

Abstract
The 3C-like protease (3CLpro) is essential for the replication and transcription of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), making it a promising target for the treatment of corona virus disease 2019 (COVID-19). In this study, a series of 2,3,5-substituted [1,2,4]-thiadiazole analogs were discovered to be able to inhibit 3CLpro as non-peptidomimetic covalent binders at submicromolar levels, with IC50 values ranging from 0.118 to 0.582 μM. Interestingly, these compounds were also shown to inhibit PLpro with the same level of IC50 values, but had negligible effect on proteases such as chymotrypsin, cathepsin B, and cathepsin L. Subsequently, the antiviral abilities of these compounds were evaluated in cell-based assays, and compound 6g showed potent antiviral activity with an EC50 value of 7.249 μM. It was proposed that these compounds covalently bind to the catalytic cysteine 145 via a ring-opening metathesis reaction mechanism. To understand this covalent-binding reaction, we chose compound 6a, one of the identified hit compounds, as a representative to investigate the reaction mechanism in detail by combing several computational predictions and experimental validation. The process of ring-opening metathesis was theoretically studied using quantum chemistry calculations according to the transition state theory. Our study revealed that the 2,3,5-substituted [1,2,4]-thiadiazole group could covalently modify the catalytic cysteine in the binding pocket of 3CLpro as a potential warhead. Moreover, 6a was a known GPCR modulator, and our study is also a successful computational method-based drug-repurposing study.
Keywords: SARS-CoV-2; 3CLpro; Covalent inhibitors; 2,3,5-substituted [1,2,4]-thiadiazole; Warhead; Ring-opening metathesis
Graphical abstract
2,3,5-substituted [1,2,4]-thiadiazoles as potent inhibitors targeting 3C-Like protease were proposed to covalently bind with the catalytic cysteine via a ring-opening metathesis reaction mechanism.
1. Introduction
Currently, the COVID-19 virus continues to ravage the earth. Although the coverage of COVID-19's vaccination has greatly improved, new virus variants are breaking through the gradually forming immune barrier [1]. Therefore, drug discovery and development against SARS-CoV-2 seems to be particularly urgent and important. The 3C-like protease (3CLpro), an essential enzyme of SARS-CoV-2 conserved in the variants, plays a key role in viral replication and transcription. The active form of 3CLpro exists as a dimer, and there are 11 cleavage sites on viral polyproteins 1a and 1ab [2]. The sequence of SARS-CoV-2 3CLpro is almost identical to that of SARS-CoV 3CLpro (96% identity) [3]. It is believed that 3CLpro is a promising drug target for the treatment of COVID-19.
The active site of 3CLpro contains a catalytic dyad consisting of Cys145 and His41. Covalent inhibitors targeting Cys145 have been used to inhibit the activity of 3CLpro. Despite potential toxicity and off-target effects, covalent inhibitors may have a longer binding time and stronger binding ability to proteins [4]. The warhead is the key to covalent bond formation for covalent inhibitors. A variety of covalent peptidomimetic inhibitors with conventional warheads (acrylamide groups, α, β-unsaturated carbonyl groups, aldehyde groups, ketone groups, and cyano groups) have been developed [2,[5], [6], [7], [8], [9]]. Some non-peptidomimetic inhibitors with diverse warheads have also been reported, including ebselen, carmofur, disulfiram and ester [2,[10], [11], [12]]. Among these inhibitors, ebselen, carmofur, and disulfiram have been shown to be nonspecific 3CLpro inhibitors. A recent study has shown that the natural product myricetin covalently binds to Cys145 by the warhead pyrogallol [13]. The warheads β-lactam and thioester have also been used to develop inhibitors of 3CLpro [14,15]. The discovery of novel warheads is critical to the development of drugs targeting 3CLpro.
Drug repositioning is an effective strategy to find new uses for approved or investigational therapeutics and to accelerate the drug discovery and development process [16]. Drug repositioning can take advantage of high efficiency and low cost to rapidly respond to sudden and rare diseases such as COVID-19 [17]. Molecular docking is an important computational approach for drug repurposing. Some potential drugs can be rapidly found by molecular docking from approved or investigational ligand databases.
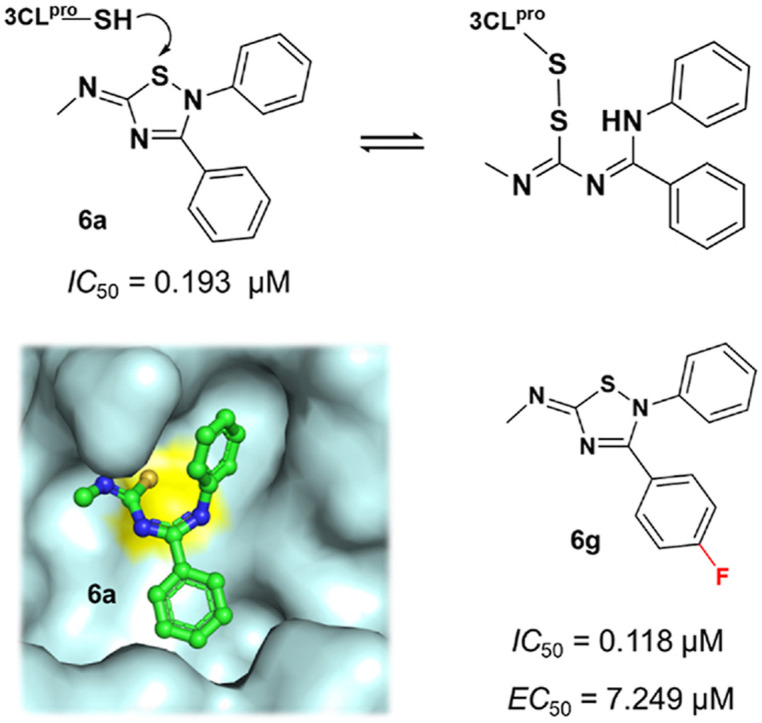
Here, we presented a non-peptidomimetic inhibitor (6a, Fig. 1 b) that can covalently bind to 3CLpro via a ring-opening metathesis reaction. 6a has been previously described as a modulator of several G protein-coupled receptors (GPCRs) [[18], [19], [20]]. Some of the 2,3,5-substituted [1,2,4]-thiadiazole analogs of 6a were designed for synthesis to enhance the inhibitory effects and antiviral activities against SARS-CoV-2 3CLpro. The most interesting part of this work is that we have identified a new type of cyclic warhead for the development of covalent inhibitors against cysteine-containing drug targets. The mechanism of covalent bond formation between the inhibitor and 3CLpro has been thoroughly investigated by a combination of computational methods and experimental techniques, shedding light on the further development of covalent 3CLpro inhibitors.
Fig. 1.

The discovery of the lead compound 6a. (a) Schematic representation of the workflow to identify the 3CLpro inhibitor. (b) 2D structure of compound 6a. (c) Representative inhibition profile for 6a against 3CLpro. Error bars represent the mean ± SD of three replicates. (d) SPR sensorgrams of compound 6a bound to 3CLpro at concentrations of 0.39–12.5 μM. Experiments were performed using Biacore 8K (GE Healthcare) and data were analyzed using Biacore 8K analysis software. (e) The predicted surface of the electrostatic potential of 6a and 3CLpro. The surface model represents 3CLpro. 6a (green) bound at the active site is shown as a stick-ball model. The residue Cys145 is depicted as a sphere.
2. Methods and materials
2.1. Discovery of hit molecules by virtual screening
The database of approximately 170,000 compounds was downloaded from the Core Technology Facility of Center for Excellence in Molecular Cell Science of Chinese Academy of Sciences. We firstly preprocessed the database using the LigPrep module in Maestro (Schrödinger 2020-1) including the prediction of protonation states and the generation of low-energetical 3D conformations. All compounds in the database were converted to their probable ionization states at pH 7.0 ± 2.0. Default settings were used for all other parameters. In this study, the compound containing more than one amide group was defined as a peptidomimetic inhibitor and filtered using the Ligand Filtering Panel in Maestro. We then created a database of non-peptidomimetic compounds.
The structure of SARS-CoV-2 3CLpro was retrieved from the Protein Data Bank (PDB code: 6LZE [5]). The complexed ligand in this structure was deleted in Maestro. Subsequently, the apo protein structure was structurally refined using the Protein Preparation Wizard [21] in Maestro to assign correct protonation states and formal charges, add missing residues, and optimize the three-dimensional conformations. The grid was centered on the centroid of residue Cys145. The binding region was defined by a 20 Å × 20 Å × 20 Å box for grid generation. Default settings were used for all other parameters. The database of compounds was first filtered using the Run QikProp option and the Prefilter by Lipinski's Rule [22,23] option in Maestro. The remaining compounds were docked in the generated grid by Glide (v8.7) [24] in Maestro. In the simulation of docking, each ligand was allowed to keep 5 poses to be written out and the rest conditions set as default. The top 1000 ranked compounds were kept for further visual inspection. Finally, we selected 65 molecules for further evaluation of their effects on 3CLpro.
2.2. Prediction of the binding mode of compounds to 3CLpro
To determine the potential binding site of 6a on the surface of 3CLpro, a global non-covalent docking simulation was performed on the entire surface of 3CLpro by Glide (v8.7) [24] in Maestro. We generated 12 grids to fully cover the entire protein structure via Receptor Grid generation in Maestro. The center of each grid was set to the centroid of a cysteine in 3CLpro (12 cysteines in total). To determine the best possible covalent binding site, 6a was docked to each grid using Glide (v8.7) in Maestro. Default settings were used for other remaining parameters. We analyzed the binding modes and docking score in detail and finally determined that Cys145 was the most likely reaction residue. We also predicted the binding mode of the ring-opened structure of 6a, named 5a, using Glide (v8.7) in Maestro.
To predict the covalent binding mode of 6a, a covalent docking simulation was performed using the Covalent Docking Panel [25] in Maestro. Since there is no ring opening reaction type, the N–S bond of 6a was deleted to match existing reaction types. We chose the Cys-145 as the reactive residue and the centroid of Cys-145 as the box center. The reaction type was disulfide formation. Default settings were used for all other parameters.
To predict the binding modes of derivatives 6b-6k, we performed molecular docking for 6b-6k based on the clustered conformation of 6a after molecular dynamics simulation. Subsequently, structural refinement of the molecules was performed to optimize the modeled complex structures by Prime MM-GBSA in Maestro and to calculate the binding free energy (ΔG binding).
2.3. DFT calculations
The enzyme-inhibitor complex was constructed by docking inhibitor 6a to the substrate-binding pocket of 3CLpro. Then, the complex model was constructed for DFT calculations with reduced cluster models of the inhibitor-enzyme system by truncating the substrate-binding pocket of the enzyme-inhibitor complex. The model included all key residues such as Thr25, His41, Cys44, Met49, Tyr54, Cys145, Met165, and Gln189 around the inhibitor. To increase efficiency of calculations, only the side chains of these residues were retained. Thus, threonine was modeled as isopropanol, histidine as 4-methyl-1H-imidazole, tyrosine as 4-methylphenol, cysteine as methanethiol, methionine as methylthioethane, and glutamine as 1-carbamoylethane. To preserve the strain of the surrounding protein environment, locations of atoms that lead to the backbones of key amino acid residues were constrained from the crystal structure. The net charge of the 6a-3CLpro complex was 0 and existed in the singlet spin state. All calculations were conducted using the Gaussian 09 software package [26]. The geometry optimization was performed utilizing the hybrid functional B3LYP [27,28] and a triple zeta 6-311G (d, p) basis set [29]. The transition states were validated to possess only one imaginary frequency corresponding to the reaction coordinate in the computed Hessians. The intrinsic reaction coordinate (IRC) approach was applied to ensure that the minimum states were associated with the corresponding transition states [30]. Dispersion effects were also investigated by using the D3 version of Grimme's dispersion with Becke-Johnson damping (GD3BJ) [31]. Single point calculations using a 6-311G (d, p) basis set containing diffuse functions were performed to improve the final energies of the optimized structures. Solvent effects were included by calculating energies in diethylether (dielectric constant 4.3) using a variation of IEFPCM - the SMD solvation model, which is the recommended choice for calculating ΔG of solvation [32]. The final Gibbs free energies were calculated taking into account zero-point vibration corrections, solvent effects, and entropy effects.
The frontier molecular orbital analysis and molecular electrostatic potential analysis were performed with the optimized structures. The results are visualized using GaussView 5.0. (The coordinates of the optimized structures can be found in Supplementary Information, section Ⅳ).
2.4. Molecular dynamics simulation
To analyze the binding modes of 3CLpro with the compounds, we performed molecular dynamics simulations. The enzyme-inhibitor complexes were built by docking ligands onto the substrate-binding pocket of 3CLpro. The ligands were optimized using the Gaussian 09 software package [26]. The partial charges of the ligands were calculated using Multiwfn [33] and the topology files of the ligands were prepared using ACPYPE [34]. The GAFF force field was used in the ligand topology files. The Amber99sb force field implemented in Gromacs 2020.3 was used throughout the simulation [[35], [36], [37]]. The initial complex structure was solvated in a cubic box and TIP3P-water was chosen as the solvent model. The Cl− and Na+ ions were added by replacing the waters. The system was relaxed with 5000 steps of steep descent minimization followed by 50,000 steps of conjugate gradient minimization. The NVT ensemble (100 ps, 2 fs step size, 300 K) was conducted to stabilize the temperature of the system and NPT ensemble (100 ps, 2 fs step size, 300 K, 1 bar) was used to stabilize the pressure of the system. Finally, 200 ns MD simulation trajectories (2-fs step size) were obtained. The Particle Mesh Ewald (PME) method implemented in Gromacs2020.3 was used to address the long-range electrostatic interactions in the production phase. The average conformations of 3CLpro with 6a or 5a during 199–200 ns were clustered with a cut-off value of 0.15.
2.5. Protein expression and purification of SARS-CoV-2 3CLpro
SARS-CoV-2 3CLpro was expressed and purified as previously described [13,38,39]. Briefly, the full-length cDNA of SARS-CoV-2 3CLpro (GenBank: MN908947.3) with an N-terminal 6 × His-SUMO2 fusion tag was cloned into the pET15b vector. The plasmid was then transformed into BL21 (DE3) cells for protein expression. The expressed protein was purified by a Ni-NTA column (GE Healthcare) and cleaved by the SUMO-specific peptidase 2 (SENP2) to remove the 6 × His-SUMO2 fusion tag. The resulting protein sample was further purified by Q-Sepharose (GE Healthcare) followed by size-exclusion chromatography (Superdex200, GE Healthcare). The eluted protein samples were kept in solution (10 mM Tris, pH 7.5) for enzymatic inhibition assay and surface plasmon resonance assay.
2.6. Enzymatic inhibition assay of SARS-CoV-2 3CLpro
The enzymatic inhibition assay against SARS-CoV-2 3CLpro was performed as previously described [13,38,39]. A fluorescence resonance energy transfer (FRET) protease assay was applied to measure the inhibitory effect of compounds against SARS-CoV-2 3CLpro. The fluorogenic substrate MCA-AVLQSGFR-Lys (Dnp)-Lys-NH2 was synthesized by GenScript (Nanjing, China). The FRET-based protease assay was performed as follows. The recombinant SARS-CoV-2 3CLpro (50 nM final concentration) was mixed with serial dilutions of each compound in 80 μL assay buffer (50 mM Tris–HCl, pH 7.3, 1 mM EDTA) and incubated for 10 min. The reaction was initiated by adding 40 μL of fluorogenic substrate at a final concentration of 10 μM. Thereafter, the fluorescence signal at 320 nm (excitation)/405 nm (emission) was immediately measured every 35 s for 3.5 min using a Bio-Tek Synergy-H1 plate reader. The initial rates of the reactions with compounds added at different concentrations were calculated in comparison to the reaction added with DMSO and used to generate inhibition profiles.
2.7. Surface plasmon resonance (SPR) analysis
All SPR experiments were performed at 25 °C in HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3.0 mM EDTA, and 0.05% (v/v) Tween-20) containing 5% DMSO with Biacore 8K (GE Healthcare). 3CLpro was immobilized on the CM5 chip at a level of 8000–10000 response units (RU). 10 mM compound stock solution was diluted with 1x HBS-EP buffer in an eight-concentration series with a final DMSO concentration of 5%. Analyte was injected and run through the chip with an association time of 240 s and a dissociation time of 200 s and washed with 50% DMSO in each cycle. Solvent correction was performed every 48th cycle. Raw data were reduced, double-referenced, and solvent-corrected using Biacore 8K Evaluation Software. The K D value of each compound was calculated using a steady-state affinity model with a constant R max.
2.8. Liquid Chromatography−Mass spectrometry
30 μL purified protein 3CLpro (1 mg/mL) was incubated with 1 μL ligand solution (50 mM) for 30 min at 37 °C. The incubation liquid was transferred into a spin column. After centrifugation at 8000g for 5 min, the flow-through fluid was discarded. The column was washed with 400 μL of PBS buffer and then treated with 200 μL of 50% methanol to elute the compounds. Then, the elution liquid was transferred to another EP tube and dried under rapid vacuum to concentrate it. Finally, the compounds were re-dissolved in 20 μL 50% methanol to do Liquid Chromatography−Mass Spectrometry (LC-MS) analysis.
2.9. Enzymatic selectivity assays
Enzymatic selectivity assays against SARS-CoV-2 PLpro (the papain-like protease) were performed as previously described [40]. Enzymatic selectivity assays against host proteases (chymotrypsin, cathepsin B and cathepsin L) were carried out as previously described [9,41].
The PLpro assay was performed as follows: 50 nM PLpro was mixed with serial dilutions of the compounds under the conditions of 50 mM HEPES, pH 7.5, 0.1 mg/mL BSA. After incubation for 30 min, the reactions were initiated by the addition of 10 μM fluorogenic peptide. The reaction was initiated by adding 40 μL Fluorogenic substrate (RLRGG-AMC) to reach a final concentration of 20 μM. Thereafter, the fluorescence signal at 360 nm (excitation)/460 nm (emission) was immediately measured every 1 min for 5 min using a Bio-Tek Synergy-H1 plate reader.
The chymotrypsin assay was performed as follows: Chymotrypsin (Sigma, catalog # SLCH1926) (5 nM final concentration) was mixed with serial dilutions of compounds in 80 μL assay buffer (phosphate buffer saline, pH 7.4) and incubated for 10 min. The reaction was started by adding 40 μL Fluorogenic substrate N-succinyl-AAPF-AMC (10 μM final concentration). After that, the fluorescence signal at 380 nm (excitation)/460 nm (emission) was immediately measured every 1 min for 10 min using a Bio-Tek Synergy-H1 plate reader.
The cathepsin B assay was performed in the reaction buffer containing 20 mM sodium acetate (pH 5.5), 1 mM EDTA, and 2 mM DTT. 0.25 units of Cathepsin B (Sigma, catalog # SLCJ4379) and the testing compound were added to each well and incubated for 30 min at ambient temperature. The enzymatic reaction was initiated by adding 40 μL FRET substrate Z-RR-AMC (1 μM final concentration). After that, the fluorescence signal at 340 nm (excitation)/440 nm (emission) was measured immediately every 1 min for 10 min with a Bio-Tek Synergy-H1 plate reader.
The cathepsin L (2 nM at a final concentration) was mixed with serial dilutions of each compound in 50 μL buffer (100 mM potassium phosphate, pH 6.8, 5 mM EDTA-Na, 0.001% Triton X-100, and 2 mM DDT). The reaction was initiated by adding 4 μL fluorogenic substrate (Z-VVR-AMC for CTSL, Z-RR-AMC for CTSB, AMC = 7-amino-4-methyl-coumarin, and Z = benzyloxycarbonyl) with a final concentration of 20 μM. After that, the fluorescence signal at 360 nm (excitation)/450 nm (emission) was immediately measured every 10 s for 10 min with a EnVision plate reader.
The rates of the reactions with compounds added at different concentrations compared to those added with DMSO were calculated and used to establish inhibition profiles.
2.10. Cell permeability
The cell permeability assay was performed by SIMM-SERVIER Joint Biopharmacy Laboratory. Caco-2 cells were cultured in high sugar DMEM medium at 37 °C and 2% CO2 with a relative humidity of 90%. Cells were subcultured every 7 days. The cells between the 40th to 60th generations were used to perform the assay. After 21 days of culture, the tightness of the cell monolayer formation should be greater than 400 Ω cm2. Test compounds were diluted three times with the transport buffer (HBSS). Then 20 μM 6a and a mixture of 6a and GF120918 were added to each corresponding position. The cells were cultured at 37 °C for 95 min. Samples were taken from the dosing side at 5 min and 95 min, respectively. The receiving chamber was sampled at 35 min and 95 min, respectively. Test and reference compounds were quantified by LC−MS/MS analysis based on the peak area ratio of analyte/IS.
The permeability coefficient (P app) is calculated from the following equation:
| (1) |
Where is the cumulative concentration of a compound in the receiver chamber as a function of time (μM/s), is the solution volume in the receiver chamber, is the area of the cell monolayer and is the donor concentration at time zero.
2.11. Cell lines and viruses
African green monkey kidney Vero E6 cells (ATCC-1586) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin antibiotics. Cells were kept at 37 °C in a 5% CO2 atmosphere. The delta strain of SARS-CoV-2 (IVCAS6.7585) was obtained from the National Virus Resource Center (Wuhan Institute of Virology, Chinese Academy of Sciences). It was propagated and titrated in Vero E6 cells.
2.12. Antiviral activities measurement
Antiviral activities measurement was performed as previously described [5]. In our study, pre-seeded Vero E6 cells to 48-well plates (50,000 cells/well) for 16–18 h, treated with maintenance medium (DMEM + 2% FBS) containing compounds at 100 μL/well for 1h. Then, the cells were inoculated with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.01 for 1 h, the supernatant was removed, PBS washed twice, and re-added fresh medium containing compounds at 200 μL/well. The cells were incubated at 37 °C for 24 h. The cell supernatant was collected; antiviral activities were evaluated by quantification of viral copy numbers in the cell supernatant via real-time fluorescence quantitative PCR (qRT-PCR). DMSO was used in the control. Primary screening of the antiviral effect of compounds 6a-6k was firstly performed at 50 μM, followed by compounds 6g, 6i and 6j with better antiviral effects were further tested. To detect the half maximal effective concentration (EC 50) of compounds 6g, 6i and 6j, cells were incubated with a medium containing gradient concentration of the three compounds at 100 μL/well for 1 h, and other operations were as described above. The inhibition rates of 6g, 6i and 6j were calculated based on the viral copy numbers, and EC 50 was calculated with GraphPad Prism software 8.0. The experiments were done in triplicates and all the infection experiments were performed at biosafety level 3 (BSL-3).
2.13. Cytotoxicity measurement
Cell viability was performed in a 96-well plate with triplicate for each concentration. All drugs were diluted 2 times with 9 gradients starting at an appropriate concentration in maintenance medium (DMEM containing 2% FBS). After 24 h incubation, the supernatant was removed, and 10 μL WST-8 (2-(2-methoxy-4-(phenyl)-3-(4-(phenyl) to 5 (2,4-sulpho benzene) −2 h-tetrazolium monosodium salt) in maintenance medium was added in medium. Plates were measured at 450 nm wavelength using a spectrophotometer (BioTek) after 0.5–1 h incubation, and cell viability was calculated.
3. Results and discussion
3.1. Identification of lead compound 6a
As shown in Fig. 1a, at the first stage of the virtual screening, those compounds containing more than one amide group were defined as peptidomimetic compounds. To create a database of non-peptidomimetic inhibitors, the peptidomimetic compounds were removed using Maestro. The non-peptidomimetic compounds were further filtered following Lipinski's Rule of Five [22,23]. Subsequently, we selected the crystal structure of SARS-CoV-2 3CLpro (PDB code: 6LZE [5]) to identify potential inhibitors. The remaining compounds were docked into the active site of 3CLpro. Finally, a visual inspection was performed to analyze the predicted binding modes between the top 1000 molecules and the protein. As a result, 65 compounds were chosen to purchase for experimental testing.
We first measured the inhibition rates of these compounds against SARS-CoV-2 3CLpro at 20 μM (Table S1). Then the top 5 compounds (6a, 48, 55, 56, and 57) were selected to further evaluate the inhibition rates at 10 μM, 1 μM, and 0.1 μM (Table S2). Among these compounds, 48, 55, 56, and 57 belonged to flavonoids (Fig. S1), such a group of compounds have been reported as the inhibitors of SARS-CoV-2 3CLpro [13,42] and hence were excluded from our subsequent studies. Eventually, 6a was selected for further study (Fig. 1b). Firstly, we determined the 50% inhibitory concentration (IC 50) value of 6a. The result indicated the excellent inhibitory effect of 6a, with an IC 50 value of 0.193 ± 0.008 μM (Fig. 1c). We also measured the representative dose-dependent response curves for 6a against 3CLpro by surface plasmon resonance (SPR) assay (Fig. 1d). SPR provided affinity values and indicated that 6a strongly interacted with 3CLpro with K D values of 0.798 ± 0.065 μM. On the other side, the SPR sensorgram presents slow association and dissociation rates of 6a against 3CLpro, indicating 6a could be a reversible covalent inhibitor of the protein. The docking result also showed that 6a could fit well into the catalytic site of 3CLpro (Fig. 1e). Moreover, the ADMET properties of 6a were also computationally assessed from the web server (https://drug.ai.tencent.com/console/cn/admet). Combining the high permeability tested by the Caco-2 permeability assay (Table S3) and the predicted lead-like properties (Table S4), 6a was demonstrated to be a potential antiviral drug lead.
3.2. Exploring the covalent-binding reaction mechanism of 6a
Previous studies have described 6a as a modulator of various GPCRs, such as opioid, adrenergic, muscarinic, dopaminergic and adenosine receptors [[18], [19], [20]]. Combined with our results, this proves the reliability and practicality of drug repositioning by molecular docking. It was suggested that the 2,3,5-substituted [1,2,4]-thiadiazole group of 6a could be nucleophilically attacked by the sulfur atom of cysteine in the protein. And this could lead to the opening of its 2,3,5-substituted [1,2,4]-thiadiazole ring [19,20]. Inspired by this finding, we investigated the reaction mechanism of 6a in the recognition of 3CLpro. As shown in Fig. 2 a, the cyclic warhead of 6a was located near the Cys145 of 3CLpro in the predicted binding mode of the complex. Intuitively, it was suggested that 6a reacts with 3CLpro by forming a disulfide bond with Cys145. Finally, the adduct (6a-3CLpro) was either recycled to 6a or converted to the free thiourea 5a in the presence of a reductant.
Fig. 2.

The proposed reaction mechanism and predicted binding modes. (a) Simplified schematic representation of the proposed reaction mechanism. (b) Predicted binding mode of 6a with the 3CLpro of SARS-CoV-2. (c) Predicted covalent binding mode of 6a to the 3CLpro. (d) Predicted binding mode of 5a with the 3CLpro. Cartoon model represents 3CLpro (palecyan). 6a (green) and 5a (blue) bound at the active sites are depicted as stick-ball models. The interaction residues are shown as sticks.
To further confirm the binding site of 6a mentioned above, we performed global docking of 6a to 3CLpro. Judging from the binding modes, the orthostatic pocket of 3CLpro was determined to be the most likely binding site (Fig. S2 and Table S5). Moreover, Cys145 at this site was obviously exposed to solvent. Therefore, the covalent binding between 6a and Cys145 should be likely. As illustrated in Fig. 2b, 6a fit well into the orthostatic pocket. The reaction group of 6a was closed to Cys145, and the C-Ph occupied the S2 pocket by π–π interaction with His41. Covalent docking was also performed to predict the binding mode of the adduct. The results suggested that 6a formed a disulfide bond with Cys145. The N-Ph of 6a rotated into the S1 pocket and formed a π–π interaction with His163 (Fig. 2c).
The inhibitory effect of 6a to 3CLpro might come from a combination of two types of binding interactions between 6a (ring-opened form 5a) and the protein, i.e., the non-bonded state of 6a and the covalent-bonded state of 5a with the protein. To prove that 6a inhibits 3CLpro by covalent interaction, a time-dependence test was conducted for 6a. The incubation time of 6a and proteins were 0, 5, 10 and 20 min. As shown in Fig. S3, the inhibition increases with time. The IC 50 values were 0.409, 0.302, 0.206 and 0.191 μM, respectively. The results indirectly prove the covalent interaction between 6a and 3CLpro.
Since the adduct form was not stable for characterization [19,43], we used Liquid Chromatography−Mass Spectrometry (LC-MS) to determine the molecular interaction mechanism of 6a. We incubated 6a with protein, washed the mixture and identified the incubation via LC-MS (Fig. 3 a). As a result, the m/z (mass-to-charge ratio) of the peaks with the highest relative abundance for Elute A and Elute B are 270.1067 and 270.1059, respectively (Fig. 3b and Fig. S4). The m/z of the 6a [M+1]+ and 5a [M+1]+ were measured as 268.0903 and 270.1059 via MS assay. The results showed that 5a was the main component of Elute A and confirmed that 6a reacted with 3CLpro, which was consistent with previous studies [19,43]. In addition, in elute B, we also found the peak at 268.0903 (related to 6a), although the signal is weak. Additionally, these findings also show that 6a has strong reactivity with 3CLpro. However, this does not explicitly prove that the reaction was promoted by the covalent bond with the protein, since the disulfide bond was easily broken. We also tested the inhibitory effect of 5a against 3CLpro. The results showed that 5a could also inhibit 3CLpro, with a much weaker binding affinity (IC 50 = 39.980 ± 6.443 μM) (Fig. 3c). As shown in Fig. 2d, the predicted binding mode of 5a is similar to that of 6a. C-Ph of 5a formed a π–π interaction with His41, and the thiourea group of 5a was located at the position of 6a′s reactive group. In addition, the MS result showed that the main component of elution B is 5a. Therefore, we hypothesized that 5a has a low affinity with 3CLpro through noncovalent interactions, while the strong inhibition of 6a may be due to the combination of noncovalent and covalent binding forms before and after the reaction.
Fig. 3.

Liquid Chromatography−Mass Spectrometry. (a) Experimental workflow of LC-MC to determine the binding motif of 6a. (b) 6a and 5a were incubated with 3CLpro from SARS-CoV-2 and analyzed by LC−MS. (c) Representative inhibition profile for 5a against 3CLpro.
Since the complex is difficult to identify experimentally, we simulated the reaction process via transition state search. In the reactant of the 6a-3CLpro complex (Fig. 4 a), the C-Ph of 6a interacts with the imidazole ring of His41 through π-π stacking. In the next step, the thiol group of Cys145 attacks the S1 atom of 6a and the proton (H1) is simultaneously transferred to the N1 atom to cleave the S1–N1 bond (Fig. 4a). In the transition state, the S2–S1 distance shrunk from 3.76 to 2.77 Å and the S1–N1 bond elongated from 1.75 to 1.90 Å. Distance changes confirmed the concerted nature of this process, whose energy barrier is calculated to be 28.9 kcal/mol (Fig. 4b). The S2–S1 distance was further shortened to 2.09 Å to form a disulfide bond in the product which was calculated to be 22.8 kcal/mol exergonic from the reactant. In addition, the S1–N1 bond is completely cleaved in this state. And the N-Ph rotated into the S1 pocket, which was consistent with the covalent docking result (Fig. 2, Fig. 4a). The π-π stacking described above was observed throughout the entire process. Therefore, all these results indicated that 2,3,5-substituted [1,2,4]-thiadiazole group was a potential ‘warhead’ targeting the catalytic cysteine Cys145.
Fig. 4.

A proposed 6a-3CLproreaction pathway. (a) Structures (in Å) of the reactant, transition state and product for the reaction of 6a with 3CLpro of SARS-CoV-2. (b) Potential energetics diagrams for the reaction of 6a with 3CLpro.
To investigate why cyclization renders 6a more susceptible to nucleophilic attack by the sulfur atom of cysteine, we depicted the Frontier Molecular Orbitals (FMOs) and Molecular Electrostatic Potentials (MEPs) of 6a and 5a via Gaussian [26]. The FMOs provide information about the reactivity and stability of molecular compounds. The highest occupied molecular orbital (HOMO) relates to electron-donating properties, while the lowest occupied molecular orbital (LUMO) reveals the electron acceptor properties [44]. In terms of the overall distribution of HOMO and LUMO, the orbital distribution area of 6a is similar to that of 5a. As shown in Fig. 5 a, the LUMO orbitals are widely located on the surface of 6a and 5a, while the HOMO orbitals on the C-Ph of 6a and 5a are minor or absent. The orbital energy gap ΔE of compound 6a is 4.226 eV, which is slightly larger (0.022 eV) than that of compound 5a (4.204 eV). Due to the similarity of the skeletons, the overall energy gap between 6a and 5a is slight. From the point of view of the local area, the HOMO lobe of 5a is significantly larger than that of 6a, indicating that the electron-donating ability of 5a is stronger. The LUMO lobe's distribution on the cyclized ‘warhead’ (2,3,5-substituted [1,2,4]-thiadiazole group) of 6a is more concentrated, thus this position may be more vulnerable to being attacked by nucleophiles such as cysteine's sulfur atom. In addition, MEPs display electron density distributions on the structures of 6a and 5a. The red area indicates the electron-rich area with electrophilic activity, while the blue area is the electron-deficient area with nucleophilic activity. As displayed in Fig. 5b, the density surface of 5a′s sulfur atom is red and thus the electrostatic potential around it is negative. In contrast, the electrostatic potential of the region for the sulfur atom on 6a is positive, providing availability for approaching negatively charged particles. The atomic charges of 6a and 5a were also calculated and exhibited in Fig. 5b. The atomic charge of S1 on 5a is −0.370, while the S1 on 6a′s cyclized ‘warhead’ is +0.286, which is coincided with the result of MEPs. Therefore, 2,3,5-substituted [1,2,4]-thiadiazole group can be applied as a potential electrophilic ‘warhead’ for the design of covalent inhibitors.
Fig. 5.

The Frontier molecular orbitals, molecular electrostatic potential, and charge distribution of compounds 6a and 5a. (a) HOMO and LUMO orbitals for compounds 6a and 5a. (b) Charge distribution and molecular electrostatic potentials of compounds 6a and 5a. The values of atomic charges are shown in red. The charge type is Mulliken charge.
To further investigate the binding modes between 3CLpro and ligands and to design derivatives, we performed 200 ns molecular dynamics simulations for 3CLpro in complex with either 6a or 5a. Based on the trajectories, we depicted the root-mean-square deviation (RMSD) plots of 3CLpro with 6a or 5a (Fig. 6 a-6b). RMSD can reveal the positional change between protein conformation and initial conformation in the simulation process. And RMSD is an important characterization to assess whether the protein conformation is stable along the trajectory. As shown in Fig. 6 a-6b, the protein conformations remained stable with approximate RMSD values within 2.0 Å of their initial coordinates until the end of the simulation. The RMSD plots of ligands 6a and 5a were also shown. The ligand 6a remained stable over during the 200 ns period, while the RMSD value of 5a mainly underwent bigger changes. The results explained why 5a showed weaker inhibitory activity against 3CLpro. We further clustered the average conformations of 3CLpro with 6a and 5a during 199–200 ns. As shown in Fig. 6c, the 2,3,5-substituted [1,2,4]-thiadiazole group of the compound 6a formed a hydrogen bond with G143 and faced the S1 pocket. Two benzene rings of 6a formed hydrophobic interactions with surrounding residues and occupied the S1′ and S2 sites, respectively. And the C-Ph of 6a also formed π–π interaction with His41. The binding of the compound of 5a was unstable (Fig. 6d). 5a formed hydrogen bonds with M165 and Q189. Similar to 6a, two benzene rings of 5a also occupied the S1’ and S2 sites. The binding free energy (ΔG binding) of 6a and 5a with 3CLpro was also calculated by MM-GBSA. 6a exhibited stronger binding affinity (−58.23 kJ/mol) than 5a (−47.57 kJ/mol).
Fig. 6.

Analysis of the binding modes by molecular dynamics simulation. (a) The root-mean-square deviation (RMSD) plots of 3CLpro with 6a or 5a (b). The proteins' curves were shown as black. The ligands' curves were shown as red. (c) The clustered structures of 3CLpro with 6a or 5a (d). Compounds 6a (green) and 5a (blue) bound to the active site were depicted as stick-ball models. The interaction residues are represented as sticks. The proteins were shown as cartoon or electrostatic potential surfaces. The MD simulations were run for 200 ns. The structures were clustered from 199 to 200 ns trajectories.
3.3. Structural optimization and synthesis
To further optimize the structure of lead compound 6a, we analyzed the binding mode between 6a and 3CLpro. The molecular dynamics simulations (Fig. 6c) showed that three aromatic groups of 6a, i.e., 2,3,5-substituted [1,2,4]-thiadiazole, C-Ph, and N-Ph were in S1, S1′ and S2 of the catalytic site, respectively. Based on the binding modes, we designed several analogs of 6a, i.e. 6b-6k. The smaller groups such as methyl, cyclopropyl, and 3,3-difluorocyclobutyl were introduced at R1 position of 6a (Scheme 1 ). At the same time, we also introduced many substituents like methoxy, ethoxy, and fluorine atoms on C-Ph and N-Ph. We also performed molecular docking for 6b-6k based on the binding mode of 6a and predicted the binding free energies (ΔG binding) by MM-GBSA. In the terms of binding modes, the poses of all derivatives except for 6j were similar to that of 6a. Due to the steric effect of the ethoxy group, the N-Ph of 6j could not occupy the S1’ site. 6b-6i and 6k formed hydrogen bonds with G143. 6g formed a hydrogen bond with E166. The predicted binding free energy results suggested that the binding affinity of 6b, 6c, 6d, and 6h was stronger than that of 6a.
Scheme 1.

Reagents and conditions: (a) ArNH2, Et3N, EtOAc, r.t, overnight (b) SOCl2, N2, 135 °C, 4h (c) NaSCN, acetone, −15 °C–0 °C (d) R1NH2, acetone, overnight (e) Br2, 0 °C–5 °C.
We also plotted the Molecular Electrostatic Potentials (MEPs) and Frontier Molecular Orbitals (FMOs) of 6b-6k to study the influence of the modified group on molecular reactivity (Fig. S5-S6). The atom S1 is attacked by nucleophiles, and atom N1 accepts protons. As shown in Fig. S5, the atomic charges of atom S1 for compounds 6c and 6e are larger than those of 6a. The atomic charges of atom N1 for compounds 6b-6k are all smaller than that of 6a, which means that atom N1 gets protons more easily. The introduction of halogen atoms (6e and 6g) makes the electrostatic potential on the molecular surface positive, making the compounds more susceptible to nucleophilic attack by the sulfur atom of cysteine. In addition, the energy of LUMO orbitals can reflect the electrophilic ability of molecules. The lower the LUMO energy, the easier the molecule is reduced by nucleophiles [45]. The LUMO energies (ELUOM) of 6b-6e, 6g, 6i and 6k are lower than that of 6a (Fig. S6), which means that electrophilic abilities of these compounds are stronger than that of 6a. While 6f, 6h and 6j have weaker electrophilic capacities due to the introduction of EDG (Electron Donating Group) groups on the aromatic ring. Although the reactivities of compounds 6f, 6h and 6j are reduced, the introduction of methoxy or ethoxy groups towards the hydrophobic site S2 may increase the affinity.
The general synthetic route of the target compound 6a-6k is depicted in Scheme 1 within 5 steps. First, intermediates 2a-k were synthesized by corresponding substituted benzoyl chloride 1a-k and substituted aniline. Subsequently, intermediates 2a-k were used as starting materials which were converted into compound 3a-k in the presence of SOCl2. Intermediates 3a-k reacted with NaSCN for the substitution of chlorine and followed by addition of the corresponding primary amines to form the thiourea compounds 5a-k in moderate yields. Finally, hydrobromide salts of desired 2,3,5-substituted [1,2,4]-thiadiazoles (6a-6k) were obtained by ring closure reaction with bromine. (General synthetic procedures are given in the Supplementary Information, Section Ⅲ).
3.4. The enzymatic activities of 2,3,5-substituted [1,2,4]-thiadiazoles
As in the case of 6a, a FRET-based protease assay was also used to measure the inhibitory activities of the other 15 derivatives of 6a against SARS-CoV-2 3CLpro (Table 1 , Fig. S7). The modified groups at R1 moiety included methyl-, cyclopropyl-, 1,1-difluorocyclobutanyl-, 4-methylphenylsulfonyl-, morpholinyl- and 4-methylpiperazinyl-. The inhibitory activities of these compounds revealed that a smaller group in the R1 position (6a, 6b) might be more appropriate. When the hydrogen of 4-Ph at R2 moiety was replaced by fluorine, the enzymatic inhibitory activity increased slightly (6d). The fluorine substitution, chlorine substitution, and methoxy substitution at R3 moiety improved inhibitory activity (6e-6g). The weaker inhibitory activity of 6j could be attributed to the ethoxy's steric effect of the ethoxy. Among these compounds, 6g exhibited the most potent inhibitory activity with an IC 50 value of 0.118 ± 0.011 μM.
Table 1.
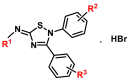 .
.| Compounds | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|
| 6a | -CH3 | H | H | 0.193 ± 0.008 |
| 6b | 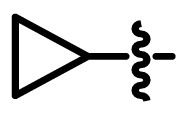 |
H | H | 0.143 ± 0.014 |
| 6c | 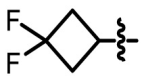 |
H | H | 0.323 ± 0.031 |
| 6d | -CH3 | 4-F | H | 0.124 ± 0.018 |
| 6e | -CH3 | H | 4-Cl | 0.136 ± 0.007 |
| 6f | -CH3 | H | 4-OMe | 0.165 ± 0.016 |
| 6g | -CH3 | H | 4-F | 0.118 ± 0.011 |
| 6h | -CH3 | 4-F | 4-OMe | 0.373 ± 0.033 |
| 6i | -CH3 | 3-F,4-F,5-F | 4-OMe | 0.582 ± 0.059 |
| 6j | -CH3 | 4-F | 4-OEt | 0.462 ± 0.037 |
| 6k | -CH3 | 4-F | 3-OMe, 4-OMe | 0.252 ± 0.017 |
| Nirmatrelvir | – | – | – | 0.078 ± 0.008 |
An enzymatic assay was also performed with the addition of 4 mM DTT. As shown in Table S7, compounds 6a and 6g lost their inhibitory activity. The results also confirm our hypothesis that compounds form a disulfide bond with 3CLpro. Disulfide bonds can be easily broken by DTT. Therefore, the compounds cannot inhibit 3CLpro after the addition of 4 mM DTT. In addition, we also performed an enzyme inhibition assay against SARS-CoV-2 PLpro (the papain-like protease), chymotrypsin, human cathepsin B, and human cathepsin L to test target selectivity. 6a and 6g were chosen to assess potential inhibition. We found that these compounds could also inhibit PLpro. The IC 50 values of 6a and 6g against PLpro are 0.655 μM and 0.448 μM, respectively. As shown in Table 2 and Table S8, both compounds showed weak inhibition at 100 μM (chymotrypsin), 50 μM (cathepsin B) or 100 μM (cathepsin L). The result suggests that 6a and 6g have excellent target selectivity towards coronavirus proteases. Here we find that PLpro is also the target of these compounds through an enzyme activity assay. This is also an example of drug repurposing. These molecules can also be cysteine sensitive as covalent inhibitors and may act on other cysteine proteases. We can convert the compound into a selective inhibitor based on its binding modes, which is the focus of our future research.
Table 2.
Inhibition IC50 (uM) of 6a and 6g against SARS-CoV-2 PLpro, chymotrypsin, human cathepsin B, and human cathepsin L.
| Compounds | SARS-CoV-2 PLpro | Chymotrypsin | Cathepsin B | Cathepsin L |
|---|---|---|---|---|
| 6a | 0.655 ± 0.063 | >100 | >50 | >100 |
| 6g | 0.448 ± 0.033 | >100 | >50 | >100 |
3.5. The antiviral activities at the cellular level
Furthermore, we measured the inhibitory activities of 6a-6k and 5a at 50 μM against SARS-CoV-2 in Vero E6 cells (Table S8). The antiviral efficacy was assessed by quantifying the number of viral copies in the cell supernatant using qRT-PCR. The dose-dependent inhibition of the three most potential compounds (6g, 6i and 6j) for SARS-CoV-2 replication was also tested. And the half-maximal effective concentrations (EC 50) were 7.249 μM,9.338 μM and 14.830 μM (Fig. 7 ). The cytotoxicity of the three compounds in Vero cell was determined by the CCK8 assay, and the half-cytotoxic concentrations (CC 50) were all >100 μM. Taken together, these data indicate that 2,3,5-substituted [1,2,4]-thiadiazoles could be potent in inhibiting SARS-CoV-2 activity.
Fig. 7.

In vitro Inhibitory activity profiles of 6g, 6i, and 6j against SARS-CoV-2. (a) Quantitative RT-PCR assays were performed to measure viral copy number in the cellular supernatant. The y-axis indicates the percentage inhibition of the virus relative to samples treated with DMSO (vehicle). (b) Cell viability test. The y-axis represents percent of cell viability relative to the sample treated with DMSO (vehicle). The y-axis indicates the percent inhibition of the virus relative to the DMSO treated sample (vehicle). Means ± SDs of three biological replicates are presented.
4. Conclusion
In this work, we found a SARS-CoV-2 3CLpro inhibitor 6a and tested the inhibitory effects against SARS-CoV-2 3CLpro and antiviral activities at cellular level of 2,3,5-substituted [1,2,4]-thiadiazole analogs of 6a. The IC 50 values of these compounds ranged from 0.118 to 0.582 μM. And the compound 6g exhibited the potent antiviral activity with an EC 50 value of 7.249 μM. The mass spectrometry assay demonstrated the occurrence of reaction between 3CLpro and 6a. The reaction process was simulated by non-covalent docking, covalent docking, transition state searchs, and molecular dynamics simulations. The FMOs and MEPs of 6a indicated that the electrophilic ability of the 2,3,5-substituted [1,2,4]-thiadiazole group could be a potential covalent warhead. Replacing and modifying of covalent warheads to improve the activity of inhibitors will benefit covalent inhibitor design.
Author contributions
PXR, SWL, QYH, and FB performed computational simulations and analysis. CYY, HL, and HL synthesized the compounds and performed NMR and MS experiments. TQN, HXS, LL and YCX prepared the protein, enzyme inhibition assay, and enzymatic selectivity tests. XLZ, ZXY, LL performed the SPR assay. RXZ and LKZ performed antiviral activities measurement in cells. WHD and JL provided suggestions for the draft. All the authors were involved in writing the manuscripts. The manuscript was written with contributions of all authors. All authors have approved to the final version of the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We acknowledge the funding support by the Science and Technology Commission of Shanghai Municipality grants (No. 20430780300 and 20QA1406400), Lingang Laboratory, Grant No. LG202102-01-03, LG202103-04-03, and LG-QS-202205-02, National Key R&D Program of China (No. 2021YFC0864900 and 2022YFC3400501), start-up package from ShanghaiTech University, and Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine at ShanghaiTech University. We are grateful for the support from HPC Platform and SIAIS Analytical Chemistry Platform of ShanghaiTech University, Core Technology Facility of Center for Excellence in Molecular Cell Science of Chinese Academy of Sciences, and SIMM-SERVIER Joint Biopharmacy Laboratory.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2023.115129.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
Data availability
Data will be made available on request.
References
- 1.Krause P.R., Fleming T.R., Longini I.M., Peto R., Briand S., Heymann D.L., Beral V., Snape M.D., Rees H., Ropero A.-M., Balicer R.D., Cramer J.P., Muñoz-Fontela C., Gruber M., Gaspar R., Singh J.A., Subbarao K., Van Kerkhove M.D., Swaminathan S., Ryan M.J., Henao-Restrepo A.-M. SARS-CoV-2 variants and vaccines. N. Engl. J. Med. 2021;385:179–186. doi: 10.1056/NEJMsr2105280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutanto F., Konstantinidou M., Dömling A. Covalent inhibitors: a rational approach to drug discovery. RSC med. chem. 2020;11:876–884. doi: 10.1039/d0md00154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.-K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B., Shen C., Mao X., Liu X., Sun W., Yang W., Ni X., Wang K., Xu L., Duan Z.L., Zou Q.C., Zhang H.L., Qu W., Long Y.H., Li M.H., Yang R.C., Liu X., You J., Zhou Y., Yao R., Li W.P., Liu J.M., Chen P., Liu Y., Lin G.F., Yang X., Zou J., Li L., Hu Y., Lu G.W., Li W.M., Wei Y.Q., Zheng Y.T., Lei J., Yang S. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L., Yang Q., Zhu Y. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 9.Ma C., Xia Z., Sacco M.D., Hu Y., Townsend J.A., Meng X., Choza J., Tan H., Jang J., Gongora M.V., Zhang X., Zhang F., Xiang Y., Marty M.T., Chen Y., Wang J. Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 2021;143:20697–20709. doi: 10.1021/jacs.1c08060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amporndanai K., Meng X., Shang W., Jin Z., Rogers M., Zhao Y., Rao Z., Liu Z.-J., Yang H., Zhang L., O'Neill P.M., Samar Hasnain S. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021;12:1–7. doi: 10.1038/s41467-021-23313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X., Duan Y., Yu J., Yang X., Yang X., Yang K., Liu X., Guddat L.W., Xiao G., Zhang L., Yang H., Rao Z. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A.K., Raghavaiah J., Shahabi D., Yadav M., Anson B.J., Lendy E.K., Hattori S.-i., Higashi-Kuwata N., Mitsuya H., Mesecar A.D. Indole chloropyridinyl ester-derived SARS-CoV-2 3CLpro inhibitors: enzyme inhibition, antiviral efficacy, structure–activity relationship, and X-ray structural studies. J. Med. Chem. 2021;64:14702–14714. doi: 10.1021/acs.jmedchem.1c01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su H., Yao S., Zhao W., Zhang Y., Liu J., Shao Q., Wang Q., Li M., Xie H., Shang W., Ke C., Feng L., Jiang X., Shen J., Xiao G., Jiang H., Zhang L., Ye Y., Xu Y. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021;12:3623. doi: 10.1038/s41467-021-23751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malla T.R., Brewitz L., Muntean D.-G., Aslam H., Owen C.D., Salah E., Tumber A., Lukacik P., Strain-Damerell C., Mikolajek H., Walsh M.A., Schofield C.J. Penicillin derivatives inhibit the SARS-CoV-2 main protease by reaction with its nucleophilic cysteine. J. Med. Chem. 2022;65:7682–7696. doi: 10.1021/acs.jmedchem.1c02214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pillaiyar T., Flury P., Krüger N., Su H., Schäkel L., Barbosa Da Silva E., Eppler O., Kronenberger T., Nie T., Luedtke S., Rocha C., Sylvester K., Petry M.R.I., McKerrow J.H., Poso A., Pöhlmann S., Gütschow M., O'Donoghue A.J., Xu Y., Müller C.E., Laufer S.A. Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: enzyme inhibition, structure–activity relationships, antiviral activity, and X-ray structure determination. J. Med. Chem. 2022;65:9376–9395. doi: 10.1021/acs.jmedchem.2c00636. [DOI] [PubMed] [Google Scholar]
- 16.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., Norris A., Sanseau P., Cavalla D., Pirmohamed M. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 17.Singh T.U., Parida S., Lingaraju M.C., Kesavan M., Kumar D., Singh R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020;72:1479–1508. doi: 10.1007/s43440-020-00155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fawzi A.B., Macdonald D., Benbow L.L., Smith-Torhan A., Zhang H., Weig B.C., Ho G., Tulshian D., Linder M.E., Graziano M.P. SCH-202676: an allosteric modulator of both agonist and antagonist binding to G protein-coupled receptors. Mol. Pharmacol. 2001;59:30–37. doi: 10.1124/mol.59.1.30. [DOI] [PubMed] [Google Scholar]
- 19.Göblyös A., de Vries H., Brussee J., Ijzerman A.P. Synthesis and biological evaluation of a new series of 2, 3, 5-substituted [1, 2, 4]-thiadiazoles as modulators of adenosine A1 receptors and their molecular mechanism of action. J. Med. Chem. 2005;48:1145–1151. doi: 10.1021/jm049337s. [DOI] [PubMed] [Google Scholar]
- 20.Lewandowicz A.M., Vepsäläinen J., Laitinen J.T. The ‘allosteric modulator’SCH‐202676 disrupts G protein‐coupled receptor function via sulphydryl‐sensitive mechanisms. Br. J. Pharmacol. 2006;147:422–429. doi: 10.1038/sj.bjp.0706624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 22.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 23.Lipinski C.A. Lead-and drug-like compounds: the rule-of-five revolution. Drug discovery today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 25.Zhu K., Borrelli K.W., Greenwood J.R., Day T., Abel R., Farid R.S., Harder E. modeling, Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014;54:1932–1940. doi: 10.1021/ci500118s. [DOI] [PubMed] [Google Scholar]
- 26.Frisch M.J., Trucks G.W., B Schlegel H., E Scuseria G., Robb M., Cheeseman J., Scalmani G., Barone V., Mennucci B., Petersson G.A.H., Nakatsuji H., Caricato M., Li X., P Hratchian H., F Izmaylov A., Bloino J., Zheng G., L Sonnenberg J., Hada M., Fox D. Gaussian, Inc.; Wallingford CT: 2009. Gaussian 09 (Revision A02) [Google Scholar]
- 27.Becke A.D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 28.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 29.Francl M.M., Pietro W.J., Hehre W.J., Binkley J.S., Gordon M.S., DeFrees D.J., Pople J.A. Self‐consistent molecular orbital methods. XXIII. A polarization‐type basis set for second‐row elements. J. Chem. Phys. 1982;77:3654–3665. [Google Scholar]
- 30.Ischtwan J., Collins M.A. Determination of the intrinsic reaction coordinate: comparison of gradient and local quadratic approximation methods. J. Chem. Phys. 1988;89:2881–2885. [Google Scholar]
- 31.Grimme S., Ehrlich S., Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 32.Marenich A.V., Cramer C.J., Truhlar D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 33.Lu T., Chen F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 34.Sousa da Silva A.W., Vranken W.F. Acpype - AnteChamber PYthon parser interfacE. BMC Res. Notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 36.Páll S., Zhmurov A., Bauer P., Abraham M., Lundborg M., Gray A., Hess B., Lindahl E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020;153 doi: 10.1063/5.0018516. [DOI] [PubMed] [Google Scholar]
- 37.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong M., Nie T., Shao Q., Li M., Su H., Xu Y. In silico screening-based discovery of novel covalent inhibitors of the SARS-CoV-2 3CL protease. Eur. J. Med. Chem. 2022;231 doi: 10.1016/j.ejmech.2022.114130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su H.X., Yao S., Zhao W.F., Li M.J., Liu J., Shang W.J., Xie H., Ke C.Q., Hu H.C., Gao M.N., Yu K.Q., Liu H., Shen J.S., Tang W., Zhang L.K., Xiao G.F., Ni L., Wang D.W., Zuo J.P., Jiang H.L., Bai F., Wu Y., Ye Y., Xu Y.C. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H., Wang Q., Su H., Shao Q., Zhao W., Chen G., Li M., Xu Y. Identification of cysteine 270 as a novel site for allosteric modulators of SARS-CoV-2 papain-like protease. Angew. Chem. Int. Ed. 2022;61 doi: 10.1002/anie.202212378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Unoh Y., Uehara S., Nakahara K., Nobori H., Yamatsu Y., Yamamoto S., Maruyama Y., Taoda Y., Kasamatsu K., Suto T., Kouki K., Nakahashi A., Kawashima S., Sanaki T., Toba S., Uemura K., Mizutare T., Ando S., Sasaki M., Orba Y., Sawa H., Sato A., Sato T., Kato T., Tachibana Y. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022;65:6499–6512. doi: 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H., Ye F., Sun Q., Liang H., Li C., Li S., Lu R., Huang B., Tan W., Lai L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzym. Inhib. Med. Chem. 2021;36:497–503. doi: 10.1080/14756366.2021.1873977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahlin J.L., Nissink J.W.M., Strasser J.M., Francis S., Higgins L., Zhou H., Zhang Z., Walters M.A. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 2015;58:2091–2113. doi: 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukrd K., Yomezawa T., Skrlnga H. A molecular orbital theory of reactivity in aromatic hydrocarbons. Chem. Phys. 1952;20:722–725. [Google Scholar]
- 45.Zhuo L.G., Liao W., Yu Z.X. A frontier molecular orbital theory approach to understanding the Mayr equation and to quantifying nucleophilicity and electrophilicity by using HOMO and LUMO energies. Asian J. Org. Chem. 2012;1:336–345. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.