

Abstract
The SCN encephalopathies are one of the rare early childhood intractable epileptic encephalopathies associated with pleomorphic seizures, cognitive decline, motor, and behavioral abnormalities that begin in early infancy. There is a dearth of data on phenotype and genotype of SCN encephalopathies from the Indian subcontinent, hence we are reporting clinical and molecular profile and outcome of SCN developmental and epileptic encephalopathies. This is a retrospective chart review of SCN developmental and epileptic encephalopathies in a tertiary care center, Bangalore, India between January 2015 and March 2020. All children with clinical features of SCN developmental and epileptic encephalopathies and confirmed with pathogenic variants were included. A total of 50 cases of SCN developmental and epileptic encephalopathies were analyzed, 31 of them were male and the mean age of presentation was 7.8 months. Precipitating factors for the first episode of seizure were fever and vaccination accounting for 33 and 8 children, respectively. Forty (80%) children had prolonged seizures and 15 (30%) had epileptic spasms. All children had a normal birth history and normal development before the onset of seizures, which was followed by developmental delay and regression. Thirty (60%) children had behavioral difficulties, notable hyperactivity, and autistic features. Neuroimaging and the initial electroencephalogram (EEG) were normal in all patients. The mean age of abnormal EEG was 14 months. The various subtypes of SCN variants were SCN1A in 31 children followed by SCN2A and SCN9A in eight children each and SCN1B in three children. Frameshift and nonsense mutations were associated with more severe phenotype and poor outcome compared with missense mutations. Thirty-four patients partially responded to treatment and the rest were refractory. The results of genetic testing were used to guide treatment; sodium channel blocking antiepileptic drugs were discontinued in 15 patients and sodium channel blocking agents were started in 3 patients with partial response. Three out of four children on stiripentol had a partial response. The SCN developmental and epileptic encephalopathies can present with epileptic spasms in addition to other types of seizures. Epileptic spasms are more common in nonsense and frameshift mutations. The outcome is poor in children with epileptic spasms compared with those without epileptic spasms. Genetic testing helps to select antiepileptic drugs that lead to seizure reduction.
Keywords: Dravet syndrome, SCN developmental and epileptic encephalopathies , SCN mutation , epileptic spasms
Introduction
The SCN gene encoding the neuronal voltage-gated sodium channel, is associated with several epilepsy syndromes. The SCN mutations represent the archetypal channelopathy associated with a wide phenotypic spectrum of epilepsies ranging from relatively milder form of febrile seizures, genetic epilepsy with febrile seizures plus (GEFS + ), to a more severe epileptic condition known as Dravet syndrome (DS) 1 or its variants called severe myoclonic epilepsy, borderline 2 and intractable childhood epilepsy with generalized tonic-clonic seizures 3 . Both of these variant types lack one or more features of the classical DS. SCN mutations are also associated with other forms of developmental and epileptic encephalopathies (DEEs) such as SCN1A -related DEE, SCN2A encephalopathy, and SCN8A epileptic encephalopathy which are distinctive and far more severe than DS. DS spectrum disorders account for the majority of the reported cases with the SCN gene mutations.
DS begins in the first year of life with tonic-clonic seizures triggered by fever followed by refractory myoclonic seizures. 4 Some drugs such as carbamazepine, lamotrigine, phenobarbital, and phenytoin can exacerbate epilepsy. 5 Early psychomotor development is normal before seizures onset, but developmental stagnation occurs later. 6
Early infantile SCN1A encephalopathy can be readily distinguished from DS by several features. It has a younger age at onset, beginning at 3 months compared with the typical seizure onset age range of 4 to 15 months in DS. It is associated with profound developmental impairment rather than the severe to mild intellectual disability usually seen in DS. Other features differentiating early infantile SCN1A encephalopathy from DS are epileptic spasms, and a severe hyperkinetic movement disorders which are not seen in DS. 7 The SCN2A encephalopathy can present as Ohtahara and West syndrome, epilepsy of infancy with migrating focal seizures, with severe developmental impairment, movement disorders, axial hypotonia, severe gastrointestinal symptoms and responds to sodium channel blockers. 8
The recent review by Mei et al 9 concluded that the clinical spectrum of the DS does not have firmly established boundaries. Recent evidence also suggests that the nature of the mutation may affect the phenotype. 4 The missense mutations are associated with less severe phenotypes as compared with truncating mutations, however, correlations between specific mutation and phenotype remain weak. Early diagnosis is important for the selection of antiepileptic medications and prognostication. There is a lack of studies in the Indian subcontinent; hence we planned this report to expand the phenotype of SCN DEEs, phenotype–genotype correlation, and outcomes in South India.
Methods
This is a retrospective chart review of children with genetically confirmed SCN DEEs in a tertiary care referral center, from the southern part of India. The medical records of children attending the pediatric neurology clinic and those who were admitted to the pediatric neurology and pediatric medicine ward from January 2015 to March 2020 were analyzed. Children with clinical features of SCN DEEs and confirmed to have variants in the SCN gene were included. Clinical notes were reviewed to determine: age of seizure onset, family history, birth history, developmental history, seizure semiology, precipitants of seizures, and apparent response to antiepileptic drugs. MRI of the brain, electroencephalogram (EEG), and targeted next-generation sequencing (NGS) genetic analysis were reviewed.
Genetic testing was done by NGS and confirmed by Sanger sequencing. DNA extracted from blood was used to perform targeted gene capture using a custom capture kit. The libraries were sequenced to mean >80–100X coverage on the Illumina sequencing platform as per the manufacturer protocol. Sequences obtained were aligned to the human reference genome (GRCh37/hg19), QC, data mapping, variant calling, and annotation of variants with external and internal data sources was achieved with a customized Genome Analysis Tool kit (GATK) framework. Gene/variant annotation was achieved using the Visual evoked potentials (VEP) program against the Ensemble release 91 human gene model. The analyzed region includes the coding exons and ± 10 base pairs of flanking intronic region on both sides of each exon. Clinically relevant mutations were annotated using variants published in the literature and a set of diseases databases—ClinVar, Online Mendelian Inheritance in Man (OMIM) (updated on November 21, 2018), Genome-wide association study (GWAS), Human Gene Mutation Database (HGMD) (v2018.3), and SwissVar. Common variants are filtered based on allele frequency in 1000Genome Phase 3, ExAC (v1.0), gnomAD (v2.1), EVS, dbSNP (v151), 1000 Japanese Genome, and Indian population database. The nonsynonymous variant effect was calculated using multiple algorithms such as PolyPhen-2, SIFT, MutationTaster2, and LRT. Only nonsynonymous and splice site variants found in the targeted gene capture were used for the clinical interpretation and synonymous deep intronic variants that were not previously reported were not included in the analysis. QIAGEN Ingenuity Variant Analysis was used to identify variants that are relevant to the clinical indication and were classified as per the ACMG guidelines. The variants were classified based on the standard guidelines of ACMG-AMP and are scored based on the evidence and strength of each criterion as described in the ACMG guidelines.
Statistical Analysis
Categorical data are presented in the form of numbers and percentages whereas continuous data are presented as mean and standard deviation. The unpaired t -test was used to compare the difference between the two groups. A p -value of less than 0.05 was considered statistically significant. Statistical analysis was performed with SPSS version 21. Ethical clearance was obtained, from the Institutional Ethics Committee.
Results
The various clinical and laboratory features are mentioned in Table 1 . Fifty patients were included in this study and the male to female ratio was 1.6:1. All children had a normal birth history and a family history of seizures was present in 14% of children. The mean age of presentation was 7.8 months and first seizure occurred within 6 months of age in 68% of children. The precipitating factors for the first seizure were fever in 66%, vaccination in 16%, and there was no triggering event in 18% of children. History of prolonged seizures with status epilepticus was present in 80% of children and 30% (15) of children presented with epileptic spasms. All children had normal development before the onset of seizures, this was followed by a delay in development and cognitive decline in all children after the onset of seizures. Thirty (60%) children developed behavioral issues notable hyperactivity and autistic features at follow-up. The initial EEG was normal in all the children while follow-up EEGs were abnormal by the mean age of 14 months. Various subtypes of SCN variants were observed at different rates with the most frequent being those in SCN1A (31/50), followed by SCN2A (8/50) and SCN9A (8/50), then SCN1B (3/50) as shown in Figs. 1 and 2 .
Table 1. Showing clinical and laboratory parameters of SCN developmental and epileptic encephalopathies .
| Parameter | Number = 50 (%) |
|---|---|
| Male: Female | 31: 19 (62:38) |
| Mean age of presentation | 7.85 mo |
| Age of onset of first seizure within 6 mo | 34 (68%) |
| Precipitating factors for seizures | |
| • Fever | 33 (66%) |
| • Vaccination | 8 (16%) |
| • None | 9 (18%) |
| Normal development prior to the onset of seizures. | 50 (100%) |
| Family history of seizures | 7 (14%) |
| Presentation with epileptic spasms | 15 (30%) |
| Status epilepticus or prolonged seizures | 40 (80%) |
| Both febrile and afebrile seizures | 50 (100%) |
| Hyperactivity with autistic features | 30 (60%) |
| Mean age of first abnormal EEG | 14 mo |
| Partial response—more than 50% reduction in monthly seizure frequency. | 34 (68%) |
Fig. 1.
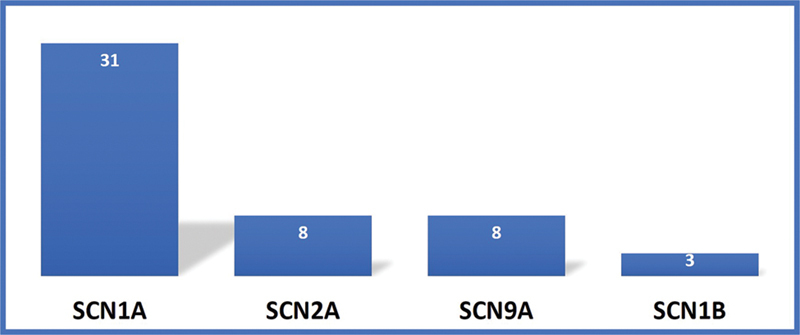
Showing various subtypes of SCN developmental and epileptic encephalopathies.
Fig. 2.
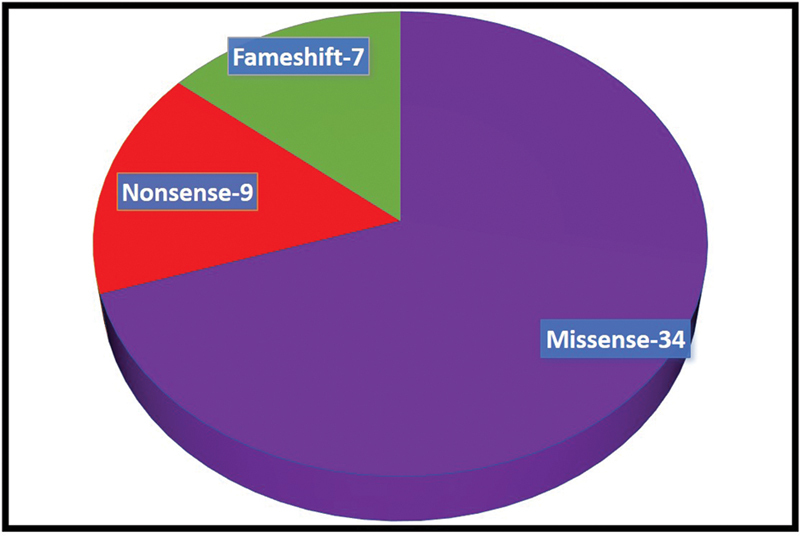
Showing type of mutations in SCN developmental and epileptic encephalopathies.
Of the four children on stiripentol three showed partial response with more than 50% reduction of seizures. Based on the results of genetic testing, sodium channel blockers were discontinued in 30% (15) of children after which a significant reduction in seizure frequency was noted ( Fig. 3 ). More than 90% reduction in seizure frequency was noted in 11 of 15 patients and more than 50% reduction in seizure frequency was noted in 4 of 15 after discontinuation of sodium channel blockers. Two children with SCN2A pathogenic variants responded to phenytoin and one child with SCN2A responded to lamotrigine with >50% reduction in seizures. Out of 15 children with epileptic spasms, 4 of the 15 (26%) children showed a response to steroids and 2 of the 15 (13%) children responded to vigabatrin. None of the children with epileptic spasms are ambulatory and all had autistic features. Frameshift and nonsense mutations are associated with more severe phenotype compared with missense mutation as shown in Table 2 , Figs. 4 and 5 .
Fig. 3.
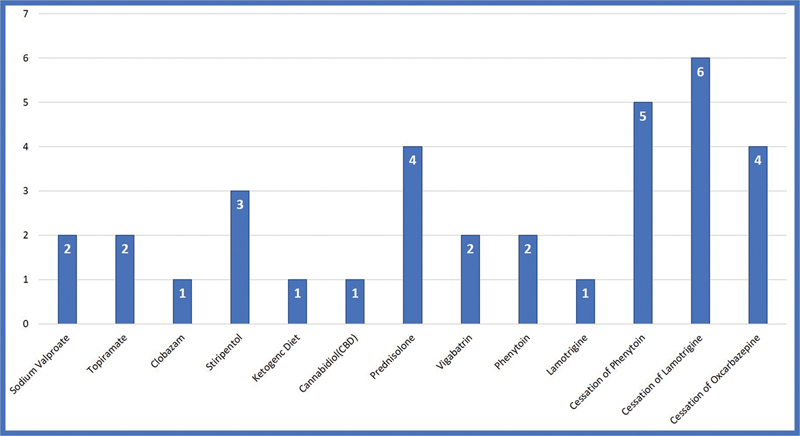
Showing response to antiepileptic drugs in SCN developmental and epileptic encephalopathies.
Table 2. Showing heterozygous variants in different subtypes of SCN developmental and epileptic encephalopathies.
| Case no (P) | Gene | Exon | Variant | ACMG classification | Mutation type |
Seizures | Treatment response |
Outcome |
|---|---|---|---|---|---|---|---|---|
| P1 | SCN2A | 2 | c.70G > A (p.Ala24Thr) |
VUS | M | FS, ES | TPM | Mod GDD, ASD |
| P2 | SCN1B | 3 | c.254G > A (p.Arg85His) |
LP | M | FS, ME | VPA | GDD, ADHD |
| P3 | SCN1B | 3 | c.265C > T (p.Arg89Cys) |
VUS (HOM) | M | FS, ME | Refractory | GDD, ADHD |
| P4 | SCN1A | 2 | c.302G > A (p.Arg101Gln) |
P | M | ME, AA LGS |
Steroids | Mild GDD |
| P5 | SCN1A | 2 | c.316_319delinsC p.Ser106_Ala107delinsPro |
VUS | M | FS, ES | Steroids | ASD, ADHD |
| P6 | SCN1A | 3 | c.442A > T (p.Ser148Cys) |
VUS | M | FS, ME | KD | GDD |
| P7 | SCN1B | 3 | c.560delG (p.Arg187ProfsTer52) |
VUS | F | FS, ME, ES | CLZ | GDD, ADHD |
| P8 | SCN9A | 4 | c.446C > G (p.Pro149Arg) |
VUS | M | FS, ME | VPA | GDD, ADHD |
| P9 | SCN1A | 4 | c.530G > A (p.Gly177Glu) |
LP | M | FS, ME | CBD | GDD |
| P10 | SCN1B | 4 | c.566C > T; (p.Thr189Met) |
VUS | M | FS, ME | LEV | GDD, ADHD |
| P11 | SCN1A | 4 | c.566C > T (p.Pro189Leu) | VUS | N | FS, ME, ES | Refractory | GDD |
| P12 | SCN1A | Intron 4 | c.602 + 1G > A | LP | F | FS, SE, ES | TPM | Severe GDD |
| P13 | SCN2A | 5 | c.605C > T (p.Ala202Val) |
LP | M | FS, ME | PHT | GDD, ADHD |
| P14 | SCN1A | 5 | c.628G > A (p.Gly210Ser) |
LP | M | FS, ME | Refractory | GDD |
| P15 | SCN9A | 5 | c.554G > A (p.Arg185His) | VUS | M | FS, ME | LEV | GDD/ASD |
| P16 | SCN2A | 6 | c.769T > C (p.Phe257Leu) |
VUS | M | Focal seizures | PHT | Mild GDD |
| P17 | SCN2A | 7 | c.781G > A (p.Val261Met) |
LP | M | FS, ME | LTB | GDD, ADHD |
| P18 | SCN2A | 7 | c.823C > T (p.Arg275Ter) |
P | N | FS, ES | VGB | Severe GDD, ASD |
| P19 | SCN1A | 6 | c.955C > T (p.Gln319Ter) |
P | N | FS, ES, ME | VPA | Mild GDD |
| P20 | SCN1A | 7 | c.983A > G (p.Glu328Gly) |
VUS | M | FS, ME | Steroids | Mod GDD |
| P21 | SCN1A | 7 | c.1006T > C (p.Cys336Arg) |
VUS | M | FS, ME | Cessation of PHT | Mild GDD |
| P22 | SCN1A | 8 | c.1082G > T (p.Gly361Val) | LP | M | FS, ME | TPM | ASD, ADHD |
| P23 | SCN1A | Intron 8 | c.1170 + 1G > A | P | N | FS, ES, ME | Cessation of PHT | Mild GDD |
| P24 | SCN1A | 10 | c.1614_1615delinsAT (p.Glu539Ter) |
P | N | FS, ME | Cessation of LTG | GDD |
| P25 | SCN1A | 11 | c.1702C > T (p.Arg568Ter) |
P | M | FS, ME | Refractory | GDD, ADHD |
| P26 | SCN1A | 11 | c.1780G > T (p.Glu594Ter) |
P | N | FS, ME | Polytherapy controlled | Severe GDD, ASD |
| P27 | SCN9A | 12 | c.1828C > A (p.Pro610Thr) |
P | M | FS, ME, ES | Cessation of PHT | GDD |
| P28 | SCN9A | 12 | c.1844A > G (p.Asn615Ser) |
VUS | M | FS, ME | Refractory | GDD, ADHD |
| P29 | SCN1A | Intron 14 | c.2505 + 3A > T | P | F | FS, ME, ES | Cessation of PHT | GDD |
| P30 | SCN1A | 14 | c.2588T > A (p.Leu863Ter) |
P | N | FS, ME, ES | Refractory | GDD, ADHD |
| P31 | SCN2A | 16 | c.2819G > A (p.Cys940Tyr) |
VUS | M | FS, ME | Refractory | GDD |
| P32 | SCN1A | 15 | c.2712dupT (p.Ala905Cysfs Ter10) |
P | F | FS, ME | Cessation of LTG | GDD, ADHD |
| P33 | SCN1A | 16 | c.3199G > A (p.Ala1067Thr) |
VUS | M | FS, ME | Cessation of LTG | GDD, ADHD |
| P34 | SCN1A | 16 | c.3199G > A (p.Ala1067Thr) |
VUS | M | FS, SE, ES | VGB | Severe GDD, ASD |
| P35 | SCN1A | 16 | c.3199G > A (p.Ala1067Thr) |
VUS | M | FS, SE, ES | STP | Severe GDD, ASD |
| P36 | SCN1A | 16 | c.5030T > C (p.Leu1677Pro) | VUS | M | Febrile Focal | Ref/none | GDD, ASD |
| P37 | SCN1A | 19 | c.3724_3725dupTA(p.Asp1243LeufsTer28) | P | F | FS, ME | VPA | Mild GDD |
| P38 | SCN9A | 21 | c.3832C > C/T (p.Leu1278Phe) |
VUS | M | FS, ME | Refractory | GDD |
| P39 | SCN1A | 21 | c.4073G > T (p.Trp1358Leu) | LP | M | FS, ME | STP | GDD, ADHD |
| P40 | SCN1A | Intron 22 | c.4338 + 1G > A | P | M | FS, ME | TPM | GDD, ASD |
| P341 | SCN1A | 19 | c.3851G > C (p.Trp1284Ser) |
P | M | FS, SE | Steroids | GDD, ASD |
| P42 | SCN2A | 23 | c.4303C > T (p.Arg1435Ter) |
P | N | FS, ME, ES | PHT | GDD, ADHD |
| P43 | SCN1A | 22 | c.4312A > T (p.Met1438Leu) |
VUS | F | FS, ME | Cessation of PHT | GDD |
| P44 | SCN1A | 22 | c.4313T > G (p.Met1438Arg) |
VUS | M | FS, ME | Refractory | GDD, ADHD |
| P45 | SCN1A | 27 | c.4796_4798delATT(p.Tyr1599del) | LP | F | FS, ME | Cessation of LTG | GDD |
| P46 | SCN1A | 26 | c.4855A > G (p.Met1619Val) |
VUS | M | FS, ME | Cessation of LTG | GDD, ADHD |
| P47 | SCN1A | 26 | c.4907G > A (p.Arg1636Gln) |
LP | M | FS, ME | Refractory | GDD |
| P48 | SCN9A | 27 | c.4999A > G (p.Met1667Val) |
VUS | M | FS, ME | Cessation of LTG | GDD, ADHD |
| P49 | SCN2A | 27 | c.5397T > G (p.Tyr1799Ter) |
VUS | N | ME, GTCS, FS | LTG | GDD, ASD |
| P50 | SCN9A | 27 | c.5734G > T; (p.Asp1924Tyr) |
VUS | M | Focal, ES | STP | GDD, ADHD |
Abbreviations: AA, Atypical absences; ACTH, Adrenocorticotropic hormone; ADHD, attention-deficit hyperactivity disorder; ASD, autistic spectrum disorder; CBD, cannabidiol; CLB, clobazam; ES, epileptic spasms; ES, epileptic spasms; F, frame shift; GDD, global developmental delay; GTCS, generalized tonic, clonic seizures; KD, ketogenic diet; LEV, levetiracetam; LP, likely pathogenic; LTG, lamotrigine; ME, myoclonic epilepsy; M, missense; N, nonsense; PHT, phenytoin; P, pathogenic; STP, stiripentol; TPM, topiramate; VGB, vigabatrin; VPA, valproic acid; VUS, variant of uncertain significance.
Note: SCN1A - NM_006920.6; NM_001353948.2; NM_001353960.2; NM_001202435.3; NM_001353955.2; NM_001165963.3; NM_001353961.2; SCN2A- NM_001040143.2; SCN9A - NM_002977.3; SCN1B - NM_199037.5; NM_001037.5.
Fig. 4.
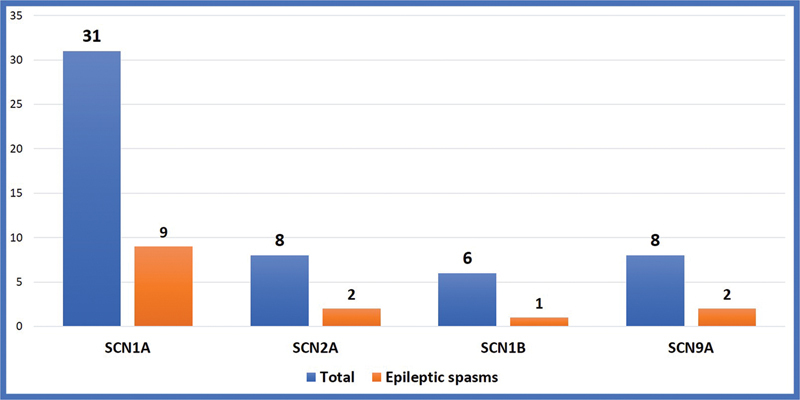
Showing epileptic spasms in various subtypes of SCN mutations in SCN developmental and epileptic encephalopathies.
Fig. 5.
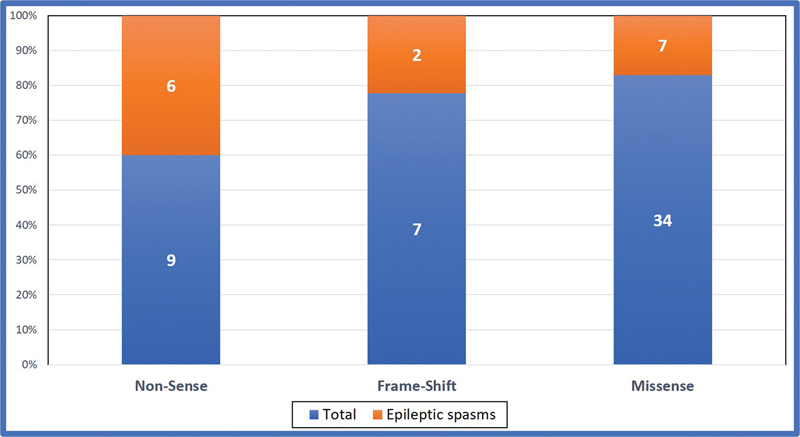
Showing epileptic spasms in various types of SCN mutations in SCN developmental and epileptic encephalopathies.
Three novel variants were noted, two in SCN1A and one in SCN9A. The NM_001165963.3 c.316_319delTCTGinsC (p.Ser106_Ala107delinsPro) variant in SCN1A was associated with epileptic spasms, refractory seizures with autistic features. The NM_006920.6 c.955C > T (p.Gln319Ter) variant was associated with mild developmental delay, and seizures controlled with valproate, while the NM_002977.3 c.446C > G (p.Pro149Arg) variant was associated with significant developmental delay and attention-deficit hyperactivity disorder (ADHD) and the seizures were controlled with valproate.
Discussion
DEEs are a heterogeneous group of rare neurodevelopmental disorders, characterized by (1) early-onset seizures that are often intractable, (2) electroencephalographic abnormalities, (3) developmental delay or regression, and (4) in some cases, early death. 10 One of the most well-characterized DEEs is DS. The SCN1A gene mutations are thought to represent a genetic form of early-onset epileptic encephalopathies with a common designation as DS. 11 Although DS is inherited in an autosomal dominant pattern most cases are the result of de novo mutations. A strong indicator for SCN gene analysis is an epileptic encephalopathy with seizure onset before 1 year of age, even if cognitive decline does not occur for several years thereafter. Diagnosis of SCN DEEs is based on age at onset, seizure types, clinical course, and confirmed by variants detected by NGS in the SCN gene. There is a lack of studies in India on SCN encephalopathies, hence we are reporting this series of SCN DEEs.
In this study out of 50 cases, males were more affected accounting for 62% of the cases with a male to female ratio of 2:1. 6 The mean (standard deviation) age of the first seizure was 7.8 (9.40) months, with a range of 2 to 10 months, and is characterized by a generalized or unilateral clonic seizure. Thirty-four (68%) children presented with first seizure within the first 6 months of age. In a study of SCN1A mutation by Cetica et al, the mean age at first seizure was 5.19 months. Of the patients who experienced their first seizure after 12 months of age none developed Dravet syndrome. 12 Fever was the most common trigger for initial seizures, followed by vaccination accounting for 66 and 16% of children, respectively. In a study of 53 patients by Caraballo and Fejerman, the first seizure was associated with febrile illness in 75.5% and vaccination in 24.5% of children 13 while vaccine triggered seizures were noted in 27% of children with DS in the Tro-Baumann et al, study. 14
A family history of epilepsy or febrile seizures was reported in approximately 25% of cases. 4 This is consistent with our findings where we show that 14% of children had a family history of seizures although none had the DS phenotype. Polymorphic seizure semiology emerges by age 2 years and may include focal and generalized myoclonus, and atypical absences. 15 Similar to previous studies, we observed diverse of seizure types, including generalized tonic, clonic seizures, myoclonic jerks, atypical absence seizures, epileptic spasms, and focal seizures. Development is normal at the onset, with a plateau and progressive decline between 1 and 4 years of age, typically occurring in the second year of life. 5 The degree of neurobehavioral impairment is reported to range from minor learning difficulty to global developmental delay. 16 Similarly, we note initial normal development followed by regression in all the children ( Table 3 ).
Table 3. Comparison of our data with previously reported studies SCN developmental and epileptic encephalopathies.
| Study | No | Age in mo. (mean) |
Clinical features | Gene | Treatment response | Outcome | Phenotype genotype correlation |
|---|---|---|---|---|---|---|---|
| Verheyen et al 17 |
33 | 9–48 (33.2 mo) | PS, GDD | SCN 1A-33 | NR | Severe GDD | Later age of walking is associated with low IQ |
| Ragona et al 18 |
37 | 3–11 (5.7 ± 2.1) |
FS, PS | SCN 1A 84% | NR | ID-33, Behavioral issues-21 | High seizure frequency associated with low IQ |
| Caraballo and Fejerman 13 | 53 | 3–12 (6 mo) |
GTCS, ME, FS, AA |
NR | KD, TPM, STP | ID-53, ADHD-45, ASD-2 |
NR |
| Kapoor et al 19 |
9 | Mean: 5 | FS, SE, PS | SCN1A-8 SCN1B-1 |
Multiple AED |
GDD-9, ASD 4, ADHD-3 | NR |
| Do et al 20 |
18 | 3–10 (7 ± 1) |
FS, PS | SCN1A-12 | CLB, TPM, VPA, LEV |
Low IQ | No correlations |
| Alame et al 21 |
8 | 6–18 | GTCS, FS, ME | SCN1A-7 | VPA, TPM, LEV, PB |
ID-4, Death-2, NA-2 | No correlation |
| Villeneuve et al 22 | 21 | Mean (6 ± 1) |
SE, GTCS, FS |
SCN1A-19 | NR | ID-19, Behavioral issues-6 | NaV1–1 dysfunction had a negative effect on IQ |
| Petrelli et al 23 |
25 | Mean (5.5 ± 2.2) |
GTCS, FS, SE | SCN1A-17 | NR | ID-16 | SCN1A-severe phenotype |
| Li et al 24 |
37 | Mean (9.3 ± 3.6) | GTCS, FS, ME, AA, | NR | TPM, VPA, CZP |
ASD-9 ID–95% |
ASD is associated with low IQ |
| Current study | 50 | Mean (8.1) |
FTS, GTCS, FS, ES ME, AA | SCN1A-31 SCN2A-8 SCN9A-8 SCN1B-3 |
VPA, CLB, TPM, STP, CBD, KD VGB, ACTH |
ID-50, ADHD-20, ASD-10 |
ES are associated with poor outcome |
Abbreviations: ACTH, adrenocorticotropic hormone; ADHD, attention-deficit hyperactivity disorder; ASD, autistic spectrum disorder; CBD, cannabidiol; CLB, clobazam; CZP, clonazepam; ES, epileptic spasms; ES, epileptic spasms; FS, febrile seizures; GDD, global developmental delay; GTCS, generalized tonic, clonic seizures; ID, intellectual disability; KD, ketogenic diet; LEV, levetiracetam; LTG, lamotrigine; ME, myoclonic epilepsy; NR, not reported; PB, phenobarbital; PHT, phenytoin; PS, pleomorphic seizures; STP, stiripentol; TPM, topiramate; VGB, vigabatrin; VPA, valproic acid.
Consistent with other studies we show that the initial EEG is usually normal, however, abnormalities become evident with recurrent seizures and include generalized, focal, or multifocal anomalies, such as multifocal spikes, spike and waves, polyspike and waves discharges, and a slowing of background activity. 25
In a recent review 26 of 58 brain MRIs in patients with SME, 22.4% of the patients showed abnormal findings consistent with cortical brain atrophy, cerebellar atrophy, hippocampal sclerosis, focal cortical dysplasia, and ventricular enlargement. Our study showed normal neuroimaging in all the children. We have not done a repeat MRI after the confirmation of the diagnosis to look for any abnormal findings at follow-up.
Around 70 to 80% of children with epileptic encephalopathy and severe myoclonic epilepsy of infancy have point mutations or gross rearrangements in the SCN1A gene. In our study, 34 (68%) of them were associated with a missense mutation. Over 1,200 variants associated with epilepsy have been reported in SCN1A . 27 In this study 31 (62%) were due to SCN1A mutations. Truncating loss-of-function variants are almost always associated with severe phenotypes, proving that haploinsufficiency of SCN1A is pathogenic, whereas missense variants are associated with a wide spectrum of phenotypes that vary from DS to much milder forms of epilepsy, such as febrile seizures and “febrile seizures plus,” which are often familial and part of GEFS + . 28 In the current study epileptic spasms are commonly seen in nonsense mutation 66.6%, and frame-shift mutation (28.5%), and less common in missense mutation (20.5%). No significant differences were noted in epileptic spasms in different subtypes of SCN mutation as shown in Fig. 4 . Less than 1% of DS patients have a homozygous mutation in SCN1B , and very few have GABRG2 or SCN2A mutations. 29 In this study, SCN1B variants were noted in 6% (3) of patients which is higher than previously reported. SCN2A and SCN8A -linked encephalopathies are associated with gain-of-function mutations and possible response to sodium channel blockers . 30 In this study, three patients with SCN2A mutations responded to sodium channel blockers. Their phenotype was more heterogeneous, began earlier usually in the neonatal period and was associated with epileptic spasms and poor outcomes compared with other mutations.
We noted three novel mutations of which p.Ser106_Ala107delinsPro in SCN1A was associated with epileptic spasms, refractory seizures with autistic features, c.955C > T (p.Gln319Ter) was associated with mild developmental delay, and seizures were well controlled with valproate, while c.446C > G (p.Pro149Arg) was associated with significant developmental delay, ADHD, and seizures were partially controlled with valproate.
The most common etiology identified in cases with the epileptic encephalopathy severe myoclonic epilepsy of infancy is de novo, heterozygous, a loss-of-function variant in SCN1A seen in 70 to 80% of children. 31 In our study SCN1A mutation was the most common and was seen in 56% of children which is comparable with other studies. DS secondary to SCN2A mutation has been reported in the literature 32 and accounted for 16% of children in our study group. Association of SCN1A pathogenic missense variant with epileptic spasms was reported by Wallace et al in 2003 33 while SCN2A and SCN8A gene mutations in association with the West syndrome were reported by Nakamura et al. 34 In our study, 15 (30%) cases presented with epileptic spasms in clusters and associated with hypsarrhythmia, the mean age of presentation of spasms was 3.5 months. The subtypes of mutations in epileptic spasms are SCN1 in nine, SCN2A in thee, SCN 9A in two, and SCN1B in one case. These children had a more severe phenotype and severe developmental delay. None of the parents in our study had the same genotype as the child. Our study highlights epileptic spasms as a presenting feature in Dravet syndrome which was seen in 30% of the children.
Common comorbidities that develop after seizure onset include intellectual disability, gait abnormalities, and behavioral concerns manifesting as ADHD or autistic traits, aggressiveness, irritability, relational difficulties, and oppositional behaviors. A study by Li et al 24 suggested that nearly one-quarter of the patients with DS had autism and almost 95% showed intellectual disability which correlates with our study showing behavioral issues in 60% of children of which, ADHD was found in 40%, autism was found in 20% of children followed by developmental regression in 100% of children after the onset of seizures.
The outcome is poor and, after 4 years of age, patients usually reach a steady state of intractable seizures, intellectual impairment, behavioral disorders, and neurological abnormalities. Myoclonic seizures usually cease and are replaced with nocturnal generalized clonic or absence seizures 4 The number of seizures is a risk factor for the degree of developmental regression. The mortality rate is approximately 16% and is related to prolonged convulsive seizures, drowning, and sudden unexpected death. 4 In our study, none of the children expired. The limitation of our study is that it is a retrospective chart review.
Conclusion
The SCN DEEs should be considered in children with refractory epilepsy, onset in infancy with recurrent febrile seizures, or febrile status epilepticus with normal birth history and development before the onset of seizures. They can present with epileptic spasms in addition to other types of seizures and the developmental outcome is poor compared with those without epileptic spasms. Epileptic spasms are common in nonsense and frameshift mutations. Genetic testing enables early identification of the disease and influences the selection of antiepileptic drugs and hence outcome.
Conflict of Interest None declared.
Authors' Contribution
V.K.G. did the supervision, guidance, and reviewed the manuscript. M.B. was involved in the management of the children and the preparation of the manuscript. H.V. was involved in the preparation of the manuscript. V.M.S. was involved in the diagnosis and the preparation of the manuscript. S.K.C. was involved in the diagnosis and the preparation of the manuscript. A.M. has given valuable inputs in the management of children. S.K.S. has given valuable inputs in the management of children. N.B. was involved in the diagnosis and the preparation of the manuscript.
References
- 1.Scheffer I E, Nabbout R. SCN1A -related phenotypes: Epilepsy and beyond . Epilepsia. 2019;60 03:S17–S24. doi: 10.1111/epi.16386. [DOI] [PubMed] [Google Scholar]
- 2.Hattori J, Ouchida M, Ono J. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia. 2008;49(04):626–633. doi: 10.1111/j.1528-1167.2007.01475.x. [DOI] [PubMed] [Google Scholar]
- 3.Fujiwara T, Sugawara T, Mazaki-Miyazaki E. Mutations of sodium channel alpha subunit type 1 ( SCN1A ) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures Brain 2003126(Pt 3):531–546. [DOI] [PubMed] [Google Scholar]
- 4.Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol. 2005;95:71–102. [PubMed] [Google Scholar]
- 5.Brunklaus A, Ellis R, Reavey E, Forbes G H, Zuberi S M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome Brain 2012135(Pt 8):2329–2336. [DOI] [PubMed] [Google Scholar]
- 6.Incorpora G. Dravet syndrome. Ital J Pediatr. 2009;35(01):27. doi: 10.1186/1824-7288-35-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parihar R, Ganesh S. The SCN1A gene variants and epileptic encephalopathies . J Hum Genet. 2013;58(09):573–580. doi: 10.1038/jhg.2013.77. [DOI] [PubMed] [Google Scholar]
- 8.Howell K B, McMahon J M, Carvill G L. SCN2A encephalopathy: a major cause of epilepsy of infancy with migrating focal seizures . Neurology. 2015;85(11):958–966. doi: 10.1212/WNL.0000000000001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia. 2019;60 03:S2–S7. doi: 10.1111/epi.16054. [DOI] [PubMed] [Google Scholar]
- 10.Nieh S E, Sherr E H. Epileptic encephalopathies: new genes and new pathways. Neurotherapeutics. 2014;11(04):796–806. doi: 10.1007/s13311-014-0301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carranza Rojo D, Hamiwka L, McMahon J M. De novo SCN1A mutations in migrating partial seizures of infancy . Neurology. 2011;77(04):380–383. doi: 10.1212/WNL.0b013e318227046d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cetica V, Chiari S, Mei D. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations . Neurology. 2017;88(11):1037–1044. doi: 10.1212/WNL.0000000000003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caraballo R H, Fejerman N. Dravet syndrome: a study of 53 patients. Epilepsy Res. 2006;70 01:S231–S238. doi: 10.1016/j.eplepsyres.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 14.Tro-Baumann B, von Spiczak S, Lotte J. A retrospective study of the relation between vaccination and occurrence of seizures in Dravet syndrome. Epilepsia. 2011;52(01):175–178. doi: 10.1111/j.1528-1167.2010.02885.x. [DOI] [PubMed] [Google Scholar]
- 15.Oguni H, Hayashi K, Osawa M. Severe myoclonic epilepsy in infancy: clinical analysis and relation to SCN1A mutations in a Japanese cohort . Adv Neurol. 2005;95:103–117. [PubMed] [Google Scholar]
- 16.Korff C, Laux L, Kelley K, Goldstein J, Koh S, Nordli D., Jr Dravet syndrome (severe myoclonic epilepsy in infancy): a retrospective study of 16 patients. J Child Neurol. 2007;22(02):185–194. doi: 10.1177/0883073807300294. [DOI] [PubMed] [Google Scholar]
- 17.Verheyen K, Wyers L, Del Felice A, Schoonjanset A-S. Independent walking and cognitive development in preschool children with Dravet syndrome. 2021;63(04):472–479. doi: 10.1111/dmcn.14738. [DOI] [PubMed] [Google Scholar]
- 18.Ragona F, Brazzo D, De Giorgi I. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients. Brain Dev. 2010;32(01):71–77. doi: 10.1016/j.braindev.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Kapoor D, Anand A, Sharma S. Dravet syndrome: a case series. Indian J Pediatr. 2021;88(01):82. doi: 10.1007/s12098-020-03383-z. [DOI] [PubMed] [Google Scholar]
- 20.Do T T, Vu D M, Huynh T T. SCN1A gene mutation and adaptive functioning in 18 Vietnamese children with Dravet syndrome . J Clin Neurol. 2017;13(01):62–70. doi: 10.3988/jcn.2017.13.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alame S, El-Houwayek E, Nava C, Sabbagh S, Fawaz A, Gillart AC, Hasbini D, Depienne C, Mégarbané A.Dravet Syndrome in Lebanon: First Report on Cases with SCN1A Mutations, Case Rep Med. 2019. Jan 21;2019:5270503. doi: 10.1155/2019/5270503. PMID: 30805006; PMCID: PMC6360541. [DOI] [PMC free article] [PubMed]
- 22.Villeneuve N, Laguitton V, Viellard M. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav. 2014;31:143–148. doi: 10.1016/j.yebeh.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 23.Petrelli C, Passamonti C, Cesaroni E. Early clinical features in Dravet syndrome patients with and without SCN1A mutations Epilepsy Res 201299(1-2):21–27. [DOI] [PubMed] [Google Scholar]
- 24.Li B M, Liu X R, Yi Y H. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 2011;21(03):291–295. doi: 10.1016/j.yebeh.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 25.Dravet C. Severe myoclonic epilepsy in infants and its related syndromes. Epilepsia. 2000;41 09:7–10. doi: 10.1111/j.1528-1157.2000.tb02210.x. [DOI] [PubMed] [Google Scholar]
- 26.Striano P, Mancardi M M, Biancheri R. Brain MRI findings in severe myoclonic epilepsy in infancy and genotype-phenotype correlations. Epilepsia. 2007;48(06):1092–1096. doi: 10.1111/j.1528-1167.2007.01020.x. [DOI] [PubMed] [Google Scholar]
- 27.Catterall W A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(01):13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 28.Meng H, Xu H Q, Yu L. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype . Hum Mutat. 2015;36(06):573–580. doi: 10.1002/humu.22782. [DOI] [PubMed] [Google Scholar]
- 29.Catterall W A. Bethesda, MD: National Center for Biotechnology Information; 2012. Sodium channel mutations and epilepsy; pp. 675–687. [PubMed] [Google Scholar]
- 30.Meisler M H, Helman G, Hammer M F. SCN8A encephalopathy: research progress and prospects . Epilepsia. 2016;57(07):1027–1035. doi: 10.1111/epi.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukuma G, Oguni H, Shirasaka Y. Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB) . Epilepsia. 2004;45(02):140–148. doi: 10.1111/j.0013-9580.2004.15103.x. [DOI] [PubMed] [Google Scholar]
- 32.Ogiwara I, Ito K, Sawaishi Y. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies . Neurology. 2009;73(13):1046–1053. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallace R H, Hodgson B L, Grinton B E. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003;61(06):765–769. doi: 10.1212/01.wnl.0000086379.71183.78. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura K, Kato M, Osaka H. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome . Neurology. 2013;81(11):992–998. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]