OO9 | Inactive | HC | TM |
| mGlu5 | 51D | CMPD-25 | 5CGC | Inactive | HC | TM |
| mGlu5 | 51E | HTL14242 | 5CGD | Inactive | HC | TM |
| mGlu5 | D7W | Fenobam | 6FFH | Inactive | HC | TM |
| mGlu5 | D8B | M-MPEP | 6FFI | Inactive | HC | TM |
| Class F | ||||||
| SMO | SNT | SANT-1 | 4N4W | Inactive | HC/CL | EC-TM |
| SMO | VIS | Vismodegib | 5L7I | Inactive | HC/CL | EC-TM |
Activation states were included from GPCRDB, the categories are defined based on interhelical Cα distances.
Site types are assigned as intrahelical – HC, conformational lock – CL, signaling interface – SI.
Site location is indicated as trans-membrane helical bundle – TM, extra-helical – EH, extracellular side – EC, intracellular side – IC.
We first applied FTMap to the above 39 structures. All non-protein atoms have been removed prior to the mapping that identified strong binding sites within 21 structures shown in Table 2. Among these 21 structures there were proteins from each of the GPCR classes, representing 15 unique receptors. Together, as shown in Figure 1, the receptors covered the range of allosteric binding sites, including intra-helical and extra-helical regions. Analyzing these results, one should consider the present limitation of FTMap that cannot identify allosteric sites located at the protein-membrane interface due to its current parametrization based on complexes of small organic molecules with soluble proteins.10 Within the set of 21 allosteric ligand structures with strong predicted hot spots, an average of 180 probe atoms were located within 3 Å of the allosteric ligand. Some structures had overlaps as high as 339 probe atoms. Thus, this step of the analysis has shown that in 21 structures the allosteric ligand overlaps with a strong binding hot spot.
Table 2.
GPCR structures and AlphaFold2 models with strong binding sites located at bound allosteric ligands
| Target | PDB ID | Number of overlapping probe atoms a | Number of overlapping probe atoms for AF2 model b | Structures with ≥ 84 overlapping probe atoms c | Maximum number of overlapping probe atoms d |
|---|---|---|---|---|---|
| Class A | |||||
| A2A | 5UIG | 170 | 144 | 283 | 263 |
| β2 | 5X7D | 129 | 76 | 34 | 178 |
| CCR2 | 5T1A | 194 | 129 | 20 | 194 |
| CCR5 | 4MBS | 339 | 320 | 320 | 384 |
| CCR7 | 6QZH | 180 | 116 | 11 | 180 |
| CCR9 | 5LWE | 169 | 76 | 47 | 186 |
| CXCR4 | 3ODU | 213 | 110 | 233 | 329 |
| CXCR4 | 3OE0 | 279 | 262 | 321 | 340 |
| FFA1 | 4PHU | 104 | 87 | 13 | 149 |
| FFA1 | 5KW2 | 296 | 174 | 47 | 296 |
| FFA1 | 5TZR | 149 | 81 | 14 | 149 |
| FFA1 | 5TZY | 178 | 174 | 49 | 286 |
| GPR52 | 6LI0 | 157 | 128 | 95 | 264 |
| M2 | 4MQT | 204 | 128 | 127 | 217 |
| PAR2 | 5NDD | 97 | 18 | 190 | 251 |
| PAR2e | 5NDZ | 70 | 92 | 1 | 95 |
| Class B | |||||
| CRF1 | 4K5Y | 169 | 0 | 6 | 169 |
| Class C | |||||
| mGlu1 | 4OR2 | 191 | 171 | 191 | 263 |
| mGlu5 | 4OO9 | 102 | 0 | 146 | 244 |
| Class F | |||||
| SMO | 4N4W | 152 | 51 | 196 | 289 |
| SMO | 5L7I | 213 | 177 | 49 | 243 |
Number of probe atoms within 3Å of the ligand from mapping the target after removing the ligand.
Number of probe atoms within 3Å of the ligand from mapping the AF2 model of the protein.
Number of GPCR structures with a strong hot spot (with over 84 probe atoms) within 3Å of the ligand copied from the target structure.
Maximum number of probe atoms overlapping with the ligand copied from the target structure among all GPCR structures.
Mapping of 5NDZ yields fewer than 84 probe atoms, but the threshold is exceeded when mapping the AF2 model.
Figure 1.
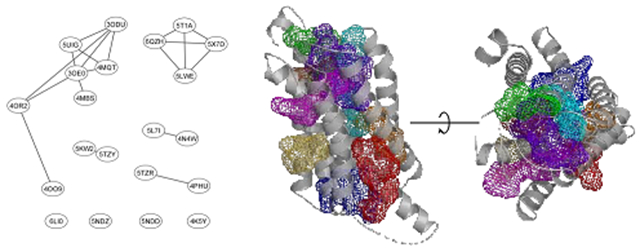
Examples of FTMap site prediction (mesh) in proteins (gray) without co-crystalized allosteric ligands. Binding site predictions were determined by selecting FTMap probe atoms within 3 Å of an allosteric ligand (green sticks) placed by structural alignment. A. Predicted binding pocket in the A2A protein (PDB 3REY) overlayed with the allosteric ligand IT1t from the CXCR4 protein (PDB 3ODU). B. Predicted binding pocket in the GPR52 protein (PDB 6LI1) overlayed with the allosteric ligand C17 from the CCR2 protein (PDB 5T1A). C. Predicted binding pocket in the DRD2 protein (PDB 6CM4) overlayed with the allosteric ligand SANT-1 from the SMO protein (PDB 4N4W). D. Predicted binding pocket in the LPAR1 protein (PDB 4Z34) overlayed with the allosteric ligand TAK-875 from the FFAR1 protein (PDB 4PHU). E. Predicted binding pocket in the P2Y12 protein (PDB 4PXZ) overlayed with the allosteric ligand Compound 1 from the FFAR1 protein (PDB 5KW2). F. Predicted binding pocket in the GLR protein (PDB 5YQZ) overlayed with the allosteric ligand CP-376395 from the CRFR1 protein (PDB 4K5Y). G. Predicted binding pocket in the AGTR1 protein (PDB 4YAY) overlayed with the allosteric ligand C17 from the FFAR1 GPR52 (PDB 6LI0). H. Predicted binding pocket in the PE2R3 protein (PDB 6AK3) overlayed with the allosteric ligand AZ3451 from the PAR2 protein (PDB 5NDZ). I. Predicted binding pocket in the CXCR4 protein (PDB 3OE8) overlayed with the allosteric ligand AZ8838 from the PAR2 protein (PDB 5NDD).
Validating FTMap on GPCRs models generated by AlphaFold2
Since the AlphaFold2 (AF2) program is capable generating high accuracy models of proteins, we considered such models for the validation of the FTMap results applied to X-ray structures. First, we calculated pairwise RMSD values between the AF2 models and X-ray structures for the transmembrane region of the 39 structures with bound allosteric modulators (Table S1). The average RMSD found was 1.006 Å. Second, we have applied FTMap to these AF models. As described earlier, FTMap detected strong binding hot spots at the allosteric site in 21 of the 39 structures (see Table 2). In contrast, FTMap found strong hot spots in the AF2 models of only 17 of these 21 structures, also shown in Table 2. The additional 4 sites that were unable to be detected are the AF2 models of CRF1 (4K5Y) with the overall RMSD of 1.975 Å, SMO (4N4W) with an RMSD of 0.445 Å, mGlu5 (4OO9) with 0.653 Å, and PAR2 (5NDD) with an RMSD of 0.716 Å. For the AF2 model of the CRF1 protein the allosteric site is occluded by helix 6. However, the model shows a low per-residue confidence score for TM2. The allosteric binding site is defined by TM2 and TM3, so it is apparent that the low accuracy of the homology model distorted the allosteric site location beyond recognition by FTMap. In the AF2 model of the SMO protein, the allosteric pocket is slightly smaller than in the X-ray structure (4N4W) and the site is detected with 51 overlapping probe atoms, which is considered a too weak hot spot. For the mGlu5 protein the AF2 model places the side chain of Trp 785 directly into the allosteric site, limiting the access of probe atoms. For the model of the PAR2 protein, a slight movement of the Lys 131 side chain in the AF2 model caused restricting the ligand binding site, and the mapping indicated a very weak hot spot (18 probe atoms) at the allosteric pocket. We also calculated pairwise RMSD values for the transmembrane region of the remaining 354 structures with AF2 models. We could not consider five receptors (Uniprot IDs: P69332, B1B1U5, Q80KM9, Q98SW5 and Q9WTK1, represented by 8 experimental structures) that have no pre-calculated AF2 models available, and were not considered in our calculations. The average RMSD was 0.85 Å for the other 346 structures.
Clustering of allosteric site locations in GPCRs with strong hot spots
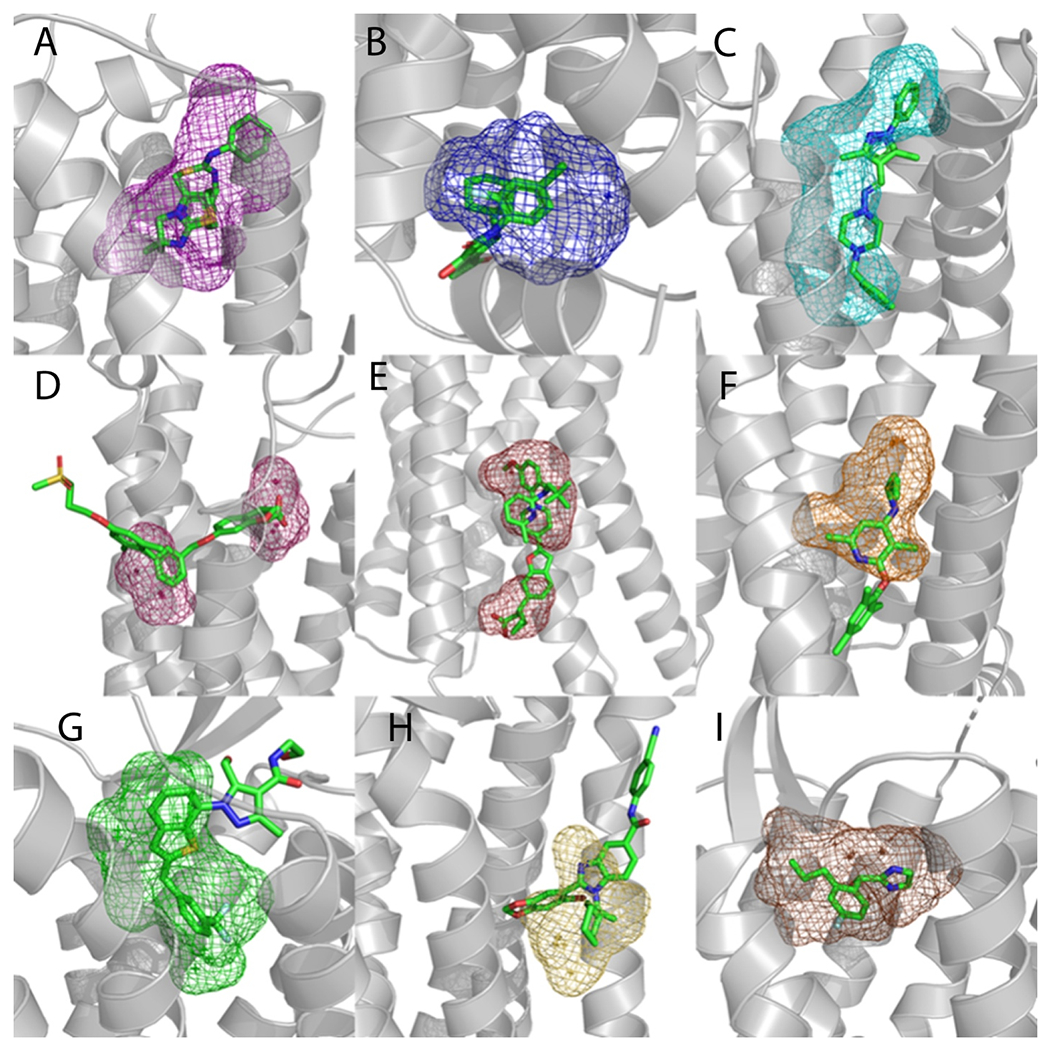
As will be shown, each location in the 21 structures with a bound ligand and strong binding site serves as a potential allosteric site in a large number of additional GPCRs. Here we investigate how the locations of the hot spots that define the 21 sites relate to each other. To determine the similarities we considered each structure with its predicted hot spot and superimposed it with all other 20 structures with their ligands included. For each structure then counted the number of probe atoms overlapping with ligands in other structures. The numbers of overlaps were used to create a non-symmetric similarity matrix shown in Table S2. As discussed in Methods, we defined a measure of similarity between the binding sites in different structures based on the predicted hot spot populations overlapping with ligands. The graph in Figure 2 shows the 21 structures as nodes, with two nodes connected if the binding sites in the two structures overlap. As shown in Figure 2A, based on this overlap measure the sites in CXCR4 (3ODU), A2A (5UIG), M2 (4MQT), a site in a different CXCR4 structure (3OE0), and CCR5 (4MBS) are close to each other and form one cluster we identify as Cluster 1 (the structures considered are shown in parenthesis). Although this overlap is predicted on the basis of the hot spots, according to Figure 3A the ligands in these structures indeed overlap. (We note that the ligand in 3OE0 is a cyclic peptide, which is much larger than the ligands in the other four structures and hence is not shown in Figure 3A). The site predicted in mGlu1 (4OR2) is further apart from these five, although the ligands still overlap, and the site in mGlu5 (4OO9) is even further away, overlapping only with the ligand of mGlu1 (4OR2). In fact, the sites in these two structures are classified as being in the trans-membrane helical bundle (TM), rather than in the trans-membrane helical bundle on the extracellular side (EC-TM) as the other five structures in Cluster 1. Based on probe overlap, the second largest cluster (Cluster 2, shown in Figure 3B) is formed by the sites in CCR2 (5T1A), CCR7 (6QZH), B2 (5X7D), and CCR9 (6LWE) that all have a site at the signaling interface (SI) on the intracellular side (IC). In addition to these clusters the mapping predicts strong sites that occur in three pairs of structures. The first pair consists of two SMO structures 5L71 and 4N4W (identified as Cluster 3 in Figure 3C), both having sites at the conformational lock at an intrahelical site (HC/CL), the second pair formed by the two FFA1 structures 5TZR and 4PHU (Cluster 4 in Figure 3D) with sites that are classified as extrahelical, extracellular and trans-membrane (EH-EC-TM), and the third pair is formed by 5KW2 and 5TZY, FFA1 structures that both have extra-helical sites (EH). Finally, structures of GPR52 (6LI0), PAR2 (5NDZ), another PAR2 structures with a different site (5NDD), and CRF1 (4K5Y) have binding sites that differ from the other sites, and hence are not in any of the clusters. Note that both 5NDZ and 5NDD are PAR2 structures but include allosteric ligands that bind at very different locations. In summary, we conclude that the strong hot spots in the 21 structures considered here map into nine distinct sites, each represented as a colored mesh in Figure 2B. As will be shown below, each of these 21 sites occur as strong hot spots and thus potential allosteric ligand binding sites in many additional GPCR structures that have no bound allosteric modulators. We emphasize that the similarity measure defined in this section is based on hot spots predicted to overlap with an allosteric site, and thus does not require a structure co-crystallized with an allosteric ligand. However, application to such structures as described here validates the methodology, since the similarity of the binding site locations is known.
Figure 2.
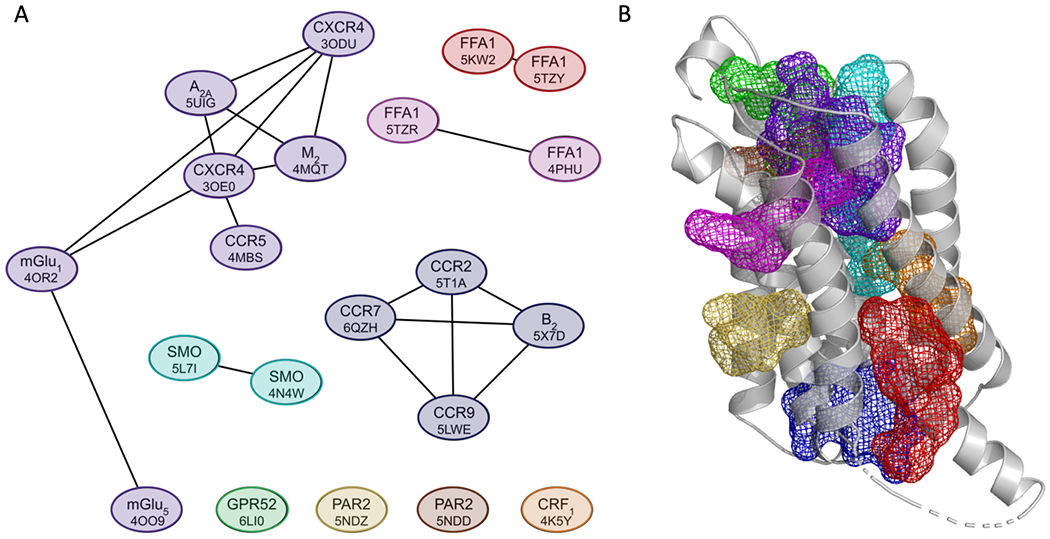
Locations of allosteric sites in structures co-crystallized with ligands. A. Similarity based clustering of the allosteric sites in the 21 structures with bound ligands and strong hot spots. The length of the edges connecting the nodes represents the level of similarity based on the measure of probe overlap, with smaller distances indicating higher numbers of overlapping probes. As shown, the 21 sites map to 9 consensus locations. B. The 9 consensus binding sites defined by the clusters shown in A. The color coding of the mesh representations is as follows: purple – Cluster 1 (3ODU, 4MQT, 4MBS, 5UIG, 3OE0, 4OR2, and 4OO9); blue – Cluster 2 (5T1A, 6QZH, 5LWE and 5X7D); cyan – Cluster 3 (5L7I and 4N4W); pink – Cluster 4 (5TZR and 4PHU); red – Cluster 5 (5KW2 and 5TZY); orange - 4K5Y; green - 6LI0; yellow - 5NDZ; and brown - 5NDD.
Figure 3.
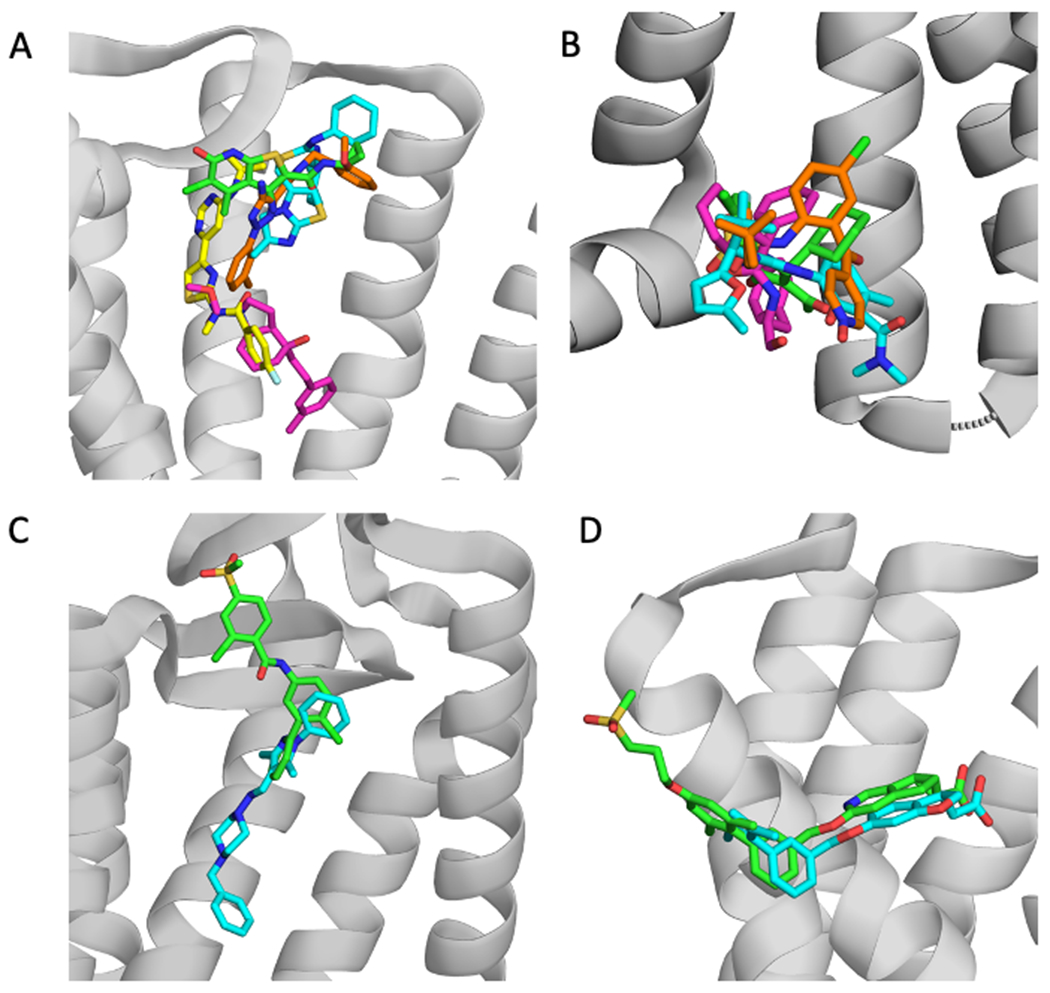
Examples of allosteric ligand clusters. PDB IDs are shown in parenthesis. A. Cluster 1: 2CU green (4MQT), ITD cyan (3ODU); FM9 yellow (4OR2), 8D1 orange (5UIG), 2U8 pink (4OO9). The grey cartoon represents the protein structure 4MQT. B. Cluster 2: VT5 green (5T1A), 8VS pink (5X7D), JLW cyan (6QZH), and 79K orange (5LWE). The cartoon shows the protein structure 5T1A. C. Cluster 3: VIS (5L7I) green, and SNT (4N4W) cyan. The cartoon shows the protein structure 5L7I. D. Cluster 4: MK6 green (5TZR), and 2YB cyan (4PHU). The protein structure shown is 5TZR.
Extending the analysis to all GPCRs X-ray structures
After mapping the GPCR structures with known allosteric binding sites we applied FTMap to the remaining 373 structures, and for each of the 21 structures with a bound allosteric ligand identified all structures that had a strong hot spot overlapping with the ligand. Each of the 21 “parent” structures, on average, had 117 “daughter” structures that had a strong hot spot (with ≥ 84 probe atoms) overlapping with the ligand in the “parent” structure. For each of the 21 “parent” structures, Table S3 lists the 10 PDB IDs of the proteins that, after superimposing the structures, have the highest number of hot spot atoms overlapping with the ligand. Analysis of the GPCRs with strong hotspots at the same location as an allosteric ligand binding site revealed that site locations can be conserved across families and classes of GPCRs. We emphasize that the hot spots in many GPCRs overlap with ligands in several of the 21 “parent” structures. In fact, as we discussed, the 21 structures map only to nine distinct sites, so all the sites found by FTMap must be located at one of these nine sites. However, even ligands that bind at overlapping hot spots may only partially overlap (see Figures 3C and 3D for examples), and considering all 21 “parent” structures rather than the 9 consensus sites provides better defined measures of site similarity. We also emphasize that for each of the 21 structures we collect GPCR structures that have hot spots overlapping with the ligand in the “parent” structure. Since some of these ligands are very large, they may overlap with hot spots from different proteins that do not overlap with each other, increasing the number of GPCRs for the “parent” structure. Thus, while a strong hot spot in such proteins is really located at a site that binds the ligand in the “parent” protein, it does not necessarily overlap with the strongest hot spot in the latter structure.
We recall that FTMap did not find strong hot spots overlapping with the co-crystallized allosteric ligand in 18 of the 39 structures. Nevertheless we checked if the other structures have strong hot spots at the locations corresponding to the ligands in these 18 structures. As shown in Table S4, no strong hot spot with >84 probes has been found for eight of the 18 structures, indicating that the site is weak in all structures. However, between 1 and 126 “daughter” structures with strong hot spots have been identified for the remaining 10 “parent” structures without strong hot spots. Since we do not consider the “daughter” structures for the 18 X-ray structures in which FTMap could not find druggable allosteric sites, based on our analysis such daughter structures are false negatives. Accordingly, while the mapping predicts conserved allosteric sites in many GPCR structures, we do not claim finding all such sites. Nevertheless, we believe that showing the high level of allosteric site conservation even among unrelated GPCRs is an interesting result.
The large number of GPCRs that have sites overlapping with each of the 21 known sites might suggest that each GPCR has many potential ligand binding sites. However, the results of mapping also show that the majority of GPCRs have three or fewer sites that are predicted to be capable of binding a ligand with high affinity (Figure 4). As we argued, in a large variety of GPCRs these sites are located at one of the nine locations we have identified in the previous section. Thus, in spite of their structural complexity and dynamical nature, it appears that GPCRs have only a limited number of locations that can serve as ligand binding sites, and that the same sites exist in many GPCRs, including receptors with low sequence similarity/homology. However, as mentioned, some of the allosteric ligands are very large and may bridge multiple binding sites.
Figure 4.
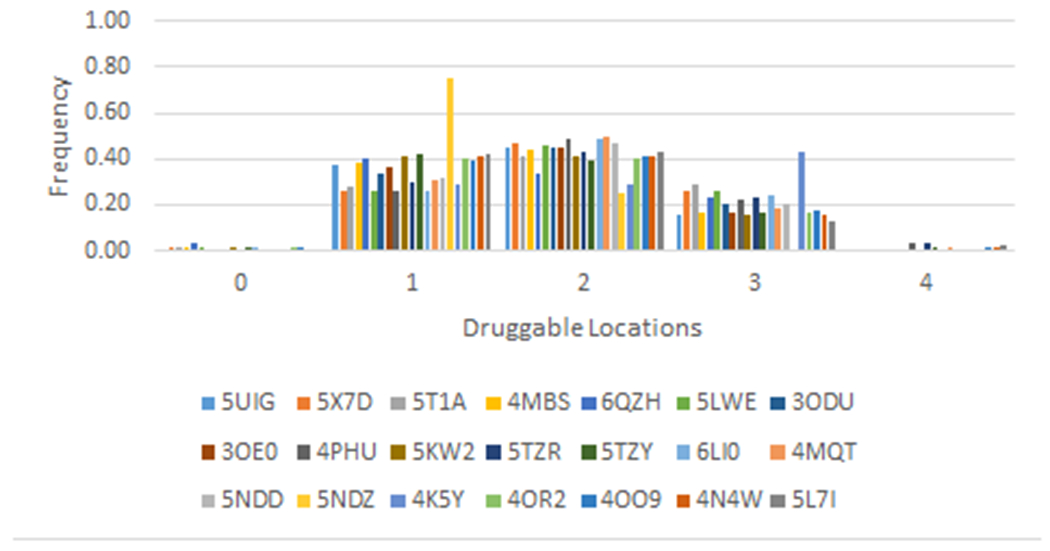
Distribution of the number of druggable sites in the clusters defined by the 21 GPCRs co-crystalized with allosteric ligands.
Validation of predicted allosteric sites
As emphasized in this paper and in our many FTMap related publications,12, 17 a strong binding hot spot indicates a potential ligand binding site. Our major hypothesis is that if a hot spot binds an allosteric ligand in one GPCR, due to the overall similarity of GPCR structures a strong binding hot spot at the same location in other GPCRs also binds some ligands that are likely to behave as allosteric modulators. This assumption can be validated in three ways. First, the best validation is to have an X-ray structure co-crystallized with an allosteric modulator that binds at the predicted site. Second, if no X-ray structure is available, docking can be used to show that a known allosteric modulator binds at the predicted site. The third option, also without a co-crystal structure, is having an allosteric ligand that binds to the second GPCR, and mutating some of the residues surrounding the predicted site to show that the mutations impact allosteric modulation.
Fully prospective validation by the first approach would require co-crystallization of some GPCR with allosteric ligands that bind at the predicted site. Since determining the X-ray structures of GPCRs is still very difficult, we have to rely on the 21 structures that already have modulators binding at strong hot spots. Each line in Figure 2A connects two structures that have both strong hot spots and bound allosteric modulators at the same location and thus provides a (retrospective) support of our main hypothesis. Accordingly, Figure 3A shows the ligands in the largest cluster (Cluster 1) in Figure 2A. The latter indicates that the hot spot of mGlu5 (4OO9) overlaps only with the ligand bound to mGlu1 (4OR2) and vice versa. The two ligands are shown in pink and yellow, respectively, in Figure 3A. Table S2 shows that the hot spot in 4OO9 overlaps with its own ligand (102 probe atoms) and the ligand in 4OR2 (122 probe atoms). The ligands in the four GPCR structures in Cluster 2 in Figure 2A show tighter overlap (Figure 3B). Similarly, each linked pair in Figure 2A defines a predicted allosteric site. We understand that predicting and confirming novel allosteric pairs based on the mapping of ligand-free structures would be a stronger validation. However, it is well recognized that confirming allostery of a ligand is far from simple. In fact, all the known NAMs were not identified as allosteric modulators but as antagonists/inverse agonists until their binding sites were determined by crystallography (except for AZ3451 in PAR2).18
For validation by docking we have performed a large scale computational study. In Table S5 we list the 278 GPCR PDB structures that have a hot spot with more than 84 probe clusters overlapping with the allosteric modulator in one of the 21 “parent” structures with a strong hot spot at the allosteric site. The first column is the PDB ID of the “parent” structure, color-coded by cluster according to those shown in Figure 2B. The second column is the PDB ID of the structure without a co-crystallized allosteric ligand (to be referred to as the “daughter” structure) that has a strong hot spot overlapping with the ligand in the “parent” structure, followed by the number of overlapping probe atoms and the family of the “daughter” GPCR. We note that this list has been filtered to only show the “parent” structure that has a ligand that overlaps with the strongest hot spot of the “daughter” structure. For each of the “daughter” structures we searched the Allosteric Database (ASD) to identify potential allosteric ligands. The first column in Table S6 shows the PDB IDs of the “daughter” and “parent” structures from Table S5, and the ASD ID of a ligand that, according to the Allosteric Database, binds to the “daughter” protein. We extracted the ligand in MOL2 format from ASD, and docked it to the “daughter” structure using Autodock Vina.19 The docking was restricted to a region defined by the union of 3.0 Å boxes around each probe atom. All Vina parameters were set to their default values. Each docking run generated 10 poses of the ligand. Columns 2 of Table S6 shows the pose ID of the docked ligand that was closest to the hot spot (consensus cluster) whose ID and the number of probe clusters is shown in column 3 of Table S5. The shortest distance between the center of mass of the docked ligand and the center of mass of the probe clusters that define the consensus site is shown in column 4 of Table S6. Column 5 shows the pose ID of the docked ligand that had the shortest distance to the allosteric modulator copied from the “parent” protein after superimposing the two structures. Columns 6 and 7 identify the 3-letter PDB code of the ligand in the “parent” structure and the distance between the center of mass of the docked ligand and the center of mass of the ligand from the “parent” structure. As shown, in most cases the distance does not exceed 3 Å, indicating that the predicted location can accommodate the known allosteric modulator. Notice that generally we consider several allosteric ligands from the ASD, and frequently not all of them dock at the predicted site, but usually at least one of the candidate ligands binds so close that it can be considered to bind in the same pocket that binds the modulator in the “parent” structure.
Demonstrating the third method of validation via mutations we consider the dopamine D2 receptor. As shown in Table S3, FTMap reveals that the D2 structure 6CM4 has a strong binding hot spot that substantially overlaps with the ligand SANT1 binding at the known allosteric site of the Smoothened receptor structure 4N4W. Although the dopamine D2 receptor has not been co-crystallized with any allosteric ligand, a recent paper describes the identification and validation of an allosteric site that binds a positive allosteric modulator (PAM) UCB compound.20 It was predicted that the site, in order of decreasing impact, is surrounded by residues Trp 100, Tyr 408, Ile 184, Glu 95, Leu 94, Thr 412, Ser 409, Trp 413, Asp 114, Phe 102, His 393, Phe 110, and Val 91.17 The validation confirmed that the compound modulated cAMP production, and involved mutating some of the above residues. 17 The mapping of D2 receptor structure 6CM4 using FTMap has detected a strong hot spot surrounded by the above residues (Figure 5A).17 We were able to use the location based on the mapping results to successfully dock the UCB compound into the known allosteric site (Figure 5A). To show that this site in 6CM4 is really the one that binds the allosteric ligand SANT1 in the Smoothened receptor structure 4N4W, we superimposed the two structures and copied the ligand SANT1 from 4N4W into 6CM4 (Figure 5B). Although SANT1 does not bind as deep in the pocket as predicted for the UCB compound, FTMap clearly predicts the same location that binds the allosteric ligand SANT1 in the “parent” structure 4N4W.
Figure 5.
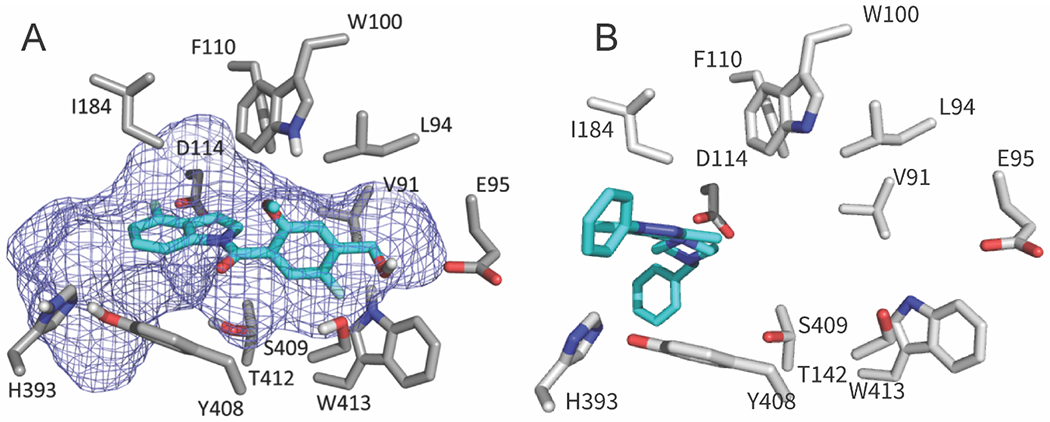
Validation of predicted allosteric sites. A. FTMap site prediction (mesh) matches the recently validated UCB compound (cyan) binding location on the D2 receptor (PDB ID 6CM4). Key residues from the D2 receptor are represented as sticks. The UCB compound was docked using the FTMap probes as the docking box for Autodock Vina. B. The ligand SANT1 copied from the Smoothened receptor structure 4N4W into the dopamine D2 receptor structure 6CM4 after superimposing the two structures.
Site conservation within a specific GPCR subtype: Muscarinic acetylcholine receptors
We started by evaluating the conservation of allosteric sites within a specific GPCR family having a single endogenous ligand (acetilcholine). For this we first looked at the class A muscarinic acetylcholine receptor family. Although in the family only one M2 structure (PDB ID 4MQT) is co-crystallized with an allosteric modulator,21 it is assumed that both the orthosteric and allosteric site locations are conserved for M1 through M5.22 Table 3 lists the structures with the most conserved allosteric sites among the muscarinic acetylcholine receptor proteins, and shows that the site is indeed conserved in all members of the family, irrespective of the activation state. As shown, while 4MQT is in the active state, the only other active-state structure 4MQS has ranked relatively low in terms of overlapping probe atoms, and all other “daughter” structures are in the inactive state. For each structure we show the root mean square deviation (RMSD) from 4MQT, sequence similarity, and pocket volume calculated by the dpocket option of the fpocket program.23, 24 In addition, we use dpocket to extract a number of pocket descriptors and form a similarity score ranging from similar (0) to dissimilar (1).
Table 3.
Analysis of structures with probe atoms overlapping the ligand PAM in the active-state muscarinic acetylcholine receptor 2, PDB ID 4MQT 21
| Receptor | PDB ID | Overlapping probe atoms | Pocket volume, Å3 | RMSD, Å | Sequence Similarity, % | Similarity score | Statea |
|---|---|---|---|---|---|---|---|
| M2 | 4MQT | 204 | 275.3 | Active | |||
| M3 | 4U14 | 136 | 128.0 | 1.35 | 87.3 | 0.267 | Inactive |
| M5 | 6OL9 | 124 | 160.9 | 1.13 | 85.7 | 0.191 | Inactive |
| M2 | 5ZKC | 110 | 141.5 | 1.53 | 99.3 | 0.203 | Inactive |
| M3 | 5ZHP | 95 | 81.2 | 1.31 | 87.3 | 0.158 | Inactive |
| M1 | 6WJC | 95 | 123.3 | 1.88 | 83.6 | 0.349 | Inactive |
| M3 | 4U15 | 93 | 147.5 | 1.46 | 87.2 | 0.205 | Inactive |
| M2 | 5ZK3 | 91 | 134.9 | 1.55 | 98.9 | 0.188 | Inactive |
| M4 | 5DSG | 84 | 117.7 | 1.14 | 95.1 | 0.238 | Inactive |
| M2 | 5YC8 | 84 | 89.9 | 1.50 | 99.3 | 0.180 | Inactive |
| M3 | 4DAJ | 80 | 108.0 | 1.28 | 87.3 | 0.183 | Inactive |
| M2 | 4MQS | 79 | 78.0 | 0.20 | 99.6 | 0.135 | Active |
| M1 | 5CXV | 79 | 98.3 | 1.71 | 84.0 | 0.290 | Inactive |
| M2 | 3UON | 72 | 62.9 | 1.46 | 99.3 | 0.220 | Inactive |
Activation states were included from GPCRDB, the categories are defined based on interhelical Cα distances.
We also created a phylogenetic tree of the 18 different muscarinic acetylcholine receptor structures based on sequence similarity and colored the nodes to represent the level of the conservation, based on whether the hot spots are close to the ligand bound in 4MQT (Figure 6). The colors vary from light yellow to dark purple to show increasing overlap of the site with the ligand 2CU bound to the “parent” protein 4MQT. Interestingly, the structures with the most conserved sites, represented by darker colors on the tree, are not necessarily the structures closest in sequence similarity to 4MQT. The GPCR with the strongest allosteric site conservation (M3 receptor, PDB ID 4U14)25 has relatively low sequence similarity to M2 (4MQT). There is no evidence that RMSD, sequence similarity, or dpocket similarity measures can be used to accurately predict the conservation level of an allosteric site.
Figure 6:
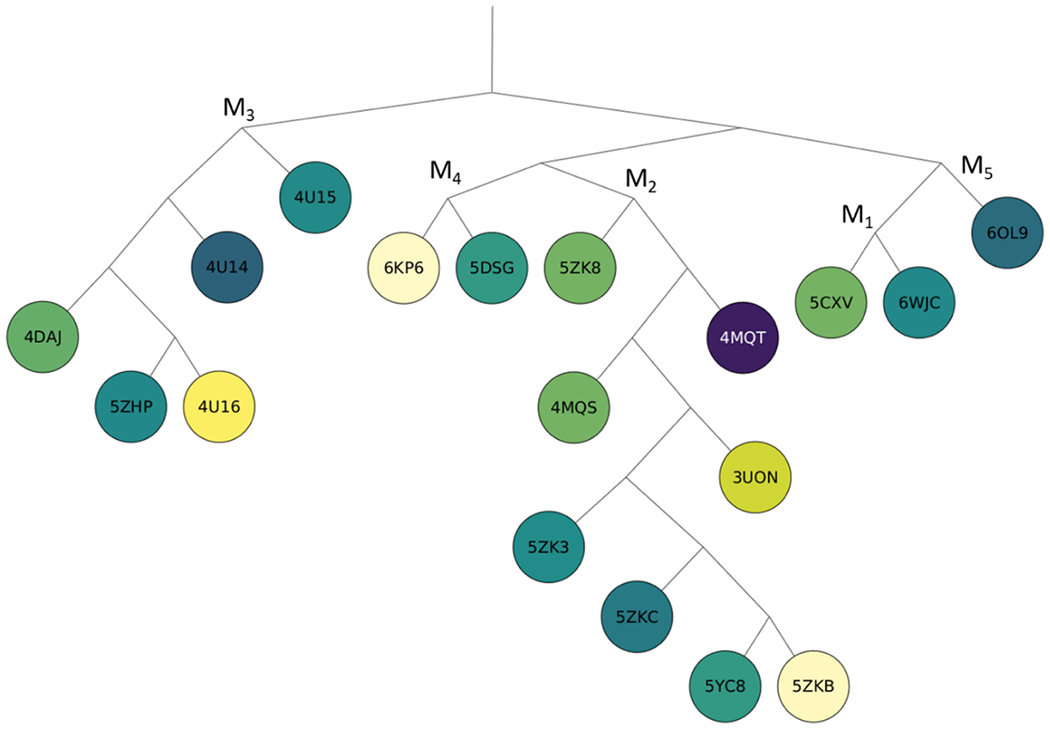
Phylogenetic tree of proteins in the muscarinic acetylcholine receptor family, colored from yellow to dark purple based on the number of probe atoms overlapping with the allosteric ligand 2CU bound in the PDB structure 4MQT after superimposing the structures.
Site conservation across a GPCR family: chemokine receptors
Next, we branched out to determine if allosteric sites are conserved within a family of GPCRs having multiple endogenous ligands with increased complexity and binding preferences. For this we chose the allosteric structure with the strongest site determined by FTMap. The site of the ligand Maraviroc in the class A chemokine receptor CCR5 structure 4MBS 26 had 339 overlapping probe atoms, indicating a very strong site. After overlapping the mapped structures with 4MBS we have found 320 structures that had 84 or more probe atoms overlapping with the bound Maraviroc. Initially we focused the evaluation of site conservation on the 14 additional chemokine receptor structures shown in Table 4 that all are in the inactive state. The chemokine receptor branch of the GPCR phylogenetic tree, shown in Figure 7, contains 14 different chemokine receptor structures, colored from light yellow to dark purple based on the level of site conservation. In 13 of the 14 structures, strong site conservation was observed. Unlike the muscarinic acetylcholine receptors, the chemokine allosteric site conservation within the family is generally correlated with sequence similarity. This is exemplified by the darkest colored nodes being on the same branch. Additionally, four of the five CCR5 structures contain the highest numbers of overlapping probe atoms. Nine of the 14 chemokine receptor structures contain one of the four unique ligands co-crystallized with the protein in the region of the allosteric site.
Table 4.
Conservation of the allosteric site within the class A chemokine receptor CCR5, PDB ID 4MBS 26
| IUPHAR Namea | PDB ID | Ligand ID | Overlapping probe atoms | Pocket volume, Å3 | Sequence similarity, % | RMSD, Å | Similarity score | State |
|---|---|---|---|---|---|---|---|---|
| CCR5 | 4MBS | MRV | 339 | 839.8 | Inactive | |||
| CCR5 | 6AKY | A4X | 384 | 796.0 | 100.0 | 0.42 | 0.169 | Inactive |
| CCR5 | 5UIW | 339 | 651.9 | 100.0 | 0.74 | 0.161 | Inactive | |
| CCR2 | 6GPX | F7N | 317 | 574.0 | 92.0 | 0.79 | 0.286 | Inactive |
| CCR5 | 6AKX | A4R | 313 | 747.6 | 100.0 | 0.25 | 0.060 | Inactive |
| CXCR4 | 3OE8 | ITD | 287 | 574.9 | 69.7 | 1.44 | 0.323 | Inactive |
| CXCR4 | 3ODU | ITD | 262 | 667.1 | 68.2 | 1.99 | 0.296 | Inactive |
| CXCR4 | 3OE0 | 248 | 624.5 | 68.1 | 1.31 | 0.334 | Inactive | |
| CXCR4 | 3OE6 | ITD | 226 | 558.9 | 70.0 | 1.86 | 0.229 | Inactive |
| CCR2 | 6GPS | F7N | 218 | 579.0 | 93.1 | 0.85 | 0.272 | Inactive |
| CXCR4 | 4RWS | 217 | 599.2 | 67.6 | 2.69 | 0.234 | Inactive | |
| CXCR4 | 3OE9 | ITD | 173 | 301.1 | 69.6 | 1.67 | 0.271 | Inactive |
| CCR2 | 5T1A | 73R | 157 | 363.4 | 89.0 | 0.93 | 0.261 | Inactive |
| CCR7 | 6QZH | 93 | 131.9 | 72.4 | 1.71 | 0.321 | Inactive | |
| CCR9 | 5LWE | 53 | 212.6 | 68.3 | 2.89 | 0.474 | Inactive |
Results for the 13 additional chemokine receptor structures are included for comparison.
Figure 7:
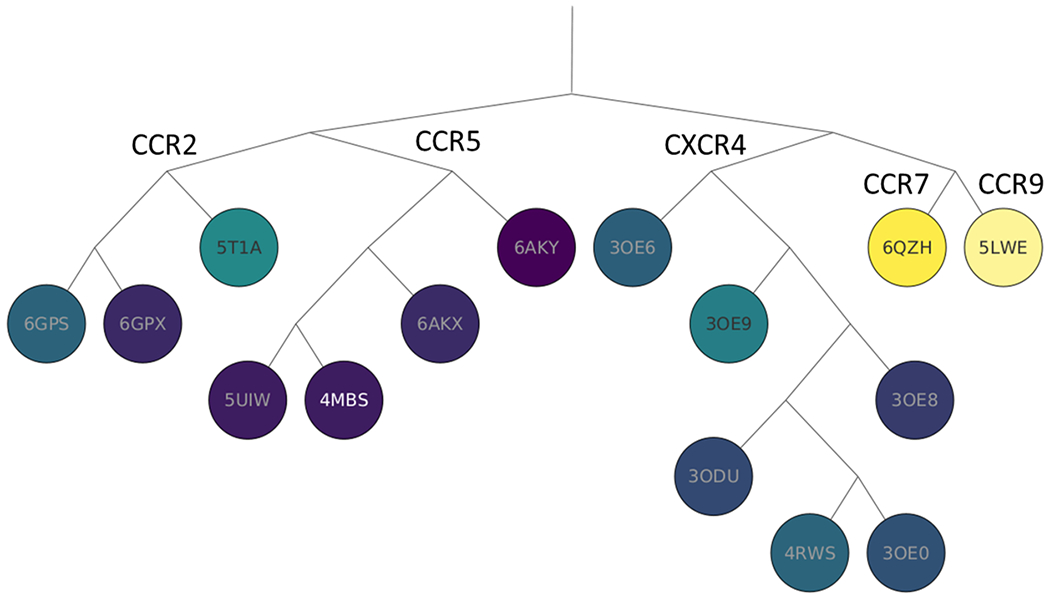
Phylogenetic tree of proteins in the chemokine family, colored from yellow to dark purple, based on the number of probe atoms overlapping with the allosteric ligand Maraviroc (MRV) bound in the PDB structure 4MBS of the CCR5 protein after superimposing the structures.
A mesh representation of the predicted allosteric binding pocket was created by encapsulating all FTMap probe atoms from consensus clusters within 4 Å of the allosteric ligand, Maraviroc (MRV). As shown in Figure 8A, the results of mapping the CCR5 structure 6AKX are consistent with the binding site of the allosteric ligand MRV from 4MBS. 6AKX is one of the nine chemokine receptor structures. As shown in Figure 8B, 6AKX is co-crystallized with the ligand A4R that overlaps with the binding pocket in 4MBS. A4R shows an example of what can be assumed to be another allosteric ligand that is highly similar to the allosteric ligand MRV bound in the “parent” CCR5 structure 4MBS. Although A4R is a structural analog of Maraviroc, due to a lack of a pharmacological profiling 6AKX is not included in the list of 39 allosteric proteins co-crystallized with allosteric ligands. Mapping results for the CCR2 structure 5T1A, shown in Figure 8C, also indicate a biding pocket at the MRV site. Additionally, 5T1A contains a co-crystalized ligand, 73R, which has partial overlap with the allosteric site (Figure 8D). It is interesting that mapping reveals an allosteric site that as large as the site binding Maraviroc in 4MBS, although the allosteric ligand 73R that actually binds to the 5T1A structure is much smaller.
Figure 8:
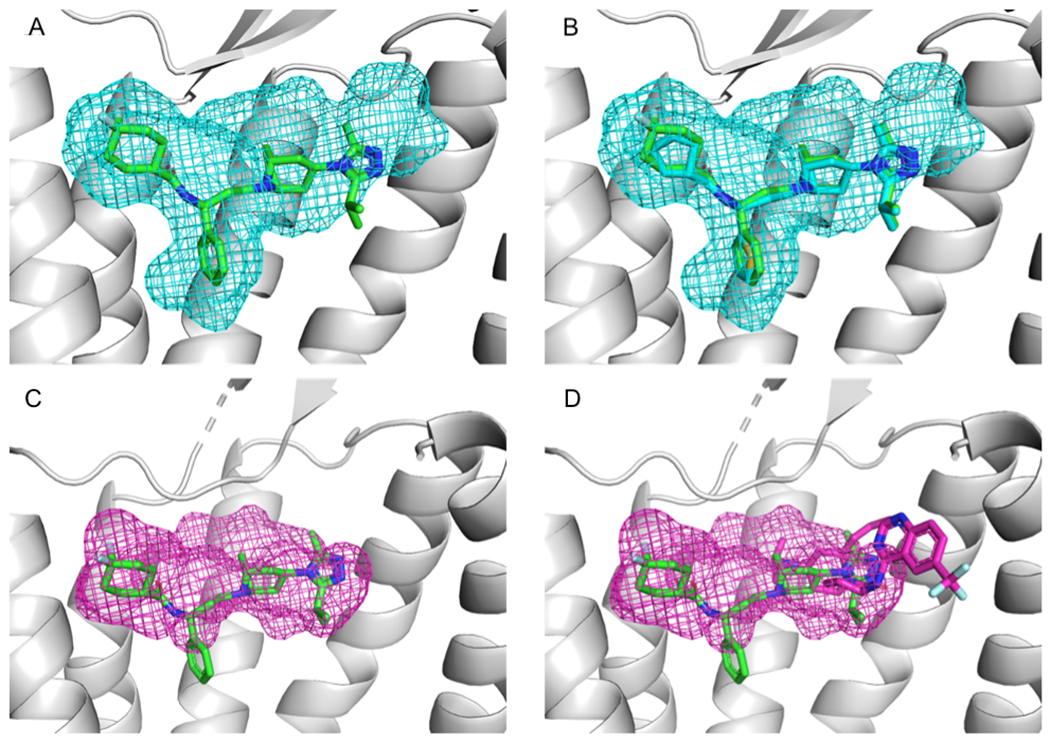
Mapping of class A chemokine receptors. A. Results of mapping the CCR5 structure 6AKX (gray), shown as a mesh, superimposed with the allosteric ligand Maraviroc (MRV, shown as green stick) from the allosteric CCR5 structure 4MBS. B. The ligand A4R (cyan sticks), co-crystalized with the 6AKX protein, binds in the location consistent with both the mapping results and the MRV binding site. C. Results of mapping the CCR2 structure 5T1A (gray), shown as mesh superimposed with the allosteric ligand MRV (green sticks) from 4MBS. Thus, the mapping results for 5T1A are consistent with the known allosteric binding site of MRV. D. The structure 5T1A contains a co-crystalized ligand, 73R (pink sticks). Note that the mapping of 5T1A reveals a binding site which is large enough to accommodate a ligand of the size of MRV, although the actual ligand, 73R, is much smaller.
Site conservation across GPCR classes: Class A C-X-C motif chemokine receptor 4 (CXCR4)
To extend our study of allosteric site conservation, we chose a C-X-C motif chemokine receptor 4 (CXCR4) structure (PDB ID 3ODU 27), co-crystalized with the allosteric ligand ITD. As shown in Table 5, FTMap strongly detected the binding site of allosteric ligand ITD; there were 213 probe atoms overlapping with the ligand. In total 232 structures had at least 84 probe atoms overlapping with the ligand copied into the other structures after superposition. These structures included proteins from multiple families including Class A (representing 96% of structures), Class B, Class C and Frizzled GPCRs, as well as multiple conformational states, with 40 active-state, 179 inactive-state, 12 intermediate structures, and one structure classified as ‘other’. Thus, the site is conserved across the different conformational states, although the top 10 structures with the strongest overlaps are all inactive-state (Table 5). Over half of the 270 structures came from only four groups of proteins: 51 adenosine receptors, 48 adrenoceptor, 11 opioid, and 20 orexin receptors.
Table 5.
Top 10 GPCR structures with the highest number of probe atoms overlapping the ligand ITD in the inactive-state, Class A chemokine receptor CXCR4, PDB 3ODU 27
| Class | IUPHAR name | PDB ID | Overlapping probe atoms | Volume, Å3 | RMSD, Å | Sequence similarity, % | Similarity score | Statea |
|---|---|---|---|---|---|---|---|---|
| A | CXCR4 | 3ODU | 213 | 403.5 | Inactive | |||
| A | DP2 | 6D26 | 329 | 380.2 | 1.8 | 57.4 | 0.368 | Inactive |
| A | DP2 | 6D27 | 286 | 409.4 | 1.8 | 56.0 | 0.410 | Inactive |
| A | A2A | 3REY | 253 | 362.7 | 5.7 | 52.3 | 0.465 | Inactive |
| A | OX1 | 4ZJ8 | 248 | 416.8 | 2.6 | 58.8 | 0.159 | Inactive |
| A | A2A | 3VG9 | 226 | 266.5 | 5.7 | 49.8 | 0.406 | Inactive |
| A | D4 | 6IQL | 219 | 340.3 | 6.1 | 55.2 | 0.327 | Inactive |
| A | OX1 | 6TP3 | 218 | 446.5 | 2.8 | 59.2 | 0.328 | Inactive |
| A | OX2 | 5WS3 | 217 | 436.8 | 2.1 | 57.8 | 0.232 | Inactive |
| A | A1 | 5UEN | 214 | 345.7 | 4.6 | 50.5 | 0.349 | Inactive |
| A | CXCR4 | 3OE8 | 211 | 253.5 | 0.6 | 99.3 | 0.170 | Inactive |
Activation states were included from GPCRDB, the categories are defined based on interhelical Cα distances.
The two prostaglandin D2 Receptor 2 (DP2 receptor) structures, 6D26 and 6D27 28 show high level of site conservation with 329 and 286 probe atoms overlapping with the ligand ITD bound to 3ODU (Table 5 and Figures 9A, 9B, and 9C). Despite low overall sequence similarities (average of 56.7 %), three of the 10 residues that comprise the allosteric site are conserved in both DP2 receptors. The conserved residues are Trp 102(3ODU)/97, Arg 183/179 and Cys 186/182 (Figures 9B and 9D). Although the two DP2 structures have co-crystalized ligands in the ITD pocket, no pharmacological data were available to confirm that this is an allosteric site, and hence the DP2 structures were also excluded from our list of GPCR structures with bound allosteric modulators. The RMSD between the 7TM domains of 3ODU and 6D26 is 1.75 Å and the RMSD between the 7TM domains of 3ODU and 6D27 is 1.80 Å, and thus the structures are not very similar. More generally, RMSD, sequence similarity, or dpocket similarity all seem to be somewhat poor predictors of allosteric site conservation.
Figure 9:
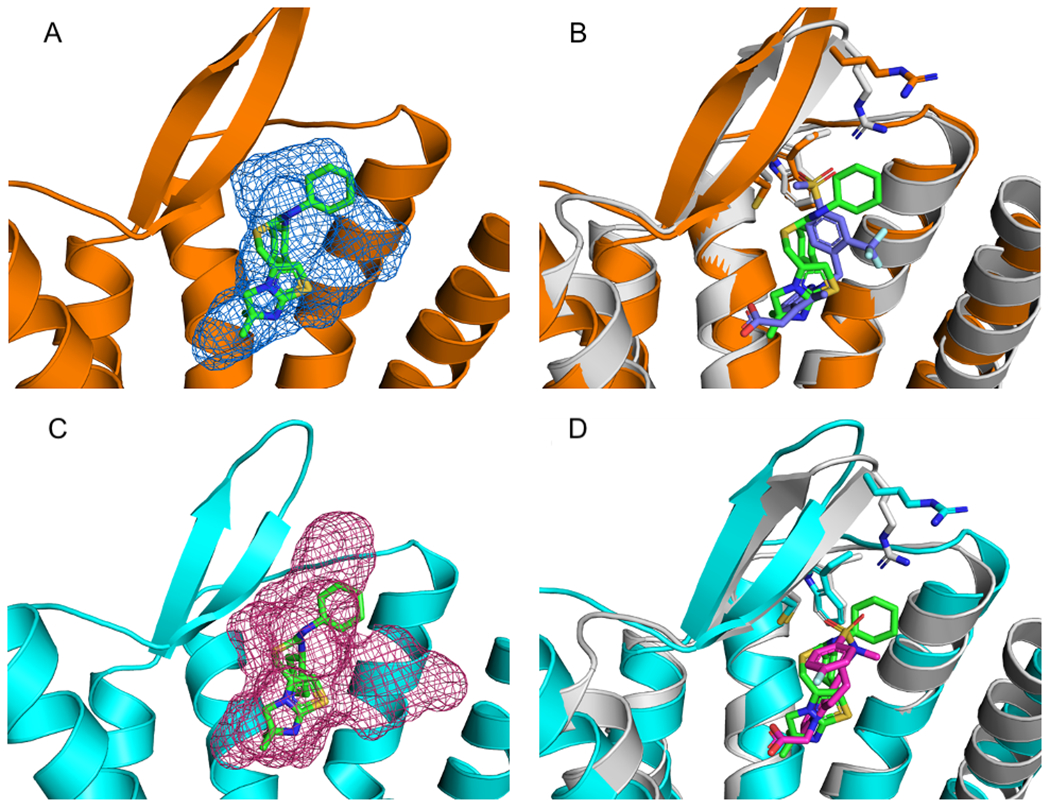
Mapping of Class A C-X-C motif chemokine receptors. A. Mapping results, represented as blue mesh, for the Class A Prostaglandin D2 Receptor 2 (DP2receptor) (PDB ID 6D26) (orange) superimposed with the allosteric ligand IT1t (PDB ID ITD) (green sticks) from Class A allosteric protein C-X-C motif chemokine receptor 4 (PDB ID 3ODU). B. 6D26 with co-crystalized ligand (PDB code FSY) (blue) superimposed with the allosteric protein, 3ODU (gray). Also shown are stick representations of three residues from the ITD binding pocket in 3ODU that were conserved in the 6D26 structure. C. Mapping results, represented as pink mesh, for the DP2 receptor structure 6D27 (cyan) with the allosteric ligand ITD (green sticks) from 3ODU. D. 6D27with co-crystalized ligand FT4 (pink sticks) superimposed with 3ODU (gray) and co-crystalized ligand ITD (green sticks). Also shown are the three residues from 3ODU’s ITD binding pocket that were conserved in the 6D27 structure.
Site conservation across GPCR classes: Class B corticotropin-releasing factor receptor 1 (CRF1)
The structure 4K5Y 29 of the class B (secretin) corticotropin-releasing factor receptor 1 (CRF1) protein is co-crystallized with the allosteric ligand 1Q5. As shown in Table 6, FTMap identified the binding site with 169 probe atoms placed within 3 Å of the allosteric ligand 1Q5 in the 4K5Y structure. Based on our criteria, the site predicted by FTMap is a strong site. There were five structures (excluding 4K5Y) that had 84 or more probe atoms within 3 Å of the superimposed allosteric ligand 1Q5, including one active-state, one intermediate and three inactive-state structures. Within the five structures with significant site conservation, as indicated by probe overlap, there were two Class A and three Class B structures. The Class A protein with the highest number of overlapping probe atoms was the C-X-C motif chemokine receptor 4 (CXCR4) structure 3OE9 27 (Figure 10A). As shown in Figure 10B, 4K5Y and 3OE9 share the following conserved residues within the allosteric site: Leu 280/208, Leu 287/216 and Tyr 327/256. Mapping results strongly indicate that the 1Q5 binding site is a highly conserved allosteric site despite a low sequence similarity of 53.4% with a high structural RMSD of 6 Å. Additionally, the dpocket similarity score was 0.218, which does not indicate substantial similarity of the binding pockets.
Table 6.
Analysis of the 10 protein structures with the highest number of overlapping probe atoms to the 1Q5 ligand in the inactive-state allosteric corticotropin-releasing factor receptor 1 protein, PDB 4K5Y 29.
| Class | IUPHAR Name | PDB ID | Overlapping Probe Atoms | Volume, Å3 | RMSD, Å | Sequence Similarity, % | Similarity score | Statea |
|---|---|---|---|---|---|---|---|---|
| B | CRF1 | 4K5Y | 169 | 325.2 | Inactive | |||
| B | Glucagon | 5YQZ | 147 | 121.6 | 3.3 | 64.4 | 0.257 | Inactive |
| B | CRF1 | 4Z9G | 113 | 247.3 | 0.8 | 100.0 | 0.091 | Inactive |
| A | CXCR4 | 3OE9 | 103 | 152.2 | 6.0 | 53.4 | 0.218 | Inactive |
| B | GLP-1 | 5NX2 | 89 | 113.7 | 4.2 | 63.6 | 0.234 | Interm. |
| A | Rhodopsin | 6FKA | 85 | 49.5 | 5.1 | 50.6 | 0.239 | Active |
| A | Rhodopsin | 6FKC | 70 | 27.3 | 4.9 | 50.6 | 0.243 | Active |
| A | Rhodopsin | 6FK6 | 63 | 36.6 | 5.1 | 50.6 | 0.302 | Active |
| A | D2 | 6LUQ | 60 | 95.6 | 6.4 | 50.2 | 0.418 | Inactive |
| A | Rhodopsin | 6FK8 | 57 | 21.0 | 5.0 | 50.6 | 0.258 | Active |
| A | D2 | 6CM4 | 56 | 111.7 | 5.4 | 49.4 | 0.280 | Inactive |
Activation states were included from GPCRDB, the categories are defined based on interhelical Cα distances. (Interm.: Intermediate)
Figure 10:
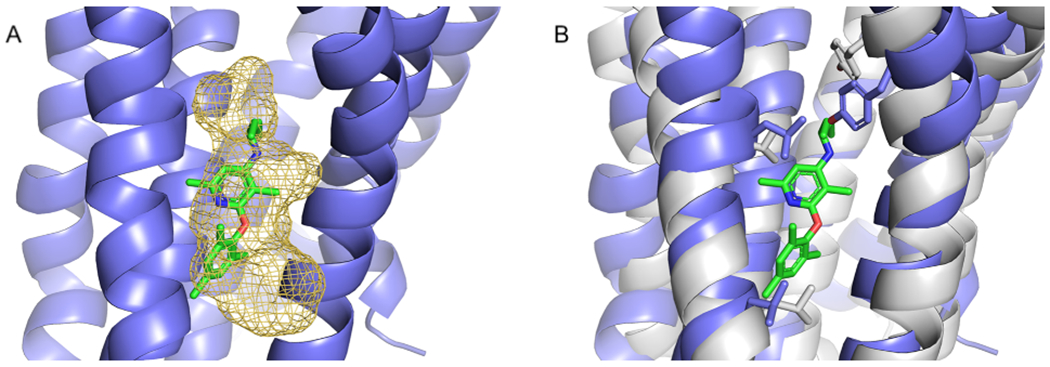
Mapping of Class B corticotropin-releasing factor receptor. A. Results of mapping the Class A C-X-C motif chemokine receptor 4, CXCR1 (PDB ID 3OE9) (blue), shown as a yellow mesh. The allosteric ligand 1QW (green) from Class B corticotropin-releasing factor receptor 1, CRFR1 (PDB ID 4K5Y) is shown for reference. B. Conserved residues (gray) of 4K5Y that are part of the 1QW binding site.
Site conservation across activation states
To get an overall picture on how the binding sites are conserved across different activation states, we have collected, for each ‘parent’ structure, the number of corresponding ‘daughter’ structures that had a conserved site with at least 84 overlapping probe atoms, grouped by the activation state (Table S7). Clearly, the conservation level of the sites varies to a great degree, from 320 matching structures (3OE0, CXCR4 chemokine receptor) to a single matching structure (5NDZ, PAR2 receptor). In the vast majority of cases, most of the matching ‘daughter’ structures are in the inactive state, but this is to be expected based on the distribution of structures (271 inactive-state vs. 88 active-state and 34 intermediate). We note that of the 18 structures in which FTMap did not detect the site, one structure was active (6N48), one was intermediate (4XNV), and the rest were inactive. Most binding sites are conserved across all activation states. Some rare exceptions are 5X7D (β2 receptor) and 5T1A (CCR2 receptor), where only inactive-state structures contain the same binding site within the overlap cutoff of ≥ 84 atoms. However, in both cases there are multiple active-state structures slightly below this cutoff, with 77-82 probe atoms overlapping. We can therefore conclude that most of the allosteric sites investigated here are robust towards the conformational changes of the GPCRs affecting the activation state.
Known allosteric ligands show limited overlap on GPCR targets
To get an overall picture of the structural and ligand coverage of the GPCR allosteric sites we have analyzed metadata from the GPCRDB database9 as well as the entries of the Allosteric Database (ASD) 8, 30, 31 adapting the methodology of Vass et al.32 Currently, 43 experimental structures with a bound allosteric ligand exist, for a total of 21 GPCRs, containing 38 unique ligands (37 small molecules and one peptide). For this study we were only interested in allosteric sites located in the 7TM domain, therefore we removed Smoothened Homolog protein from our set, resulting in 39 allosteric structures co-crystalized with an allosteric ligand. By comparison, the total number of structures is 183 for these 21 receptors and according to GPCRDB, the current (2020 September) number of all GPCR X-ray structures is 394 for 77 unique receptors. Thus, even though slightly less than 10% of all GPCR structures contain an allosteric ligand, close to 30% of the structurally explored receptors have at least one PDB entry with an allosteric ligand bound. These numbers hint at the generality of allosteric modulation among GPCRs, despite the respective structural efforts still being at a relatively early stage (the most well studied receptor, mGluR5 has 5 available structures with allosteric modulators, while the typical case for the rest of the receptors is one single structure).
The Allosteric Database (ASD)8 is to our knowledge the most comprehensive collection of allosteric ligands, merging reported experimental results from web resources like IUPHAR 33 and Drugbank 34 as well as patent files. Here, ASD has constituted the basis of retrieving allosteric ligand information for the respective GPCRs, the results are summarized in Table 7. For the 21 GPCRs, there are 14,158 unique ligands in total, out of which 145 are peptides. This set covers weak binders as well, since there is currently no option in ASD to filter the ligands based on binding affinity or bioactivity. Notably, over 80% (11,817) of these ligands are reported for three GPCRs: cannabinoid receptor 1 (CB1), GABA receptor type B (GABAB) and metabotropic glutamate receptor 5 (mGluR5). Many of these entries come from patents, without an exact bioactivity value reported. In addition, over 100 allosteric ligands are reported for the M2, GLP-1R, GCGR, mGluR1 and Smoothened receptors (Table 7). Interestingly, there is a very small number of ASD ligands (274 ligands, representing less than 2% of the dataset) that are chemically similar to the co-crystallized ligands of the respective receptors, suggesting a large chemical space available for targeting the allosteric sites. Similarly, there is very little overlap between the ligand sets of different receptors (472 ligands, less than 4% of the dataset). Most notably, the glucagon receptor GCGR and the glucagon-like peptide receptor GLP-1R share 135 allosteric ligands (31%), while 28 allosteric modulators are shared between metabotropic glutamate receptors 1 and 5 (14%). Most of the overlaps are with closely related receptors, e.g., bioactivities of the 75 M2 ligands (28%) are, without exception, on other muscarinic acetylcholine receptors. Since allosteric sites are generally considered to be more specific than orthosteric pockets, the limited overlap of ligand chemotypes is not unexpected. Consequently, we can conclude that not much information can be retrieved or implied from the allosteric ligand data regarding the conservation of allosteric sites.
Table 7.
Coverage of GPCRs in terms of the number of reported allosteric ligands (ASD database), experimental structures containing allosteric ligands (GPCRDB), as well as the overlap between the respective ligand sets, quantified according to various criteria
| Receptor | Structuresa | Allo. ligands (Xray)b | Allo. ligands (ASD)c | Allo. ligands (ASD) similar to X-ray ligandsd | Allo. ligands (ASD) similar to X-ray ligands of other GPCRse | Allo. ligands (ASD) of other GPCRs similar to X-ray ligandsf | Allo. ligands active at other GPCRs (ASD) |
|---|---|---|---|---|---|---|---|
| All (21/419) | 223 | 36 (1) | 14158 (145) | ||||
|
| |||||||
| Class A (14/299) | 150 | 22 (1) | 2447 (78) | ||||
|
| |||||||
| Aminergic (2/37) | 45 | 5 | 292 (23) | ||||
| M2 | 11 | 2 | 269 (11) | 4 | 0 | 62 | 75 |
| β2 | 34 | 3 | 23 (12) | 2 | 0 | 4 | 1 |
|
| |||||||
| Peptide (2/77) | 6 | 4 | 4 | ||||
| C5a1 | 3 | 2 | 3 | 0 | 0 | 3 | 1 |
| PAR2 | 3 | 2 | 1 | 0 | 0 | 0 | 0 |
|
| |||||||
| Protein (5/29) | 28 | 6 (1) | 92 (54) | ||||
| CCR2 | 3 | 1 | 1 | 0 | 0 | 0 | 0 |
| CCR5 | 13 | 1 | 34 | 0 | 1 | 13 | 2 |
| CCR7 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| CCR9 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| CXCR4 | 10 | 2 (1) | 56 (54) | 0 | 0 | 0 | 2 |
|
| |||||||
| Lipid (2/37) | 15 | 4 | 1961 (1) | ||||
| CB1 | 11 | 1 | 1944 (1) | 57 | 1 | 32 | 6 |
| FFA1 | 4 | 3 | 17 | 1 | 0 | 37 | 0 |
|
| |||||||
| Nucleotide (2/12) | 52 | 2 | 98 | ||||
| A2A | 49 | 1 | 42 | 0 | 0 | 2 | 3 |
| P2Y1 | 3 | 1 | 56 | 8 | 0 | 1 | 1 |
|
| |||||||
| Orphan (1/81) | 4 | 1 | 0 | ||||
| GPR52 | 4 | 1 | 0 | 0 | 0 | 0 | 0 |
|
| |||||||
| Class B (3/21) | 36 | 5 | 937 (67) | ||||
| CRF1 | 6 | 1 | 68 (63) | 1 | 0 | 0 | 0 |
| GLP-1R | 19 | 3 | 435 (4) | 101 | 3 | 156 | 136 |
| GCGR | 11 | 1 | 434 | 48 | 159 | 0 | 135 |
|
| |||||||
| Class C (3/23) | 26 | 8 | 10638 | ||||
| GABAB | 13 | 2 | 1284 | 3 | 2 | 0 | 2 |
| mGluR1 | 2 | 1 | 765 | 16 | 1 | 29 | 109 |
| mGluR5 | 11 | 5 | 8589 | 33 | 22 | 30 | 166 |
|
| |||||||
| Class F (1/11) | 11 | 1 | 136 | ||||
| Smoothened | 11 | 1 | 136 | 0 | 0 | 26 | 0 |
X-ray, electron microscopy and NMR structures according to GPCRDB and ASD.
Unique allosteric ligands appearing in at least one structure. Peptide ligands (MW > 800 Da) are indicated in brackets.
Unique allosteric ligands in ASD. Peptide ligands (MW > 800 Da) are indicated in brackets.
ASD ligands that are similar (≥ 0.4 ECFP4 or ≥ 0.8 MACCS Tanimoto similarity) to at least one of the X-ray ligands of the same receptor.
ASD ligands of the specific receptor that are similar (≥ 0.4 ECFP4 or ≥ 0.8 MACCS Tanimoto similarity) to at least one of the X-ray ligands of other receptors.
ASD ligands of other GPCRs that are similar (≥ 0.4 ECFP4 or ≥ 0.8 MACCS Tanimoto similarity) to at least one of the X-ray ligands of the specific receptor.\
DISCUSSION
We used the protein mapping program FTMap to identify binding hot spots in GPCRs, i.e., energetically important regions capable of ligand binding. Our goal has been to investigate potential allosteric sites. For soluble proteins such analysis generally involves benchmark sets that include both the ligand-bound and ligand-free structures of the proteins. Mapping is applied to both, and the expectation is that the ligand binding site is also found in the ligand-free structure. The bound structures can be used for the validation of the results, as the predicted hot spots should overlap with the bound ligand. However, no such benchmark can be obtained for GPCRs. Although the number of GPCR structures has been increasing, only 39 structures include allosteric ligands, and only in four cases has the same GPCR been solved with and without an allosteric ligand. We first applied FTMap to the 39 structures after removing the ligands, and found the allosteric sites strong enough to be considered druggable in 21 cases. However, in contrast to soluble proteins, we cannot show that the method can also identify the sites in ligand-free structures of the same proteins, since such structures are not available. Instead, we set out to investigate whether the same locations have strong ligand binding sites in other GPCRs, and hence FTMap was applied to all 394 GPCRs with X-ray structures available.
The analysis revealed that for each of the 21 structures that have strong sites with bound allosteric ligands there exist a number of GPCR structures that have a strong site at the same location. As expected, most such additional structures belong to the same GPCR type. However, sites at the same location can be also found for GPCRs that are of different types or even belong to different families. This result would not be surprising if each GPCR had many different sites capable of ligand binding. However, our results also show that this is not the case, as most GPCR structures have at most three but most frequently only two strong binding sites. Thus, in spite of the complexity of the GPCR structure with seven transmembrane helices and many areas that can be expected to accommodate drug-sized molecules, in each GPCR the number of locations that are suitable for binding ligands with relatively high affinity is very small, and such locations are conserved among many GPCRs, sometimes with very moderate structure and sequence similarity. The analysis of ligands known to bind to such GPCRs reveals that having allosteric sites at the same location implies neither the similarity of the ligands, nor the similarity of the residues forming the sites, although in some cases the same residues may occur in both. Thus, these sites are not identifiable based strictly on sequence similarity, RMSD or ligand similarities. Somewhat related or even stronger conclusions have been reached in a recent paper concerning cholesterol binding sites in GPCRs.16 Analyzing the available GPCR structures in the PDB it was shown that he vast majority of bound cholesterol molecules are found in 12 spatially distinct allosteric binding pockets that, however, lack consensus cholesterol-binding geometry or residues. Thus, even the same ligand binds in very different local environments.
We admit that our analysis has three important caveats. First, our findings are based on the analysis of the available X-ray structures, and no attempts were made to account for conformational changes by running molecular dynamics (MD) simulations. Long enough MD simulations may generate conformational diversity creating binding sites that are not among the nine identified in the X-ray structures.35, 36 In particular, the available structures do not account for the possibility of cryptic allosteric sites, although the mapping generally finds hot spots near such sites even without well-formed pockets.37 Second, some of the allosteric ligands co-crystallized with GPCRs are very large, and may overlap with distinct hot spots in multiple proteins that themselves do not overlap. In spite of these caveats, the nine distinct sites we identified are clearly important and accommodate allosteric ligands in many different GPCRs. Third, some of the GPCR structures have low resolution, which may affect the accuracy of the mapping results and even the exact location of the ligands. While these limitations may somewhat impact the exact results presented in this paper, we are confident that the major conclusions remain unchanged.
METHODS
Collection of structural data
GPCR structures and corresponding data, including activation state classification, were downloaded from the GPCRDB database.9 At the time of downloading (August 31, 2020), there were 394 published X-ray crystallography structures, including 39 that have been co-crystallized with ligands binding at allosteric sites within the 7TM domain (Table 1). The 7TM region of each structure was determined by using the Protein Domain Parser. 38 PyMOL (Schrödinger, LLC.) was used to perform structure-based alignments and to calculate root mean square deviations (RMSDs). Sequence similarities were calculated using the sequence similarity method from the OEChem Toolkit (OpenEye Scientific Software).
Collection of allosteric ligand data
Receptor complexes containing allosteric ligands were collected based on the GPCRDB database9 and from primary scientific literature. The Allosteric Database (ASD) 8, 30, 31 was used for collecting data on allosteric modulators: briefly, the offline version of the database was downloaded and parsed with custom Python scripts. Ligands with less than six heavy atoms were ignored, and those with a molecular weight over 800 Da were considered to be peptides. Adapting the ligand similarity analysis developed for GPCR ligands,32 we identified pairs of “similar” ligands if the Tanimoto similarity of MACCS or Morgan39 fingerprints was over 0.8 or 0.4, respectively. The RDKit package was used for fingerprint and similarity calculations.40 Data on the effects of mutations on allosteric ligand binding/affinity were looked up from the GPCRDB database.9
Identification of allosteric sites by FTMap
The 7TM domain of each structure was mapped using the FTMap algorithm, implemented in the FTMap server.11, 12 The server considers only the protein structure, as all hetero atoms, including water molecules, included in the structure file, are removed prior to mapping. FTMap places thousands of copies of 16 small organic molecules as probes on a dense grid around the protein surface, finds favorable positions for each probe type, clusters the positions of the bound probes, and ranks the probe clusters based on their average energy. For each probe type the six lowest energy clusters are retained, and are clustered with the clusters of other probe types to form consensus clusters. The consensus clusters are considered as the predicted binding hot spots, ranked by the number of probe clusters contained. We note that we have used the command line implementation of the FTMap algorithm called ATLAS,41 which in some cases yields slightly different results from those produced by the FTMap server.12 The original set of GPCRs with co-crystalized allosteric ligands was filtered into a subset of 21 proteins where FTMap was able to predict a strong binding site for the ligand. For comparison of the FTMap results for the 394 proteins and the 21 allosteric sites, the protein structures with the predicted hot spots were aligned to the protein structures co-crystallized with allosteric ligands. To determine binding site conservation, we counted the number of probe atoms within 3 Å of the ligand.
Based on our results, for each GPCR co-crystalized with an allosteric ligand we searched for structures that had strong hot spots overlapping with the ligand copied from the “parent” structure. In previous findings, FTMap hot spots that contained 16 or more probe clusters were shown to be likely druggable, with sufficiently high affinity for ligand binding.12, 17, 42 The average FTMap probe molecule has 5.25 heavy atoms. Therefore, site conservation was defined by 5.25 x 16 84 or more probe atoms overlapping with the ligand from the “parent” structure.17 For each structure we also determined the number of binding sites predicted to be druggable, and the results were visualized with a histogram. FTMap results underwent an additional round of clustering with a radius of 0.7 Å prior to the counting of druggable sites. The Clustal Omega tool, Multiple Sequence Alignment,43 was used to create a phylogenetic tree based on the 7TM domains of the GPCR structures. The tree was converted to graphml and visualized with Cytoscape.44
Pocket volumes were also calculated for each GPCR using the dpocket algorithm from the fpocket suite.23 Fpocket is based on the concept of alpha spheres. Each alpha sphere is a sphere that contacts four atoms on its boundary and contains no internal atom. For a protein, very small spheres are located within the protein, large spheres at the exterior, and clefts and cavities correspond to spheres of intermediate radii. The ensemble of alpha spheres defined from the atoms of a protein were filtered using the default minimal and maximal radii values in fpocket. Once the alpha spheres are selected, to calculate pocket volume the dpocket algorithm defines a box containing all atoms and vertices situated within 4Å of the reference ligand. Each of the 21 co-crystalized allosteric ligands was used as the reference ligand. The pocket volume was calculated using a Monte Carlo algorithm. The default settings were used except for the number of iterations performed when running the Monte Carlo algorithm (–v) option which was set to 500,000.
The dpocket program was also used to extract 15 pocket descriptors, including the number of alpha spheres, the density of the cavity, the polarity score, the mean local hydrophobic density, the proportion of apolar alpha spheres, the maximum distance between two alpha spheres, the hydrophobicity score, the charge score, the volume score, and the pocket volume 24. We ran dpocket on a total of 21 x 394 pockets. This resulted in 21 separate tables which each contained 15 dpocket descriptor columns and 394 rows. The absolute difference between the “parent” allosteric protein’s pocket descriptors and each of the 394 protein pocket descriptors were calculated. This resulted in 21 separate difference tables, each with 15 columns of pocket descriptors and 394 rows with the absolute difference between protein’s pocket and the allosteric protein’s pocket. Then, the differences for each pocket descriptor were scaled from 0 to 1 by subtracting the minimum descriptor value for that column and dividing by the maximum descriptor value for that column. This resulted in 21 separate tables containing 15 x 394 scaled differences. The 15 values in each row was added together to get a single difference in pockets (maximum value of 15), which resulted in 21 tables containing 394 differences. The difference column was then scaled from 0 to 1 for the final dpocket similarity score.
Validation by docking
We extracted the ligand in MOL2 format from the ASD (Allosteric Database) and docked them to the respective sites using Autodock Vina.19 The docking was restricted to a region defined by the union of 3.0 Å boxes around each probe atom. All Vina parameters were set to their default values. Each docking run generated 10 poses of the ligand.
Constructing a binding site similarity matrix based on predicted hot spot populations
As mentioned, to determine the similarities among the binding site locations of the 21 structures with bound allosteric ligands and strong hot spots we considered each structure with its predicted hot spot and superimposed it with all other 20 structures with their ligands included. For each structure we then counted the number of probe atoms overlapping with the ligands, and considered these numbers as measures of similarity. Results are shown in Table S2. The second column of the table lists the 21 structures we have mapped, each identified by a number from 1 to 21. In each row of the table, we show the number of probe atoms obtained by the mapping when considerations are restricted to probes that are within 3 Å of the ligand copied from the structure identified by the number of the particular column. For example, all numbers in the first row of Table S2 are based on the mapping of the structure 3ODU (also identified as structure 1). The number 213 in column 3 of this row shows that 213 probes overlap with the ligand (ITD) bound in 3ODU. The next number, 172, shows that 172 probe atoms placed by the mapping of 3ODU overlap with the ligand PRD copied from structure 2 (3OE0) after superposing the structures. The number 16 in the next column shows that the 3ODU hot spot includes only 16 probe atoms that overlap with the ligand 1Q5 from the structure 4K5Y, identified as structure number 3. According to the next column in the same row the overlap between the 3ODU hot spot and the ligand MRV from structure 4 (4MBS) includes 262 probes. Thus, based on these results we can conclude that the hot spots of 3ODU overlap not only with its own bound ligand, but also the ligands copied from 3OE0 and 4MBS. However, the hot spot of 3ODU barely overlaps with the ligand bound to 4K5Y. Conversely, the numbers in the third column of Table S1 show the overlap between the hot spots of each of the 21 structures and the ligand copied from 3ODU identified as structure 1. This column reveals that the hot spots in structures 3ODU, 3OE0, and 4BMS all have many probes overlapping with the ligand from 3ODU, and hence we conclude that these structures have overlapping binding hot spots at the site binding the allosteric ligand in 3ODU. As shown in Table 1, in all three structures the allosteric site is intrahelical (HC) and is located in the transmembrane region on the extracellular side (TM_EC). The similarity measure based on the overlap of probes with the ligand from a different GPCR structures is not commutative. For example, while the mapping of 3ODU yields 262 probe atoms that overlap with the ligand from 4MBS, the mapping of 4MBS yields only 83 probe atoms that overlap with the ligand from 3ODU. In fact, the ligand in 3ODU (PDB code ITD) is much smaller that the ligand Maraviroc (PDB code MRV) bound to 4MBS. More generally, if we regard Table S1 as a 21x21 matrix A, then A(i,j) ≠ A(j,i). Therefore we assumed that the mapping results suggest overlapping ligand binding sites only when both A(i,j) > 84 and A(j,i) > 84, thus the site in each structure substantially overlaps with the ligand from the other structure. For such sites we calculate the measure of overlap as [A(i,j) +A(j,i)]/2, thereby make the overlap matrix symmetric.
Supplementary Material
Table S1. RMSD between X-ray structures and AF2 models of the 39 structures with bound allosteric ligands
Table S2. Overlapping probe atoms among the allosteric sites of the 21 GPCR structures with ligand and strong hot spot
Table S3. The 10 proteins with the highest level of hot spot overlap with the allosteric ligand bound to 21 GPCRs with strong hot spots at the ligand binding site. Each of the 21 “parent” structures with the bound ligand is listed in bold
Table S4. “Parent” structures without strong hot spots at the allosteric site
Table S5. “Parent” and “daughter” structures for validation by docking
Table S6. Docking results
Table S7. Number of structures with conserved sites, grouped by activation state, for each ‘parent’ structure listed in Table 1.
ACKNOWLEDGMENTS
This investigation was supported by the grant R35GM118078 from the National Institute of General Medical Sciences and by the National Brain Research Program (2017-1.2.1-NKP-2017-00002). D.B. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and the ÚNKP- 21-5 New National Excellence Program of the Ministry for Innovation and Technology.
Footnotes
DATA AND SOFTWARE AVAILABILITY
PDB structures were downloaded from the RCSB Protein Data Bank (https://www.rcsb.org). Binding data were downloaded from the GPCRdb database (https://gpcrdb.org). AlphaFold models were downloaded from the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk). The FTMap algorithm is free for academic and governmental use and can be accessed through the FTMap server (https://ftmap.bu.edu). The command-line implementation of FTMap named ATLAS can be licensed from Acpharis Inc. (https://acpharis.com). The PyMOL Molecular Graphics System can be licensed from Schrodinger (https://pymol.org/2/). An academic license for OEChem Toolkit was obtained through OpenEye Scientific Software (https://www.eyesopen.com). The open source cheminformatics software RDKit was freely obtained (https://www.rdkit.org). The open source protein pocket detection algorithm Fpocket was freely downloaded (https://github.com/Discngine/fpocket). The docking program AutoDock Vina is freely available from https://vina.scripps.edu.
Supporting Information
The Supporting Information is available free of charge at
The authors declare no competing financial interest.
REFERENCES
- (1).Fredriksson R; Lagerstrom MC; Lundin LG; Schioth HB, The G-protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol 2003, 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- (2).Alexander SPH; Christopoulos A; Davenport AP; Kelly E; Mathie A; Peters JA; Veale EL; Armstrong JF; Faccenda E; Harding SD; Pawson AJ; Sharman JL; Southan C; Davies JA; Collaborators C, The Concise Guide to Pharmacology 2019/20: G protein-Coupled Receptors. Br. J. Pharmacol 2019, 176 Suppl 1, S21–S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hauser AS; Attwood MM; Rask-Andersen M; Schioth HB; Gloriam DE, Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat Rev Drug Discov 2017, 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Christopoulos A, Advances in G protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol 2014, 86, 463–478. [DOI] [PubMed] [Google Scholar]
- (5).Ma N; Nivedha AK; Vaidehi N, Allosteric Communication Regulates Ligand-Specific GPCR Activity. FEBS J 2021, 288, 2502–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).May LT; Leach K; Sexton PM; Christopoulos A, Allosteric Modulation of G protein-Coupled Receptors. Annu. Rev. Pharmacol. Toxicol 2007, 47, 1–51. [DOI] [PubMed] [Google Scholar]
- (7).Wootten D; Christopoulos A; Sexton PM, Emerging Paradigms in GPCR Allostery: Implications for Drug Discovery. Nat Rev Drug Discov 2013, 12, 630–644. [DOI] [PubMed] [Google Scholar]
- (8).Huang Z; Zhu L; Cao Y; Wu G; Liu X; Chen Y; Wang Q; Shi T; Zhao Y; Wang Y; Li W; Li Y; Chen H; Chen G; Zhang J, Asd: A Comprehensive Database of Allosteric Proteins and Modulators. Nucleic Acids Res. 2011, 39, D663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pandy-Szekeres G; Munk C; Tsonkov TM; Mordalski S; Harpsoe K; Hauser AS; Bojarski AJ; Gloriam DE, Gpcrdb in 2018: Adding GPCR Structure Models and Ligands. Nucleic Acids Res 2018, 46, D440–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wakefield AE; Mason JS; Vajda S; Keseru GM, Analysis of Tractable Allosteric Sites in G protein-Coupled Receptors. Sci. Rep 2019, 9, 6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Brenke R; Kozakov D; Chuang GY; Beglov D; Hall D; Landon MR; Mattos C; Vajda S, Fragment-Based Identification of Druggable ‘Hot Spots’ of Proteins Using Fourier Domain Correlation Techniques. Bioinformatics 2009, 25, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kozakov D; Grove LE; Hall DR; Bohnuud T; Mottarella SE; Luo L; Xia B; Beglov D; Vajda S, The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat Protoc 2015, 10, 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wakefield AE; Yueh C; Beglov D; Castilho MS; Kozakov D; Keseru GM; Whitty A; Vajda S, Benchmark Sets for Binding Hot Spot Identification in Fragment-Based Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6612–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Zidek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D, Highly Accurate Protein Structure Prediction with Alphafold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Varadi M; Anyango S; Deshpande M; Nair S; Natassia C; Yordanova G; Yuan D; Stroe O; Wood G; Laydon A; Zidek A; Green T; Tunyasuvunakool K; Petersen S; Jumper J; Clancy E; Green R; Vora A; Lutfi M; Figurnov M; Cowie A; Hobbs N; Kohli P; Kleywegt G; Birney E; Hassabis D; Velankar S, Alphafold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res 2022, 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Taghon GJ; Rowe JB; Kapolka NJ; Isom DG, Predictable Cholesterol Binding Sites in Gpcrs Lack Consensus Motifs. Structure 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kozakov D; Hall DR; Napoleon RL; Yueh C; Whitty A; Vajda S, New Frontiers in Druggability. J Med Chem 2015, 58, 9063–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wu Y; Tong J; Ding K; Zhou Q; Zhao S, GPCR Allosteric Modulator Discovery. Adv Exp Med Biol 2019, 1163, 225–251. [DOI] [PubMed] [Google Scholar]
- (19).Trott O; Olson AJ, Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ciancetta A; Gill AK; Ding T; Karlov DS; Chalhoub G; McCormick PJ; Tikhonova IG, Probe Confined Dynamic Mapping for G Protein-Coupled Receptor Allosteric Site Prediction. ACS Cent Sci 2021, 7, 1847–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kruse AC; Ring AM; Manglik A; Hu J; Hu K; Eitel K; Hubner H; Pardon E; Valant C; Sexton PM; Christopoulos A; Felder CC; Gmeiner P; Steyaert J; Weis WI; Garcia KC; Wess J; Kobilka BK, Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature 2013, 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Burger WAC; Sexton PM; Christopoulos A; Thal DM, Toward an Understanding of the Structural Basis of Allostery in Muscarinic Acetylcholine Receptors. J. Gen. Physiol 2018, 150, 1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Le Guilloux V; Schmidtke P; Tuffery P, Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC bioinformatics 2009, 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Schmidtke P; Barril X, Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J Med Chem 2010, 53, 5858–5867. [DOI] [PubMed] [Google Scholar]
- (25).Thorsen TS; Matt R; Weis WI; Kobilka BK, Modified T4 Lysozyme Fusion Proteins Facilitate G Protein-Coupled Receptor Crystallogenesis. Structure 2014, 22, 1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tan Q; Zhu Y; Li J; Chen Z; Han GW; Kufareva I; Li T; Ma L; Fenalti G; Li J; Zhang W; Xie X; Yang H; Jiang H; Cherezov V; Liu H; Stevens RC; Zhao Q; Wu B, Structure of the CCR5 Chemokine Receptor-Hiv Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wu B; Chien EY; Mol CD; Fenalti G; Liu W; Katritch V; Abagyan R; Brooun A; Wells P; Bi FC; Hamel DJ; Kuhn P; Handel TM; Cherezov V; Stevens RC, Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang L; Yao D; Deepak R; Liu H; Xiao Q; Fan H; Gong W; Wei Z; Zhang C, Structures of the Human Pgd2 Receptor Crth2 Reveal Novel Mechanisms for Ligand Recognition. Mol. Cell 2018, 72, 48–59 e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hollenstein K; Kean J; Bortolato A; Cheng RK; Dore AS; Jazayeri A; Cooke RM; Weir M; Marshall FH, Structure of class B GPCR Corticotropin-Releasing Factor Receptor 1. Nature 2013, 499, 438–443. [DOI] [PubMed] [Google Scholar]
- (30).Huang Z; Mou L; Shen Q; Lu S; Li C; Liu X; Wang G; Li S; Geng L; Liu Y; Wu J; Chen G; Zhang J, Asd V2.0: Updated Content and Novel Features Focusing on Allosteric Regulation. Nucleic Acids Res 2014, 42, D510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shen Q; Wang G; Li S; Liu X; Lu S; Chen Z; Song K; Yan J; Geng L; Huang Z; Huang W; Chen G; Zhang J, Asd V3.0: Unraveling Allosteric Regulation with Structural Mechanisms and Biological Networks. Nucleic Acids Res 2016, 44, D527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Vass M; Kooistra AJ; Yang D; Stevens RC; Wang MW; de Graaf C, Chemical Diversity in the G Protein-Coupled Receptor Superfamily. Trends Pharmacol Sci 2018, 39, 494–512. [DOI] [PubMed] [Google Scholar]
- (33).Harmar AJ; Hills RA; Rosser EM; Jones M; Buneman OP; Dunbar DR; Greenhill SD; Hale VA; Sharman JL; Bonner TI; Catterall WA; Davenport AP; Delagrange P; Dollery CT; Foord SM; Gutman GA; Laudet V; Neubig RR; Ohlstein EH; Olsen RW; Peters J; Pin JP; Ruffolo RR; Searls DB; Wright MW; Spedding M, Iuphar-Db: The Iuphar Database of G protein-Coupled Receptors and Ion Channels. Nucleic Acids Res 2009, 37, D680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wishart DS; Knox C; Guo AC; Cheng D; Shrivastava S; Tzur D; Gautam B; Hassanali M, Drugbank: A Knowledgebase for Drugs, Drug Actions and Drug Targets. Nucleic Acids Res 2008, 36, D901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ivetac A; McCammon JA, Mapping the Druggable Allosteric Space of G-protein Coupled Receptors: A Fragment-Based Molecular Dynamics Approach. Chem Biol Drug Des 2010, 76, 201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Miao Y; Nichols SE; McCammon JA, Mapping of Allosteric Druggable Sites in Activation-Associated Conformers of the M2 Muscarinic Receptor. Chem. Biol. Drug Des 2014, 83, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Beglov D; Hall DR; Wakefield AE; Luo L; Allen KN; Kozakov D; Whitty A; Vajda S, Exploring the Structural Origins of Cryptic Sites on Proteins. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E3416–E3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Alexandrov N; Shindyalov I, Pdp: Protein Domain Parser. Bioinformatics 2003, 19, 429–430. [DOI] [PubMed] [Google Scholar]
- (39).Rogers D; Hahn M, Extended-Connectivity Fingerprints. J Chem Inf Model 2010, 50, 742–754. [DOI] [PubMed] [Google Scholar]
- (40).Landrum G, RDKit: A Software Suite for Cheminformatics, Computational Chemistry, and Predictive Modeling. http://rdkit.sourceforge.net.
- (41).Hall DR; Enyedy IJ, Computational Solvent Mapping in Structure-Based Drug Design. Future Med Chem 2015, 7, 337–353. [DOI] [PubMed] [Google Scholar]
- (42).Kozakov D; Hall DR; Chuang GY; Cencic R; Brenke R; Grove LE; Beglov D; Pelletier J; Whitty A; Vajda S, Structural Conservation of Druggable Hot Spots in Protein-Protein Interfaces. Proc Natl Acad Sci U S A 2011, 108, 13528–13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sievers F; Higgins DG, Clustal Omega for Making Accurate Alignments of Many Protein Sequences. Protein Sci. 2018, 27, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T, Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RMSD between X-ray structures and AF2 models of the 39 structures with bound allosteric ligands
Table S2. Overlapping probe atoms among the allosteric sites of the 21 GPCR structures with ligand and strong hot spot
Table S3. The 10 proteins with the highest level of hot spot overlap with the allosteric ligand bound to 21 GPCRs with strong hot spots at the ligand binding site. Each of the 21 “parent” structures with the bound ligand is listed in bold
Table S4. “Parent” structures without strong hot spots at the allosteric site
Table S5. “Parent” and “daughter” structures for validation by docking
Table S6. Docking results
Table S7. Number of structures with conserved sites, grouped by activation state, for each ‘parent’ structure listed in Table 1.