

Hereditary spastic paraplegia (HSP) is a rare and genetically heterogeneous disease. 1 Presenilin‐1 (PSEN1) mutations are responsible for both early‐onset familial Alzheimer's disease (AD) 2 and HSP. 3 We present a case of spastic paraplegia (SP) and cognitive impairment due to a novel de novo pathogenic variant in PSEN1.
A 32‐year‐old woman was referred to our unit in November 2021 because of SP of unknown etiology. Her disease debuted with difficulties when running at the age of 27. Several months later she began to drag her feet when walking associated with stiffness of the legs. At the age of 30, the family reported loss of balance with falls, subtle memory problems, and urinary urgency. In the following 2 years, both cognitive and motor problems worsened quickly making the patient dependent for most of the daily living activities. There was no family history of SP or early onset dementia.
On examination, she presented slight dysarthria and word‐finding difficulties. Ocular pursuit was clearly saccadic, but there was no nystagmus or ophthalmoparesis. She showed slight bilateral dysmetria and dysdiadochokinesia in the upper‐limbs (Video 1) that was not assessable in the lower‐limbs because of spasticity. There was proximal weakness in the legs, especially in the left one. Vibration sense was mildly decreased on both feet. Lower‐limbs were markedly spastic (Ashworth 3) with inward deviation of the left foot and hyperextension of the left leg. Deep tendon reflexes were markedly brisk, mainly on the left side. Bilateral Babinski and Hoffmann signs, as well as inexhaustible ankle clonus were present. Standing up or walking was impossible without support because of unsteadiness with a tendency to fall backward. Assisted gait was markedly spastic (Video 1). Spastic Paraplegia Rating score was 31/52.
Video 1.
The video shows the presence of pyramidal signs with generalized hyperreflexia involving mainly the left limbs and bilateral ankle clonus; ataxia, with slight upper‐limbs dysmetria and dysdiadochokinesia; and gait alteration because of spasticity with inward deviation of the left foot and loss of balance.
Serum and cerebrospinal fluid (CSF) work‐up was negative for acquired SP. Brain magnetic resonance imaging (MRI) showed the presence of diffuse cortical atrophy involving mainly both parietal lobes (Fig. 1A,B). Spinal MRI was normal. Neuropsychology showed a multidomain cognitive dysfunction involving mainly visuoperceptive and visuoconstructive tasks, attention, calculation, executive functions, memory, verbal fluency and comprehension, and ideomotor praxias. Montreal Cognitive Assessment scored 11/30.
FIG. 1.
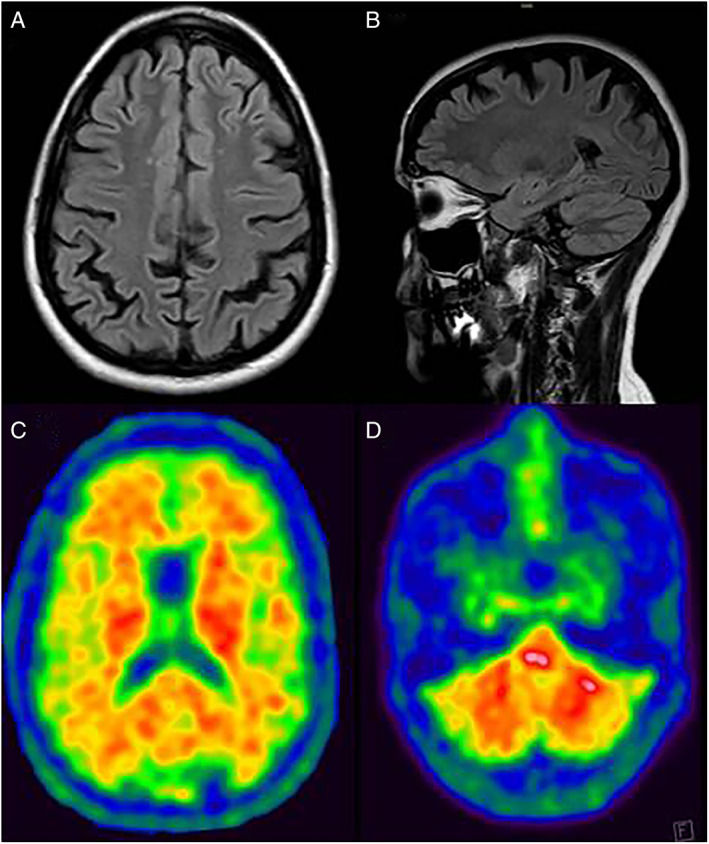
Brain MRI on FLAIR sequences in the axial (A) and sagittal (B) planes showing cortical atrophy involving mainly the parietal cortices.18F‐flutemetamol PET scan showing abnormal amyloid deposition in the cerebral cortex and basal ganglia (C), and the cerebellum (D). MRI, magnetic resonance imaging, FLAIR, fluid attenuated inversion recovery; PET, positron emission tomography.
Whole exome sequencing disclosed a novel heterozygous variant, c.1261A>G (p.Thr421Ala), in the PSEN1. Segregation analysis by Sanger sequencing confirmed this variant in the patient, but not in her parents, in whom the possibility of false parenthood was also ruled out. 18F‐flutemetamol brain positron emission tomography (PET) showed increased cortical uptake in the posterior cingulum, precuneus and temporal, frontal and parietal cortices. Increased uptake was also present in the basal ganglia and cerebellar gray matter (Fig. 1C,D). CSF study showed low levels of β‐amyloid 1–42 (239 pg/mL) and high levels of tau (620 pg/mL) and phosphorylated tau protein (123 pg/mL).
SP happens in ~13.7% of patients with PSEN1 mutations. In 7.5% of them, pure SP at onset is followed by cognitive decline later in the disease. 3 In our case, SP was associated to the presence of ataxic signs and symptoms. This could be related to the extensive cerebellar involvement because of amyloid deposition as the brain PET scan disclosed. Amyloid PET studies in PSEN1 mutation carriers have shown pathologic uptake in nearly every cortical region, basal ganglia, and cerebellum, even a decade or more before the estimated age of onset. 4 , 5
PSEN1 mutations responsible for HSP are mainly located in or beyond exon 8. 3 In our patient, the variant is at exon 12 and has not previously been described. In silico studies suggest this variant has an impact on functional and structural integrity of the protein because it is located in a transmembrane domain. Underlying pathogenic mechanisms by which PSEN1 mutations can induce HSP are not well known yet. HSP related genes and mutated PSEN1 could share some pathological pathways involved in motor neuron development and survival. 2 Additionally, the catalytic action of ϒ‐secretase could be important not only for APP processing, but for other substrates of particular relevance for motor neurons. 3
“Cotton wool” plaques (CWPs) are a characteristic neuropathological finding in AD‐linked PSEN‐1 mutations. In patients with HSP, CWPs and tau deposition are found mainly in the frontal motor cortices. 6 , 7 In these cases, the cerebellum does not contain CWPs, but shows a particularly extensive amyloid angiopathy. Although we do not have pathological confirmation, abnormal cortical amyloid deposition in the PET scan together with altered β‐amyloid and tau protein CSF levels also support the pathogenicity of the variant we have found.
Recent studies suggest that de novo mutations in PSEN1, as in our case, are not unusual in sporadic early‐onset AD. 8 For this reason, we suggest to search for PSEN1 mutations not only in patients with familial SP and early AD, but also in patients with apparently sporadic SP, mainly if secondary causes have reasonably been ruled out and they have rapid clinical progression or develop early cognitive impairment
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
E.M.: 1A, 1B, 1C, 3A.
M.J.: 1C, 3A, 3B.
J.G.: 1B, 3B.
Y.C.: 1A, 1B, 3B.
A.P.: 1C, 3B.
M.A.: 1C, 3B.
N.F.: 3B.
L.R.: 1C, 3B.
R.S.: 1A, 1B, 1C, 3B.
Disclosures
Ethical Compliance Statement: The approval of an institutional review board was not required for this work. Informed patient consent was obtained. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: No specific funding was received for this work and the authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Acknowledgments
We thank Berta Muñoz‐Martín for her technical support to the edition of the video.
Relevant disclosures and conflict of interest are listed at the end of this article.
[Correction added on 9 November, 2022, after first online publication: CC‐BY‐NC‐ND copyright line updated.]
References
- 1. Hedera P. Hereditary spastic paraplegia overview. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle WA: University of Washington, Seattle; 1993. –2022. [PubMed] [Google Scholar]
- 2. Alzheimer's Disease Collaborative Group . The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet 1995;11:219–222. [DOI] [PubMed] [Google Scholar]
- 3. Chelban V, Breza M, Szaruga M, et al. Spastic paraplegia preceding PSEN1‐related familial Alzheimer's disease. Alzheimer's Dement 2021;13:e12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sala‐Llonch R, Falgàs N, Bosch B, et al. Regional patterns of 18F‐florbetaben uptake in presenilin 1 mutation carriers. Neurobiol Aging 2019;81:1–8. [DOI] [PubMed] [Google Scholar]
- 5. Ghisays V, Lopera F, Goradia DD, et al. PET evidence of preclinical cerebellar amyloid plaque deposition in autosomal dominant Alzheimer's disease‐causing Presenilin‐1 E280A mutation carriers. Neuroimage Clin 2021;31:102749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verkkoniemi A, Kalimo H, Paetau A, Somer M, Iwatsubo T, Hardy J, Haltia M. Variant Alzheimer disease with spastic paraparesis: neuropathological phenotype. J Neuropathol Exp Neurol 2001;60:483–492. [DOI] [PubMed] [Google Scholar]
- 7. Karlstrom H, Brooks WS, Kwok JB, et al. Variable phenotype of Alzheimer's disease with spastic paraparesis. J Neurochem 2008;104:573–583. [DOI] [PubMed] [Google Scholar]
- 8. Lanoiselée HM, Nicolas G, Wallon D, et al. APP, PSEN1, and PSEN2 mutations in early‐onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 2017;14:e1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]