

Abstract
Knobloch syndrome (KS) is an autosomal recessive disorder caused by biallelic pathogenic variants in COL18A1 . KS clinically manifests with the typical eye findings (high myopia, vitreoretinal degeneration, retinal detachment, and lens subluxation), variable neurological findings (occipital encephalocele, polymicrogyria, cerebellar malformations, epilepsy, and intellectual disability), and the other uncommon clinical manifestations. Literature review of all KS patients (source PubMed) was done with special reference to cerebellar abnormalities. Here, we report two siblings with typical KS with posterior fossa malformations and novel cerebellar midline cleft abnormality analyzed by whole exome sequencing. Known pathogenic homozygous variant c.2908C > T; (p.Arg970Ter) in exon 26 of COL18A1 was found as a cause for KS. These two siblings presented with early-onset severe ocular manifestations, facial dysmorphism, and variable central nervous system manifestations along with novel cerebellar midline cleft abnormality. The presence or absence of structural brain malformations and genotypes does not absolutely predict cognitive functions in KS patients. However, the presence of posterior fossa abnormality may be predictive for the development of ataxia in later life and needs further studies.
Keywords: Knobloch syndrome, occipital encephalocele, lens subluxation, vitreoretinal degeneration, cerebellar vermis midline cleft
Introduction
Knobloch syndrome (KS) (MIM #267750) is an autosomal recessive disorder characterized by early-onset ocular manifestation often associated with complications progressing to blindness, and variable central nervous system (CNS) manifestations which may or may not affect cognitive functions. 1 2 3 4 5 6 7 Other rarely involved organs could be the lungs, kidneys, and heart. 8 9 Fertility seems to be unaffected in some KS patients. 7 External physical examination might show facial dysmorphism and other minor malformations. 8 9 10
Background
Syndromic genetic disorders need an accurate diagnosis to screen, prognosticate, treat (medically and/or surgically), and facilitate the follow-up of associated/progressive/late-onset clinical manifestations. KS is one of the single gene disorders requiring both medical and surgical treatments.
KS is caused by biallelic pathogenic variants in COL18A1 localized to chromosome 21q22.3. 8 COL18A1 encodes for three isoforms with the use of two different promoters and alternate splicing of the third exon. These three isoforms differ in sizes at the N-terminal region and are named as N-303 (short), N-453 (medium), and N-738 (long). 1 All are expressed in basement membranes with selective predominant expression of isoforms in certain tissues. COL18A1 proteins play a critical role in the development and maintenance of eye and brain. The three isoforms are cleaved at the C-terminal end to give rise to endostatin. Endostatin has antiangiogenic property and governs other endothelial cell cellular activities as well.
COL18A1 is the only gene reported as the cause for KS. Most of the homozygous or compound heterozygous pathogenic variants reported are located between exons 32 and 42, most commonly in the exon 41, and majority produces a truncated protein affecting all the isoforms, including endostatin formation. 11 Only two families of KS (from Brazil sharing common haplotypes) have been described with mutations located at the N-terminal region, and most of these patients were found to have a relatively less severe ocular manifestations, as they result in deficiency of only short isoform. 1 Pathogenic variants located toward C-terminal and/or affecting all the isoforms including endostatin have been associated with severe ocular manifestations. There are no predictors of CNS manifestations based on pathogenic variants in COL18A1 ; however, it is believed that deficiency of all isoforms (due to pathogenic variants located at C-terminal end) and abnormal neuronal migration is more likely to be associated with epilepsy. 1 12 c3514-3515delCT (nucleotide position is based on isoform 2) represents recurrent common small deletion hotspot mutation in COL18A1 , and has been reported in homozygous/compound heterozygous state in many patients from different ethnic backgrounds. 1 3 6
Eye Manifestations in KS
Ocular features in KS are present in all children at an early age, and include high myopia and vitreoretinal degeneration (clinically presenting with nystagmus, strabismus, affected visual acuity, and/or cataracts). Recurrent retinal detachment, lens subluxation (commonest temporal), distinctive lens opacities, glaucoma, and pigment dispersion syndrome are the associated clinical complications, require careful monitoring as they may or may not be present at young age, and may manifest as the age advance. 1 4 6 9 13 Other anterior segment abnormalities described are absent iris crypts, iris transillumination, poor pupillary dilatation, persistent pupillary membrane, and lens opacities. 6 Most of the KS individuals are blind before the age of 20 years. 1
CNS Manifestations in KS
CNS manifestations are highly variable among patients with KS. The most common CNS lesion is occipital encephalocele seen in 50 to 90% of KS patients. 1 4 6 8 Encephaloceles are not mandatory for the diagnosis, as in the later clinical reports, many patients were reported to have no occipital defect. 7 Occipital lesion can range from patchy hypopigmented hairs, focal alopecia with or without soft tissue swelling, atretic encephalocele, to frank occipital encephalocele. Other variable CNS manifestations/malformations are polymicrogyria (frontal and temporal), heterotopic gray matter, agenesis of the septum pellucidum, enlarged cisterna magna, cerebellar malformations, epilepsy (17%), developmental delay, intellectual disability (20%), learning difficulties, ataxia, and sometimes cognitive decline. 3 5 6 7 Some of the CNS clinical and radiological findings could be late age onset/findings (after four decades)—cognitive decline, brain stem and supratentorial volume loss, and slender spinal cord. 3
Prognosis and Outcome in KS
Severity of organ involvement prognosticates and guides clinicians about prognosis and management, especially eye and CNS malformations. Other rarely involved organs should be looked and monitored for anomalies based on the findings: kidney for duplex renal system, hydronephrosis, and renal dysfunction; heart for structural heart malformations; and skin for delayed wound healing. 9
Patient Details
Two male siblings born to nonconsanguineous parents were referred with a history of decreased visual acuity in both the eyes, noted at the age of 2 to 3 years. The elder sibling (patient 1) attended regular school although with some learning difficulties. The younger sibling (patient 2) did not have learning difficulties; however, at the age of 8 to 9 years, he was operated for small occipital encephalocele diagnosed at birth. No surgical records were available for review.
Patient 1
He was seen by us at the age of 13 years 10 months with decreased vision in both eyes. He had a best corrected visual acuity (BCVA) of 6/120, N24 in the right eye 2/60p, N24p in the left eye. Orthoptic evaluation showed left exotropia. Anterior segment examination showed bilateral smooth cryptless irides with no transillumination, aphakia in the right eye (no history of any intraocular surgery) and nasal subluxation of lens in the left eye ( Fig. 1A ). The intraocular pressure (IOP) was normal 17 mm Hg, with the I-care in both the eyes. Fundoscopy showed myopic disc with tessellations in both eyes and chorioretinal atrophic patches over the posterior pole and white fibrillary vitreous condensations in the form of vitreous veils. In addition, right eye showed posterior lens dislocation. The child was diagnosed with bilateral axial myopia, chorioretinal scars, posterior dislocation of lens with deprivational amblyopia in the right eye, and ectopia lentis with refractive amblyopia in the left eye.
Fig. 1.
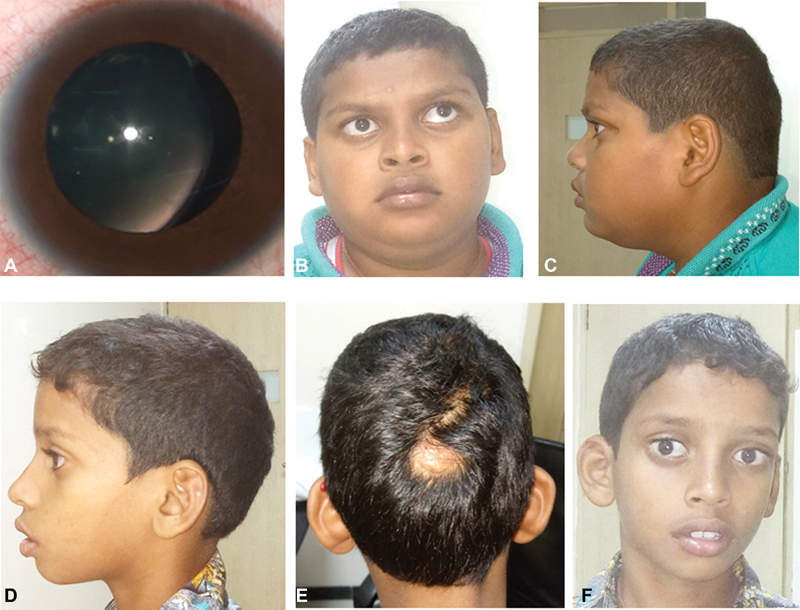
( A ) Patient 1—smooth cryptless irides with nasal subluxation of the crystalline lens in the left eye. ( B, C ) Patient 1 face—wide eyes with left exotropia, mid-face retrusion, short philtrum, thick lips, micrognathia, brachycephalic head shape, and short neck. ( D–F ) Patient 2—scaphocephaly head shape, focal alopecia with scar measuring 1.5 × 2 cm in the occipital region, and face—wide eyes with exotropia, anteverted low set ears, short philtrum, thick lips, and microretrognathia.
On examination, facial features included wide eyes, left eye exotropia, mid-face retrusion, short philtrum, thick lips, and micrognathia. Other physical features included brachycephalic head shape, generalized obesity, and short neck ( Fig. 1B, 1C ). He had a head circumference of 51 cm (25th–50th centiles), a height of 146 cm (10th–25th centiles), and arm span of 149 cm. Otherwise, systemic examination was unremarkable.
Magnetic resonance imaging (MRI) of the brain images demonstrated a small 2 mm defect in the occipital bone below the lambdoid suture. Hypointense strands in the subcutaneous soft tissues, extending through the bone defect and passing through the split adjacent dural sinus were noted in T1 and T2 images. Cisterna magna was relatively large. There was a midline cleft in the mid-part of the vermis, extending around medially on the right side, not reaching the fourth ventricle. Dislocation of the right lens which was seen in the posterior part of the eye globe was also observed ( Fig. 2A–F ).
Fig. 2.

Patient 1—( A ) Midline sagittal MRI, T1-weighted image demonstrates an occipital bone defect (angled arrow). Cisterna magna is enlarged (Star). ( B ) Coronal view of T1-weighted image shows a 2-mm bone defect below the lambdoid suture (zigzag arrow). ( A, C ) There is a deep fissure in the vermis (open arrows). ( C ) Axial T2-weighted image shows the lens (white arrow) dislocated in the posterior segment. ( D ) Axial T2-weighted image at a higher level show stranding of subcutaneous tissues and splitting of the distal part of superior sagittal sinus. ( E, F ) Axial T-weighted and T1-weighted images demonstrate a focal area of cortical heterotopia (arrow) along the lateral wall of left lateral ventricle. Large cistern magna is redemonstrated. Rest of the brain is normal. Patient 2—( G, H ) Axial and sagittal MRI, T1-weighted images demonstrate a large midline bone defect in occipital bone (white arrow). There are few hypointense contents in the adjacent subcutaneous soft tissues. ( H–J ) Vermis is defective in the midline with the defect extending to the roof of the fourth ventricle (open arrows). ` T2-weighted image demonstrates deformed eye balls, absent lens in the right eyeball (zigzag arrow). In the posterior fossa, there is a gross enlargement of cisterna magna (star).
He underwent lensectomy with anterior vitrectomy in the left eye to achieve aphakia for visual rehabilitation. Vitrectomy for the dislocated lens in the right eye was deferred in view of the quiet eye with absence of any inflammation and normal IOP. Conservative management with glasses was advised. It was decided that surgery could be planned for the right eye if the IOP increased significantly or the visual acuity deteriorated further. After surgery, he was given glasses of +5.50Ds for distance and +2.50Ds in both eyes. His BCVA improved to 6/96 for distance and N36 for near in both eyes.
Patient 2
He is a 10-year 6-month-old patient presented with decreased vision in both eyes and was diagnosed with bilateral subluxation of lens. He had undergone lensectomy in the right eye elsewhere. He had a BCVA of 6/60, N12p in the right eye with +5.0Ds for distance and +2.5Ds addition for near and 6/120p, N24 in the left eye. Orthoptic evaluation showed left exotropia. Anterior segment examination showed bilateral smooth cryptless irides with no transillumination, aphakia in the right eye, and inferotemporal subluxation of the crystalline lens in the left eye. The IOP was normal, 10 and 13 mm Hg in the right eye and left eye, respectively, using the I-care in both the eyes. Fundoscopy showed myopic disc with tessellations and chorioretinal atrophic patches over the macula. The child was diagnosed with bilateral axial myopia and macular scar with aphakia with deprivational amblyopia in the right eye and ectopia lentis with refractive amblyopia in the left eye.
On examination, his physical features included scaphocephaly head shape, focal alopecia with scar measuring 1.5 × 2 cm in the occipital region; facial features included wide eyes, anteverted low set ears, short philtrum, thick lips, and microretrognathia ( Fig. 1D–F ). He had a head circumference of 52.2 cm (50th–75th centiles), a height of 135 cm (25th–50th centiles), and arm span of 136 cm. Systemic examination was unremarkable. Neuroimaging with MRI showed a bony defect in the midline in the occipital bone, just below the lambdoid suture. There were T1 and T2 hypointense strands within the soft tissues at the defect and in the adjacent subcutaneous region. Posterior fossa evaluation revealed large cisterna magna. There was a midline defect in the vermis at the mid-part which was communicating to the fourth ventricle. A tiny area of cortical heterotopia was also noted along the lateral wall of atria of the left lateral ventricle. Rest of the intracranial structures was within normal limits. The absence of a lens was also noted in both eyeballs ( Fig. 2G–J ).
He underwent lensectomy with anterior vitrectomy in the left eye for visual rehabilitation. After surgery, his BCVA improved to 6/60 for distance with +4.75Ds and N18 for near with an addition of +2.5Ds in the left eye resulting in alternating exotropia ( Fig. 1F ). Both siblings were given training for further visual rehabilitation and referred to appropriate centers closer to their hometown for computer training.
Molecular Methods and Results
Molecular Testing
After proper informed consent, exome sequencing was performed for the proband only. The exonic and the flanking genomic regions were captured using Agilent SureSelect Human All Exon V6 kit. Exome sequencing was performed as described earlier with average coverage of 100 to 130X, 95% of bases covered at a minimum of 20X with 90% sensitivity. 14 15 For analysis of the data obtained from exome sequencing, the raw data were retrieved in FASTQ format and aligned to GRCh37 assembly using Burrows–Wheeler Aligner (v0.7.15) and our in-house pipeline based on Genome Analysis Toolkit Best Practices. These data were annotated by ANNOVAR and our in-house scripts. 16 The called variants were analyzed based on variant prioritization and filtering strategy as outlined in Supplementary Table S1 . The list of rare homozygous variants observed in the proband is represented in Supplementary Table S2 . Validation of the disease-causing variant in the family was done by Sanger sequencing.
Results
A known stop-gain variant (ClinVar accession ID: VCV000438061.1; ClinVar variant ID: 438061), c.2908C > T; (p.Arg970Ter) in exon 26 of COL18A1 (NM_030582.4) was identified in homozygous state in the proband. Sanger sequencing and segregation analysis confirmed the carrier status of parents, while proband's similarly affected sibling carried the same variant in homozygous state as seen in Fig. 3 . This variant has not found reported in population databases such as gnomAD, nor in our in-house database of 654 exomes. COL18A1 has three isoforms; the sequence variant is found in all the isoforms and is described in Supplementary Table S3 . Multiple in silico analysis tools (MutationTaster, SIFT, FATHMM) are predicting the variant to be disease causing. This sequence variant is either predicted to trigger nonsense-mediated mRNA decay or result in truncated protein production, and also predicting to be pathogenic for the endostatin functioning. This stop-gain variant was classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines for classifying sequence variants. 17 The clinical features observed in the siblings are in concordance with KS.
Fig. 3.

Pedigree showing inheritance pattern and individual partial Sanger sequences (below) showing biallelic variant c.2908C > T in exon 26 of COL18A1 in the affected siblings (III.1, III.2) (indicated by short black arrows). The variant is observed in heterozygous state in both the parents (II.3, II.4) (indicated by long black arrows).
Discussion
Both brothers had high axial myopia, vitreoretinal degeneration, lens subluxation/dislocation, and smooth cryptless irides. Fundoscopy showed myopic disc with tessellations in both eyes and chorioretinal atrophic patches. Both of them never had any regular formal ophthalmic checkup resulting in deprivational and refractive amblyopia. They underwent surgery as described earlier along with visual rehabilitation to restore the best possible vision as a part of their management. They required careful monitoring for associated comorbidities to prevent and/or manage further vision loss.
Although both siblings had almost similar eye findings, their CNS manifestations were clinically variable. The elder sibling showed learning difficulties without occipital encephalocele and milder neuroradiological findings––atretic encephalocele, large cisterna magna, and cerebellar vermis midline cleft, whereas the younger sibling showed normal cognitive functions and relatively severe neurological findings––occipital encephalocele, large cisterna magna, cerebellar abnormality, and tiny cortical heterotopia. These distinct CNS findings among the brothers of this report evidence an intrafamilial variability and suggest that the presence or severity of the structural brain malformations does not always correlate with a low cognitive function. Similar instances of KS patients with normal cognitive functions in spite of structural brain malformations has been reported. 2 13 18 However, both of them require follow-up close monitoring for seizures, ataxia, and cognitive decline. Up to date, four patients (two adults, one child, and one fetus) have been reported with cerebellar lesions (cerebellar atrophy, cerebellar vermis atrophy, complete agenesis of cerebellar vermis, and abnormal cerebellar hemispheres). 3 5 10 Two adults with cerebellar atrophy had late-onset ataxia; however, it is not clear whether cerebellar atrophy was congenital or if show age-dependent progression, and thus, this needs further studies on more adults. 3 Nystagmus was reported in one of the four patients with KS (NG1426-1—13 years old female) in the absence of cerebellar lesions, whereas the other with cerebellar vermis atrophy had no cerebellar signs at the age of 13 years. 5 Cerebellar vermian (midline cleft) observations in our patients appear unique since changes are focal and limited to around mid-vermian region. These changes may be at the lesser end of severity compared with the cases in earlier reports. 5
Frequently reported facial features in KS include midface retrusion, depressed nasal bridge, epicanthic folds, and micrognathia, 8 10 and were found in the siblings reported here, except for epicanthic folds.
Our patients showed the homozygous pathogenic variant c.2908C > T in exon 26 of COL18A1 , previously reported by Carss et al (2017); however, no clinical details are clearly mentioned for this mutation in the article, other than inherited retinal dystrophy. 19 Although parental consanguinity is not present in this family (as we expect more frequent homozygous mutations in probands born to consanguineous parents), homozygous mutation here might represent isolated population and high inbreeding practices among different population. Similar situation in a family with homozygous recurrent hotspot mutation in COL18A1 born to nonconsanguineous parents from North India has been reported. 3 Pathogenic variant c.2908C > T mutation, like most of other mutation reported in KS, results in truncated protein and predicted to affect all the isoforms including endostatin, explaining severe ocular manifestations in both siblings. The chance for the parents of having another child with KS is 25%. The presence or absence of structural brain malformations and genotypes does not absolutely predict cognitive functions. 2 18 However, posterior fossa abnormalities may be predictive of development of ataxia at the later age and needs further studies.
Conclusion
In conclusion, we report two siblings with KS due to known homozygous pathogenic variant in COL18A1 with early-onset severe ocular manifestations, variable CNS manifestations, and facial dysmorphism.
Acknowledgments
We are thankful to the family for the participation in the study. We are thankful to Indian Council of Medical Research for funding.
Funding Statement
Funding This study was funded by Indian Council of Medical Research (File No 5/7/1508/2016).
Footnotes
Conflict of Interest None declared.
References
- 1.Suzuki O T, Sertié A L, Der Kaloustian V M. Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am J Hum Genet. 2002;71(06):1320–1329. doi: 10.1086/344695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kliemann S E, Waetge R T, Suzuki O T, Passos-Bueno M R, Rosemberg S. Evidence of neuronal migration disorders in Knobloch syndrome: clinical and molecular analysis of two novel families. Am J Med Genet A. 2003;119A(01):15–19. doi: 10.1002/ajmg.a.20070. [DOI] [PubMed] [Google Scholar]
- 3.Paisán-Ruiz C, Scopes G, Lee P, Houlden H. Homozygosity mapping through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(07):993–997. doi: 10.1002/ajmg.b.30929. [DOI] [PubMed] [Google Scholar]
- 4.Khan A O, Aldahmesh M A, Mohamed J Y, Al-Mesfer S, Alkuraya F S. The distinct ophthalmic phenotype of Knobloch syndrome in children. Br J Ophthalmol. 2012;96(06):890–895. doi: 10.1136/bjophthalmol-2011-301396. [DOI] [PubMed] [Google Scholar]
- 5.Caglayan A O, Baranoski J F, Aktar F. Brain malformations associated with Knobloch syndrome--review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatr Neurol. 2014;51(06):806–8.13E10. doi: 10.1016/j.pediatrneurol.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hull S, Arno G, Ku C A. Molecular and clinical findings in patients with Knobloch syndrome. JAMA Ophthalmol. 2016;134(07):753–762. doi: 10.1001/jamaophthalmol.2016.1073. [DOI] [PubMed] [Google Scholar]
- 7.Corbett M A, Turner S J, Gardner A. Familial epilepsy with anterior polymicrogyria as a presentation of COL18A1 mutations. Eur J Med Genet. 2017;60(08):437–443. doi: 10.1016/j.ejmg.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Sertié A L, Sossi V, Camargo A A, Zatz M, Brahe C, Passos-Bueno M R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome) Hum Mol Genet. 2000;9(13):2051–2058. doi: 10.1093/hmg/9.13.2051. [DOI] [PubMed] [Google Scholar]
- 9.Balikova I, Sanak N S, Fanny D. Three cases of molecularly confirmed Knobloch syndrome. Ophthalmic Genet. 2020;41(01):83–87. doi: 10.1080/13816810.2020.1737948. [DOI] [PubMed] [Google Scholar]
- 10.Keren B, Suzuki O T, Gérard-Blanluet M. CNS malformations in Knobloch syndrome with splice mutation in COL18A1 gene. Am J Med Genet A. 2007;143A(13):1514–1518. doi: 10.1002/ajmg.a.31784. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki O, Kague E, Bagatini K. Novel pathogenic mutations and skin biopsy analysis in Knobloch syndrome. Mol Vis. 2009;15:801–809. [PMC free article] [PubMed] [Google Scholar]
- 12.Charsar B A, Goldberg E M. Polymicrogyria and intractable epilepsy in siblings with Knobloch syndrome and homozygous mutation of COL18A1. Pediatr Neurol. 2017;76:91–92. doi: 10.1016/j.pediatrneurol.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Passos-Bueno M R, Marie S K, Monteiro M. Knobloch syndrome in a large Brazilian consanguineous family: confirmation of autosomal recessive inheritance. Am J Med Genet. 1994;52(02):170–173. doi: 10.1002/ajmg.1320520209. [DOI] [PubMed] [Google Scholar]
- 14.Girisha K M, Shukla A, Trujillano D. A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet. 2016;90(06):536–539. doi: 10.1111/cge.12762. [DOI] [PubMed] [Google Scholar]
- 15.Shukla A, Hebbar M, Srivastava A. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017;62(07):723–727. doi: 10.1038/jhg.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ACMG Laboratory Quality Assurance Committee . Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White R J, Wang Y, Tang P, Montezuma S R. Knobloch syndrome associated with polymicrogyria and early onset of retinal detachment: two case reports. BMC Ophthalmol. 2017;17(01):214. doi: 10.1186/s12886-017-0615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NIHR-BioResource Rare Diseases Consortium . Carss K J, Arno G, Erwood M. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100(01):75–90. doi: 10.1016/j.ajhg.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]