

Abstract
Defining the genome-wide chromatin landscape has been a goal of experimentalists for decades. Here we review highlights of these efforts, from seminal experiments showing discontinuities in chromatin structure related to gene activation to extensions of these methods elucidating general features of chromatin related to gene states by exploiting deep sequencing methods. We also review chromatin conformational capture methods to identify patterns in long-range interactions between genomic loci.
1. Basic chromatin structure
Assembly of eukaryotic genomes into the hierarchical structures collectively known as chromatin fulfills the opposing functions of compacting ~2 meters of DNA to fit within the confines of the cell nucleus (about 10 μm), while providing appropriate access to the genetic material for processes such as gene expression. The basic repeating subunit of chromatin, the nucleosome, is comprised of an octamer of the core histones H2A, H2B, H3 and H4, which form a spool onto which ~147 base pairs of DNA is tightly wrapped in ~1 ¾ superhelical turns, and a shorter segment of unwrapped linker DNA of variable length, [1]. Long arrays of nucleosomes are typically condensed into higher-order structures mediated by nucleosome-nucleosome interactions, punctuated by less compacted chromatin [2,3]. Factors that influence chromatin structure and ultimately biological function include nucleosome occupancy, binding of transcription factors, chromatin remodeling complexes, histone modifying enzymes, and posttranslational modifications [3–5]. In order to better understand how changes to chromatin structure are brought about and the consequences of different chromatin states, it is necessary to map the locations of chromatin modifying factors, nucleosome locations, and higher order chromatin structures relative to specific functional DNA elements such as genes, enhancers, and promoters. Here we review the powerful techniques that have been developed to delineate these various aspects of chromatin structure and to further understanding of how chromatin structure impacts gene expression.
2. Identification of nucleosome-depleted regions
In pioneering experiments, treatment of chromatin with nucleases revealed that transcriptionally active regions are more susceptible to enzymatic digestion [6–10] For example, active globin genes were found to be preferentially excised by DNase I [9,10], and the 5’ ends of Drosophila heat shock genes were found to be more sensitive to DNase I digestion upon heat shock, indicating specific changes in chromatin structure leading to increased exposure of genomic DNA are associated with active genes [6–8]. Similarly, studies in S. cerevisiae using DNase I and MNase digestion found that expression of PHO5 is associated with changes in the positioning and occupancy of nucleosomes within the promoter region [11–13]. A breakthrough in establishing a functional role of chromatin structure in the regulation of gene expression came from studies from the Grunstein lab demonstrating that abrupt H4 depletion and thus disruption of the canonical nucleosome structure resulted in basal PHO5 transcription in the absence of activator signals [14,15]. These landmark studies demonstrated that nucleosomes are an essential component in the regulation of gene expression, maintaining PHO5 genes in a fully repressed state and that the disruption of promoter nucleosomes is a prerequisite for transcriptional activation.
With the advent of next generation sequencing (NGS) methods, a number of traditional chromatin-probing techniques were adapted to investigate the chromatin structure and function throughout entire genomes. For example, the ability of DNase I to cut at selected accessible sites in active chromatin combined with next-generation sequencing has allowed genome-wide mapping of nuclease-accessible chromatin regions [16,17] (Fig 1A). A complementary non-enzymatic method is formaldehyde assisted isolation of regulatory elements (FAIRE) which also efficiently maps open regions of chromatin [18,19]. FAIRE takes advantage of the fact that DNA-associated chromatin proteins, primarily histones, are depleted in DNase I-accessible active promoters and enhancers, and interrogates chromatin by using formaldehyde to crosslink proteins associated with DNA. As histones contain proportionally a large number of lysines in close proximity to DNA, they represent the preponderance of crosslinked proteins. Because the vast majority of the genomic DNA is assembled into nucleosomes, following phenol-chloroform extraction, much of genomic DNA resides in the organic phase while any DNA sufficiently reduced or lacking crosslinks segregates to the aqueous phase [20–22]. DNA recovered from the aqueous phase is then sequenced and mapped to the reference genome in order to identify DNA segments depleted of histones (Fig. 1B). The open chromatin regions identified in FAIRE map well with other hallmarks of active chromatin such as DNase I hypersensitive sites, histone acetylation, and RNAPII occupancy [21]. Because FAIRE does not rely on enzymatic digestion, it has the benefit of avoiding the sequence bias of enzyme-based techniques [23–27]. However, FAIRE does exhibit higher background than enzyme-based techniques [28].
Figure 1: Common methods for mapping chromatin accessibility.
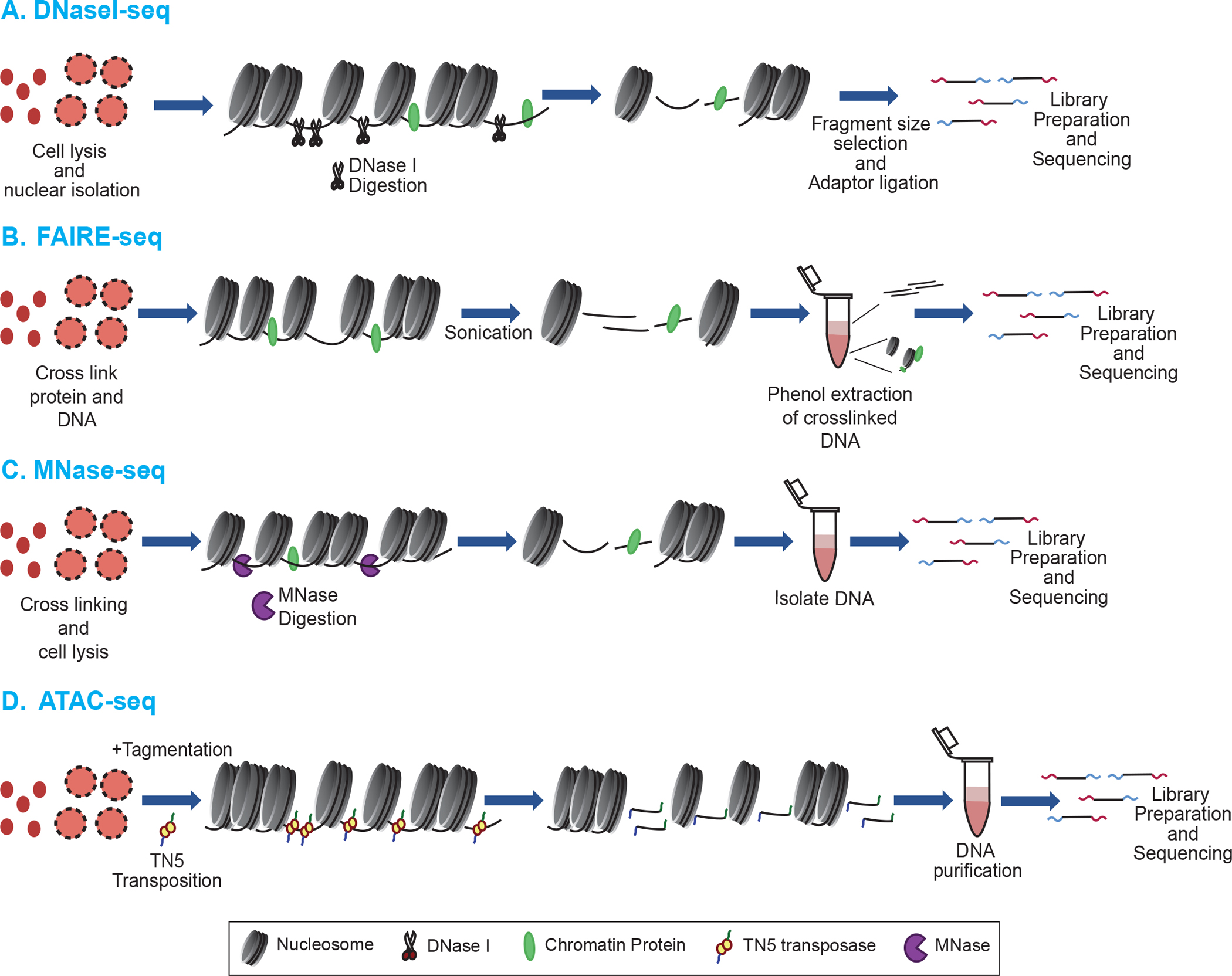
The first step in each technique is cell lysis and nuclear isolation. A. DNase-seq: Utilizes cleavage by DNaseI endonuclease to identify the open chromatin regions. Cleavage is attenuated where chromatin proteins (Green and yellow ovals) are bound to DNA, including nucleosomes. Short DNA fragments resulting from the DNaseI cleavage are sequenced. B. FAIRE-seq: Chromatin proteins are crosslinked to the DNA using formaldehyde and chromatin is fragmented by sonication. Protein-free DNA fragments remain in the aqueous phase during phenol:chloroform extraction and are sequenced. C. MNase-seq: MNase is used to digest away linker DNA regions and nucleosomal DNA is sequenced. D. ATAC-seq: A hyperactive Tn5 transposase simultaneously cleaves and inserts DNA adapters at accessible regions of the genome, which are then used for sequencing.
Due to its endo- and exo-nuclease activities, micrococcal nuclease (MNase) has long been used to probe native chromatin structure and excise nucleosome subunits [29], in a manner similar to endogenous nucleases originally used to expose the repeating structure of chromatin [30,31]. Indeed, the nucleosome core particle (consisting of ~147 ± 5 bp of DNA and the core histone octamer) was originally defined as a product of MNase digestion [32]. Typically, isolated nuclei are digested with MNase, which endonucleolytically cuts linker DNA between nucleosome cores then exonucleolytically trims linker DNA until reaching the edge of a nucleosome core region, which is digested at a slower rate. Early investigations took advantage of MNase, but were limited to interrogation of specific genomic loci [7,8,23]. Expansion of these studies with the advent of tiling arrays allowed for greater understanding of nucleosome occupancy and positioning across the genome [33–35]. More recently, combining MNase with NGS technologies has led to genome-wide mapping of nucleosomes for a number of organisms via MNase-seq (Fig. 1C) [36–40]. MNase-seq has also been combined with standard chromatin immunoprecipitation (ChIP, see below) to map histone variants, histone posttranslational modifications (PTMs) and nonhistone proteins as the presence or absence of these factors has consequences on chromatin structure [41–43].
MNase experiments have also provided new insights into the occupancy and positioning of nucleosomes within gene regulatory elements. Generally, deep sequencing of nucleosome core particle DNA shows that nucleosomes are loosely aligned with respect to transcription start sites (TSSs), with the greatest positioning found for the “+1” nucleosome [44]. Interestingly, a ‘nucleosome-depleted region’ (NDR) is found upstream of TSSs for active genes in metazoans and for most genes in yeast [41,45]. Given that the core histones provide only a kinetic block to MNase trimming, nucleosome cores will ultimately be destroyed by MNase, in a manner dependent on the enzyme’s preference for A/T rich DNA sequences and the inherent stability of the nucleosome, which may be affected by specific posttranslational modifications and incorporation of histone variants. Recent studies suggest that some genes have an apparent ‘fragile’ nucleosome within their promoters which is rapidly destroyed upon MNase digestion [46–48]. Whether NDRs are occupied by fragile nucleosomes or nonhistone chromatin proteins is an area of active debate, nevertheless, it is clear that the extent of MNase digestion can influence the ability to map nucleosomes [43,47,48]. Indeed, titrating MNase activity revealed classes of nucleosomes whose occupancy increases or decreases with digestion, demonstrating that accessibility and occupancy are distinct properties [49]. Moreover, more ‘accessible’ nucleosomes were generally found associated with open chromatin marks such as DNase I hypersensitivity, histone acetylation, and H3 K4 methylation, while more ‘inaccessible’ nucleosomes were associated with closed chromatin marks including histone H3 K9 and K27 trimethylation [49]. However, a quantitative study found that restriction enzyme cleavage rates were nearly identical in euchromatin and heterochromatin, indicating that overall promoter DNA accessibility is not a primary determinant of gene regulation, and suggesting that nucleosome stability determined by MNase may be largely due to sequence preference of the enzyme [50].
An MNase-independent technique for identifying open chromatin regions employs a hyperactive Tn5 transposase, and is known as Assay for Transposase Accessible Chromatin using SEQuencing (ATAC-seq). Regions of the chromatin that are nucleosome dense and compact are more resistant to Tn5 cleavage and insertion of NGS adapters, while more accessible nucleosome depleted regions (i.e. promoters and enhancers) are preferentially mapped by this method (Fig. 1D). ATAC-seq has the advantage of ligating the NGS adapters onto the digested chromatin for sequencing, eliminating error-introducing library preparation steps [51]. Extensive ATAC-seq can also be used to map nucleosome positions and transcription factor binding changes during cellular development [51–56]. Perhaps the greatest advantage of ATAC-seq is that it requires significantly less cells (105) as opposed to a minimum of 106-107 cells for other techniques such as FAIRE and MNase-seq [51]. ATAC-seq has also been used in single-cell analyses to investigate rare cell populations and cell-to-cell variation [52,57,58]. While FAIRE-seq and ATAC-seq have been highly effective in identifying open, protein depleted regions of chromatin, MNase-seq and related techniques are more often used to investigate the position and composition of nucleosomes.
3. Mapping and analysis of chromatin binding proteins
A now widely used method by which binding sites of specific proteins are identified is Chromatin immunoprecipitation (ChIP), which involves formaldehyde crosslinking of chromatin proteins to DNA, followed by fragmentation, typically by sonication, but sometimes by nuclease digestion (Fig. 2A) [59–63]. (However, it is possible to perform nucleosome-based ChIP under native (i.e. uncrosslinked) conditions.) Antibody specific to a protein of interest is used to pull down the protein-DNA complex, and the DNA is subjected to NGS. Importantly, antibodies of interest, especially posttranslational modification specific antibodies, should be characterized thoroughly for cross-reactivity by dot-blot, Western blot, and peptide competition assays, etc. as many commercial antibodies suffer from off-target recognition, interfence of epitope recognition by neighboring PTMs, and inability to distinguish between mono, di, and trimethylation modification status [64–70]. Several resources are available to check commonly used antibodies such as http://www.histoneantibodies.com [71] and site http://proteincapture.org [72]. ChIP-seq methods are now widely used to map core histones and their variants, specific histone posttranslational modifications, and nonhistone proteins [61,73–76].
Figure 2: Methods to assess genomic occupancy of chromatin proteins.
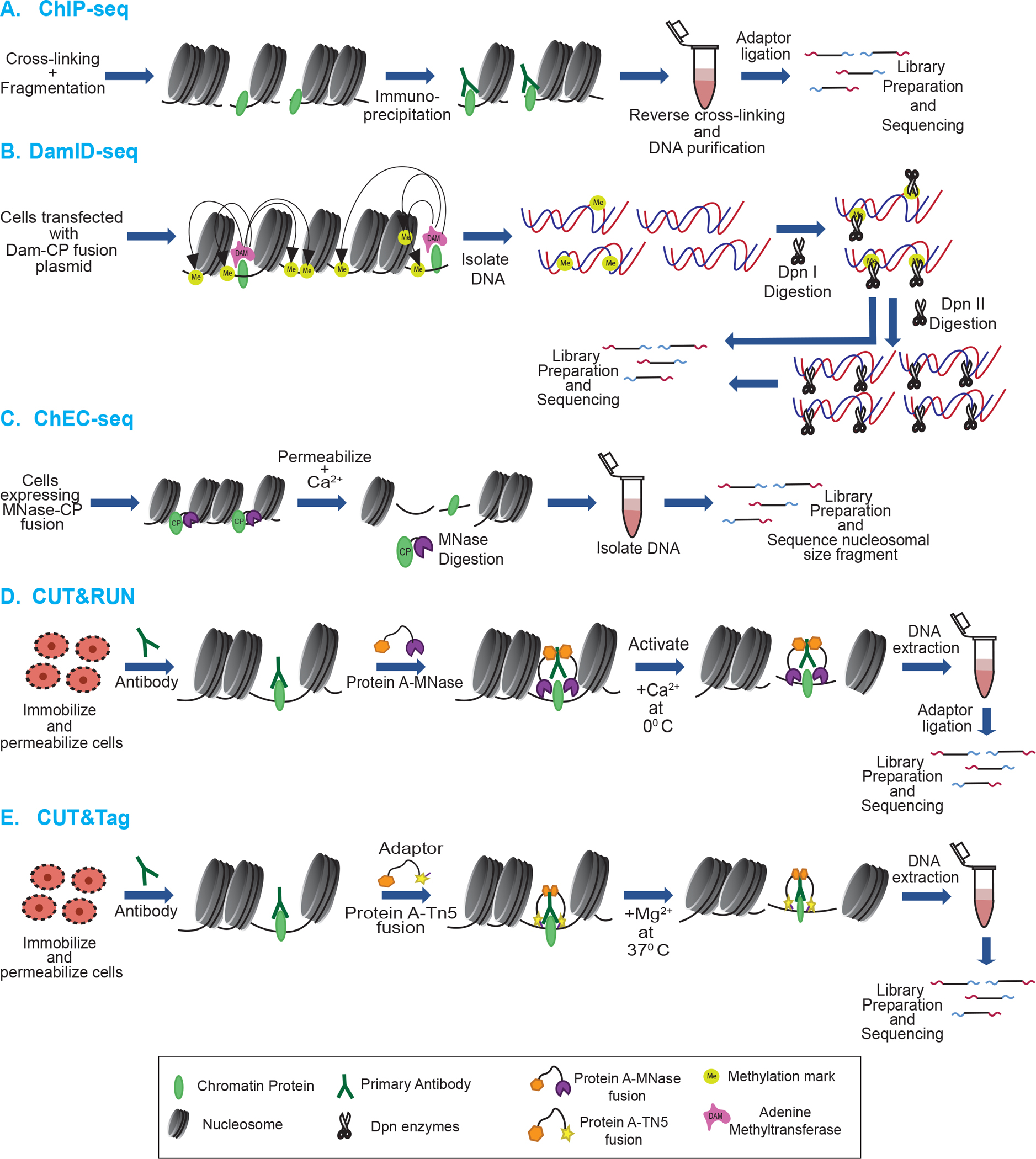
A. ChIP-seq: Proteins of interest are crosslinked to DNA, complexes pulled down via a specific antibody, and associated DNA is sequenced and mapped to the reference genome. B. DamID-seq: A chromatin protein of interest is expressed in cells in a DNA adenine methyltransferase (Dam) fusion. The fusion protein localizes to the appropriate genomic regions resulting in methylation of nearby adenines in Dam cognate sites. The DNA is digested with the methylation-dependent restriction enzyme DpnI. Part of the DpnI digested product is further digested by isoschizomer DpnII enzyme which cleaves the unmethylated DNA. DNA from both digestions is subjected to sequencing. C. ChEC-seq: The chromatin protein of interest is expressed in a fusion with MNase, direting MNase to digest nearby DNA. Released DNA fragments are sequenced and mapped back to the reference genome.
D. CUT&RUN: Protein A-MNase fusion is used to bind specific antibody against protein of interest. MNase is activated by the addition of Ca2+, cleaving the DNA nearby, releasing the fragments out of the nucleus, which are sequenced. E. CUT&Tag: Protein A-Tn5 fusion is used to bind specific antibody against a protein of interest. Addition of Mg2+ activates Tn5 and integrates adapters at cleavage sites. The released DNA is purified and subjected to sequencing.
The related ChIP-exo technique combines ChIP with exonuclease trimming to map crosslinks between individual histones and DNA at single-nucleotide resolution. This technique has been used to identify noncanonical nucleosome structures, which may lack one or more histone proteins [47,48,77–80]. Close examination of ChIP-exo patterns indicated that contacts to DNA by all four histones – and especially H2A/H2B – were significantly reduced on the promoter-proximal sides of nucleosomes in the upstream end of gene bodies [77]. This data suggests that significant populations of asymmetric nucleosomes exist, perhaps due to RNAP II-dependent transient displacement of histone-DNA contacts, or interactions with remodeler proteins, such as RSC [48,79,81], and is consistent with greater transcription-dependent displacement of H2A/H2B [82].
Similar to ChIP based techniques, the method of DNA adenine methyltransferase identification (DamID) utilizes the power of Dam to mark the genomic location of a protein of interest by expression of a plasmid-based chromatin protein-Dam fusion in the cell (Fig. 2B). The fusion protein binds to specific genomic loci where it methylates adenines within nearby GATC cognate sequences. The methylations are easily identified in the genomic DNA since DpnI digestion requires methylated GATC sites, a modification that is absent in most eukaryotic genomes. This technique was initially performed in Drosophila melanogaster, where binding sites of HP1, GAGA and Sir2p were identified genome-wide [83].
A modification of DamID is Nucleosome Occupancy and Methylome sequencing (NOMe-seq). This technique takes advantage of de novo GpC methyltransferase enzyme (M.CviPI) to artificially methylate GpC dinucleotides of chromatin that are left unprotected by DNA binding proteins or nucleosomes. This results in high resolution footprint of nucleosome occupancy as well as provides information on DNA methylation status of a particular gene locus since methylated GpC residues, which are uncommon in mammalian genomes, can easily be distinguished from common endogenous CpG methylation by bisulphite sequencing [84,85].
Similar to DamID, a method known as Chromatin Endogenous Cleavage (ChEC-seq), involves genetically fusing a chromatin binding protein to micrococcal nuclease generally in yeast, which allows for direct mapping of the protein of interest [86–88]. Because of the general endo/exo nuclease activity of MNase, ChEC-seq provides significantly greater resolution than DamID, which depends on a nearby target sequence [86,87,89]. The MNase fusion protein is expressed and is activated upon the introduction of calcium. Indeed, the Ca2+ -dependence of MNase has allowed ChEC-seq to be used for kinetic binding analyses of specific factors as well as identification of high and low affinity binding sites by examining the extent of chromatin digestion [86] (Fig. 2C). Unlike a standard ChIP-seq experiment which can hinder findings if the epitope is occluded as a result of protein-protein crosslinking, ChEC-seq can be performed under native conditions (i.e. no crosslinking). Given that many chromatin-binding proteins exist in multi-protein complexes, ChEC-seq has been able to identify binding sites not found in ChIP-seq experiments [86,90].
A related technique is cleavage under targets and release using nuclease (CUT&RUN), which can be used with lower cell numbers (~100 (histones) to ~1,000 (transcription factors)) [91,92]. In this technique, cells are lysed and nuclei are immobilized on lectin coated magnetic beads, incubated with an antibody selectively recognizing a chromatin protein of interest, and MNase is directed to the antibody by incubation with a Protein A-MNase fusion which releases targeted protein-DNA complexes into the supernatant, and the DNA is sequenced (Fig. 2D). However, the original CUT&RUN technique had limitations including problematic purification of the untagged protein A-MNase fusion, and variable digestion of chromatin, requiring a spike-in of heterologous DNA to calibrate samples [93]. An improved method employs His-tagged-Protein A-Protein G hybrid-MNase that is easily purified by metal-affinity chromatography, and allows efficient binding to a broad range of antibodies (Protein A often binds poorly to mouse antibodies). The modified protocol also eliminates spike-in of heterologous DNA as the carry-over of E. coli DNA from purification of the MNase fusion was found to be sufficient for calibration [93].
Most recently, CUT&RUN has been modified to leverage ATAC-seq technology, in a method termed cleavage under targets and tagmentation (CUT&Tag), in which Protein A or ProteinA/G is fused to Tn5 transposase, which tags the chromatin with adaptors for NGS at the location of the targeted protein of interest [94] (Fig. 2E). Moreover, CUT&Tag can be applied to small cell numbers (100–1,000) like CUT&RUN and ATAC-seq [94]. Studies conducted with these techniques have been carried out in cell types ranging from yeast to human. A recent modification in the CUT&Tag technique involves tethering Tn5 to antibodies targeting histone PTMs associated with open chromatin. This method is known as Cleavage Under Targeted Accessible Chromatin (CUTAC), and produces maps of accessible sites similar to ATAC-seq, but with much lower cell numbers [95]. A summary of the techniques described above is presented in Table 1.
Table 1:
Summary of common techniques used to study chromatin accessibility and genome-wide chromatin protein distribution
| FAIRE-seq | MNase-seq | ATAC-seq | DNaseI Seq | DamID-seq | ChIP-seq | CUT & RUN | CUT & Tag | |
|---|---|---|---|---|---|---|---|---|
| Sample prep * | ~3 days | ~2 days | ~4 h | ~1–3 days | ~3–5 days | 3–4 days | 1–2 days | 1–2 days |
| Fragmentation Method | Sonication | MNase Digestion | Tn5-based tagmentation | DNase I digestion | DpnI Digestion | Sonication | MNase Digestion | Tn5-based tagmentation |
| Cell Number Required | ~1–50 million | ~10,000–100,000 | ~50,000 | ~1–50 million | ~10,000 | 1–10 million | As low as 100–1000 Usually 500,000 | Usually 5,000–500,000 |
| Target | Access Chrom | Access Chrom/Nuc Pos | Access Chrom | Access Chrom | Targ Prot Bndg Site | Targ Prot Bndg Site | Targ Prot Bndg Site | Targ Prot Bndg Site |
| Single Cell? | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Resolution | High | High | Very High | High | Low | High | Very High | Very High |
4. Analysis of inter-nucleosomal interactions
A family of techniques has been developed to map direct and indirect inter-nucleosomal interactions mediated by chromatin binding proteins, providing tremendous insight into the large-scale three-dimensional architecture of chromatin. The earliest of these is known as chromosome conformation capture (3C), which allows for assessment of interactions between two distinct loci in chromatin [96] (Fig. 3). 3C requires formaldehyde crosslinking of nuclei, resulting in covalent linkage between closely juxtaposed loci. Although the exact nature of the crosslinks holding loci together is not clear, these likely involve capture of long-range inter-nucleosome interactions mediated by the core histone tail domains. The crosslinked chromatin is fragmented by treatment with a restriction enzyme and then is ligated in a manner that favors ligation of crosslinked loci. Next, crosslinking is reversed, proteins removed, and PCR used to quantify the association of interacting loci. A limitation of 3C is that one needs to interrogate the potential association of two regions of interest with specific PCR primers [96–98]. Early 3C studies identified interactions between regulatory elements within the beta-globin locus linearly spaced ~50 kb away from each other [99,100] and identified chromatin looping as a dynamic mechanism that regulates gene expression by facilitating interactions between spatially distanced regulatory elements such as promoters and enhancers [101,102] and promoter and terminators in yeast [103].
Figure 3: Overview of chromosome conformation capture techniques:
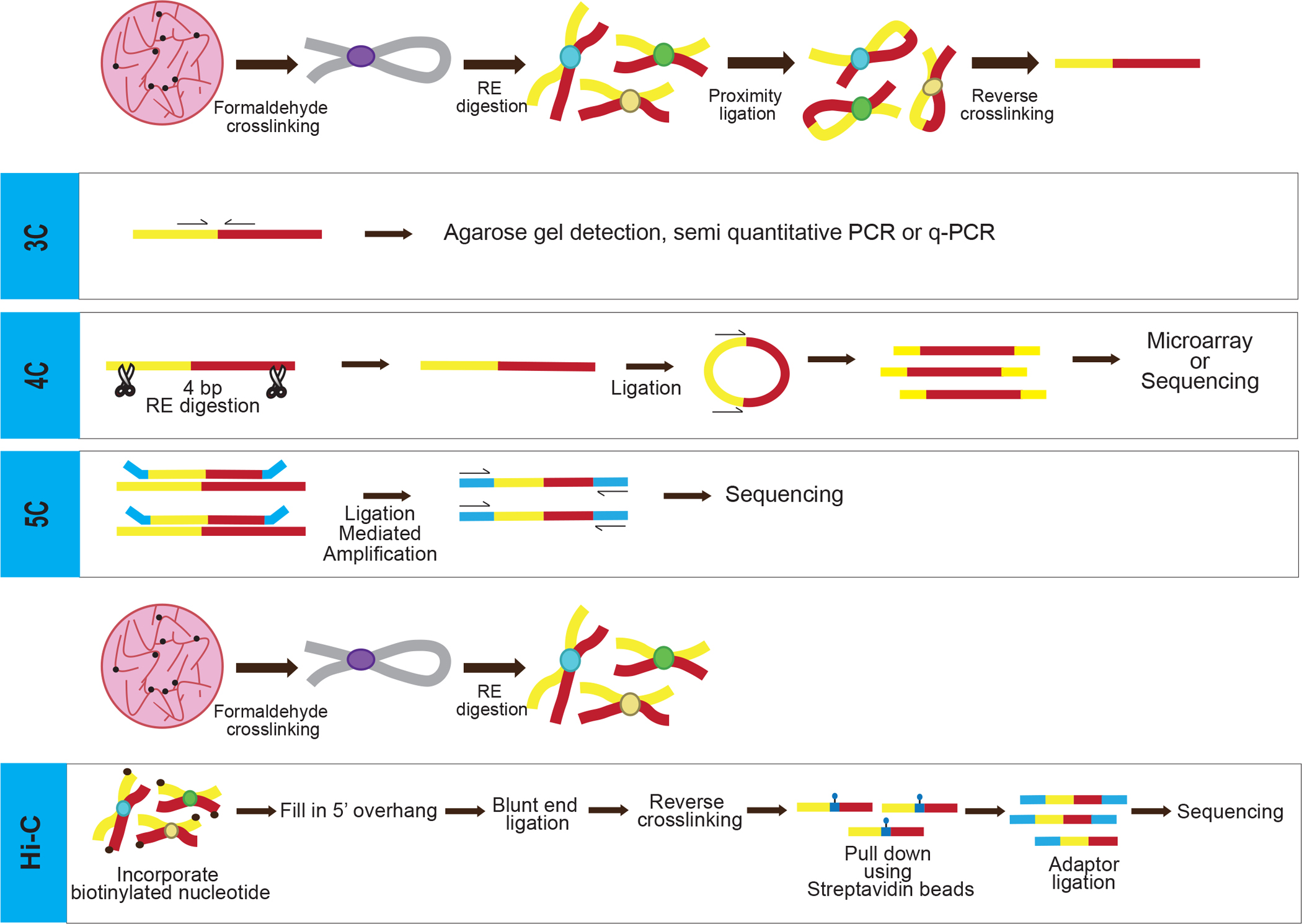
All methods share the common step of formaldehyde crosslinking in situ, capturing interaction between different chromatin segments. For 3C, 4C and 5C, the DNA is fragmented by restriction enzyme digestion then the free ends ligated, capturing ends brought in close proximity by chromatin structure, crosslinking is reversed, and the DNA purified. The DNA is either used for PCR with specific primers (3C), or is further digested by a second restriction enzyme, followed by religating shorter fragments and inverse PCR (4C). The PCR product is subjected to microarray hybridization or deep sequencing. In 5C, all the steps are same as 3C except the specific PCR primers tagged with universal T7 and T3 primer sequences are annealed to sequences abutting the ligation site and ligated, and PCR products are sequenced. In Hi-C, the free DNA ends in the crosslinked, digested chromatin is filled with biotinylated nucleotides before ligation, crosslinking reversed, and ligated DNA is isolated and subjected to deep-sequencing.
The advancement of next generation sequencing technologies spurred derivatives of 3C, known as chromosome conformation capture-on-ChIP (4C) and chromosome conformation capture carbon copy (5C), which allowed for much wider interrogation of genomic interactions between chromatin regions. Like 3C, in 4C the chromatin is crosslinked, digested with restriction enzyme(s) and digested fragments ligated to join the DNAs from two discrete loci (Fig. 3). However, prior to the final ligation step, the DNA is subjected to digestion with a second restriction enzyme, then ligated to generate smaller DNA circles consisting of the crosslinked loci [104,105]. Identification is achieved by performing inverse PCR and then sequencing the amplified DNA fragments. Unlike 3C, 4C requires primers to only a single region of interest, which allows for identification of interactions between a defined region and other (unspecified) loci [104,105].
5C allows for the simultaneous detection of many chromatin interactions by combining 3C with multiplexed ligation-mediated amplification of thousands of primers from candidate interacting regions in one reaction [106] (Fig. 3). Typically, thousands of primers are introduced to a 3C library that are designed to include portions of the 3C restriction site and adjoining genomic sequence throughout two candidate interacting regions of interest. Pairs of primers annealing next to each other (due to ligation of the restriction site in the 3C reaction) are ligated together, then the ligated primers are amplified, and sequenced using universal PCR primers [106] (Fig. 3). 5C improves the ability to identify chromatin looping interactions between promoters and enhancer elements mediated by architectural proteins [107,108]. These technologies have also helped identify and characterize topologically associating domains (TADs), which are spatially separated genomic loci but are in close proximity due to their transcriptional state (i.e. active or inactive) [109,110].
An unbiased chromosome conformation capture technique known as Hi-C allows for probing interactions throughout the genome at the megabase scale [111,112]. This technique involves pairing spatial proximity ligation with deep sequencing allowing for identification of long-range chromatin interactions rather than specified loci like its predecessors [111]. Similar to other chromatin capture techniques, the crosslinked DNA is digested by restriction enzyme. However, during ligation, the overhangs generated by restriction digestion are filled with dNTPs, including biotinylated dNTPs which are selectively retained via streptavidin affinity purification to reduce background from sequencing [113,114] (Fig. 3). Because of its unbiased, expansive interrogation, Hi-C has the limitation of requiring a substantial amount of reads and is limited in resolution [114]. However, there are adaptations to this method that work around this roadblock such as the use of a sequence capture step to enrich for regions of interest allowing for deeper sequencing at these loci [115–117]. The method has been adapted for single cells [118].
Micrococcal nuclease chromosome conformation assay (Micro-C) is a modified form of Hi-C that uses MNase digestion to yield mononucleosomes in crosslinked chromatin instead of restriction enzymes to give smaller, nucleosome-sized fragments, making Micro-C better suited for short range analysis of chromatin folding. This technique was used for the first time in budding and fission yeast to elucidate contacts between nucleosomes [113,119]. Later it was extended to mammalian systems [120,121].
Many inventive variations on the chromatin conformation capture techniques have been developed. For example, Chromatin Interaction Analysis by Paired-End Tag sequencing (ChIA-PET) was developed to detect de novo oestrogen receptor alpha-bound global chromatin interactions in oestrogen treated MCF-7 cells, leading to the generation of first specific human chromatin interactome map [122]. ChIA-PET takes advantage of ChIP, 3C, and high-throughput sequencing techniques to determine genome-wide de novo long-range chromatin interactions via a protein of interest. After formaldehyde crosslinking, chromatin is sonicated and pulled-down using specific antibodies of interest and fragments ligated with a linker containing MmeI restriction endonuclease site, proximity ligated, and then digested with MmeI followed by paired-end tag sequencing. [122,123]. A further improvement introduced Tn5 transposase-based tagmentation to randomly fragment nuclear proximity ligation products having a biotin linker and to simultaneously add adaptors to cleavage sites, leading to improved sequence alignment accuracy and enabling haplotype-specific mapping of chromatin interactions [124]. However, this is an antibody-based method specific for a protein of interest and thus does not reveal global protein interactions [125].
An approach that features base-pair level interrogation is Radiation-induced spatially-correlated cleavage of DNA with sequencing (RICC-seq), developed by the Greenleaf laboratory in 2017 to study the local structure of chromatin at a 50–500 bp scale [126]. This method exploits correlated cleavages initiated by ionizing radiation that generate short DNA fragments. The short fragments are sequenced and points of cleavage mapped back to the genome. RICC-seq reveals a striking pattern of in situ intra- and inter-nucleosome contacts showing close apposition of linker DNAs reflecting histone H1 binding and signatures consistent with 2-start cross-linker folding of the chromatin fiber, especially in heterochromatin regions, with less nucleosome-nucleosome contact in active regions. These results support existence of two-start chromatin fiber structures in native interphase nuclei [126]. Major techniques used to study nucleosome-nucleosome interactions are summarized in Table 2.
Table 2:
Summary of chromatin conformation capture technique and derivatives
| Technique | Interactions Detected |
Resolution Limit | Number Cells Req’d | Timeline | Reference |
|---|---|---|---|---|---|
| 3C | One vs One | 1kb to 100kb | ~5–10 million | ~3 days | [96] |
| 4C | One Vs All | 1kb to few 100 kb | ~5 million | ~4 days | [105] |
| 5C | Many vs Many | 1kb | ~10 million | ~ 6 days | [106] |
| Hi-C | All vs All | kb to mb | ~10 million | ~ 4 days | [111] |
| Micro-C | All vs All | ~200bp-4kb | ~1 million | ~3–4 days | [113] |
| ChIA-PET | All vs All (Protein) | ~100 bp | ~100 million | ~5 days | [122] |
For further reading see [139]
5. Histone protein surface accessibility
Most of the methods discussed above rely upon DNA accessibility or DNA-based reagents for assessing changes in chromatin structure. As previously discussed, these findings have provided much information but discrepancies in analysis can arise from the amount of chromatin fragmentation, and digestion bias for particular DNA nucleotides and sequences. Further, there is evidence that differences in DNA accessibility between euchromatic and heterochromatic regions is limited raising the question of whether this is a reliable means of assessing chromatin structure [39,50,127].
Recently a technique has been devised that gages accessibility of histone proteins rather than the DNA [128,129]. Structural studies of oligonucleosomes show that considerable occlusion of the protein surface occurs when nucleosome arrays are condensed [130–132]. The proof-of-principle experiment explored the accessibility of a cysteine placed on the exposed flat protein surface of the nucleosome to a cysteine specific reagent, to measure the extent of nucleosome packing in chromatin genome-wide in isolated yeast nuclei. The study found that yeast nucleosome surfaces are generally equivalently accessible genome-wide, consistent with a globally uncompacted chromatin structure lacking substantial higher-order organization. However, a much more modest difference was found in accessibility that correlates with the presence of chromatin remodelers but not level of transcription, suggesting chromatin poised for transcription is more accessible than actively transcribed or intergenic regions. In contrast, an internal site reporting on disruption of the H3-H3 interface, previously reported to be accessible [133], was not found to be accessible in native yeast chromatin, suggesting that if such non-canonical nucleosome species are generated during transcription they are rapidly and efficiently converted to canonical nucleosomes and thus are not widely present in native chromatin.
Histone-histone interactions between nucleosomes have been mapped in vitro using cysteine-dependent crosslinking [134,135]. Most recently a multiplexed version of such an experiment, termed ICNN (identification of closest neighbor nucleosomes) has been developed wherein crosslinking between nucleosomes containing bar-coded DNA and the histone mutants H2A E64C and H4 V21C are defined for an entire nucleosome array [131]. This methodology reveals preferential interactions between nucleosome N and N+2, supporting cross-linker models for the partially and full-folded chromatin fiber [131]. One may speculate that the same powerful technique could be applied in vivo to test whether local order actually exists in folded/compacted nucleosome arrays [136].
Perspectives.
Understanding the genome-wide variations in chromatin accessibility, modifications, and interactions have been invaluable in our understanding of the mechanism of gene regulation and diseased states associated with dysregulated gene expression.
We review many of the techniques devised to study genome-wide chromatin structure and protein interactions, highlighting each techniques’ benefits and limitations.
Historically, most of the genome-wide techniques require large number of cells. However, number of cells are limited from patient samples or primary cell cultures. In future, it would be interesting to see techniques utilizing single cells or only a very small number of cells to provide high resolution data.
Funding
The work was supported by National Institutes of Health Grants R01GM052426 (to JJH), and T32GM068411 (to LTM).
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Van Holde KE: Chromatin. New York: Springer-Verlag; 1989. [Google Scholar]
- 2.Schlick T, Hayes J, Grigoryev S: Toward convergence of experimental studies and theoretical modeling of the chromatin fiber. J Biol Chem 2012, 287:5183–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepenella S, Murphy KJ, Hayes JJ: Intra- and inter-nucleosome interactions of the core histone tail domains in higher-order chromatin structure. Chromosoma 2014, 123:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishra LN, Pepenella S, Rogge R, Hansen JC, Hayes JJ: Acetylation Mimics Within a Single Nucleosome Alter Local DNA Accessibility In Compacted Nucleosome Arrays. Sci Rep 2016, 6:34808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishra LN, Hayes JJ: A nucleosome-free region locally abrogates histone H1-dependent restriction of linker DNA accessibility in chromatin. J Biol Chem 2018, 293:19191–19200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu C: The 5’ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 1980, 286:854–860. [DOI] [PubMed] [Google Scholar]
- 7.Wu C, Wong YC, Elgin SC: The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell 1979, 16:807–814. [DOI] [PubMed] [Google Scholar]
- 8.Wu C, Bingham PM, Livak KJ, Holmgren R, Elgin SC: The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell 1979, 16:797–806. [DOI] [PubMed] [Google Scholar]
- 9.Weisbrod S, Weintraub H: Isolation of a subclass of nuclear proteins responsible for conferring a DNase I-sensitive structure on globin chromatin. Proc Natl Acad Sci U S A 1979, 76:630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weintraub H, Groudine M: Chromosomal subunits in active genes have an altered conformation. Science 1976, 193:848–856. [DOI] [PubMed] [Google Scholar]
- 11.Almer A, Horz W: Nuclease hypersensitive regions with adjacent positioned nucleosomes mark the gene boundaries of the PHO5/PHO3 locus in yeast. EMBO J 1986, 5:2681–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almer A, Rudolph H, Hinnen A, Horz W: Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J 1986, 5:2689–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid A, Fascher KD, Horz W: Nucleosome disruption at the yeast PHO5 promoter upon PHO5 induction occurs in the absence of DNA replication. Cell 1992, 71:853–864. [DOI] [PubMed] [Google Scholar]
- 14.Han M, Grunstein M: Nucleosome loss activates yeast downstream promoters in vivo. Cell 1988, 55:1137–1145. [DOI] [PubMed] [Google Scholar]
- 15.Han M, Kim UJ, Kayne P, Grunstein M: Depletion of histone H4 and nucleosomes activates the PHO5 gene in Saccharomyces cerevisiae. EMBO J 1988, 7:2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE: High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, Ni B, Sklar J, Przytycka TM, Childs R, et al. : Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528:142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD: Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol Cell 2004, 14:667–673. [DOI] [PubMed] [Google Scholar]
- 19.Reinke H, Horz W: Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell 2003, 11:1599–1607. [DOI] [PubMed] [Google Scholar]
- 20.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD: FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 2007, 17:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogan GJ, Lee CK, Lieb JD: Cell cycle-specified fluctuation of nucleosome occupancy at gene promoters. PLoS Genet 2006, 2:e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagy PL, Cleary ML, Brown PO, Lieb JD: Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc Natl Acad Sci U S A 2003, 100:6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dingwall C, Lomonossoff GP, Laskey RA: High sequence specificity of micrococcal nuclease. Nucleic Acids Res 1981, 9:2659–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horz W, Altenburger W: Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res 1981, 9:2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGhee JD, Felsenfeld G: Another potential artifact in the study of nucleosome phasing by chromatin digestion with micrococcal nuclease. Cell 1983, 32:1205–1215. [DOI] [PubMed] [Google Scholar]
- 26.Green B, Bouchier C, Fairhead C, Craig NL, Cormack BP: Insertion site preference of Mu, Tn5, and Tn7 transposons. Mob DNA 2012, 3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodge JK, Weston-Hafer K, Berg DE: Transposon Tn5 target specificity: preference for insertion at G/C pairs. Genetics 1988, 120:645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsompana M, Buck MJ: Chromatin accessibility: a window into the genome. Epigenetics Chromatin 2014, 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noll M: Internal structure of the chromatin subunit. Nucleic Acids Res 1974, 1:1573–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewish DR, Burgoyne LA: Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem Biophys Res Commun 1973, 52:504–510. [DOI] [PubMed] [Google Scholar]
- 31.Hewish DR, Burgoyne LA: The calcium dependent endonuclease activity of isolated nuclear preparations. Relationships between its occurrence and the occurrence of other classes of enzymes found in nuclear preparations. Biochem Biophys Res Commun 1973, 52:475–481. [DOI] [PubMed] [Google Scholar]
- 32.Yager TD, McMurray CT, van Holde KE: Salt-induced release of DNA from nucleosome core particles. Biochemistry 1989, 28:2271–2281. [DOI] [PubMed] [Google Scholar]
- 33.Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ: Genome-scale identification of nucleosome positions in S. cerevisiae. Science 2005, 309:626–630. [DOI] [PubMed] [Google Scholar]
- 34.Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C: A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 2007, 39:1235–1244. [DOI] [PubMed] [Google Scholar]
- 35.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, et al. : Nucleosome organization in the Drosophila genome. Nature 2008, 453:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esnault C, Magat T, Garcia-Oliver E, Andrau JC: Analyses of Promoter, Enhancer, and Nucleosome Organization in Mammalian Cells by MNase-Seq. Methods Mol Biol 2021, 2351:93–104. [DOI] [PubMed] [Google Scholar]
- 37.Yu J, Xiong C, Zhuo B, Wen Z, Shen J, Liu C, Chang L, Wang K, Wang M, Wu C, et al. : Analysis of Local Chromatin States Reveals Gene Transcription Potential during Mouse Neural Progenitor Cell Differentiation. Cell Rep 2020, 32:107953. [DOI] [PubMed] [Google Scholar]
- 38.Gao W, Lai B, Ni B, Zhao K: Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq. Nat Protoc 2020, 15:68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chereji RV, Bryson TD, Henikoff S: Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol 2019, 20:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pajoro A, Muino JM, Angenent GC, Kaufmann K: Profiling Nucleosome Occupancy by MNase-seq: Experimental Protocol and Computational Analysis. Methods Mol Biol 2018, 1675:167–181. [DOI] [PubMed] [Google Scholar]
- 41.Wen Z, Zhang L, Ruan H, Li G: Histone variant H2A.Z regulates nucleosome unwrapping and CTCF binding in mouse ES cells. Nucleic Acids Res 2020, 48:5939–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alonso A, Bernstein E, Hasson D: Histone Native Chromatin Immunoprecipitation. Methods Mol Biol 2018, 1832:77–104. [DOI] [PubMed] [Google Scholar]
- 43.Chereji RV, Ocampo J, Clark DJ: MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Mol Cell 2017, 65:565–577 e563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo D, Kato D, Nogami J, Ohkawa Y, Kurumizaka H, Kono H: MNase, as a probe to study the sequence-dependent site exposures in the +1 nucleosomes of yeast. Nucleic Acids Res 2018, 46:7124–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui K, Zhao K: Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol Biol 2012, 833:413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xi Y, Yao J, Chen R, Li W, He X: Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res 2011, 21:718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knight B, Kubik S, Ghosh B, Bruzzone MJ, Geertz M, Martin V, Denervaud N, Jacquet P, Ozkan B, Rougemont J, et al. : Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev 2014, 28:1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kubik S, Bruzzone MJ, Jacquet P, Falcone JL, Rougemont J, Shore D: Nucleosome Stability Distinguishes Two Different Promoter Types at All Protein-Coding Genes in Yeast. Mol Cell 2015, 60:422–434. [DOI] [PubMed] [Google Scholar]
- 49.Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, et al. : MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 2016, 7:11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chereji RV, Eriksson PR, Ocampo J, Prajapati HK, Clark DJ: Accessibility of promoter DNA is not the primary determinant of chromatin-mediated gene regulation. Genome Res 2019, 29:1985–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ: Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013, 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ: Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523:486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daugherty AC, Yeo RW, Buenrostro JD, Greenleaf WJ, Kundaje A, Brunet A: Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res 2017, 27:2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marinov GK, Shipony Z: Interrogating the Accessible Chromatin Landscape of Eukaryote Genomes Using ATAC-seq. Methods Mol Biol 2021, 2243:183–226. [DOI] [PubMed] [Google Scholar]
- 55.Wu X, Lu M, Yun D, Gao S, Chen S, Hu L, Wu Y, Wang X, Duan E, Cheng CY, et al. : Single cell ATAC-Seq reveals cell type-specific transcriptional regulation and unique chromatin accessibility in human spermatogenesis. Hum Mol Genet 2021, 10.1093/hmg/ddab006. [DOI] [PubMed] [Google Scholar]
- 56.Willcockson MA, Healton SE, Weiss CN, Bartholdy BA, Botbol Y, Mishra LN, Sidhwani DS, Wilson TJ, Pinto HB, Maron MI, et al. : H1 histones control the epigenetic landscape by local chromatin compaction. Nature 2021, 589:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pott S, Lieb JD: Single-cell ATAC-seq: strength in numbers. Genome Biol 2015, 16:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. : The chromatin accessibility landscape of primary human cancers. Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cole HA, Ocampo J, Iben JR, Chereji RV, Clark DJ: Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res 2014, 42:12512–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lorzadeh A, Bilenky M, Hammond C, Knapp D, Li L, Miller PH, Carles A, Heravi-Moussavi A, Gakkhar S, Moksa M, et al. : Nucleosome Density ChIP-Seq Identifies Distinct Chromatin Modification Signatures Associated with MNase Accessibility. Cell Rep 2016, 17:2112–2124. [DOI] [PubMed] [Google Scholar]
- 61.Park PJ: ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 2009, 10:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Orsi GA, Kasinathan S, Zentner GE, Henikoff S, Ahmad K: Mapping regulatory factors by immunoprecipitation from native chromatin. Curr Protoc Mol Biol 2015, 110:21 31 21–21 31 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Healton SE, Pinto HD, Mishra LN, Hamilton GA, Wheat JC, Swist-Rosowska K, Shukeir N, Dou Y, Steidl U, Jenuwein T, et al. : H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc Natl Acad Sci U S A 2020, 117:14251–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weller MG: Ten Basic Rules of Antibody Validation. Anal Chem Insights 2018, 13:1177390118757462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marcon E, Jain H, Bhattacharya A, Guo H, Phanse S, Pu S, Byram G, Collins BC, Dowdell E, Fenner M, et al. : Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat Methods 2015, 12:725–731. [DOI] [PubMed] [Google Scholar]
- 66.Voskuil J: Commercial antibodies and their validation. F1000Res 2014, 3:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parseghian MH: Hitchhiker antigens: inconsistent ChIP results, questionable immunohistology data, and poor antibody performance may have a common factor. Biochem Cell Biol 2013, 91:378–394. [DOI] [PubMed] [Google Scholar]
- 68.Peach SE, Rudomin EL, Udeshi ND, Carr SA, Jaffe JD: Quantitative assessment of chromatin immunoprecipitation grade antibodies directed against histone modifications reveals patterns of co-occurring marks on histone protein molecules. Mol Cell Proteomics 2012, 11:128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Couchman JR: Commercial antibodies: the good, bad, and really ugly. J Histochem Cytochem 2009, 57:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mishra LN, Gupta N, Rao SM: Mapping of post-translational modifications of spermatid-specific linker histone H1-like protein, HILS1. J Proteomics 2015, 128:218–230. [DOI] [PubMed] [Google Scholar]
- 71.Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, Krajewski K, Guo AH, Shanle EK, Josefowicz SZ, Fuchs SM, Allis CD, et al. : An Interactive Database for the Assessment of Histone Antibody Specificity. Mol Cell 2015, 59:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Venkataraman A, Yang K, Irizarry J, Mackiewicz M, Mita P, Kuang Z, Xue L, Ghosh D, Liu S, Ramos P, et al. : A toolbox of immunoprecipitation-grade monoclonal antibodies to human transcription factors. Nat Methods 2018, 15:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson DS, Mortazavi A, Myers RM, Wold B: Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 74.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K: High-resolution profiling of histone methylations in the human genome. Cell 2007, 129:823–837. [DOI] [PubMed] [Google Scholar]
- 75.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. : Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 2007, 4:651–657. [DOI] [PubMed] [Google Scholar]
- 76.Mishra LN, Shalini V, Gupta N, Ghosh K, Suthar N, Bhaduri U, Rao MRS: Spermatid-specific linker histone HILS1 is a poor condenser of DNA and chromatin and preferentially associates with LINE-1 elements. Epigenetics Chromatin 2018, 11:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rhee HS, Bataille AR, Zhang L, Pugh BF: Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 2014, 159:1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramachandran S, Zentner GE, Henikoff S: Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res 2015, 25:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brahma S, Henikoff S: RSC-Associated Subnucleosomes Define MNase-Sensitive Promoters in Yeast. Mol Cell 2019, 73:238–249 e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rossi MJ, Lai WKM, Pugh BF: Simplified ChIP-exo assays. Nat Commun 2018, 9:2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM: Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell 2002, 9:541–552. [DOI] [PubMed] [Google Scholar]
- 82.Thiriet C, Hayes JJ: Histone dynamics during transcription: exchange of H2A/H2B dimers and H3/H4 tetramers during pol II elongation. Results Probl Cell Differ 2006, 41:77–90. [DOI] [PubMed] [Google Scholar]
- 83.van Steensel B, Delrow J, Henikoff S: Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet 2001, 27:304–308. [DOI] [PubMed] [Google Scholar]
- 84.Taberlay PC, Statham AL, Kelly TK, Clark SJ, Jones PA: Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res 2014, 24:1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA: Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 2012, 22:2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zentner GE, Kasinathan S, Xin B, Rohs R, Henikoff S: ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun 2015, 6:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmid M, Durussel T, Laemmli UK: ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 2004, 16:147–157. [DOI] [PubMed] [Google Scholar]
- 88.Grunberg S, Zentner GE: Genome-wide Mapping of Protein-DNA Interactions with ChEC-seq in Saccharomyces cerevisiae. J Vis Exp 2017, 10.3791/55836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vogel MJ, Peric-Hupkes D, van Steensel B: Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc 2007, 2:1467–1478. [DOI] [PubMed] [Google Scholar]
- 90.Tourigny JP, Saleh MM, Schumacher K, Devys D, Zentner GE: Mediator Is Essential for Small Nuclear and Nucleolar RNA Transcription in Yeast. Mol Cell Biol 2018, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Skene PJ, Henikoff JG, Henikoff S: Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 2018, 13:1006–1019. [DOI] [PubMed] [Google Scholar]
- 92.Skene PJ, Henikoff S: An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meers MP, Bryson TD, Henikoff JG, Henikoff S: Improved CUT&RUN chromatin profiling tools. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S: CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 2019, 10:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henikoff S, Henikoff JG, Kaya-Okur HS, Ahmad K: Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dekker J, Rippe K, Dekker M, Kleckner N: Capturing chromosome conformation. Science 2002, 295:1306–1311. [DOI] [PubMed] [Google Scholar]
- 97.Dekker J: Mapping in vivo chromatin interactions in yeast suggests an extended chromatin fiber with regional variation in compaction. J Biol Chem 2008, 283:34532–34540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miele A, Bystricky K, Dekker J: Yeast silent mating type loci form heterochromatic clusters through silencer protein-dependent long-range interactions. PLoS Genet 2009, 5:e1000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W: Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 2002, 10:1453–1465. [DOI] [PubMed] [Google Scholar]
- 100.Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W: CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 2006, 20:2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W: The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet 2003, 35:190–194. [DOI] [PubMed] [Google Scholar]
- 102.O’Sullivan JM, Tan-Wong SM, Morillon A, Lee B, Coles J, Mellor J, Proudfoot NJ: Gene loops juxtapose promoters and terminators in yeast. Nat Genet 2004, 36:1014–1018. [DOI] [PubMed] [Google Scholar]
- 103.Hampsey M: Molecular biology. A new direction for gene loops. Science 2012, 338:624–625. [DOI] [PubMed] [Google Scholar]
- 104.Simonis M, Klous P, Homminga I, Galjaard RJ, Rijkers EJ, Grosveld F, Meijerink JP, de Laat W: High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nat Methods 2009, 6:837–842. [DOI] [PubMed] [Google Scholar]
- 105.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W: Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 2006, 38:1348–1354. [DOI] [PubMed] [Google Scholar]
- 106.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. : Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 2006, 16:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sanyal A, Lajoie BR, Jain G, Dekker J: The long-range interaction landscape of gene promoters. Nature 2012, 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. : CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 2011, 43:630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G: Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148:458–472. [DOI] [PubMed] [Google Scholar]
- 110.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. : Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. : Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Belton JM, Dekker J: Hi-C in Budding Yeast. Cold Spring Harb Protoc 2015, 2015:649–661. [DOI] [PubMed] [Google Scholar]
- 113.Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ: Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 2015, 162:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sati S, Cavalli G: Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma 2017, 126:33–44. [DOI] [PubMed] [Google Scholar]
- 115.Dryden NH, Broome LR, Dudbridge F, Johnson N, Orr N, Schoenfelder S, Nagano T, Andrews S, Wingett S, Kozarewa I, et al. : Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res 2014, 24:1854–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jager R, Migliorini G, Henrion M, Kandaswamy R, Speedy HE, Heindl A, Whiffin N, Carnicer MJ, Broome L, Dryden N, et al. : Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nat Commun 2015, 6:6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Belaghzal H, Dekker J, Gibcus JH: Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 2017, 123:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ramani V, Deng X, Qiu R, Lee C, Disteche CM, Noble WS, Shendure J, Duan Z: Sci-Hi-C: A single-cell Hi-C method for mapping 3D genome organization in large number of single cells. Methods 2020, 170:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hsieh TS, Fudenberg G, Goloborodko A, Rando OJ: Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods 2016, 13:1009–1011. [DOI] [PubMed] [Google Scholar]
- 120.Krietenstein N, Abraham S, Venev SV, Abdennur N, Gibcus J, Hsieh TS, Parsi KM, Yang L, Maehr R, Mirny LA, et al. : Ultrastructural Details of Mammalian Chromosome Architecture. Mol Cell 2020, 78:554–565 e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hsieh TS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, Darzacq X: Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol Cell 2020, 78:539–553 e538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. : An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, et al. : ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol 2010, 11:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X, Luo OJ, Wang P, Zheng M, Wang D, Piecuch E, Zhu JJ, Tian SZ, Tang Z, Li G, et al. : Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nat Protoc 2017, 12:899–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang J, Poh HM, Peh SQ, Sia YY, Li G, Mulawadi FH, Goh Y, Fullwood MJ, Sung WK, Ruan X, et al. : ChIA-PET analysis of transcriptional chromatin interactions. Methods 2012, 58:289–299. [DOI] [PubMed] [Google Scholar]
- 126.Risca VI, Denny SK, Straight AF, Greenleaf WJ: Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature 2017, 541:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen L, Widom J: Mechanism of transcriptional silencing in yeast. Cell 2005, 120:37–48. [DOI] [PubMed] [Google Scholar]
- 128.Marr LT, Clark DJ, Hayes JJ: A method for assessing histone surface accessibility genome-wide. Methods 2020, 184:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marr LT, Ocampo J, Clark DJ, Hayes JJ: Global histone protein surface accessibility in yeast indicates a uniformly loosely packed genome with canonical nucleosomes. Epigenetics Chromatin 2021, 14:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boopathi R, Dimitrov S, Hamiche A, Petosa C, Bednar J: Cryo-electron microscopy of the chromatin fiber. Curr Opin Struct Biol 2020, 64:97–103. [DOI] [PubMed] [Google Scholar]
- 131.Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Le Roy A, Skoufias DA, Bednar J, Hamiche A, et al. : Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol Cell 2018, 72:902–915 e907. [DOI] [PubMed] [Google Scholar]
- 132.Adhireksan Z, Sharma D, Lee PL, Davey CA: Near-atomic resolution structures of interdigitated nucleosome fibres. Nat Commun 2020, 11:4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Walker J, Chen TA, Sterner R, Berger M, Winston F, Allfrey VG: Affinity chromatography of mammalian and yeast nucleosomes. Two modes of binding of transcriptionally active mammalian nucleosomes to organomercurial-agarose columns, and contrasting behavior of the active nucleosomes of yeast. J Biol Chem 1990, 265:5736–5746. [PubMed] [Google Scholar]
- 134.Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ: Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 2004, 306:1571–1573. [DOI] [PubMed] [Google Scholar]
- 135.Sinha D, Shogren-Knaak MA: Role of direct interactions between the histone H4 Tail and the H2A core in long range nucleosome contacts. J Biol Chem 2010, 285:16572–16581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maeshima K, Rogge R, Tamura S, Joti Y, Hikima T, Szerlong H, Krause C, Herman J, Seidel E, DeLuca J, et al. : Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J 2016, 35:1115–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sajan SA, Hawkins RD: Methods for identifying higher-order chromatin structure. Annu Rev Genomics Hum Genet 2012, 13:59–82. [DOI] [PubMed] [Google Scholar]
- 138.Klein DC, Hainer SJ: Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res 2020, 28:69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jerkovic I, Cavalli G: Understanding 3D genome organization by multidisciplinary methods. Nat Rev Mol Cell Biol 2021, 22:511–528. [DOI] [PubMed] [Google Scholar]