

PURPOSE
Precision medicine approaches, including germline pharmacogenetics (PGx) and management of drug-drug interactions (DDIs), are likely to benefit patients with advanced cancer who are frequently prescribed multiple concomitant medications to treat cancer and associated conditions. Our objective was to assess the potential opportunities for PGx and DDI management within a cohort of adults with advanced cancer.
METHODS
Medication data were collected from the electronic health records for 481 subjects since their first cancer diagnosis. All subjects were genotyped for variants with clinically actionable recommendations in Clinical Pharmacogenetics Implementation Consortium guidelines for 13 pharmacogenes. DDIs were defined as concomitant prescription of strong inhibitors or inducers with sensitive substrates of the same drug-metabolizing enzyme and were assessed for six major cytochrome P450 (CYP) enzymes.
RESULTS
Approximately 60% of subjects were prescribed at least one medication with Clinical Pharmacogenetics Implementation Consortium recommendations, and approximately 14% of subjects had an instance for actionable PGx, defined as a prescription for a drug in a subject with an actionable genotype. The overall subject-level prevalence of DDIs and serious DDIs were 50.3% and 34.8%, respectively. Serious DDIs were most common for CYP3A, CYP2D6, and CYP2C19, occurring in 24.9%, 16.8%, and 11.7% of subjects, respectively. When assessing PGx and DDIs together, approximately 40% of subjects had at least one opportunity for a precision medicine–based intervention and approximately 98% of subjects had an actionable phenotype for at least one CYP enzyme.
CONCLUSION
Our findings demonstrate numerous clinical opportunities for germline PGx and DDI management in adults with advanced cancer.
INTRODUCTION
Pharmacogenetics (PGx) and management of drug-drug interactions (DDIs) are two aspects of precision medicine that have the potential to optimize medication therapy in oncology and other therapeutic disciplines. PGx-guided approaches have been shown to enhance drug efficacy and safety, including within results from prospective clinical trials.1-5 Accordingly, the US Food and Drug Administration (FDA) currently includes PGx information within the labels for nearly 300 medications.6 Moreover, clinical practice guidelines that include PGx-guided recommendations have been published by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and prominent discipline-specific professional organizations (eg, the National Comprehensive Cancer Network) for over 100 medications.7,8 Similarly, DDIs are known to contribute to adverse drug events,9,10 and strategies to manage DDIs have been shown to improve patient outcomes.11 Given their important clinical implications, recommendations to manage DDIs are included both in FDA drug development guidance to industry12 and in numerous clinical practice guidelines.13,14
CONTEXT SUMMARY
Key Objective
How prevalent are opportunities for precision medicine–based clinical interventions, including germline pharmacogenetics and management of drug-drug interactions (DDIs), in patients with advanced cancer seen at our institutional molecular tumor board?
Knowledge Generated
Our findings indicate that approximately one in seven patients with advanced cancer had an opportunity for actionable pharmacogenetics during the study period (median follow-up: 2.9 years). In addition, approximately one in three patients had a potentially serious DDI affecting a major cytochrome P450 enzyme, and one in 14 patients had a potentially serious DDI involving a tyrosine kinase inhibitor and acid-reducing medication.
Relevance
Our findings suggest that patients with advanced cancer are a high-value population for clinical implementation of precision medicine.
The clinical utility of precision medicine is expected to be especially high for patients with advanced cancer given that drug therapy is commonly used not only to treat cancer but also to manage both cancer treatment–related adverse events (eg, nausea and vomiting) and comorbid conditions associated with cancer (eg, psychiatric conditions and pain syndromes). As a result, polypharmacy, typically defined as the concomitant use of five or more drugs, is exceedingly common in patients with advanced cancer.15 Polypharmacy carries an increased risk for DDIs,16 and, predictably, multiple investigations have identified serious DDIs in advanced cancer that affect patient outcomes.17 PGx-guided approaches also offer the ability to optimize therapy for numerous anticancer medications on the basis of somatic and germline genetic biomarkers. While molecular tumor boards have effectively harnessed somatic genome-guided treatment approaches to improve patient outcomes,18 germline PGx biomarkers can enhance medication safety with agents such as fluoropyrimidine and thiopurine chemotherapies.19,20 Additionally, PGx-guided approaches have been shown to enhance both efficacy and safety of selective serotonin reuptake inhibitors, tricyclic antidepressants, and opioid analgesics that are often prescribed for comorbid conditions prevalent in cancer.21-23 Given these abundant PGx opportunities in patients with cancer, it has been suggested that pre-emptive testing for PGx variants at first cancer diagnosis may be an effective clinical strategy to optimize patient outcomes.24 Furthermore, recent advancements in bioinformatics technology have enhanced the feasibility of PGx approaches in cancer through the creation of methods to extract PGx information from existing germline sequencing data generated during the clinical workflow of molecular tumor boards.25,26
Although past studies have characterized opportunities for DDI management and PGx-guided approaches in patients with advanced cancer, we are not aware of any work that has simultaneously investigated both approaches to provide a comprehensive assessment of the potential for precision medicine. Therefore, the objective of this study was to determine composite opportunities for precision medicine, incorporating both PGx-guided and DDI management strategies, within a cohort of adults with advanced solid cancers. By analyzing the potential for PGx-guided interventions since each subject's respective date of first cancer diagnosis, we also directly investigate the potential clinical utility of pre-emptively obtaining PGx information when patients are first diagnosed with cancer.
METHODS
Subject Enrollment and Eligibility
This study involved prospective genotyping and retrospective electronic health record (EHR) review of patients with solid cancers at Indiana University Health in Indianapolis, IN. Subjects were eligible to participate in the study if they had been seen in the Indiana University Health Precision Genomics clinic and enrolled in the accompanying Indiana University Total Cancer Care Protocol (part of the Oncology Research Information Exchange Network-wide Total Cancer Care initiative)27 and agreed to submit a blood sample for genotyping. Subjects were enrolled at clinic visits from February 2015 to February 2018. This research protocol and the parent Total Cancer Care Protocol were approved by Indiana University's Institutional Review Board. All subjects provided written informed consent.
Study Design and Data Collection
The purpose of this study was to investigate potential opportunities for precision medicine interventions, including PGx and management of DDIs, within a cohort of 481 adults seen at our institutional precision oncology clinic and associated solid tumor board. Demographic and clinical data, including medication prescriptions, were collected from the EHRs of all institutions participating in the Indiana Health Information Exchange, a statewide EHR data repository that includes 38 health care systems. Demographic data included age, sex, and race. Clinical data included first oncologic diagnosis and all inpatient and outpatient prescriptions. Genotyping of clinically actionable variants within major pharmacogenes was performed at the College of American Pathologists–accredited Indiana University Pharmacogenomics Laboratory using a laboratory-developed assay on the basis of the OpenArray Platform (ThermoFisher; Waltham, MA). The genes included on the genotyping platform, along with the number of variants tested for each gene, were as follows: CYP2B6 (two), CYP2C19 (six), CYP2C9 (six), CYP2D6 (11, including copy number targeting exon 9), CYP3A4 (two), CYP3A5 (three), CYP4F2 (one), DPYD (two), G6PD (two), IFNL3 (one), SLCO1B1 (two), TPMT (two), and VKORC1 (one). Detailed genotyping methods are provided in the Data Supplement, and a complete list of tested variants is available in the Data Supplement.
Precision Medicine Analyses
Detailed information about the specific methods used in the PGx, DDI, and composite precision medicine analyses, including the medication inclusion criteria and assumptions made, is available in the Data Supplement. Briefly, the PGx analyses assessed the prevalence of (1) prescriptions for medications with CPIC recommendations and (2) instances for actionable PGx, defined as prescription of a medication with a CPIC recommendation for a subject with a CPIC-defined actionable genotype-predicted phenotype. The DDI analyses assessed the prevalence of concomitant administration of perpetrator drugs with sensitive substrates of CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, and CYP3A and of coadministration of acid reducers and tyrosine kinase inhibitors. The composite precision medicine analyses compiled results from our PGx and DDI analyses and assessed the prevalence of CYP inhibitor–mediated phenoconversion for CYP2B6, CYP2C19, CYP2C9, CYP2D6, and CYP3A4.
Statistical Analyses
Data were analyzed using descriptive statistics (counts and percentages) using JMP Pro v.15.0.0.
RESULTS
Subject Demographic, Clinical, and Medication Data
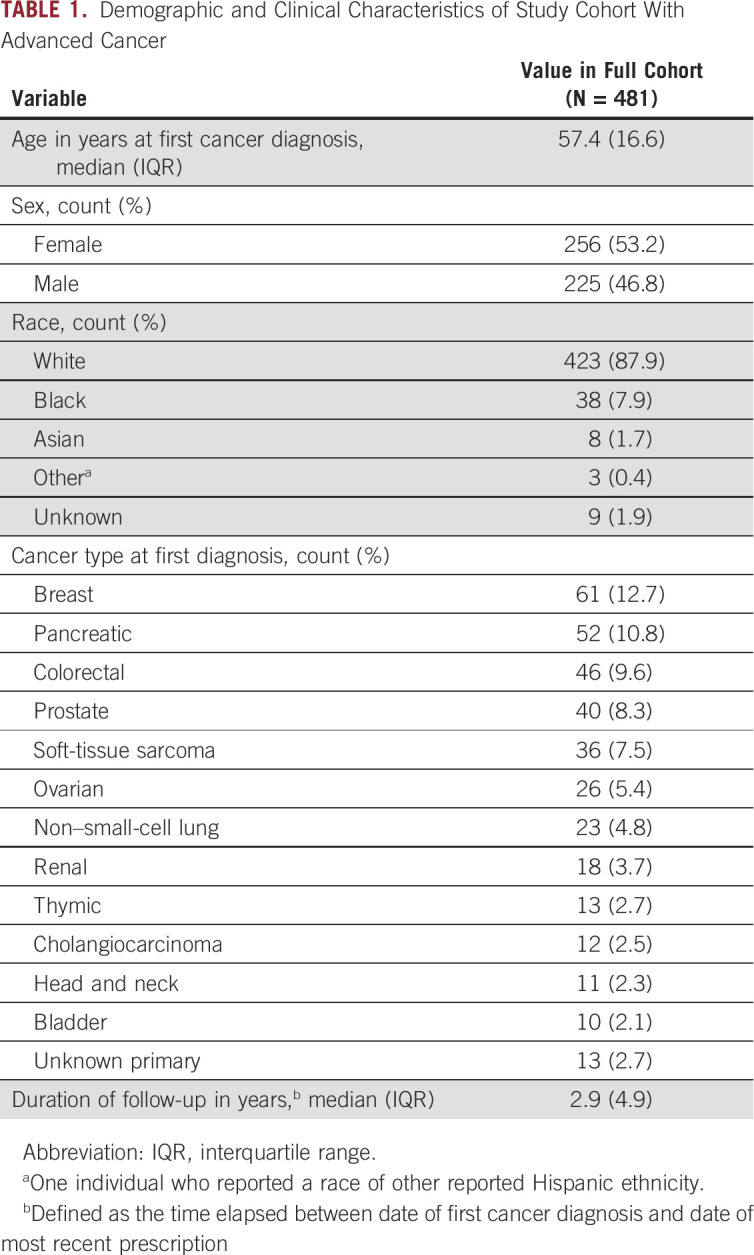
Demographic and clinical characteristics of the 481 study subjects with advanced cancer included are summarized in Table 1. The median age of our cohort was 57 ± 16.6 (median ± interquartile range) years, and most subjects were White (87.9%) and female (53.2%). The most common types of cancers at first diagnosis included breast (12.7%), pancreatic (10.8%), and colorectal (9.6%). The median duration of follow-up, defined as the time between the date of first cancer diagnosis and the date of last prescription, was 2.9 ± 4.9 (median ± interquartile range) years.
TABLE 1.
Demographic and Clinical Characteristics of Study Cohort With Advanced Cancer
Extracted medication data contained ≥ 1 prescription for 469 of 481 (97.5%) subjects. Filtering to include only prescriptions since each subject's respective date of first cancer diagnosis yielded 158,188 unique prescriptions that were assessed within our precision medicine analyses (schematic of filtering results shown in the Data Supplement). Since first cancer diagnosis, our cohort had a total of 7,074 unique prescriptions for medications contained within a CPIC guideline (herein called PGx medications) and a total of 22,642 unique prescriptions for medications that were defined as inducers, inhibitors, or sensitive substrates of CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, and/or CYP3A; acid reducers; or tyrosine kinase inihibitors (TKIs; herein called DDI medications).
PGx Analyses
Distributions of genotype-predicted phenotypes within our cohort for all pharmacogenes are displayed in Table 2. When defining actionable phenotypes as those with clinically actionable recommendations within CPIC guidelines for at least one medication, the rates of actionable phenotypes were highest for CYP2C19 (59.5%) and VKORC1 (52.4%) and lowest for TPMT (7.3%), G6PD (1.5%), and DPYD (1.0%).
TABLE 2.
Distribution of Genotype-Predicted Phenotypes Within Study Cohort for Major Pharmacogenes

Of 469 analyzed subjects, 282 (60.1%) were prescribed at least one PGx medication. These included a total of 1,045 unique PGx medications (ie, prescription of a unique PGx medication for a unique subject), with an average of 2.2 ± 2.4 (mean ± standard deviation) PGx medications/subject and a maximum of 12 PGx medications in one subject. When considering both prescribed medications and genotype-predicted phenotypes, we identified a total of 81 unique opportunities for actionable PGx. Instances of actionable PGx occurred for 67 subjects (14.3%), with 56 subjects having instances of actionable PGx involving one medication, eight subjects having actionable PGx involving two medications, and three subjects having actionable PGx involving three medications.
The prevalence of instances of actionable PGx, when stratified by the drug-gene pairs involved, are summarized in Table 3. For PGx medications prescribed in at least five subjects, the rates of actionable PGx were highest for warfarin (87.5%), amitriptyline (58.3%), and clopidogrel (42.9%). Conversely, capecitabine, fluorouracil, sertraline, and celecoxib had no instances for actionable PGx. For warfarin, subjects were considered to have actionable PGx recommendations on the basis of CYP2C9, CYP4F2, and VKORC1 genotype-based phenotypes in 20.8%, 58.3%, and 50.0% of cases, respectively. For amitriptyline, subjects were considered to have actionable PGx recommendations on the basis of CYP2C19 and CYP2D6 genotype-based phenotypes in 16.7% and 50.0% of cases, respectively.
TABLE 3.
Prevalence of PGx Medications Prescribed in Subjects With Clinically Actionable Genotype-Predicted Phenotypes on the Basis of CPIC Recommendations

DDI Analyses
Of 469 analyzed subjects, the prevalence of ≥ 1 prescription for an inducer, inhibitor, or substrate of any CYP enzyme was 49.0%, 58.0%, and 64.0%, respectively. The Data Supplement displays the prevalence of subjects with prescriptions for inducers, inhibitors, and substrates across the six enzyme systems that were assessed. Prescriptions for CYP inducers were most common for CYP2C19, CYP2C9, and CYP3A, occurring in 49.0% of subjects. Prescriptions for inhibitors were most common for CYP2D6 (occurring in 53.3% of subjects), CYP2C9 (35.0%), CYP3A (33.9%), and CYP2C19 (31.8%) while prescriptions for sensitive substrates were most common for CYP3A, CYP2D6, and CYP2C19 (prescribed in 60.3%, 59.9%, and 48.2% of subjects, respectively).
When assessing concomitant prescription of both a relevant perpetrator (inducer or inhibitor) and victim (sensitive substrate) drug, 236 subjects (50.3%) had a DDI affecting at least one CYP enzyme system. Given the frequent use of corticosteroids to treat and manage treatment-related complications for many types of cancer,29 we also performed DDI analyses excluding corticosteroids, which are potent inducers of CYP2C19, CYP2C9, and CYP3A; 225 subjects (48.0%) had a DDI affecting at least one major CYP enzyme when excluding corticosteroids. As summarized in Table 4, the prevalence of DDIs in our cohort was highest for CYP2D6 (affecting 45.2% of subjects; average of 1.5 DDIs/subject), followed by CYP3A (29.9%; 0.8 DDIs/subject), CYP2C19 (23.9%; 0.5 DDIs/subject), CYP2C9 (11.7%; 0.2 DDIs/subject), CYP2B6 (0.2%), and CYP2C8 (0%). When excluding corticosteroids, the prevalence of DDIs for CYP2C19, CYP2C9, and CYP3A was reduced to 10.2%, 7.0%, and 20.3%, respectively (Table 4). The most common drug-drug pairs contained within observed DDIs, stratified by enzyme, are available in the Data Supplement. The subject-level prevalence for serious DDIs, which were classified by the substrates involved, was 34.8% for any CYP enzyme when including corticosteroids and 29.4% when excluding corticosteroids (Table 4). Serious DDIs were most common for CYP3A, occurring in 24.9% of subjects and including sensitive substrates such as fentanyl, midazolam, and tramadol. In contrast, serious DDIs were less common for CYP2C19 (11.7% of subjects; sensitive substrates included escitalopram, sertraline, and citalopram), CYP2C9 (4.7% of subjects; substrates included warfarin, dronabinol, and phenytoin), and CYP2D6 (16.8% of subjects; substrates included tramadol, sertraline, and mirtazapine). When adjusting the prevalence of CYP enzyme-mediated DDIs on the basis of subject genotype (ie, excluding DDIs involving inducer or inhibitor drugs in subjects who are genotype-predicted poor metabolizers), the subject-level prevalence is as follows: CYP2B6: 0.2%; CYP2C19: 23.9%; CYP2C8: 0%; CYP2C9: 11.7%; CYP2D6: 44.1%; and CYP3A: 29.6% (adjusted on the basis of CYP3A4 genotype).
TABLE 4.
Number and Prevalence of Unique DDIs (ie, unique coprescription of a relevant drug-drug pair in a unique subject) by the Enzyme Involved in n = 469 Subjects Prescribed ≥ 1 Medication, Including (left) and Excluding (right) DDIs Involving Corticosteroids

TKIs have emerged as first-line treatments for many cancers, but recent investigations have described clinically significant DDIs with orally administered TKIs and acid-reducing agents, including antacids, histamine-2 receptor antagonists (H2RAs), and proton pump inhibitors (PPIs), that reduce TKI bioavailability and affect treatment outcomes.30-32 Accordingly, we characterized the prevalence of DDIs involving TKIs and acid reducers in our study population. Within our cohort, 68 subjects (14.5%) were prescribed at least one TKI, with pazopanib (prescribed in 17 subjects), sunitinib (10), and crizotinib (nine) being the most commonly prescribed. Of the 68 subjects prescribed a TKI, 33 (48.5%) had a concomitant prescription of at least one acid reducer. The most common acid reducer classes involved in DDIs were PPIs (perpetrator drug in 34 DDIs), followed by H2RAs (10) and antacids (six).
Composite Precision Medicine Analyses
To assess the prevalence of composite opportunities for precision medicine interventions, we aggregated findings from our actionable PGx, serious CYP-mediated DDI, and acid reducer TKI DDI analyses at the subject level. As shown in Figure 1, 186 subjects (39.7%) had at least one opportunity for a precision medicine intervention. Sixty-eight subjects (14.5%) had opportunities for more than one type of precision medicine intervention, with nine of these subjects (1.9%) having opportunities for PGx and management of both CYP-mediated and acid reducer TKI DDIs.
FIG 1.

Subject-level prevalence for composite precision medicine opportunities, including actionable PGx, management of serious CYP-mediated DDIs, and management of DDIs including acid reducers and TKIs. CYP, cytochrome P450; DDI, drug-drug interaction; PGx, pharmacogenetics; TKI, tyrosine kinase inihibitors.
Finally, we assessed the prevalence of CYP inhibitor–mediated phenoconversion, the process by which coadministration of a strong inhibitor functionally converts those with any genotype to a poor metabolizer phenotype, for CYP2B6, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. As shown in Figure 2, CYP inhibitor–mediated phenoconversion enhanced the number of subjects with actionable phenotypes for CYP2C19, CYP2C9, CYP2D6, and CYP3A4, increasing the prevalence from 59.5% to 72.8%, 33.3% to 55.9%, 44.7% to 76.3%, and 8.9% to 38.9%, respectively. In contrast, CYP inhibitor–mediated phenoconversion only slightly changed the number of actionable phenotypes for CYP2B6 (prevalence increased from 48.4% to 49.1%) because of the low prevalence of prescription of CYP2B6 inhibitors within our cohort. When considering all five investigated CYPs together, nearly every subject in our cohort (98.3%) had an actionable phenotype (either genotype-predicted or from CYP inhibitor–mediated phenoconversion) for at least one CYP since their date of first cancer diagnosis. In addition, 47 subjects (9.8%) had genotype-predicted or phenoconverted actionable phenotypes for all five CYP enzymes.
FIG 2.

Subject-level prevalence of clinically actionable phenotypes for major CYP enzymes on the basis of genotype and because of CYP inhibitor–mediated phenoconversion. CYP, cytochrome P450.
DISCUSSION
We provide quantitative evidence to support the immense clinical opportunities for precision medicine approaches, including germline PGx and management of DDIs, in a cohort of patients with advanced cancer. Our findings indicate that approximately 14% of subjects had opportunities for actionable PGx and that approximately 35% and approximately 7% of subjects had potentially serious DDIs involving major CYP enzymes and acid reducers coprescribed with TKIs, respectively. When incorporating both PGx and DDIs, we found that approximately 40% of subjects had at least one opportunity for a precision medicine–based intervention, and nearly all subjects (approximately 98%) had an actionable phenotype for ≥ 1 CYP enzyme. On the basis of our findings, implementation of precision medicine approaches at first cancer diagnosis is likely to provide clinical benefit to a significant proportion of patients. Although a limited number of other studies have addressed similar topics, our investigation has significant methodological advantages, including a larger cohort (N = 481), a broader PGx analysis consisting of 13 CPIC-actionable pharmacogenes, and utilization of a statewide data repository to enable more comprehensive collection of medication data.
Previous investigations have demonstrated the potential clinical impact of PGx approaches in patients with advanced cancer. Nichols et al found that 65% of patients were taking at least one PGx medication (ie, those with a CPIC guideline) and, on the basis of allele frequencies, estimated that 7.1% of patients could benefit from at least one PGx intervention.33 A study by Hertz et al34 found that 2.6% of 115 adult and pediatric patients with cancer could have benefited from PGx interventions involving CYP2C19, DPYD, and TPMT. Kasi et al35 also predicted abundant opportunities for PGx within patients with advanced cancer on the basis of genotypes for major CYP450 enzymes, although they did not analyze medication data. Many of our findings are similar to those reported in past investigations. For instance, the prevalence of prescription of PGx medications in our study was remarkably similar to that of Nichols et al when considering both prescriptions for any PGx medication and for specific PGx drugs such as ondansetron, capecitabine, and simvastatin.33 Our findings related to the distribution of actionable phenotypes are also consistent with those from past investigations33,35 and from large analyses of population allele frequencies.36 In contrast, our finding for the prevalence of subjects with potential PGx interventions (14.3%) is higher than those reported by Nichols et al (7.1%) or Hertz et al (2.6%).33,34 These differences are likely attributable to the facts (1) that we investigated the potential for PGx interventions across a wider array of pharmacogenes and (2) that we used a statewide repository to enhance the richness of our collected medication data.
Multiple investigations have also characterized the opportunities for DDI management strategies in adults with advanced cancer, estimating DDIs to occur in 27%-78% of patients.37-42 The large differences in these estimates are likely due to methodological differences among studies. For instance, studies that included both pharmacokinetic- and pharmacodynamic-based DDIs and studies that used medication lists from the EHR (rather than those verified by patients during medication reconciliation) had higher rates of potential DDIs.38,39 Our DDI prevalence of approximately 52% falls in the middle of those reported by past investigations. In terms of methodology, extracting medication data from the EHR and not being able to resolve medication days supply or whether medications were prescribed on an as-needed basis likely resulted in a higher DDI prevalence in our study. However, our DDI prevalence was likely conservative relative to other studies on the basis of other elements in our methodology, including that we excluded DDIs with pharmacodynamic mechanisms and those involving drugs commonly coadministered as cancer treatment regimens (eg, corticosteroids with docetaxel).
Our findings are impactful since they demonstrate abundant clinical opportunities for PGx in patients with advanced cancer and support preemptive genotyping at first cancer diagnosis. Our results corroborate those from other studies that identified significant opportunities for PGx in patients with cancer, including via optimization of selective serotonin reuptake inhibitor, tricyclic antidepressants, opioid, and antiemetic therapies.33,35 Relative to past investigations quantifying PGx opportunities within general medical populations, our findings suggest that patients with advanced cancer may be more likely to be prescribed drugs with CPIC recommendations and, relatedly, more likely to have opportunities for PGx interventions, although differences in study methodologies limit direct comparisons.43,44 Advances in technology have improved PGx feasibility by reducing the cost of obtaining genetic information and enabling repurposing of genetic information obtained from molecular tumor boards.26 Economic analyses have demonstrated cost savings because of toxicity sparing for both DPYD and TPMT testing,45,46 and there is currently clinical momentum to standardize PGx markers for fluoropyrimidine and thiopurine chemotherapies.47,48
We found that over half of study subjects had a DDI affecting at least one major CYP enzyme. This finding is important given that DDIs have been associated with reduced efficacy and increased adverse drug events in patients with cancer. CYP-mediated DDIs have been shown to increase adverse events attributable to both cancer therapies and concomitant medications in patients with cancer.49,50 Additionally, several studies have demonstrated clinically significant DDIs between acid-reducing agents and TKIs, evidenced by reduced progression-free and overall survival.30-32 Our findings demonstrate that these DDIs are common in patients with advanced cancer, occurring in nearly half of subjects in our study prescribed a TKI. However, it is possible that the providers told the patient to discontinue the acid reducers while taking the TKIs. Finally, to our knowledge, our work is one of the first to assess the prevalence of potential drug-drug-gene interactions (DDGIs; ie, CYP inhibitor–mediated phenoconversion) within a clinical cohort. While the strategies to manage DDGIs borrow from both PGx and DDI management approaches, consideration of DDGIs may provide critical information that modifies the risk of adverse drug events predicted from consideration of either approach in isolation.51 As demonstrated by our composite study findings that approximately 40% of subjects had at least one opportunity for precision medicine intervention and approximately 98% of subjects had an actionable phenotype for ≥ 1 CYP enzymes, PGx information and concomitant drug lists should be used in tandem to most accurately inform approaches to optimize medication therapy.
We acknowledge several limitations of our study. First, we did not mine the EHR to identify prescribing decisions or health outcomes related to the instances for precision medicine identified within our analyses. Next, our extracted medication data did not include ways to conclusively determine whether medications were prescribed on an as-needed basis or ascertain days supply to assess temporal overlap between perpetrator and victim drugs within our DDI analyses. To compensate for these limitations, we used conservative methods in our DDI analyses to estimate days supply for each prescription. Our medication data also did not consistently contain information about the medication dose. As a result, our analysis may have overestimated the prevalence of instances for actionable PGx with amitriptyline since current CPIC guidelines do not recommend clinical action at daily doses under 50 mg.21 In addition, our panel-based genotyping method only tested for relatively common functional variants in the assessed genes within our primary ethnic and racial populations. Additionally, advances in knowledge since study initiation limited our ability to assess variants with newly established relevance to pharmacotherapy (eg, HapB3 in DPYD). Finally, our genotyping panel also did not assess every pharmacogene included in CPIC guidelines but did cover genes serving as the basis for over 80% of CPIC recommendations.7
In conclusion, our work provides quantitative evidence of the vast clinical opportunities for precision medicine approaches in patients with advanced cancer, demonstrating the clinical utility of both germline PGx and DDI management strategies. Given their established clinical benefits and the abundant opportunities for their use demonstrated by our results, precision medicine approaches are likely to improve medication outcomes in patients with cancer and may provide clinical benefit if incorporated into the workflow of molecular tumor boards.
Milan Radovich
Employment: Caris Life Sciences
Leadership: Caris Life Sciences
Stock and Other Ownership Interests: LifeOmic, Macrogenics, Immunomedics, Tyme Technologies
Honoraria: Lilly
Research Funding: Lilly (Inst), Boston Biomedical (Inst), Foundation Medicine (Inst), Epic Sciences (Inst), Pfizer (Inst), Genentech (Inst)
Patents, Royalties, Other Intellectual Property: Combination therapy for the treatment of TNBC with a PI3K pathway inhibitor that targets PI3KDelta and PI3KGamma (Inst)
Travel, Accommodations, Expenses: LifeOmic, Caris Life Sciences
Victoria M. Pratt
Stock and Other Ownership Interests: Quest Diagnostics, LabCorp
Other Relationship: Avalon Heathcare
John T. Callaghan
Employment: IU health
Stock and Other Ownership Interests: Lilly, Abbott/AbbVie, Stryker (I)
Bryan P. Schneider
Honoraria: Lilly, Research to Practice
Research Funding: Genentech/Roche, Pfizer, Foundation Medicine, Exact Sciences
Todd C. Skaar
Honoraria: Tabula Rasa Healthcare
Consulting or Advisory Role: Indiana University Health
Travel, Accommodations, Expenses: Tabula Rasa Healthcare
Other Relationship: NIH
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented as a virtual poster at the American Society for Clinical Pharmacology and Therapeutics Annual Meeting, March 8-17, 2021.
SUPPORT
Supported by NIGMS R35GM131812 (awarded to T.C.S.) and funds from the Indiana University Grand Challenge to support the institutional Precision Health Initiative (provides salary and research support to T.S., R.C.L., E.J.R., S.P., M.R., B.P.S., and T.C.S.).
T.S. and R.C.L. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Tyler Shugg, Reynold C. Ly, Mustafa A. Hyder, Zeruesenay Desta, Todd C. Skaar
Financial support: Todd C. Skaar
Provision of study material or patients: Victoria M. Pratt, Bryan P. Schneider
Collection and assembly of data: Tyler Shugg, Reynold C. Ly, Mustafa A. Hyder, Milan Radovich, Marc B. Rosenman, Bryan P. Schneider, Todd C. Skaar
Data analysis and interpretation: Tyler Shugg, Reynold C. Ly, Elizabeth J. Rowe, Santosh Philips, Mustafa A. Hyder, Victoria M. Pratt, John T. Callaghan, Bryan P. Schneider, Todd C. Skaar
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Milan Radovich
Employment: Caris Life Sciences
Leadership: Caris Life Sciences
Stock and Other Ownership Interests: LifeOmic, Macrogenics, Immunomedics, Tyme Technologies
Honoraria: Lilly
Research Funding: Lilly (Inst), Boston Biomedical (Inst), Foundation Medicine (Inst), Epic Sciences (Inst), Pfizer (Inst), Genentech (Inst)
Patents, Royalties, Other Intellectual Property: Combination therapy for the treatment of TNBC with a PI3K pathway inhibitor that targets PI3KDelta and PI3KGamma (Inst)
Travel, Accommodations, Expenses: LifeOmic, Caris Life Sciences
Victoria M. Pratt
Stock and Other Ownership Interests: Quest Diagnostics, LabCorp
Other Relationship: Avalon Heathcare
John T. Callaghan
Employment: IU health
Stock and Other Ownership Interests: Lilly, Abbott/AbbVie, Stryker (I)
Bryan P. Schneider
Honoraria: Lilly, Research to Practice
Research Funding: Genentech/Roche, Pfizer, Foundation Medicine, Exact Sciences
Todd C. Skaar
Honoraria: Tabula Rasa Healthcare
Consulting or Advisory Role: Indiana University Health
Travel, Accommodations, Expenses: Tabula Rasa Healthcare
Other Relationship: NIH
No other potential conflicts of interest were reported.
REFERENCES
- 1.Claassens DMF, Vos GJA, Bergmeijer TO, et al. : A genotype-guided strategy for oral P2Y(12) inhibitors in primary PCI. N Engl J Med 381:1621-1631, 2019 [DOI] [PubMed] [Google Scholar]
- 2.Mallal S, Phillips E, Carosi G, et al. : HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 358:568-579, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Pirmohamed M, Burnside G, Eriksson N, et al. : A randomized trial of genotype-guided dosing of warfarin. N Engl J Med 369:2294-2303, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Papastergiou J, Quilty LC, Li W, et al. : Pharmacogenomics guided versus standard antidepressant treatment in a community pharmacy setting: A randomized controlled trial. Clin Transl Sci 14:1359-1368, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenblat JD, Lee Y, McIntyre RS: The effect of pharmacogenomic testing on response and remission rates in the acute treatment of major depressive disorder: A meta-analysis. J Affect Disord 241:484-491, 2018 [DOI] [PubMed] [Google Scholar]
- 6.US Food and Drug Administration : Table of Pharmacogenomic Biomarkers in Drug Labeling. US Food and Drug Administration, Silver Spring, MD, 2020 [Google Scholar]
- 7.Clinical Pharmacogenetics Implementation Consortium: Guidelines. Clinical Pharmacogenetics Implementation Consortium, Palo Alto, CA, 2020 [Google Scholar]
- 8.Shugg T, Pasternak AL, London B, et al. : Prevalence and types of inconsistencies in clinical pharmacogenetic recommendations among major U.S. sources. NPJ Genom Med 5:48, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bates DW, Cullen DJ, Laird N, et al. : Incidence of adverse drug events and potential adverse drug events. Implications for prevention. ADE Prevention Study Group. JAMA 274:29-34, 1995 [PubMed] [Google Scholar]
- 10.Wright A, Feblowitz J, Phansalkar S, et al. : Preventability of adverse drug events involving multiple drugs using publicly available clinical decision support tools. Am J Health Syst Pharm 69:221-227, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Arnold RJG, Tang J, Schrecker J, et al. : Impact of definitive drug-drug interaction testing on medication management and patient Care. Drugs Real World Outcomes 5:217-224, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.US Food and Drug Administration : Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions: Guidance for Industry. US Food and Drug Administration, Silver Spring, MD, 2020 [Google Scholar]
- 13.Department of Health and Human Services Office of AIDS Research Advisory Council : Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Washington, DC, US Department of Health and Human Services, 2018 [Google Scholar]
- 14.Grundy SM, Stone NJ, Bailey AL, et al. : 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation 139:e1082-e1143, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeBlanc TW, McNeil MJ, Kamal AH, et al. : Polypharmacy in patients with advanced cancer and the role of medication discontinuation. Lancet Oncol 16:e333-e341, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Guthrie B, Makubate B, Hernandez-Santiago V, et al. : The rising tide of polypharmacy and drug-drug interactions: Population database analysis 1995-2010. BMC Med 13:74, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma M, Vadhariya A, Chikermane S, et al. : Clinical outcomes associated with drug-drug interactions of oral chemotherapeutic agents: A comprehensive evidence-based literature review. Drugs Aging 36:341-354, 2019 [DOI] [PubMed] [Google Scholar]
- 18.Kato S, Kim KH, Lim HJ, et al. : Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun 11:4965, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Relling MV, Schwab M, Whirl-Carrillo M, et al. : Clinical Pharmacogenetics Implementation Consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther 105:1095-1105, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amstutz U, Henricks LM, Offer SM, et al. : Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther 103:210-216, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hicks JK, Sangkuhl K, Swen JJ, et al. : Clinical Pharmacogenetics Implementation Consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin Pharmacol Ther 102:37-44, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crews KR, Gaedigk A, Dunnenberger HM, et al. : Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther 95:376-382, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hicks JK, Bishop JR, Sangkuhl K, et al. : Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther 98:127-134, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffman JM, Haidar CE, Wilkinson MR, et al. : PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am J Med Genet C Semin Med Genet 166c:45-55, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Lee M, Allard WG, Bollen S, et al. : Repurposing of diagnostic whole exome sequencing data of 1,583 individuals for clinical pharmacogenetics. Clin Pharmacol Ther 107:617-627, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Numanagic I, Malikic S, Ford M, et al. : Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat Commun 9:828, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oncology Research Information Exchange Network (ORIEN). https://www.oriencancer.org/
- 28.Johnson JA, Caudle KE, Gong L, et al. : Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin Pharmacol Ther 102:397-404, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lossignol D: A little help from steroids in oncology. J Transl Int Med 4:52-54, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YM, Lai CH, Chang HC, et al. : Antacid use and de novo brain metastases in patients with epidermal growth factor receptor-mutant non-small cell lung cancer who were treated using first-line first-generation epidermal growth factor receptor tyrosine kinase inhibitors. PLoS One 11:e0149722, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu MP, Ghosh S, Chambers CR, et al. : Gastric Acid suppression is associated with decreased erlotinib efficacy in non-small-cell lung cancer. Clin Lung Cancer 16:33-39, 2015 [DOI] [PubMed] [Google Scholar]
- 32.Mir O, Touati N, Lia M, et al. : Impact of concomitant administration of gastric acid-suppressive agents and pazopanib on outcomes in soft-tissue sarcoma patients treated within the EORTC 62043/62072 trials. Clin Cancer Res 25:1479-1485, 2019 [DOI] [PubMed] [Google Scholar]
- 33.Nichols D, Arnold S, Weiss HL, et al. : Pharmacogenomic potential in advanced cancer patients. Am J Health Syst Pharm 76:415-423, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hertz DL, Glatz A, Pasternak AL, et al. : Integration of germline pharmacogenetics into a tumor sequencing program. JCO Precis Oncol 2:1-15, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasi PM, Koep T, Schnettler E, et al. : Feasibility of integrating panel-based Pharmacogenomics testing for chemotherapy and supportive Care in patients with colorectal cancer. Technol Cancer Res Treat 18:1533033819873924, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Ingelman-Sundberg M, Lauschke VM: Worldwide distribution of cytochrome P450 alleles: A meta-analysis of population-scale sequencing projects. Clin Pharmacol Ther 102:688-700, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Cheung WY: Potential drug interactions in patients with a history of cancer. Curr Oncol 21:e212-20, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ismail M, Khan S, Khan F, et al. : Prevalence and significance of potential drug-drug interactions among cancer patients receiving chemotherapy. BMC Cancer 20:335, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korucu FC, Senyigit E, Köstek O, et al. : A retrospective study on potential drug interactions: A single center experience. J Oncolog Sci 4:80-84, 2018 [Google Scholar]
- 40.van Leeuwen RW, Brundel DH, Neef C, et al. : Prevalence of potential drug-drug interactions in cancer patients treated with oral anticancer drugs. Br J Cancer 108:1071-1078, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Leeuwen RW, Swart EL, Boven E, et al. : Potential drug interactions in cancer therapy: A prevalence study using an advanced screening method. Ann Oncol 22:2334-2341, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Riechelmann RP, Del Giglio A: Drug interactions in oncology: How common are they? Ann Oncol 20:1907-1912, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Chanfreau-Coffinier C, Hull LE, Lynch JA, et al. : Projected prevalence of actionable pharmacogenetic variants and level A drugs prescribed among US Veterans Health Administration pharmacy users. JAMA Netw Open 2:e195345, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hicks JK, El Rouby N, Ong HH, et al. : Opportunity for genotype-guided prescribing among adult patients in 11 US health systems. Clin Pharmacol Ther 110:179-188, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarca K, Durand-Zaleski I, Loriot MA, et al. : Modeling the outcome of systematic TPMT genotyping or phenotyping before azathioprine prescription: A cost-effectiveness analysis. Mol Diagn Ther 23:429-438, 2019 [DOI] [PubMed] [Google Scholar]
- 46.Fragoulakis V, Roncato R, Fratte CD, et al. : Estimating the effectiveness of DPYD genotyping in Italian individuals suffering from cancer based on the cost of chemotherapy-induced toxicity. Am J Hum Genet 104:1158-1168, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weitzel KW, Smith DM, Elsey AR, et al. : Implementation of standardized clinical processes for TPMT testing in a diverse multidisciplinary population: Challenges and lessons learned. Clin Transl Sci 11:175-181, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hertz DL, Sahai V: Including DPYD on cancer genetic panels to prevent fatal fluoropyrimidine toxicity. J Natl Compr Canc Netw 18:372-374, 2020 [DOI] [PubMed] [Google Scholar]
- 49.Agergaard K, Mau-Sørensen M, Stage TB, et al. : Clopidogrel-paclitaxel drug-drug interaction: A pharmacoepidemiologic study. Clin Pharmacol Ther 102:547-553, 2017 [DOI] [PubMed] [Google Scholar]
- 50.Shah HR, Ledbetter L, Diasio R, et al. : A retrospective study of coagulation abnormalities in patients receiving concomitant capecitabine and warfarin. Clin Colorectal Cancer 5:354-358, 2006 [DOI] [PubMed] [Google Scholar]
- 51.Malki MA, Pearson ER: Drug-drug-gene interactions and adverse drug reactions. Pharmacogenomics J 20:355-366, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]