

Abstract
Mass-spectrometry-based phosphoproteomics has become indispensable for understanding cellular signaling in complex biological systems. Despite the central role of protein phosphorylation, the field still lacks inexpensive, regenerable, and diverse phosphopeptides with ground-truth phosphorylation positions. Here, we present Iterative Synthetically Phosphorylated Isomers (iSPI), a proteome-scale library of human-derived phosphoserine-containing phosphopeptides that is inexpensive, regenerable, and diverse, with precisely known positions of phosphorylation. We demonstrate possible uses of iSPI, including use as a phosphopeptide standard, a tool to evaluate and optimize phosphorylation-site localization algorithms, and a benchmark to compare performance across data analysis pipelines. We also present AScorePro, an updated version of the AScore algorithm specifically optimized for phosphorylation-site localization in higher energy fragmentation spectra, and the FLR viewer, a web tool for phosphorylation-site localization, to enable community use of the iSPI resource. iSPI and its associated data constitute a useful, multi-purpose resource for the phosphoproteomics community.
Mass-spectrometry-based phosphoproteomics has become a powerful technique for understanding intracellular signaling pathways at a systems level. Advancements over the past two decades have made proteome-scale profiling of phosphorylation events routine. However, despite the power and ubiquity of phosphoproteomics, its ability to accurately identify and localize phosphorylation sites remains incompletely characterized, with uncertain implications for the phosphoproteomics data acquired to date1–3. Specifically, the lack of precise control on false localization rates (FLRs), or the rate at which phosphopeptides are correctly sequenced but incorrectly localized, has led to the unacceptable accumulation of false positives within phosphorylation-site databases4,5.
Many groups have attempted to measure phosphopeptide false identification rates and FLRs using a variety of methods, including generating synthetic peptide libraries6–8, bioinformatic techniques9–12, and machine learning13. However, each of these methods has its drawbacks. Many synthetic libraries of biologically derived peptides are limited in size owing to the cost of phosphopeptide synthesis. Other, larger libraries are generated by the semi-random synthesis of structurally similar peptides, which capture only a small portion of phosphoproteome diversity (see Marx et al.6 and Supplementary Note 1). Many bioinformatic techniques for determining FLR were evaluated on relatively small numbers of synthetic phosphopeptides and have not been evaluated at a proteome scale. Machine-learning methods require large amounts of training data, yet proteome-scale libraries of phosphopeptides with known phosphorylation positions (also called ‘ground truth’ phosphopeptides) are lacking. An inexpensive, regenerable, and proteome-scale library of phosphopeptides whose phosphorylated residues are precisely known would be an indispensable tool for optimization and benchmarking of multiple stages in phosphoproteome analysis, from data acquisition to phosphopeptide identification and site localization.
Recent improvements to the Phosphoserine Orthogonal Translation System (pSerOTS), which can incorporate phosphoserine into proteins as a non-standard amino acid, now allow for cost-effective and regenerable phosphoserine-containing peptide synthesis in Escherichia coli at the proteome scale (Supplementary Fig. 1a)14,15. With this technology, the synthetic human serine phosphoproteome (SHSP)16 was created. The SHSP consists of 110,139 phosphorylation sites that have previously been observed in humans from PhosphoSitePlus1 that have been converted to constructs of ≤31 amino acids for expression in E. coli (Supplementary Fig. 1b). However, in the original iteration, all phosphorylation positional isomers for a given tryptic peptide were present in the same pool, complicating analysis of FLR. To address this issue, we have separated the SHSP into ten subpools, each constructed to place phosphopeptide positional isomers in separate pools (Supplementary Fig. 1b,c). We call this new version of SHSP the Iterative Synthetically Phosphorylated Isomers (iSPI). When using iSPI and the latest pSerOTS15, about 500 μg of phosphorylated peptides can be produced per liter of E. coli culture, representing ~11,000 unique phosphopeptides with precisely known phosphorylation positions in each subpool (Supplementary Fig. 1d,e and Supplementary Table 1).
iSPI is a multi-purpose resource that has many phosphoproteomics applications (Fig. 1a). To illustrate, we present our workflow for investigating FLR under various collision modes and peptide labeling states (Fig. 1b). Three subpools of ~11,000 phosphorylation sites (~33,000 unique phosphosites total, ~28,000 with tryptic peptide length of at least 7 residues) were generated and investigated as both label-free and TMTpro-labeled peptides under three collision modes: collision-induced dissociation (CID), higher energy collision-induced dissociation (HCD), and CID with multi-stage activation (MSA)17. Because the true position of every phosphorylation site was known, we could directly observe exact FLRs (Fig. 1b and Supplementary Note 2).
Fig. 1 |. iSPI is a multi-purpose, regenerable, proteome-scale human phosphopeptide resource for phosphoproteoimcs.
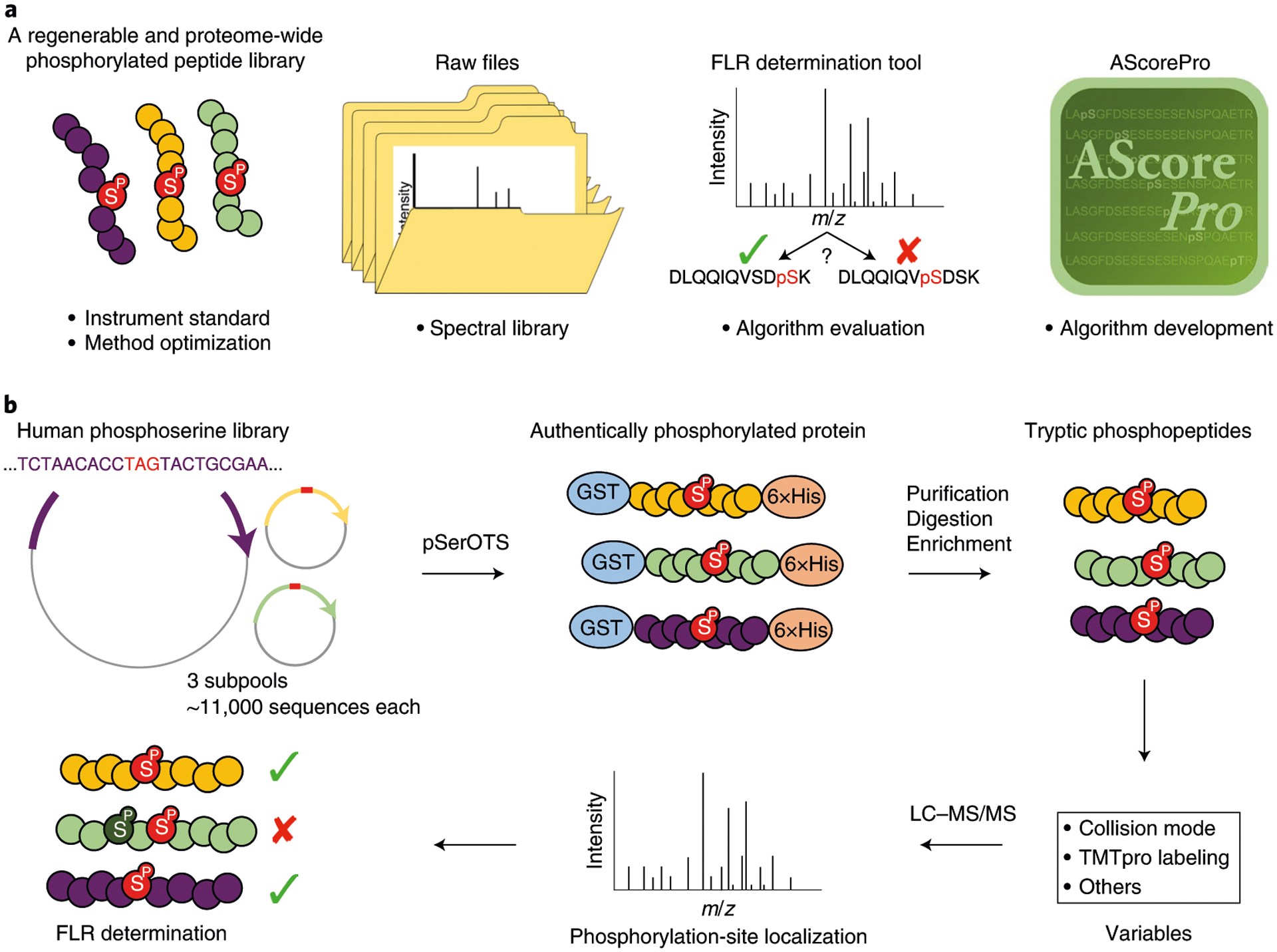
a, iSPI generates authentically phosphorylated peptides derived from the human phosphoserine proteome that can be used as standards and for instrumental method optimization. The raw data files can be used to generate ground-truth phosphopeptide spectral libraries. The FLR determination tool enables researchers to investigate their own data and data processing workflows. AScorePro is an improved version of AScore, with improved phosphorylation-site localization. b, Workflow for determining FLR using iSPI. For this report, 3 library subpools of ~11,000 phosphorylated peptides were expressed using the Phosphoserine Orthogonal Translation System (pSerOTS), which, after purification, digestion, and enrichment, generated 3 pools of peptides with known phosphorylation positions. Note that, for clarity, each subpool never contained the same phosphopeptide phosphorylated on two sites (no positional isomers). Tryptic phosphorylated peptides were analyzed under various collision modes with and without TMTpro labeling, and search-engine outputs were compared with the known positions to determine FLR. LC–MS/MS, liquid chromatography–tandem mass spectrometry.
When this workflow was applied to the AScore localization algorithm18, the number of phosphorylation-site identifications using HCD at an empirical 5% FLR cutoff was 7.2% and 21% lower for label-free and TMTpro-labeled samples, respectively, than for MSA (Fig. 2a). The FLR at 10,000 sites was 3.3% higher for the HCD collision mode than for MSA for label-free samples, and a similar 4.8% increase in FLR was observed at 7,000 sites for TMTpro-labeled samples (Fig. 2a). These data highlight the usefulness of the iSPI phosphopeptide library in evaluating and comparing phosphorylation-site identification and localization across instrument methods.
Fig. 2 |. Using iSPI to evaluate collision modes and labeling states, as well as to improve the AScore algorithm.
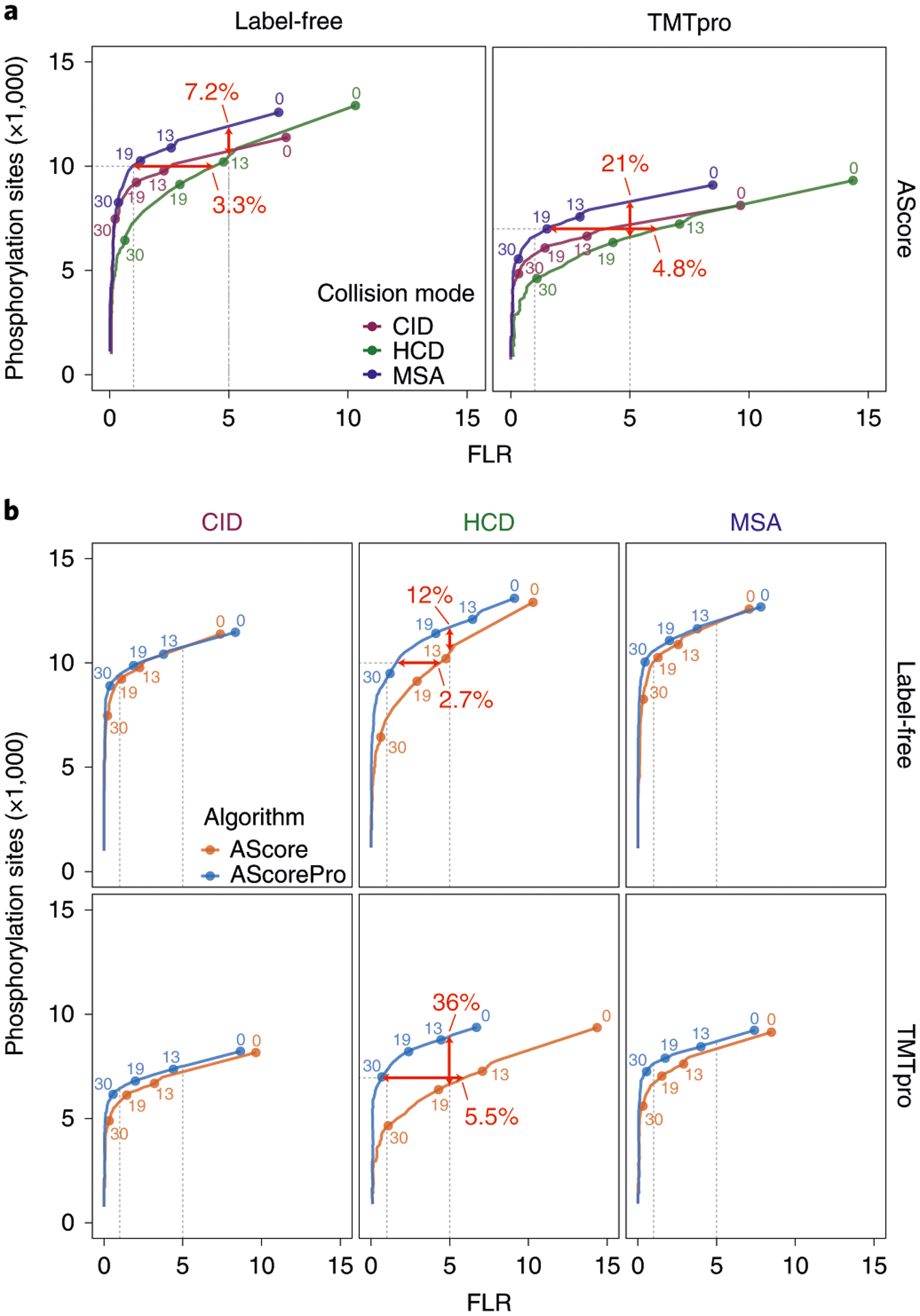
Receiver operating curves were determined; the y axis represents the total number of phosphorylation sites across the three subpools from Figure 1, and the x axis represents the precise FLR. Vertical gray dotted lines represent empirical FLRs of 0.01 and 0.05; horizontal lines represent 10,000 and 7,000 phosphorylation sites for label-free and TMTpro-labeled peptides, respectively. Labeled points represent discrete AScore cutoffs. Bold red numbers and arrows represent the differences in FLR (horizontal arrow) or identified sites (vertical arrow) between curves. For clarity, the combined totals of the three pools are plotted. a, Comparison of collected phosphopeptide spectra using CID, HCD, or MSA collision modes under label-free (left) and TMTpro-labeled (right) states. b, Comparison of AScore and AScorePro algorithms. AScorePro specifically improves FLR for data collected using HCD fragmentation.
Although the AScore algorithm was originally developed for low-resolution CID data, given the recent prevalence of high-resolution HCD methods for phosphoproteomics analysis, we sought to improve the AScore algorithm (see Supplementary Note 3 and Supplementary Fig. 2). Using iSPI as a benchmark, we developed a new cross-platform application termed AScorePro. AScorePro incorporates the option to consider −98-Da neutral loss product ions (Supplementary Fig. 3), which greatly improves the performance of the algorithm on HCD data (Supplementary Fig. 4). We further implemented the maximum of binomials (MOB) scoring algorithm to avoid peak depth selection and to make use of the increased number of fragment ions (Supplementary Fig. 3)19. The incorporation of MOB scoring further increased the number of correctly localized phosphorylation sites and decreased FLR for all conditions (Supplementary Fig. 4). Although using neutral losses for localization is phosphorylation-specific, MOB scoring may improve the localization for other post-translational modifications as well. AScorePro can accept common spectral formats, including mzXML20 and mzML21, and is available as an open-source project for integration into proteomic pipelines with both a shared method library and a command-line application.
The enhancements in AScorePro markedly improved site localization accuracy. In the high-resolution HCD dataset, at a fixed dataset size of 10,000 sites for label-free samples, AScorePro reduced the number of falsely localized sites by more than half (from 4.6% to 1.8%) (Fig. 2b). Alternatively, when label-free samples are viewed at a fixed empirical FLR of 5%, AScorePro returns an extra 1,200 sites (12%) over AScore (Fig. 2b). In the high-resolution HCD dataset for TMTpro-labeled samples, the improvement with AScorePro is even greater, removing 5 out of 6 previously falsely localized sites (from ~6% FLR to ~1% FLR) at a fixed dataset size of 7,000 sites. Alternatively, AScorePro increases the number of sites identified at a fixed empirical FLR of 5% by 2,359 (36%) (Fig. 2b). AScorePro also improved FLR for high-resolution MSA data by more than half, while the FLR for high-resolution CID data remained similar. However, both CID and MSA data showed improvements in the number of phosphorylation-site identifications at a fixed empirical 5% FLR, with label-free and TMTpro-labeled peptides increasing by 5% and 8%, respectively (Fig. 2b). Notably, including neutral loss peaks as part of AScorePro for CID and MSA data did not penalize the FLR or number of identifications, as observed in other algorithms22. We observed similar, if not greater, improvements in the number of confidently localized phosphorylation sites when analyzing low-resolution MS2 (lrMS2 collected in the ion trap) data with AScorePro (Supplementary Fig. 5), or when re-analyzing previously published biological datasets (Supplementary Fig. 6). These improvements to AScore highlight the utility of iSPI for evaluating and improving phosphorylation localization algorithms. In total, we identified ~15,700 unique phosphorylation sites of the possible ~28,000 across all conditions (resolution, labeling state, and collision mode), representing ~56% of possible identifications (Supplementary Table 2); this result is similar to the 57% of possible phosphopeptides identified in Marx et al.6 and is better than the 32.9% of phosphopeptides directly observed in SHSP16.
Although previous studies have attempted to compare different algorithms, they have either been limited to a relatively small number of synthetic phosphopeptides or compared biological datasets for which the phosphorylation localization was not precisely known12,23–26. Because ‘ground truth’ knowledge of phosphorylation-site positions across this library is known, iSPI provides a unique opportunity to compare performance across different data analysis pipelines. This comparison includes the number of site identifications at a given empirical FLR, which is especially useful as the calculated localization scores in different pipelines are not always directly comparable. Thus, the search engine Comet27 with localization by both AScore and AScorePro was compared with two common analysis pipelines: Andromeda with PTM score in the MaxQuant 2.0.3 software28, and Sequest HT29 plus MS Amanda30 with PhosphoRS22 in Proteome Discoverer 2.4. Comet with AScorePro performed similarly for label-free phosphopeptides in all three collision modes in terms of FLR and the number of sites identified at an empirical 5% FLR (Extended Data Fig. 1a,b). However, for TMTpro-labeled peptides, Comet with AScorePro outperformed both the MaxQuant and Proteome Discoverer pipelines, especially for the CID and MSA datasets (Extended Data Fig. 1a,c). We also compared AScore and AScorePro to the Fragpipe v17.1 pipeline31–33 at the peptide-spectrum match (PSM) level, and observed similar results (Supplementary Fig. 7).
iSPI is a useful tool for phosphoproteomics, as demonstrated through its use as (1) a standard to evaluate instrumental acquisition methods, (2) a ‘ground truth’ dataset to evaluate and optimize localization algorithms, and (3) a benchmark to compare analysis pipelines. iSPI can be generated by simple E. coli culture and protein purification, with strains and plasmids now available through Addgene. The mass spectrometry raw files generated in this study (PXD031171) are available for researchers to evaluate their own computational tools. Furthermore, to facilitate the community’s use of iSPI, we have created the FLR viewer for phosphorylation-site localization as a free web-based tool (http://wren.hms.harvard.edu/iSPI/). This tool can calculate the FLR for any search-engine output using iSPI (whether the user analyzes iSPI on their own instruments or uses the data in this study with their own analysis pipelines), given a list of localized phosphorylation sites and their associated localization scores (Supplementary Fig. 8). This tool can aid researchers in using the library for various applications.
In addition to the applications shown here, iSPI may be used to optimize search engines and PSM filtering; to identify optimal collision energies that balance peptide identification, phosphosite localization, TMTpro quantitation, and benchmark peptide and peptide-spectrum match identifications; and to generate ground-truth spectral libraries for machine learning, DIA analysis, or targeted proteomics methods (see Supplementary Note 3). In the future, with proper DNA encoding, the library will be expanded to include multiple phosphorylation sites14,34. Although iSPI is currently limited to phosphoserine, orthogonal translation systems capable of incorporating phosphothreonine, phosphotyrosine, or other post-translationally modified amino acids (such as acetyl-K) at proteome scales are under development, which would enable further expansion of iSPI.
Actual FLRs in biological datasets may be higher than those calculated when evaluating individual library subpools because of the presence of co-eluting positional isomers that generate chimeric spectra, as well as the presence of falsely identified phosphopeptides, which we have largely filtered out (<1%). However, by carefully designing experiments, iSPI could be used to investigate how often chimeric or co-isolating positional isomers occur, and how different analysis pipelines are affected by these spectra. iSPI could also be used to validate claims that alternative separation methods (such as TIMS or FAIMS) can distinguish between positional isomers35 at the proteome scale. We hope that iSPI, with the help of the FLR Viewer and AScorePro, will enable rigorous optimization, standardization, and benchmarking of key steps in phosphoproteomics workflows and ensure that we map the phosphoproteome with maximal confidence and coverage.
Methods
E. coli culture and protein expression
The strain rEcoli XpS15 was grown in luria broth (LB) at 37 °C in a laboratory shaker. Then, 20 mL of XpS E. coli cells containing the pSerOTS (V70)15 were grown to an optical density at 600 nm (OD600) of 0.4 and were pelleted by centrifugation and washed twice with ice-cold water. Cells were resuspended in 50 μL of ice-cold water, mixed with 100 ng subpool library plasmid, and electroporated. Cells were immediately resuspended in 1 mL LB and incubated for 1 hour at 37 °C while being shaken. At least 1 × 107 colony forming units were obtained by combining multiple electroporations, as calculated by plating serial dilutions. Recovered cells were inoculated into 100 mL LB supplemented with antibiotics and were grown overnight at 37 °C. Cultures were then back diluted to 0.15 OD, and grown to an OD of 0.7–0.8m after which protein expression was induced by 0.2% arabinose. Cultures were grown for 4 hours at 37 °C, followed by collection by centrifugation and freezing at −80 °C.
Protein purification
Bacterial cell pellets from 1 L cultures were lysed in 20 mL lysis buffer (50 mM Tris pH 7.4, 500 mM NaCl, 500 μM EDTA, 500 μM EGTA, 10% glycerol, 1 mM DTT, 50 mM NaF, 1 mM NaVO4, 1 mg/mL lysozyme (Alpha Aesar), 1 protease inhibitor mini tablet (Pierce)). Cells were sonicated on ice using an Active Motif sonicator with an 1/8-inch probe with 10 seconds on and 40 seconds off and 70% amplitude for 2 minutes total. Lysates were then passed over an equilibrated 1 mL bed volume Ni-NTA resin (Qiagen) by gravity, and washed with 10 mL wash buffer (50 mM Tris pH 7.4, 500 mM NaCl, 500 μM EDTA, 500 μM EGTA, 10% glycerol, 1 mM DTT, 50 mM NaF, 1 mM NaVO4, 20 mM imidazole). Proteins were eluted with 5 mL elution buffer (50 mM Tris pH 7.4, 500 mM NaCl, 500 μM EDTA, 500 μM EGTA, 10% glycerol, 1 mM DTT, 50 mM NaF, 1 mM NaVO4, 250 mM imidazole), and buffer exchanged into 200 mM EPPS, pH 8.5, using a 10,000-MWCO filter (Millipore), and concentrated to ~500 μL.
Phosphoproteomic sample preparation
Protein concentration was determined by BCA assay, and diluted to approximately 1 mg/mL, followed by digestion with 1:100 LysC (Wako) overnight at room temperature and 1:100 trypsin for 6 hours. Samples were desalted using SepPak vacuum cartridges (Waters). Each subpool sample was divided into three aliquots, each of which was passed through a High-Select Fe-NTA enrichment column (Pierce) for phosphopeptide enrichment, using the manufacturer’s recommendations. Samples were eluted with 200 μL elution buffer into 100 μL 10% formic acid and dried in a rotary evaporator. Samples were reconstituted in 5% acetonitrile with 5% formic acid, and peptides were quantified by BCA assay. Then, 25 μg of each subpool was placed in a vial for label-free LC–MS/MS analysis. An additional 25 μg of each subpool was dried in a rotary evaporator and reconstituted in 25 μL 200 mM EPPS. After the addition of anhydrous acetonitrile to ~30%, phosphopeptides were labeled with TMTpro reagent. One percent of the sample was removed and analyzed to ensure labeling was complete. TMTpro reactions were quenched with the addition of 0.3% hydroxylamine. Labeled peptides were dried in a rotary evaporated briefly to remove ACN, then desalted using in-house C18 stage tips. After drying in a rotary evaporator, samples were reconstituted in 20 μL for LC–MS/MS analysis.
Cost analysis
A 1.0 L culture of E. coli generates approximately 600 μg of phosphopeptides, at a cost of approximately US $175, which is about US $0.28/μg or US $0.02/peptide. This estimate does not include the one-time cost of a few hundred dollars to obtain the library from Addgene.
Liquid chromatography–tandem mass spectrometry analysis
Mass spectra were collected on an Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 35-cm in-house packed column (inner diameter 100 μm, Accucore, 2.6 μm, 150 Å) using a 150-minute gradient (from 5%–30% acetonitrile with 0.1% formic acid) at 500 nl/minute. MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 4× 105, 400–1,400 m/z). Determined charge states between 2 and 5 were required for sequencing, and a 90-second dynamic exclusion window was used. MS2 scans consisted of collision-induced dissociation (CID), multi-stage activation CID at 97.9763 Da, or higher-energy-collision-induced dissociation (HCD) of precursors using a Top10 method. High-resolution MS2 scans were collected in the Orbitrap with an isolation window of 0.7 Da, q value of 0.25, AGC target of 50,000, resolution of 50,000, and maximum injection time of 86 ms. Low-resolution MS2 scans were collected in the ion trap with an isolation window of 1.3 Da, q value of 0.25, AGC target of 7,500, and maximum ion injection time of 60 ms. Supplementary Table 3 contains the metadata on all included raw files (including labeling state, collision mode, resolution, FAIMS CVs, and so on).
Peptide identification and phosphorylation-site localization for iSPI samples.
For the Comet/AScore and Comet/AScorePro pipelines, raw files were converted to mzXML, and monoisotopic peaks were re-assigned using Monocle36. Searches were performed using the Comet search algorithm (2019.01 rev. 5)27. The protein database included common contaminant proteins (for example, trypsin and keratins), scrambled human protein entries from Uniprot (11/2018 release), E. coli entries from EcoCyc1737, and the Synthetic Human Serine Phosphoproteome16. The database was then concatenated with one composed of all protein sequences in the reversed order. We used a 20 ppm precursor ion tolerance and 0.02 Da product ion tolerance for MS2 scans collected in the Orbitrap and 1.0005 Da product ion tolerance for MS2 scans collected in the ion trap. Up to two missed cleavages were allowed. Oxidation of methionine residues (+15.9949 Da), deamidation of asparagine and glutamine residues (+0.9840 Da), phosphorylation of serine, threonine, and tyrosine residues (+79.9663 Da) were set as variable modifications. TMTpro on lysine residues and peptide N-termini (+304.2071 Da) were set as static modifications for TMTpro-labeled samples. A minimum peptide length of seven amino acids was required. PSMs were adjusted to a 1% false discovery rate (FDR)38. PSM filtering was performed using linear discriminant analysis (LDA), as described previously39, while considering the following parameters: Comet log expect, different sequence delta Comet log expect (percent difference between the first hit and the next hit with a different peptide sequence), missed cleavages, peptide length, charge state, precursor mass accuracy, number of modifications, and fraction of ions matched. Only phosphorylated peptides were considered in LDA. Each run was filtered separately. The protein-level FDR was subsequently estimated. The posterior probabilities reported by the LDA model for each peptide were multiplied to give a protein-level probability estimate. Proteins were filtered to the target 1% FDR level for each run individually using the Picked FDR method40. Phosphorylation-site localization was determined using the AScore18 or AScorePro algorithm. We implemented several optimizations in the AScore algorithm and termed the improved phosphorylation-site localization algorithm AScorePro. In the AScorePro algorithm, both peptide scores and AScores were calculated using the MOB scoring function19 with the consideration of −97.9769-Da neutral loss fragments for phosphorylated serine and threonine residues. The MOB scoring function takes advantage of more peaks in the spectrum without having to pick a specific peak depth or calculating probabilities for each peak separately. AScorePro is implemented in C# using the.NET Core framework (version 3.1) and is open source under the GPL-3.0 license (https://opensource.org/licenses/GPL-3.0). Project files are available as a library and a command-line executable that can be built for either Windows or Linux environments, allowing for integration into proteomic pipelines. The source code can be found here: https://github.com/gygilab/MPToolKit. AScorePro can work with any mass spectrometry platform capable of conversion to mzXML or mzML files. Mass tolerances of 0.6 Da and 20 ppm were used for low-resolution and high-resolution MS2 scans, respectively. Maximum peak depth was set as 15.
For the Andromeda/PTM score pipeline in MaxQuant (version 2.0.3), Andromeda was used to search against the same database, with the same set of static and variable modifications as those in the Comet/AScore pipeline. Other parameters were kept as similar as possible and included a minimum peptide length of seven residues and a maximum of two missed cleavages. Additional parameters included a minimum score of 40 and minimum delta score of 6 for modified peptides, 20 ppm MS/MS tolerance, and 12 peaks per 100-Da MS/MS interval, and the match between runs feature was not used.
For the SequestHT&MS Amanda/PhosphoRS pipeline in Proteome Discoverer (version 2.4), SequestHT and MS Amanda were used to search against the same database with the same set of static and variable modifications as those in the Comet/AScore pipeline. Other parameters were kept as similar as possible, and include a minimum peptide length of seven residues and a maximum of two missed cleavages. Default parameters were selected for SequestHT (precursor mass tolerance 10 ppm, fragment mass tolerance 0.02 Da) and MS Amanda (5ppm MS1 tolerance, 0.02 Da MS2 tolerance). The default IMP-ptmRS settings were also used, including using PhosphoRS mode, 0.03-Da fragment mass tolerance, and a maximum peak depth of 8, and automatic consideration of neutral loss fragments. We also used a Site Probability Threshold of 25 in the PSM grouper node, and report only PTMs true, only master proteins false, and a motif radius of 6 in the modification sites node.
For the Fragpipe (v17.1) pipeline, data were searched with MSFragger using the same database with the same set of static and variable modifications as those in the Comet/AScore pipeline, with a precursor tolerance of 20 ppm, and a fragment mass tolerance of 20 pm. Phosphosites were localized using PTM-Shepherd with a precursor tolerance of 0.01 Da and a fragment mass tolerance of 20 ppm.
False localization rate determination.
The iSPI sequence library was first in silico digested to generate theoretical tryptic phosphorylated peptides using the R package ‘cleaver’ (version 1.30.0). The cut occurred after each lysine and arginine. Up to two missed cleavages were allowed. A minimum peptide length of seven amino acids was required. Although the subpools were designed such that positional isomers were distributed in different subpools, a few sequences still generated tryptic phosphorylated peptides that were isomeric within the same subpool. Generally, these instances had unique 31-base oligomer sequences; however, the tryptic peptide itself was isomeric. These tryptic sequences (24–75 sequences in each subpool) were excluded in the subsequent FLR determination. There were instances where the fully tryptic peptide for a particular site was isomeric and removed, but the missed cleaved peptide was included because it is fully unique.
For FLR determination, we first removed the falsely identified peptides to the largest extent possible to exclude the effect of different search engines and peptide-spectrum-match filtering steps on the outcome (<1% of dataset). Phosphorylation sites that originated from multiply phosphorylated peptides (only singly phosphorylated sites were encoded in iSPI), reversed sequences, contaminant proteins (even if the phosphopepetide was encoded in iSPI), scrambled human sequences, and E. coli sequences were removed. Phosphorylation sites whose backbone peptide sequences were not in the intended subpool were removed. Phosphorylation sites whose peptide sequences were among the few positional isomers (see above) within the respective subpool were also removed. For the Proteome Discoverer output, the ModSites output was additionally filtered to include only unique phosphopeptides. The remaining phosphorylation sites were kept as total phosphorylation sites. Then, the FLR was calculated as the number of phosphorylation sites correctly assigned by the localization algorithm divided by the number of total phosphorylation sites (Supplementary Table 4). Receiver operating curves were generated in R v4.1.0, with the help of the R package ‘tidyverse’ (version 1.3.0).
Peptide identification and phosphorylation-site localization for published datasets.
Raw files from four previously published phosphorylation datasets were downloaded from ProteomeXchange (PXD024275 (ref.41), PXD016491 (ref.42), PXD015575 (ref.43), PXD024298 (ref.44); see Supplementary Table 5 for details). Each raw file was analyzed individually from search to protein FDR filtering (PXD024298) or phosphorylation-site quantification (PXD024275, PXD016491, PXD015575). Raw files were converted to mzXML and monoisotopic peaks were re-assigned using Monocle36. Searches were performed using the Comet search algorithm27. The human protein entries were downloaded from Uniprot in February 2020. The mouse protein entries were downloaded from Uniprot in July 2020. The yeast protein entries were downloaded from Uniprot in March 2020. The protein database included common contaminant proteins (for example, trypsin and keratins) and the entries from Uniprot for the respective species. The database was then concatenated with one composed of all protein sequences in the reversed order. We used a 50 ppm precursor ion tolerance and 0.02-Da product ion tolerance for MS2 scans collected in the Orbitrap, and 1.0005-Da product ion tolerance for MS2 scans collected in the ion trap. Oxidation of methionine residues (+15.9949 Da) and phosphorylation of serine, threonine, and tyrosine residues (+79.9663 Da) were set as variable modifications. TMTpro or TMT on lysine residues and peptide amino termini (+304.2071 Da for TMTpro, +229.1629 Da for TMT) was set as a static modification for TMTpro or TMT-labeled samples. Peptide-spectrum matches (PSMs) were adjusted to a 1% FDR38. PSM filtering was performed using linear discriminant analysis (LDA) as described previously39, and the following features were included in the LDA model: Comet log expect, different sequence delta Comet log expect (percent difference between the first hit and the next hit with a different peptide sequence), missed cleavages, peptide length, charge state, precursor mass accuracy, and fraction of ions matched. Only phosphorylated peptides were considered in LDA. Protein-level FDR was subsequently estimated. The posterior probabilities reported by the LDA model for each peptide were multiplied to give a protein-level probability estimate. Proteins were filtered to the target 1% FDR level using the Picked FDR method37. Each raw file was filtered separately. Phosphorylation-site localization was determined using the AScore18 or AScorePro algorithm. Mass tolerances of 0.6 Da and 20 ppm were used for low-resolution and high resolution MS2 cans, respectively. Maximum peak depth was set to 15. A minimum AScore or AScorePro of 13 was required. We used a fixed score cutoff of 13 here, as FLRs are generally lower than 5% for all conditions at 13 (Supplementary Figs. 4 and 5). Raw files in the PXD024298 dataset were reported at unique peptide level. Raw files in the PXD024275, PXD016491, and PXD015575 datasets were reported at quantified phosphorylation-site level (isolation specificity ≥ 0.7, reporter ion signal-to-noise ≥ 10 per channel).
Extended Data Figure 1 and Supplementary Fig 1d, e were generated using Prism 9.3.1. The figures were assembled in Adobe Illustrator 2022.
Extended Data
Extended Data Fig. 1 |. Using the iSPI to compare phosphoproteomics pipelines.
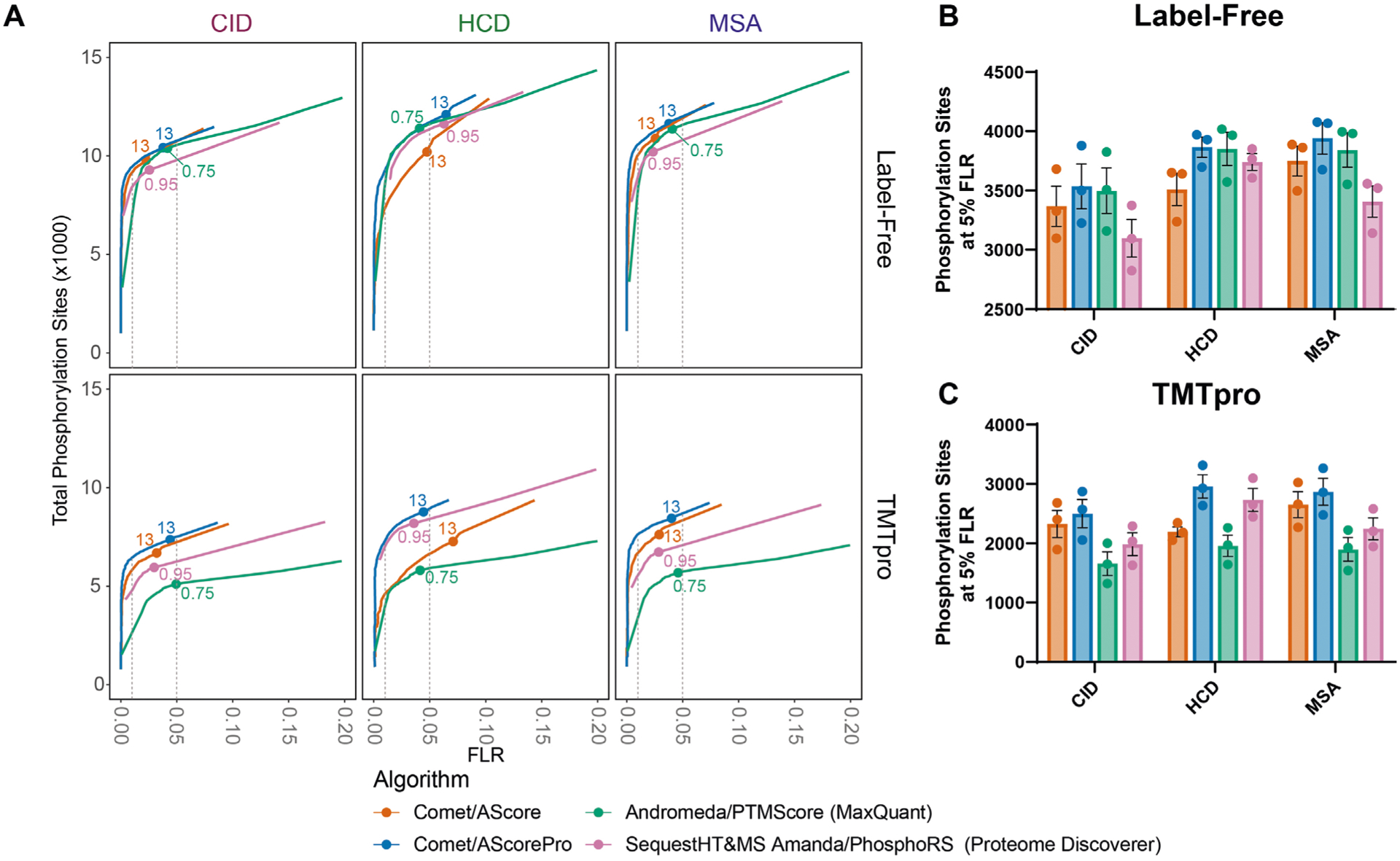
The datasets for the three subpools were analyzed using four different pipelines. Each pipeline included a database searching algorithm and a site localization tool. a) Similar to Fig. 2, receiver operating curves are shown displaying the number of combined sites from the three pools on the y-axis as a function of the FLR on the x-axis. Vertical grey dotted lines represent empirical FLR’s of 0.01 and 0.05 respectively. Labelled points represent commonly used localization cutoffs: 13 for AScore and AScorePro, 0.75 localization probability for MaxQuant, and 0.95 site probability for proteome discoverer. At an empirical FLR of 0.05, differences are apparent. b) The number of phosphorylation sites per pool is shown for label free peptides at an empirical 5% FLR. c) The number of phosphorylation sites per pool is shown for TMTpro-labeled peptides at an empirical 5% FLR. Bar represents the mean. Error bars represent the standard error of the mean for n = 3 subpools.
Supplementary Material
Acknowledgements
We would like to thank members of the Gygi Lab at Harvard Medical School for productive discussions, and S. Rogulina for technical assistance. This work was funded in part NIH/NIGMS grants GM132129 (J. A. P.), GM117230 (J. R.) and GM67945 (S. P. G). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
The iSPI library is covered under patent EP3755798A4 (pending, inventor: J. R., assignee: Yale University, Agilent Technologies Inc), and pSerOTS is covered under patent US7723069B2 (active, inventor: J. R., assignee: Yale University). The other authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41592-022-01638-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41592-022-01638-5.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592-022-01638-5.
Data Availability
The mass spectrometry data generated in this work have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD031171 (Fig. 2, Extended Data Fig. 1 and Supplementary Figs. 2, 4, 5, and 7). Previously published datasets re-analyzed in this work (Supplementary Fig. 6) are also available through ProteomeXchange via their dataset identifiers listed in Supplementary Table 5. Publicly available databases used include EcoCyc v17 (https://biocyc.org/organism-summary?object=ECOLI) and the Uniprot Human Database (11/2018 release, https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2018_11/).
Code Availability
The AScorePro software package is available as a standalone application from Github at https://github.com/gygilab/MPToolKit. The FLR viewer for phosphorylation site localization is available as a web application at http://wren.hms.harvard.edu/iSPI/. The source code for the FLR viewer is available from Github at https://github.com/gygilab/iSPI_Viewer.
References
- 1.Hornbeck PV et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu K et al. QPhos: a database of protein phosphorylation dynamics in humans. Nucleic Acids Res. 47, D451–D458 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krug K et al. A curated resource for phosphosite-specific signature analysis. Mol. Cell. Proteom 18, 576–593 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochoa D et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol 38, 365–373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalyuzhnyy A et al. Profiling the human phosphoproteome to estimate the true extent of protein phosphorylation. J. Proteome Res 21, 1510–1524 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marx H et al. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol 31, 557–564 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Ferries S et al. Evaluation of parameters for confident phosphorylation site localization using an orbitrap fusion tribrid mass spectrometer. J. Proteome Res 16, 3448–3459 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Cui L & Reid GE Examining factors that influence erroneous phosphorylation site localization via competing fragmentation and rearrangement reactions during ion trap CID-MS/MS and-MS(3.). Proteomics 13, 964–973 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Wiese H et al. Comparison of alternative MS/MS and bioinformatics approaches for confident phosphorylation site localization. J. Proteome Res 13, 1128–1137 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Suni V et al. SimPhospho: a software tool enabling confident phosphosite assignment. Bioinformatics 34, 2690–2692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma K et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Ramsbottom KA et al. Method for independent estimation of the false localization rate for phosphoproteomics. J. Proteome Res 21, 1603–1615 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W et al. Deep-learning-derived evaluation metrics enable effective benchmarking of computational tools for phosphopeptide identification. Mol. Cell. Proteom 20, 100171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirman NL et al. A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat. Commun 6, 8130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohler K, Moen J, Rogulina S & Rinehart J Principles for systematic optimization of an orthogonal translation system with enhanced biological tolerance. Preprint at bioRxiv 10.1101/2021.05.20.444985 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barber KW et al. Encoding human serine phosphopeptides in bacteria for proteome-wide identification of phosphorylation-dependent interactions. Nat. Biotechnol 36, 638–644 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF & Coon JJ A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem 76, 3590–3598 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Beausoleil SA, Villén J, Gerber SA, Rush J & Gygi SP A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 24, 1285–1292 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Mintseris J & Gygi SP High-density chemical cross-linking for modeling protein interactions. Proc. Natl Acad. Sci. USA 117, 93–102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedrioli PGA et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol 22, 1459–1466 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Martens L et al. mzML — a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110.000133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taus T et al. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res 10, 5354–5362 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Hogrebe A et al. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun 9, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potel CM, Lemeer S & Heck AJR Phosphopeptide fragmentation and site localization by mass spectrometry: an update. Anal. Chem 91, 126–141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verheggen K et al. Anatomy and evolution of database search engines—a central component of mass spectrometry based proteomic workflows. Mass Spectrom. Rev 39, 292–306 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Locard-Paulet M, Bouyssié D, Froment C, Burlet-Schiltz O & Jensen LJ Comparing 22 popular phosphoproteomics pipelines for peptide identification and site localization. J. Proteome Res 19, 1338–1345 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Eng JK, Jahan TA & Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Cox J et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 10, 1794–1805 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Tabb DL The SEQUEST family tree. J. Am. Soc. Mass. Spectrom 26, 1814–1819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dorfer V et al. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res 13, 3679–3684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D & Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu F et al. Identification of modified peptides using localization-aware open search. Nat. Commun 11, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geiszler DJ et al. PTM-shepherd: Analysis and summarization of post-translational and chemical modifications from open search results. Mol. Cell. Proteomics 20, 100018 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amiram M et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol 33, 1272–1279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muehlbauer LK, Hebert AS, Westphall MS, Shishkova E & Coon JJ Global phosphoproteome analysis using high-field asymmetric waveform ion mobility spectrometry on a hybrid orbitrap mass spectrometer. Anal. Chem 92, 15959–15967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rad R et al. Improved monoisotopic mass estimation for deeper proteome coverage. J. Proteome Res 20, 591–598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keseler IM et al. The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 45, D543–D550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Huttlin EL et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savitski MM, Wilhelm M, Hahne H, Kuster B & Bantscheff M A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol. Cell. Proteom 14, 2394–2404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J et al. TMTpro-18plex: the expanded and complete set of TMTpro reagents for sample multiplexing. J. Proteome Res 20, 2964–2972 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 17, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Paulo JA, Nusinow DP, Huttlin EL & Gygi SP Investigation of proteomic and phosphoproteomic responses to signaling network perturbations reveals functional pathway organizations in yeast. Cell Rep. 29, 2092–2104.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Popow O, Liu X, Haigis KM, Gygi SP & Paulo JA A compendium of murine (phospho)peptides encompassing different isobaric labeling and data acquisition strategies. J. Proteome Res 20, 3678–3688 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data generated in this work have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD031171 (Fig. 2, Extended Data Fig. 1 and Supplementary Figs. 2, 4, 5, and 7). Previously published datasets re-analyzed in this work (Supplementary Fig. 6) are also available through ProteomeXchange via their dataset identifiers listed in Supplementary Table 5. Publicly available databases used include EcoCyc v17 (https://biocyc.org/organism-summary?object=ECOLI) and the Uniprot Human Database (11/2018 release, https://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2018_11/).