

Abstract
Acetaminophen (APAP) overdose causes liver injury in animals and humans. Although well-studied in animals, limited longitudinal data exist on cytokine release after APAP overdose in patients. The purpose of this study was to quantify concentrations of cytokines in APAP overdose patients to determine if early cytokine or complement measurements can distinguish between surviving and non-surviving patients. Plasma was obtained from healthy controls, APAP overdose patients with no increase in liver transaminases, and surviving and non-surviving APAP overdose patients with severe liver injury. Interleukin-10 (IL-10), and CC chemokine ligand-2 (CCL2, MCP-1) were substantially elevated in surviving and non-surviving patients, whereas IL-6 and CXC chemokine ligand-8 (CXCL8, IL-8) had early elevations in a subset of patients only with liver injury. Day 1 IL-10 and IL-6 levels, and Day 2 CCL2, levels correlated positively with survival. There was no significant increase in IL-1α, IL-1β or TNF-α in any patient during the first week after APAP. Monitoring cytokines such as CCL2 may be a good indicator of patient prognosis; furthermore, these data indicate the inflammatory response after APAP overdose in patients is not mediated by a second phase of inflammation driven by the inflammasome.
Keywords: Acetaminophen, Liver inflammation, Complement, Neutrophils, Monocytes, Acute liver failure
1. Introducton
Acetaminophen (APAP) is an effective analgesic and antipyretic drug for adults and children. It is considered a safe drug when used at therapeutic doses. However, an overdose can cause severe liver injury (Jaeschke, 2015) and, in some cases, acute liver failure (Stravitz and Lee, 2019). During the last 40 years, the mechanisms of APAP-induced cell death and liver injury have been mainly investigated in mice. The initial discovery that the toxicity depends on the formation of a reactive metabolite (Mitchell et al., 1973), N-acetyl-p-aminobenzoquinone imine (NAPQI), which can react with cysteine residues of glutathione and of proteins (Jollow et al., 1973), led to the development of N-acetylcysteine (NAC) as effective antidote against APAP overdose (Rumack and Bateman, 2012). During the last 2 decades, the central role of mitochondria came into focus (Jaeschke et al., 2012a; Ramachandran and Jaeschke, 2019, 2020). Importantly, many aspects of the mechanism of APAP toxicity in mice have been verified in patients, and in human hepatocytes, including GSH depletion, and extensive protein adduct formation, oxidant stress, activation of c-jun N-terminal kinase (JNK), mitochondrial dysfunction and damage, nuclear DNA fragmentation, and ultimately necrotic cell death (Antoine et al., 2012; Davern et al., 2006; McGill et al., 2011, 2012, 2014; Xie et al., 2014). The newer insight into the pathophysiology of APAP toxicity in humans led to the consideration of fomepizole (4-methylpyrazole) as an adjunct therapy to NAC (Akakpo et al., 2022).
Cellular necrosis is characterized by cell and organelle swelling and the release of cell contents. Among the cellular constituents passively released by hepatocytes are a class of endogenous macromolecules termed damage-associated molecular patterns (DAMPs), that can bind to pattern recognition receptors (e.g., toll-like receptors) on macrophages, and trigger the transcriptional activation of pro-inflammatory genes (Kubes and Mehal, 2012; Woolbright and Jaeschke, 2017). During APAP-induced liver injury, the release of DAMPs such as high mobility group box 1 (HMGB1) protein, nuclear DNA fragments, and mitochondrial DNA (mtDNA) has been reported in the mouse model (Antoine et al., 2009; Martin-Murphy et al., 2010; McGill et al., 2012). APAP overdose triggers formation of cytokines (James et al., 2003b; Lawson et al., 2000), which was suggested to involve signaling through toll-like receptors (TLRs) by DAMPS, and activation of the inflammasome in mice (Imaeda et al., 2009; Williams et al., 2010b). As such, APAP-induced cellular necrosis triggers formation of pro-inflammatory mediators, and consequently, initiates recruitment of neutrophils and monocytes into the liver (Cover et al., 2006; Dambach et al., 2002; Holt et al., 2008; Lawson et al., 2000). Because of the cytotoxic potential of these infiltrating phagocytes (Jaeschke, 2006; Laskin et al., 2011), it is not surprising that this has triggered a controversial discussion about the relevance of this inflammatory response, with one side arguing that inflammation aggravates the initial injury and the other side concluding that the recruitment of inflammatory cells is important for tissue repair and removal of necrotic debris (Jaeschke et al., 2012b; Jaeschke and Ramachandran, 2020). Although there is clear evidence for release of DAMPs, including HMGB1, nuclear DNA fragments, and mtDNA in humans after APAP (Antoine et al., 2012; McGill et al., 2012), there is only very limited information on cytokine formation (Bonkovsky et al., 2018; James et al., 2005). In particular, the potential roles of the inflammasome and importance of IL-1β versus IL-1α, which are controversial in the murine model (Imaeda et al., 2009; Williams et al., 2010b, 2011; Zhang et al., 2018), remain unclear in the human pathophysiology. In addition, information about activation of complement is unknown in humans despite information in the mouse that suggests it may be a relevant event (Kataoka et al., 2014; Singhal et al., 2012). Prior data in humans demonstrate early differences between control patients and APAP overdose patients in cytokines such as IL-6, IL-10 and CCL2, but there are limited data outside the first 48 h of hospitalization (Antoniades et al., 2006; Berry et al., 2010; James et al., 2005). Because many of these cytokines remain elevated during this period, it is poorly understood which cytokines are increased during the extended resolution phase of the injury. Thus, the primary objective of this study was to assess cytokine and complement levels over an extended period of time and compare these data to animal studies and previous patient data. Recognizing the limited number of patients per group, a secondary objective was to compare data of survivors and non-survivors to identify potential candidates for predictive biomarkers of negative outcome.
2. Materials and methods
2.1. Acetaminophen-induced acute liver injury patients
Patient samples and data for all studies were acquired under informed consent and approved by Institutional Review Boards and adhere to the 1975 Declaration of Helsinki. Plasma samples were obtained from APAP overdose patients at the University of Kansas Hospital in Kansas City, KS, USA, and the Banner Good Samaritan Medical Center in Phoenix, AZ, USA. APAP overdose as a diagnosis was made by a physician based on standard clinical criteria including reported history of APAP overdose, detectable serum APAP levels, and/or peak amino-transferase level of ≥ 1000 IU/L. For the majority of patients, same sample ALT values were used, but in the cases where same sample ALT was not available, the nearest ALT value was used instead, which was typically < 8 h from the sample in question. One patient was missing ALT values in the SV group and not all patients had a complete time course with some starting 1–2 days into their hospitalization. The no liver transaminase group (NLT) was defined as a subgroup with limited ALT elevation after APAP overdose (ALT<100) but confirmed APAP overdose. These were patients who presented early after an overdose (Xie et al., 2015). Blood samples were collected in heparinized collection tubes at the time of study admission by hospital staff and approximately every 24 h thereafter to establish a time course, until patient death or discharge. The whole blood was centrifuged (1000 x g for 10 min) to obtain plasma. Control samples were collected from a population of randomly selected healthy volunteers around the University of Kansas Medical Center. ALT was measured in a subset of these patients and found to be within normal ranges. Informed written consent was obtained from each patient or next of kin.
2.2. Clinical data
All clinical parameters (ALT, bilirubin, creatinine) were measured in clinical laboratories at the participating hospitals using standardized clinical methods. While measurements for either INR or PT times were performed in these patients, the standard operating procedure of each site was different, and thus neither INR nor PT times were available for all patients. As such, we report the PT and INR values for each site separately. See Table 1 for definitive clinical data.
Table 1.
Patient Data.
| Survivors | Non-Survivors | NLT | Controls | |
|---|---|---|---|---|
| Age (years) | 38 ± 3 | 40 ± 6 | 34 ± 5 | 34 ± 3 |
| % Female | 85 | 88 | 80 | 75 |
| Peak ALT (U/L) | 3943 ± 605 | 5729 ± 741 | 41 ± 13 | 35 ± 3 |
| Peak Total Bilirubin (mg/dL) | 6.1 ± 1.4 | 13.1 ± 2.9 | N.D. | N.D. |
| Peak Creatinine (mg/dL) | 2.4 ± 0.5 | 2.8 ± 0.4 | N.D. | N.D. |
| Peak INR * | 4.4 ± 1.0 (n = 9) | 9.1 ± 0.7 (n = 3) | N.D. | N.D. |
| Peak PT (s) ** | 66.6 ± 46.9 (n = 3) | 84.2 ± 10 (n = 6) | N.D. | N.D. |
INR, international normalized ratio; PT, prothrombin time; N.D. not determined. Data represent mean ± SE of n = 12 survivors, n = 9 non-survivors, n = 10 NLT (overdose patients with no liver transaminase increase), n = 8 controls (healthy volunteers)
Only applicable for KU samples;
Only applicable for Phoenix samples
2.3. Cytokine analysis
Plasma cytokine levels were measured using a Millipore (Billerica, MA, USA) custom multi-plex cytokine array according to manufacturer’s instruction on a Luminex 200 (Austin, TX, USA) using 25 μL of sample as suggested by the assay. The limit of detection for each of the cytokines based on the lowest point of the calibration curve was 3.2 pg/ml. All sample values were within the range of the calibration curve. Values for Gro-alpha were not obtained for a small subset of patients. All values measured performed within expectations of manufacturer provided quality control standards. Concentrations of each analyte were determined by comparing samples to a standard curve of known concentrations.
2.4. Complement ELISA
Complement C3 was measured using a commercially available ELISA from Abcam (Abcam, Cambridge, United Kingdom) according to manufacturer’s instructions. Briefly, selected samples or standards were incubated with a biotinylated antibody against complement C3 and incubated for 2 h. A streptavidin conjugate was added after washing and the samples were visualized using a chromogen provided in the kit. Samples were measured on a Bio-Tek microplate reader using Gen 3 Software (Bio-Tek, Winooski, VT).
2.5. Statistics
Normality was assessed using the Shapiro-Wilk test and all patient data were found to be non-normal. For cytokine analysis, differences between paired groups were tested with the Mann-Whitney U test. Time course data across all groups was assessed using one-way ANOVA on ranks with Dunn’s post hoc test against control values, or against no liver transaminase increase patient group values in the case of ALT. Receiver operating characteristic (ROC) curve analysis was used to associate values with outcome. For complement C3 analysis, Student’s t-test or the Mann-Whitney U test were used depending on whether data was found to be normal or not. In all cases, p < 0.05 was considered significant. Table 2.
Table 2.
Patient Populations by Day and Group.
| Day 0 | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | |
|---|---|---|---|---|---|---|
| HC | n = 8 | |||||
| NLT | n = 10 | |||||
| SV | n = 12 | n = 13 | n = 14 | n = 11 | n = 10 | n = 9 |
| NS | n = 7 | n = 7 | n = 6 | n = 6 | n = 5 | n = 4 |
Group sizes for healthy volunteers or APAP overdose patients recruited into the study. Patients were removed upon death, liver transplantation, or discharge from the hospital. HC – healthy controls. NLT – overdose patients with no liver transaminase increase. SV – surviving overdose patients. NS – non-surviving overdose patients.
3. Results
To assess cytokine formation over time in APAP overdose patients, a small array of cytokines that were previously indicated to have a potential role in APAP toxicity in animals were selected and measured simultaneously on day of admission and for the 5 days following patient admittance. These cytokines included interleukin-1ß (IL-1ß), interleukin-1α (IL-1α), tumor necrosis factor-α (TNF-α), CXC chemokine ligand 1 (CXCL1, GRO), CXC chemokine ligand 8 (CXCL8, IL-8), interleukin-6 (IL-6), interleukin-10 (IL-10), monocyte chemoattractant protein-1 (CCL2, MCP-1), and granulocyte colony stimulating factor (G-CSF). Day 0 (D0) refers to the day the patient was initially admitted into the study. Patient data are present in Table 1. The time course of injury as measured by ALT is depicted in Fig. 1. Of note, non-surviving patients presented with a substantial shift in the injury profile with peak injury occurring on the day after study admission, whereas, surviving patients presented with peak ALT at day of admission (Fig. 1).
Fig. 1. :
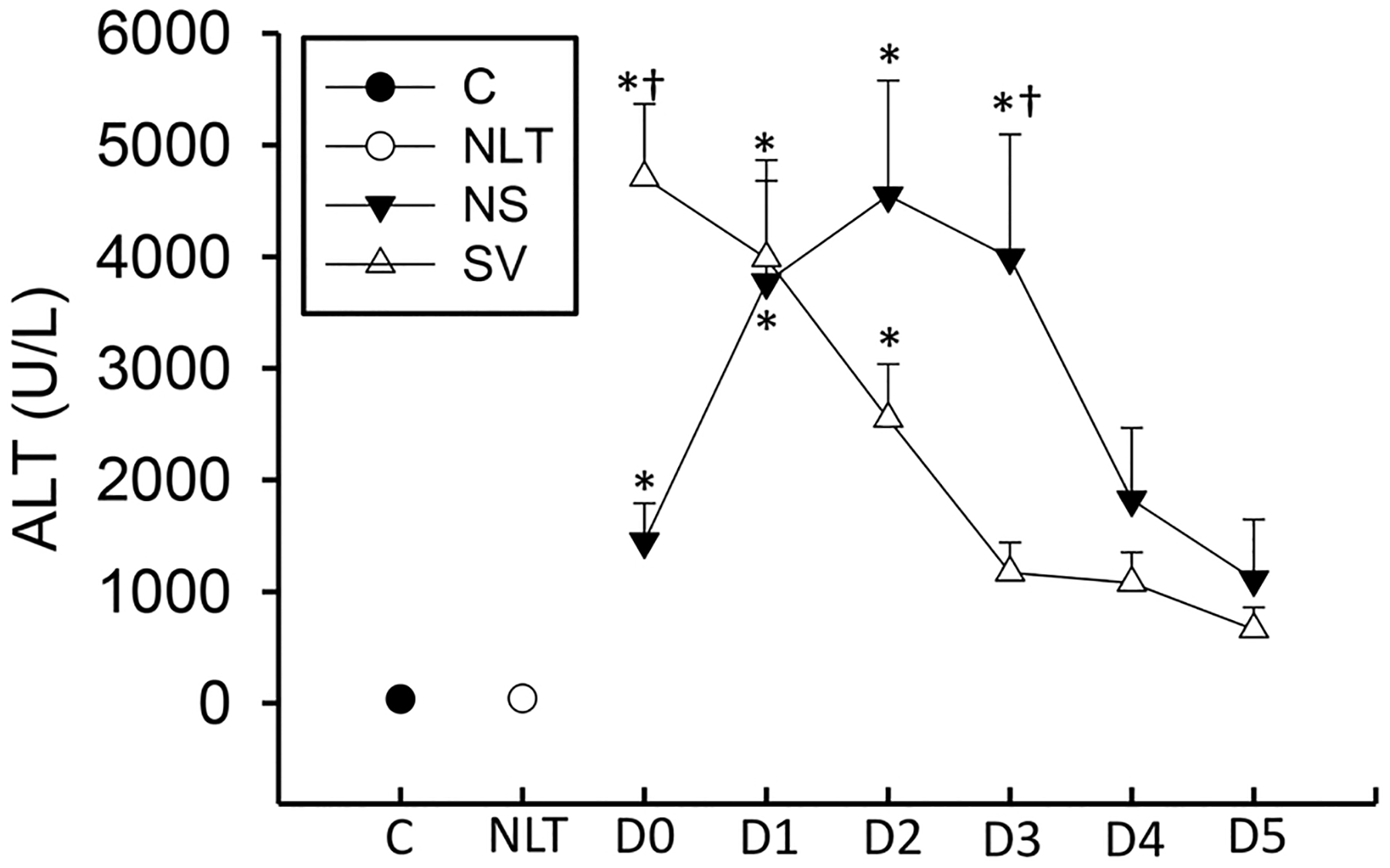
Plasma ALT activities were measured by clinical staff at Kansas University Hospital or Banner Good Samaritan Hospital over the course of the patient’s hospital stay. Control patients were acquired as in McGill et al. (2012). Data represent mean ± SE. Relevant n values for each group and time point are listed in Table 2. *p < 0.05 (compared to control or NLT – ANOVA/Dunn). †p < 0.05 (compared to matched SV group sample – t-test).
Plasma levels of IL-1α, IL-1ß, and TNF-α were measured in surviving and non-surviving APAP overdose patients with liver injury over six days to determine if there was an increase in classical pro-inflammatory cytokines (Fig. 2). The patients with liver injury were compared to control patients not exposed to APAP, and overdose patients with no increase in liver transaminases (McGill et al., 2012). TNF-α, IL-1α, and IL-1ß levels were not different between any group at any point over the first six days of the study (Fig. 2) indicating that any potential inflammatory infiltrate occurs independent of these mediators, consistent with prior results in children and adolescents with APAP overdose (James et al., 2001).
Fig. 2. :
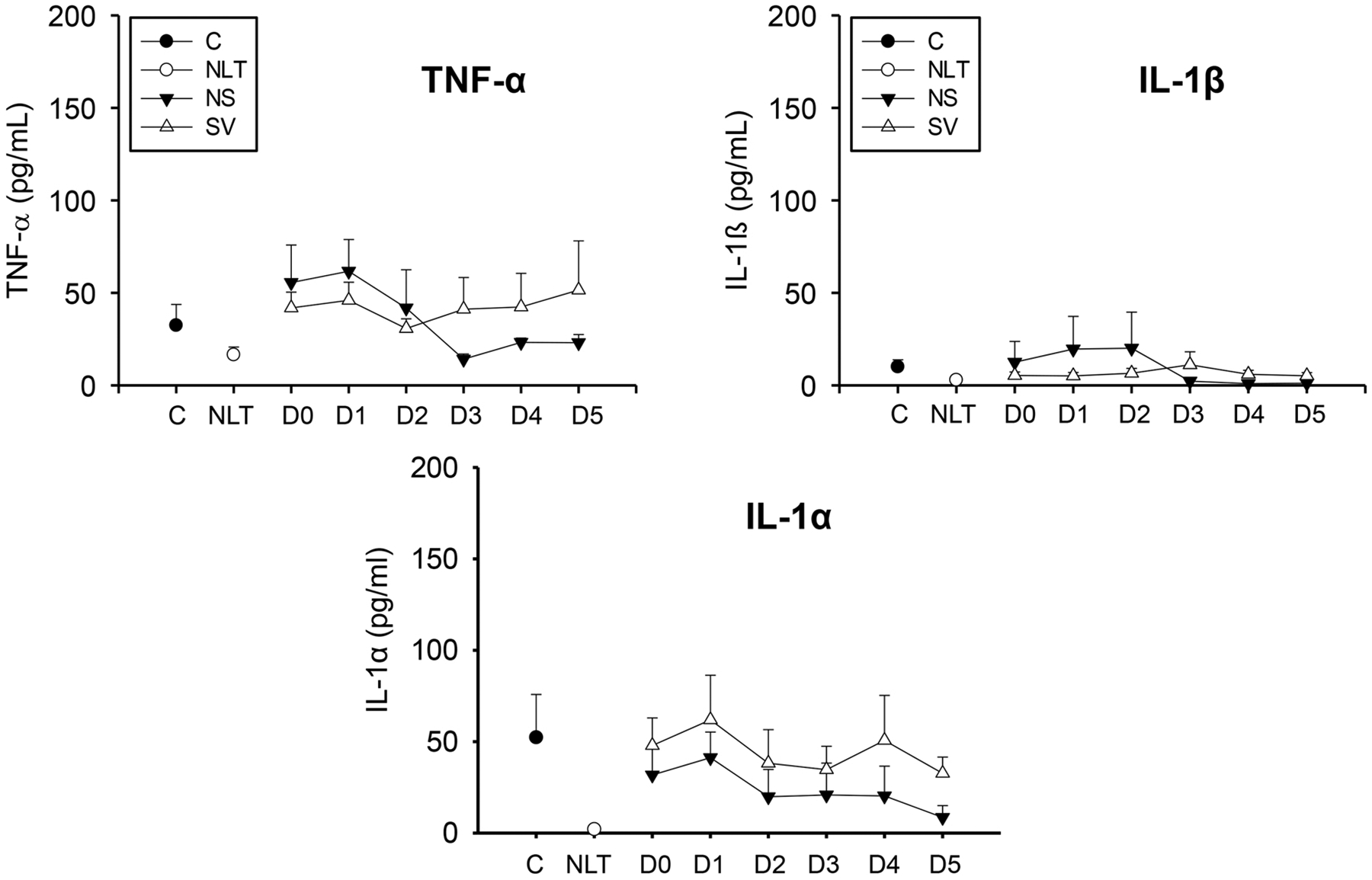
Cytokines TNF-α, IL-1α, and IL-1β were measured in a cohort of surviving (S), and non-surviving (NS) patients with APAP overdose, control patients (C), or patients with no increase in liver transaminases (NLT) over 6 days (D0 – D5). Data represent mean ± SE. Relevant n values for each group and time point are listed in Table 2.
CXCL8 and CXCL1 are potent neutrophil chemoattractants in humans (Yoshimura et al., 1987; Matsushima et al., 2022). These cytokines were measured over the first six days following admission in these patients. CXCL8 levels were elevated over both control and patients with no increase in liver transaminase values for the first six days following APAP overdose but did not reach statistical significance (Fig. 3A). This was more pronounced in the non-surviving group, although non-surviving values were not significantly greater than their surviving counterparts. CXCL1 levels were not differently elevated at any point as assessed by ANOVA analysis across all time points (Fig. 3B), however, direct comparison of surviving and non-surviving groups only indicated a trend to higher levels in the first two days of the time course, similar to other studies (James et al., 2005). Neutrophil recruitment in the mouse model occurs largely during these periods (Lawson et al., 2000), and neutrophil activation in blood is observed during this time (Williams et al., 2014); thus, these data support a model where neutrophil recruitment after APAP overdose may be mediated partially through CXCL8, the primary chemoattractant for neutrophils in humans.
Fig. 3. :
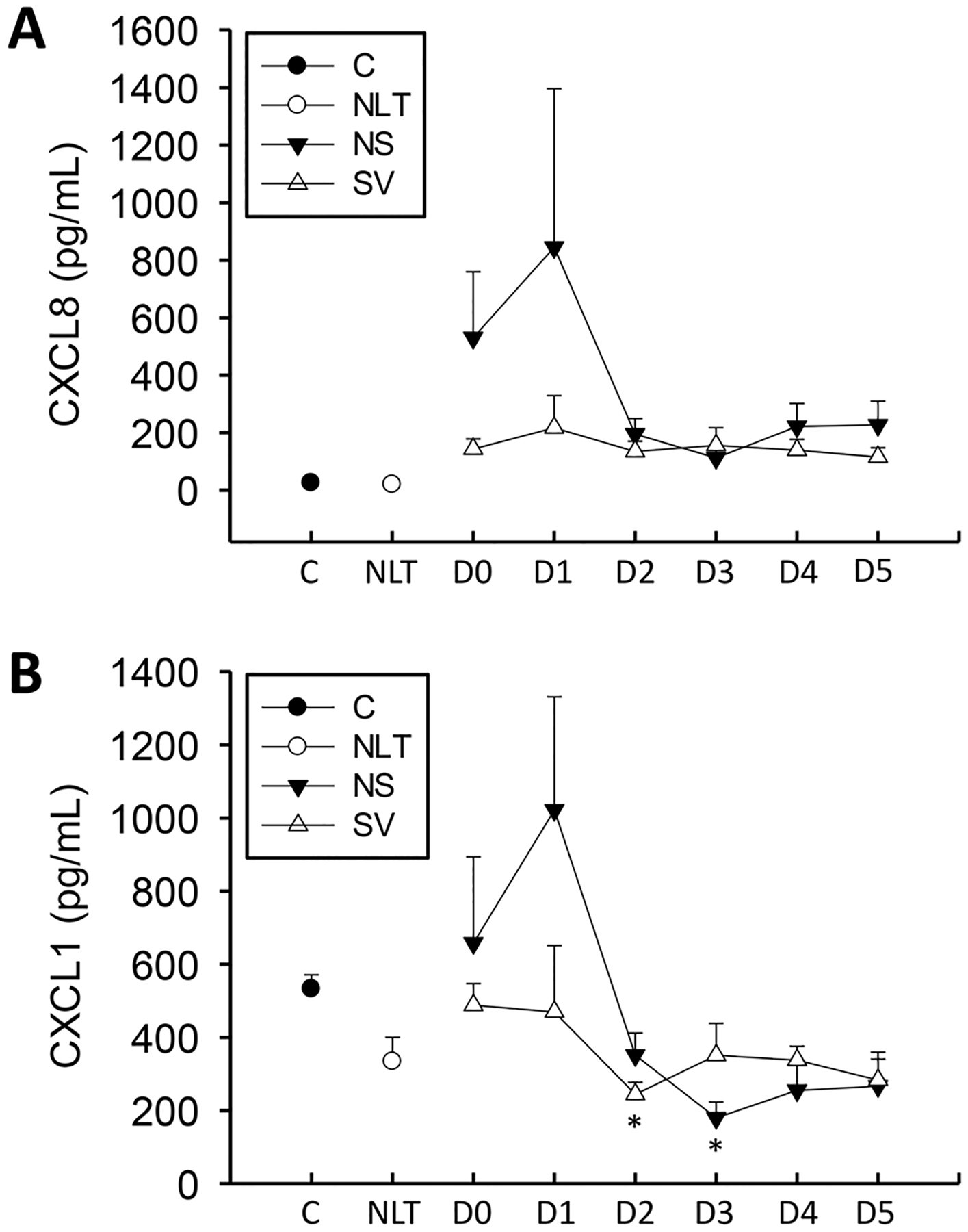
Chemokines CXCL8 (IL-8) (A) and CXCL1 (GRO) (B) were measured in a cohort of surviving (S), and non-surviving (NS) patients with APAP overdose, control patients (C), or patients with no increase in liver transaminases (NLT) over 6 days. Data represent mean ± SE. Relevant n values for each group and time point are listed in Table 2. *p < 0.05 (compared to control or NLT – ANOVA/Dunn).
CCL2 and IL-10 levels were dramatically elevated above both controls and patients with no increase in liver transaminases at both the initial point and throughout the time course in the non-surviving patients (Fig. 4). Non-surviving patients’ IL-10 levels were also significantly elevated above surviving patients at Day 1, and non-surviving patients’ CCL2 levels were elevated above surviving patients at Day 2. Of note, values at Day 2 (Fig. 4) were highly indicative of future outcome (area under the ROC curve = 0.88) for CCL2 and at Day-1 for IL-10 (area under the ROC curve = 0.91 (Table 3). Differences were not observed at other time points and thus AUROC was not performed for these time points. Thus, there is a sustained increase in CCL2 in both surviving and non-surviving APAP overdose patients that likely stimulates monocyte recruitment throughout the later points of injury and during recovery. IL-10 levels are elevated more specifically in non-surviving patients.
Fig. 4. :
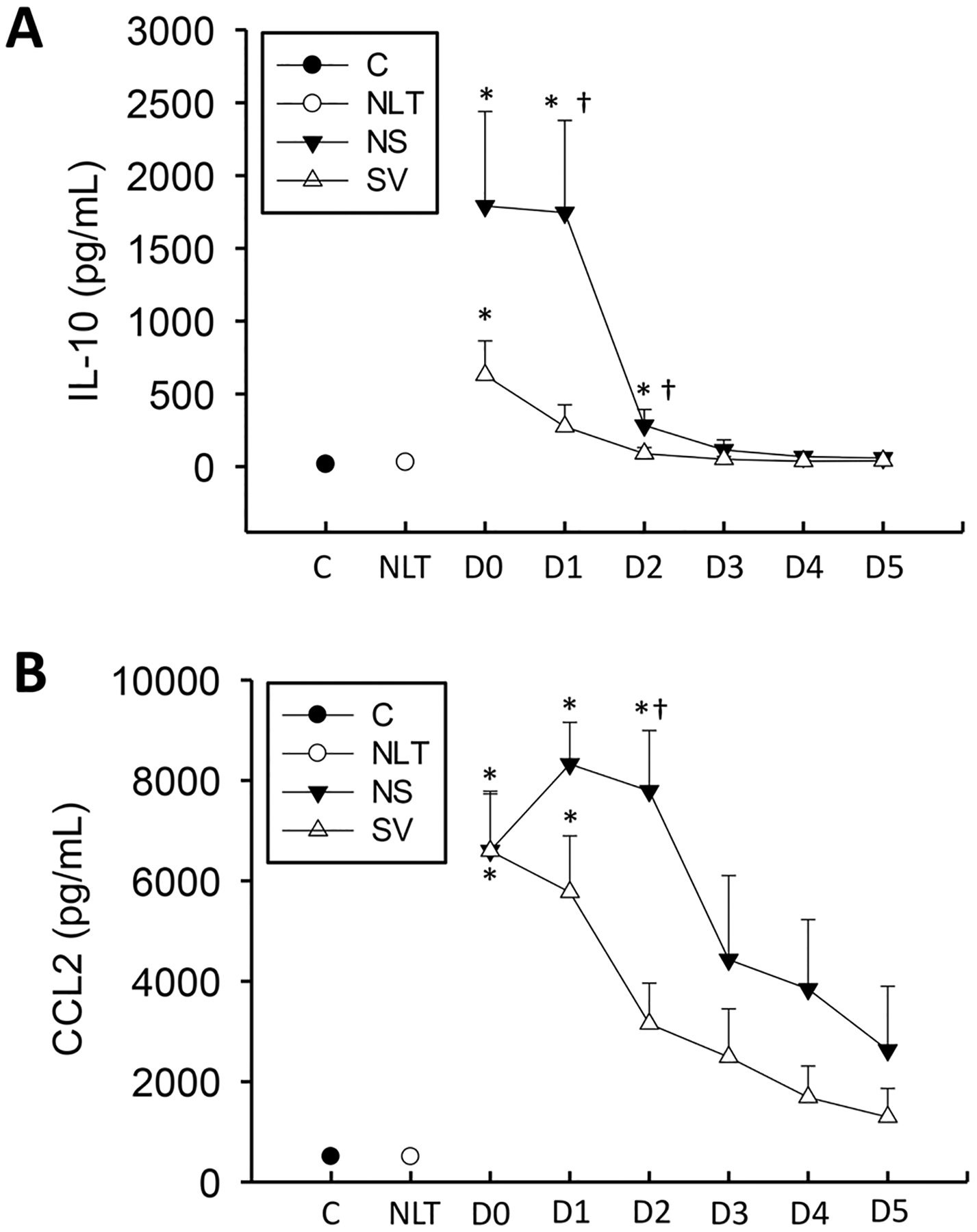
IL-10 (A) and CCL2 (B) were measured in a cohort of surviving (S), and non-surviving (NS) patients with APAP overdose, control patients (C), or patients with no increase in liver transaminases (NLT) over 6 days. Data represent mean ± SE. Relevant n values for each group and time point are listed in Table 2. *p < 0.05 (compared to control or NLT – ANOVA/Dunn). †p < 0.05 (compared to matched SV group sample – t-test).
Table 3.
Receiver Operator Characteristic Curves for Cytokines.
| Time point | p value | AUROC | |
|---|---|---|---|
| IL-10 | Day 1 | < 0.05 | 0.91 |
| CCL2 | Day 2 | < 0.05 | 0.88 |
| G-CSF | Day 1 | < 0.05 | 0.85 |
| G-CSF | Day 0 | < 0.05 | 0.78 |
Area under the receiver operator characteristic curve (AUROC) for a cytokine’s ability to predict mortality. AUROC analysis was assessed comparing the capacity of each individual cytokine to predict future mortality over the first three days of stay. No other tested cytokine values were found to be statistically significant.
p < 0.05.
IL-6 and G-CSF levels were elevated in non-surviving patients over both controls and the no increase in liver transaminase patient groups in this study during the first 3 days but did not reach statistical significance (Fig. 5). Largely due to the high variability in these samples, these values were not significantly elevated above patients with no increase in liver transaminase except D1 (IL-6). Furthermore, values in non-surviving patients were predictive of outcome at D0 and D1 (G-CSF) (Table 3). The increases in these cytokines were largely mediated by extreme changes in a small subset of patients, indicating larger sample sizes may be necessary to fully understand these changes though as this may be more related to active infection or other factors underlying the primary liver dysfunction.
Fig. 5. :
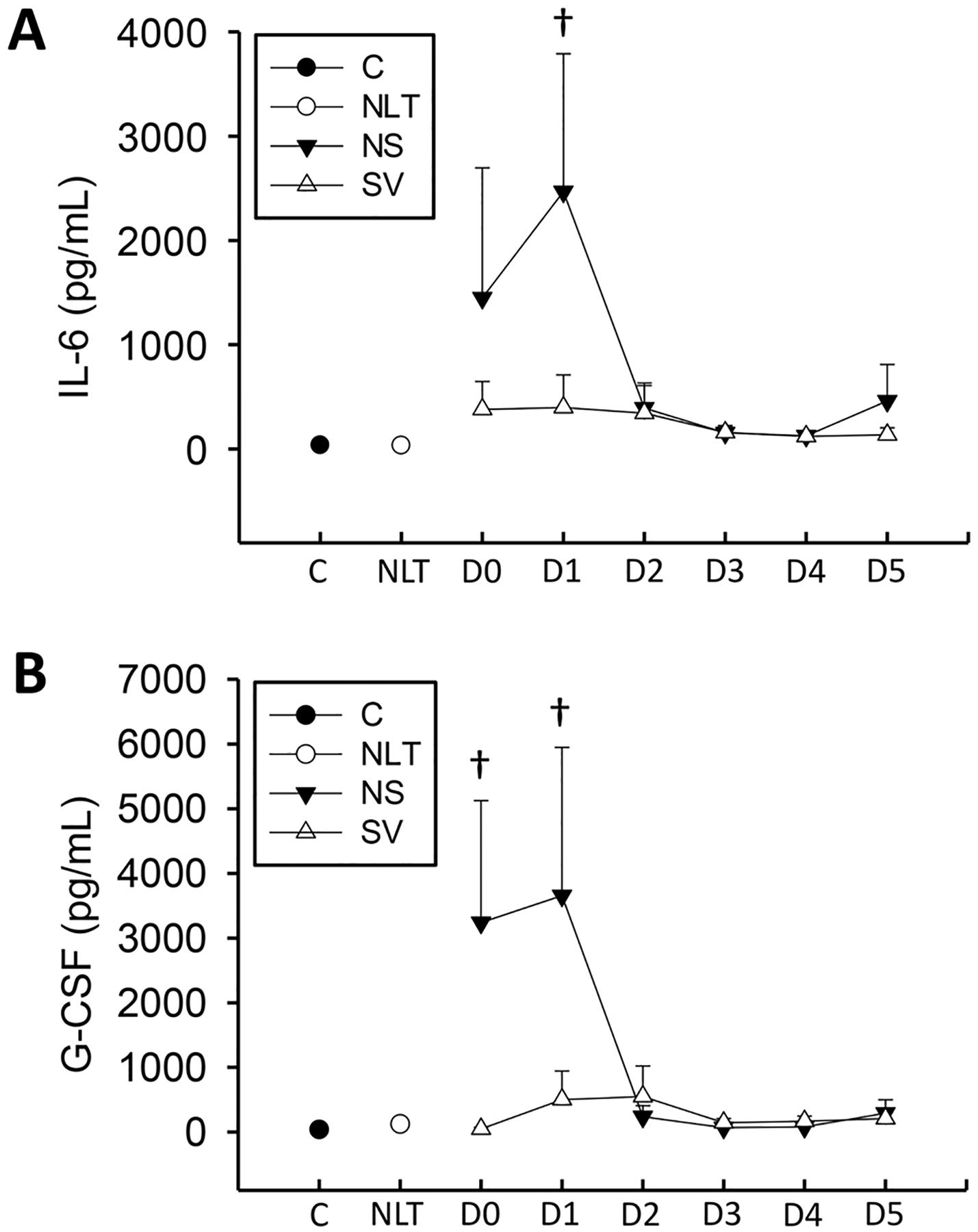
Cytokines IL-6 (A) and G-CSF (B) were measured on a cohort of surviving (S), and non-surviving (NS) patients with APAP overdose, control patients (C), or patients with no increase in liver transaminases (NLT) over 6 days. Data represent mean ± SE. Relevant n values for each group and time point are listed in Table 2. *p < 0.05 (compared to control or NLT – ANOVA/Dunn). †p < 0.05 (compared to NLT – t-test).
Previous studies have shown that complement C3 depletion in the murine model of APAP overdose resulted in a reduced neutrophil infiltrate, although there is conflicting information over whether this is also associated with protection (Kataoka et al., 2014; Singhal et al., 2012). APAP overdose in the mouse is also associated with depletion of complement C3 from plasma (Singhal et al., 2012). To assess whether there were differences in complement levels we measured complement C3 depletion in these patients. Complement C3 was depleted versus control patients or patients with no increase in liver transaminases at earlier time points in both surviving and non-surviving APAP overdose patients (Fig. 6). Of note, this value recovered over time in surviving patients, but did not recover in non-surviving patients. As such, complement C3 might be a valuable indicator of liver function recovery in patients with APAP overdose.
Fig. 6. :
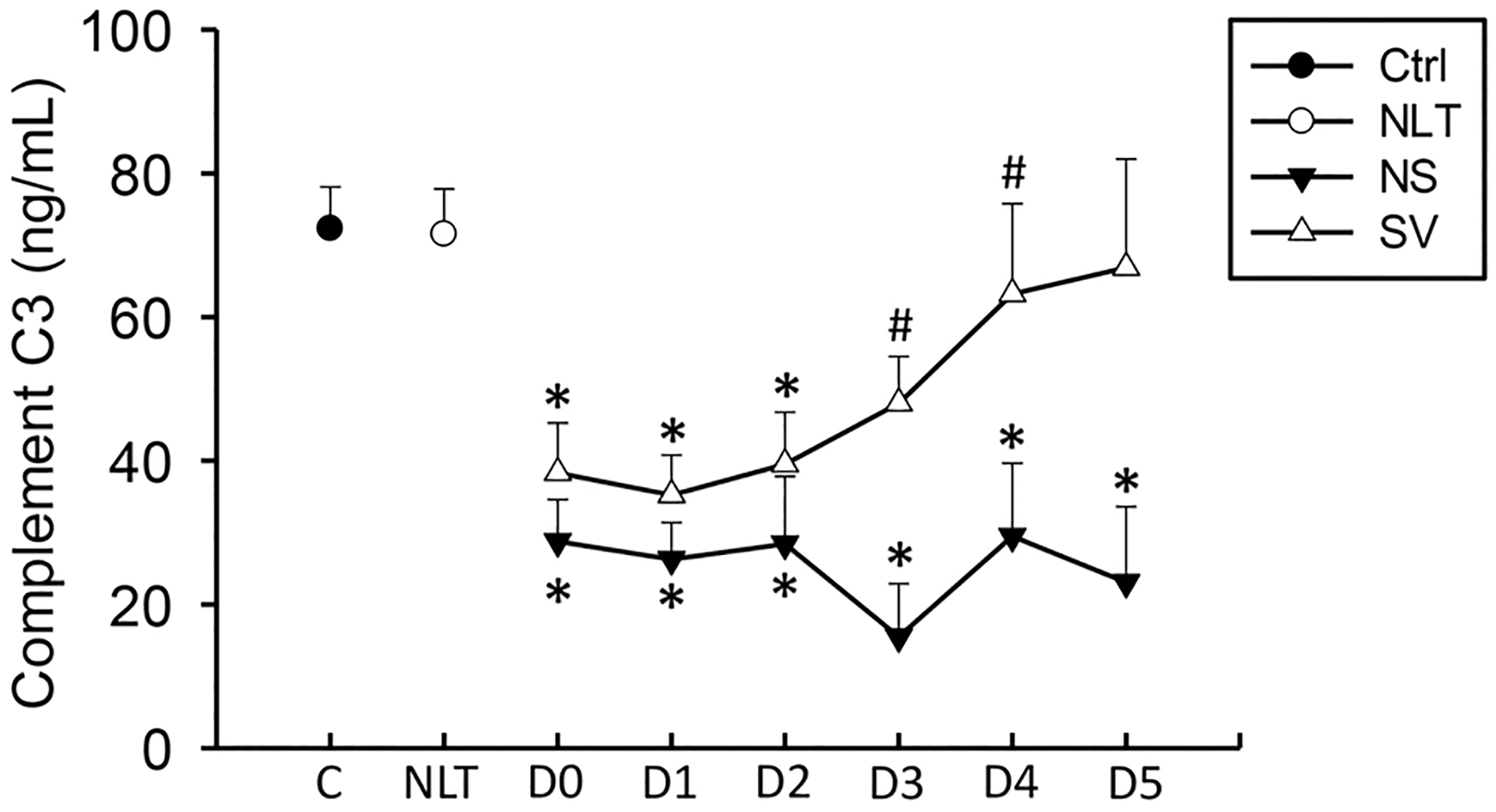
Complement C3 levels were measured on a cohort of surviving (S), and non-surviving (NS) patients with APAP overdose, control patients (C), or patients with no increase in liver transaminases (NLT) over 6 days. Data represent mean ± SE. Relevant n values for each group and time point are listed in Table 2. *p < 0.05 (compared to control or NLT – ANOVA/Dunn). †p < 0.05 (compared to NS matched pair – t-test).
In total, these data support the idea that non-surviving patients have a significant increase in the formation of specific cytokines. This altered cytokine response extends for multiple days in the case of a majority of cytokines, and potentially up to a week in some patients’ cytokine levels, such as CCL2. As such, cytokine levels may have the potential for being prognostic biomarkers in APAP overdose patients, and further studies may lead to a better understanding of the role of inflammation in APAP overdose patients.
4. Discussion
The pathophysiological relevance of the sterile inflammatory response that occurs after the onset of necrosis during APAP-induced liver injury has been the subject of considerable debate in the mouse model (Jaeschke et al., 2012b; Jaeschke and Ramachandran, 2020). This study was initiated to better determine which cytokines were present during the first week after APAP overdose in humans, and if there were sustained differences between surviving and non-surviving patients. In addition, we also measured complement C3 to determine if it might also be involved in APAP induced liver injury. Moreover, we sought to determine if the inflammatory infiltrate was largely pro-inflammatory, consistent with a secondary inflammatory injury phase, or anti-inflammatory, consistent with a secondary phase of inflammation to promote regeneration. Studies in mice have led to conflicting results with some studies pointing towards a pathological inflammatory component (Cai et al., 2014; Huebener et al., 2015; Imaeda et al., 2009; Liu et al., 2006) and other studies pointing towards inflammation being a requisite component of the resolution and regeneration phase (Chauhan et al., 2020; Nguyen et al., 2022; Stutchfield et al., 2015; Williams et al., 2010a; b, 2014; Yang et al., 2019). Results from this study indicate that there are minimal increases in traditional pro-inflammatory cytokines such as IL-1α, IL-1ß, CXCL1, and TNF-α. In contrast, significant increases were found in IL-6, CXCL8, IL-10, and CCL2 in non-surviving patients compared to surviving patients. Some of these values may be predictive of future outcome in patients, particularly Day 1 IL-10 values and Day 2 CCL2 values. Given these data, future studies investigating the role of these cytokines in larger cohorts of human patients may be warranted.
4.1. Inflammasome activation in APAP overdose patients
One of the central questions remaining in APAP toxicity is what role inflammation plays in the toxicity in patients. Necrotic hepatocytes in the murine APAP overdose model release several DAMPs that could potentially prime or activate the inflammasome including HMGB1, nuclear DNA fragments, mitochondrial DNA and ATP (Antoine et al., 2009; Hoque et al., 2012; Marques et al., 2012; Martin-Murphy et al., 2010; McGill et al., 2012). Mice deficient in inflammasome components or TLRs responsible for promoting pro-IL-1ß expression levels (Cai et al., 2014; Cavassani et al., 2013; Imaeda et al., 2009; Marques et al., 2012) were reported to be protected against APAP overdose with the proposed mechanism in each case being a reduction in associated inflammation, with an emphasis on recruited neutrophils. As activation of pro-IL-1ß to IL-1ß by caspase-1 is the primary result of inflammasome activation, it was hypothesized that IL-1ß-mediated neutrophil recruitment, and thus inhibition of IL-1ß through these varied interventions, was the mechanism behind the protection (Cai et al., 2014; Imaeda et al., 2009; Marques et al., 2012). However, this hypothesis has been challenged at multiple points. First, pan-caspase inhibitors, which also effectively inhibit caspase-1, do not reduce neutrophil infiltration and do not protect against APAP hepatotoxicity (Jaeschke et al., 2006; Lawson et al., 1999; Williams et al., 2010b). Second, specific interventions against neutrophils do not reduce APAP-induced liver injury (Cover et al., 2006; James et al., 2003a; Lawson et al., 2000; Williams et al., 2010a). Third, and most importantly, the absolute formation of IL-1β after APAP overdose was very limited in all studies (Cai et al., 2014; Imaeda et al., 2009; Williams et al., 2010b; Zhang et al., 2018) with plasma concentrations well below levels that would affect neutrophil activation (Williams et al., 2010b). The current findings in human APAP overdose patients are consistent with the observations in mice. IL-1ß levels were not significantly elevated at any point after APAP overdose in either survivors or non-survivors (Fig. 2) versus control patients. Prior studies in children with or without liver injury after APAP overdose found a similar lack of increase in IL-1ß (James et al., 2001). Thus, the total lack of any consistently measurable increase in IL-1β over a week after APAP ingestion reduces the likelihood that inflammasome activation plays an important role in human APAP overdose patients. IL-1α, which is generated independently of the inflammasome, has also been suggested to be the central mediator of the neutrophil-induced injury in the APAP hepatotoxicity model in the mouse (Zhang et al., 2018). Similar to IL-1β, there were no relevant increases in IL-1α levels in any APAP overdose patient. Furthermore, concentrations of TNF-α, another major pro-inflammatory mediator, were not significantly different between groups during the course of the study in every patient population (Fig. 2), which largely corroborates prior data indicating near-admission TNF-α values had no predictive value in APAP-induced ALF patients (Berry et al., 2010) and no significant increase in plasma TNF-α levels were detected in children or adolescents with APAP overdose (James et al., 2001) or in adults (Bonkovsky et al., 2018). However, the previous reports are based mainly on single time points or pooled samples. These human data are also consistent with observations in mice where animals deficient in TNF-α or TNF receptor-1 did not show any protection against APAP-induced liver injury (Boess et al., 1998; Chiu et al., 2003). Thus, the lack of significant pro-inflammatory cytokine formation including IL-1β, IL-1α and TNF-α, indicates a limited inflammasome activation and a limited pro-inflammatory response. These findings are consistent with the fact that neutrophil activation in peripheral blood of overdose patients occurs after the injury phase during regeneration (Williams et al., 2014). Together these data do not support the hypothesis that a pro-inflammatory response may aggravate the initial injury in APAP overdose patients.
4.2. Sustained cytokine increases in APAP overdose patients
CCL2, IL-6, and IL-10 have previously been measured in APAP patients at or around day of admission (Antoniades et al., 2006; Berry et al., 2010; James et al., 2005) and generally found to be more elevated in non-surviving patients. Data in this study largely confirm this observation with a few minimal differences likely attributable to the relatively small patient population. Because the primary goal of this study was to identify which cytokines are significantly elevated during the hospital course, the study has fewer patients than some contemporary studies powered to detect differences in survival. Moreover, we have found that these values largely remain elevated for two to five days after the initial admission. Of note, most of these cytokines are either considered anti-inflammatory as in IL-10 (Moore et al., 2001) or promote recruitment of late-stage M2 monocytes as in CCL2 (Antoniades et al., 2012; Dambach et al., 2002; Holt et al., 2008). Monocytes in patients with ALF express a largely anti-inflammatory profile (Antoniades et al., 2012, 2014) and are recruited in an CCL2 dependent manner (Antoniades et al., 2012). The sustained expression of CCL2 in non-surviving patients present in this study, and the presumed subsequent sustained recruitment of type 2 anti-inflammatory macrophages, indicates a lack of resolution to the injury in non-surviving patients. A timely start in regeneration is critical for recovery in both human patients (Schmidt and Dalhoff, 2005) and overdosed mice (Bhushan et al., 2014). This regenerative process is stimulated both by phagocytosis of necrotic debris (Chazaud, 2014; Laskin et al., 2011) and production of cytokines from recruited inflammatory cells (Antoniades et al., 2014) or damaged hepatocytes (Dambach et al., 2002). Notably, there was only a minimal increase in all cytokines in patients with no increase in liver transaminases (NLT). These were mainly early presenting patients who were treated with NAC within 8 h of an overdose and therefore never developed liver injury (Xie et al., 2015). The direct protective effect of NAC prevented the release of DAMPs from hepatocytes and thus limited the formation of any cytokines (James et al., 2003b).
One potential weakness of the paper is the lack of any comparator group from another liver disease to determine relative increases in cytokines. Previous studies from our lab have assessed cytokine values in patients with hypoxic hepatitis, another type of acute liver injury that presents similarly to APAP overdose (Weemhoff et al., 2017). The cytokine profiles of these patients were similar to APAP overdose patients, with a substantial spike followed by normalization. As that study had only a small number of mortalities in the population, direct comparisons to this study are difficult to draw with regards to the sustained increase in cytokines we observed in non-surviving APAP overdose patients. Moreover, this was a pilot study with a small number of patients that took place in multiple settings to acquire a prolonged time course. These data should be confirmed in larger trials.
4.3. Cytokines as predictive biomarkers of survival after APAP overdose
Levels of both IL-6 and IL-10 at presentation have been indicated to be highly predictive of outcome (Antoniades et al., 2006; Berry et al., 2010). In this paper, we also looked at these levels over time. Of note, while we did not see a predictive capacity at presentation with either IL-6 or IL-10, we did see a significant increase in non-surviving patients, and we did find that Day 1 IL-10 values were predictive of outcome (area under the ROC = 0.91, p < 0.05). Our data largely corroborate the fact that IL-6 and IL-10 are significantly elevated in non-surviving patients, while also extending this time course significantly. Previous studies have indicated that CCL2 levels spike in patients with elevations in ALT (James et al., 2005) which also occurred in this population. By separating the surviving and non-surviving patients, it was determinable that prolonged formation of CCL2 was associated with non-survival. CCL2 levels were predictive of outcome at early time points after presentation with substantially elevated levels in non-surviving patients even out to 6 days. It should be noted that hepatic recruitment of monocyte-derived macrophages in APAP overdose patients (Antoniades et al., 2006, 2012) and in animal models of APAP hepatotoxicity (Dambach et al., 2002; Holt et al., 2008) is associated with recovery. CCL2 is generated by hepatocytes in the centrilobular region and by liver-recruited macrophages in mice and in humans (Antoniades et al., 2012; Dambach et al., 2002). Thus, the higher CCL2 levels in non-surviving patient would suggest prolonged CCL2 formation likely due to the delayed recovery and impaired regeneration in these patients. On the other hand, the rapid removal of dead cells by macrophages and the replacement of necrotic hepatocytes by regeneration leads to the gradual resolution of the inflammatory response with declining formation of CCL2 and less macrophage recruitment. One shortcoming in this study is the lack of traditional measures of outcome in patients, such as the King’s College Criteria (KCC). KCC requires the use of clinical scores that were not consistently available for all patients. Because of the lack of KCC or other validated outcome-based scores, AUROC values should be interpreted cautiously in this study.
Importantly, the primary purpose of this study was not the discovery and validation of cytokines as biomarkers for prognosis of patient outcomes, but rather an assessment of the relative cytokine levels after an APAP overdose longitudinally. Future studies looking at the potential predictive role of cytokines, or cytokine panels will be designed with inclusion of these criteria for an accurate assessment and powered sufficiently to more accurately define which biomarkers are most relevant. Cytokines may prove as a useful means for patient prognosis though, as potential point-of-care assays could be generated that would allow for a clinician to make a rapid decision about patient prognosis.
Active components of the complement cascade, e.g., C3a and C5a, the cleaved forms of complement factor C3 and C5, are potent neutrophil chemoattractants and activators contributing to liver injury after hepatic ischemia (Jaeschke et al., 1993). In addition to cytokine formation, declining levels of the complement component C3 were observed early after APAP overdose. These observations, which are consistent with findings in the mouse model (Singhal et al., 2012), suggest extensive complement activation in these patients. It is well established that severe liver damage with release of cell contents can activate complement, which contributes to a sterile inflammatory response (Jaeschke et al., 1993). Interestingly, the initial complement activation appears to be similar in surviving and non-surviving patients, which is consistent with a similar degree of liver injury in both patient groups. However, plasma C3 levels started to recover after day 3 in surviving patients but not in non-survivors. Since complement components are synthesized in hepatocytes, the recovering C3 levels are consistent with a functional recovery of the liver parenchyma during the regeneration phase.
4.4. Conclusions
Inflammation during APAP-induced liver injury in patients is likely a pro-resolution, pro-regenerative event according to data gained in this study. There was minimal evidence for increases in plasma cytokines associated with pro-inflammatory liver injury, with considerable and sustained increases in cytokines responsible for continued recruitment of anti-inflammatory type macrophages. In addition, some of these mediators may be useful as early predictors of negative outcome in APAP overdose patients. Thus, future studies should be directed at verifying these data in larger patient cohorts with potentially a greater number of cytokines.
Acknowledgments
This work was funded in part by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01 DK102142 (HJ) and National Institute of General Medicine (NIGMS) funded Liver Disease COBRE grants P20 GM103549 (HJ) and P30 GM118247 (HJ). Additional support came from the “Training Program in Environmental Toxicology” T32 ES007079-26A2 (to B.L.W., M.R.M) from the National Institute of Environmental Health Sciences.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Akakpo JY, Ramachandran A, Curry SC, Rumack BH, Jaeschke H, 2022. Comparing N-acetylcysteine and 4-methylpyrazole as antidotes for acetaminophen overdose. Arch. Toxicol 96, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK, 2012. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol 56, 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Antoine DJ, Williams DP, Kipar A, Jenkins RE, Regan SL, Sathish JG, Kitteringham NR, Park BK, 2009. High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicol. Sci 112, 521–531. [DOI] [PubMed] [Google Scholar]
- Antoniades CG, Berry PA, Davies ET, Hussain M, Bernal W, Vergani D, Wendon J, 2006. Reduced monocyte HLA-DR expression: a novel biomarker of disease severity and outcome in acetaminophen-induced acute liver failure. Hepatology 44, 34–43. [DOI] [PubMed] [Google Scholar]
- Antoniades CG, Khamri W, Abeles RD, Taams LS, Triantafyllou E, Possamai LA, Bernsmeier C, Mitry RR, O’Brien A, Gilroy D, Goldin R, Heneghan M, Heaton N, Jassem W, Bernal W, Vergani D, Ma Y, Quaglia A, Wendon J, Thursz M, 2014. Secretory leukocyte protease inhibitor: a pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology 2014 (59), 1564–1576. [DOI] [PubMed] [Google Scholar]
- Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, Possamai LA, Bruce M, McPhail M, Starling C, Wagner B, Barnardo A, Pomplun S, Auzinger G, Bernal W, Heaton N, Vergani D, Thursz MR, Wendon J, 2012. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 56, 735–746. [DOI] [PubMed] [Google Scholar]
- Berry PA, Antoniades CG, Hussain MJ, McPhail MJ, Bernal W, Vergani D, Wendon JA, 2010. Admission levels and early changes in serum interleukin-10 are predictive of poor outcome in acute liver failure and decompensated cirrhosis. Liver Int. 30, 733–740. [DOI] [PubMed] [Google Scholar]
- Bhushan B, Walesky C, Manley M, Gallagher T, Borude P, Edwards G, Monga SP, Apte U, 2014. Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am. J. Pathol 184, 3013–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boess F, Bopst M, Althaus R, Polsky S, Cohen SD, Eugster HP, Boelsterli UA, 1998. Acetaminophen hepatotoxicity in tumor necrosis factor/lymphotoxin-alpha gene knockout mice. Hepatology 27, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Bonkovsky HL, Barnhart HX, Foureau DM, Steuerwald N, Lee WM, Gu J, Fontana RJ, Hayashi PJ, Chalasani N, Navarro VM, Odin J, Stolz A, Watkins PB, Serrano J, US Drug-Induced Liver Injury Network and the Acute Liver Failure Study Group, 2018. Cytokine profiles in acute liver injury - results from the US drug-induced liver injury network (DILIN) and the acute liver failure study group. PLoS One 13, e0206389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Huang H, Whelan S, Liu L, Kautza B, Luciano J, Wang G, Chen G, Stratimirovic S, Tsung A, Billiar TR, Zuckerbraun BS, 2014. Benzyl alcohol attenuates acetaminophen-induced acute liver injury in a Toll-like receptor-4-dependent pattern in mice. Hepatology 60, 990–1002. [DOI] [PubMed] [Google Scholar]
- Cavassani KA, Moreira AP, Habiel D, Ito T, Coelho AL, Allen RM, Hu B, Raphelson J, Carson WF 4th, Schaller MA, Lukacs NW, Omary MB, Hogaboam CM, Kunkel SL, 2013. Toll like receptor 3 plays a critical role in the progression and severity of acetaminophen-induced hepatotoxicity. PLoS One 8, e65899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Sheriff L, Hussain MT, Webb GJ, Patten DA, Shepherd EL, Shaw R, Weston CJ, Haldar D, Bourke S, Bhandari R, Watson S, Adams DH, Watson SP, Lalor PF, 2020. The platelet receptor CLEC-2 blocks neutrophil mediated hepatic recovery in acetaminophen induced acute liver failure. Nat. Commun 11, 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazaud B, 2014. Macrophages: supportive cells for tissue repair and regeneration. Immunobiology 219, 172–178. [DOI] [PubMed] [Google Scholar]
- Chiu H, Gardner CR, Dambach DM, Durham SK, Brittingham JA, Laskin JD, Laskin DL, 2003. Role of tumor necrosis factor receptor 1 (p55) in hepatocyte proliferation during acetaminophen-induced toxicity in mice. Toxicol. Appl. Pharmacol 193, 218–227. [DOI] [PubMed] [Google Scholar]
- Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, Jaeschke H, 2006. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol 216, 98–107. [DOI] [PubMed] [Google Scholar]
- Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL, 2002. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology 35, 1093–1103. [DOI] [PubMed] [Google Scholar]
- Davern TJ 2nd, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, Lalani E, Munoz S, Shakil AO, Lee WM, 2006. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 130, 687–694. [DOI] [PubMed] [Google Scholar]
- Holt MP, Cheng L, Ju C, 2008. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol 84, 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque R, Sohail MA, Salhanick S, Malik AF, Ghani A, Robson SC, Mehal WZ, 2012. P2X7 receptor-mediated purinergic signaling promotes liver injury in acetaminophen hepatotoxicity in mice. Am. J. Physiol. Gastrointest. Liver Physiol 302, G1171–G1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ, Schwabe RF, 2015. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest 125, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ, 2009. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Invest 119, 305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, 2006. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol. Gastrointest. Liver Physiol 290, G1083–G1088. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, 2015. Acetaminophen: dose-dependent drug hepatotoxicity and acute liver failure in patients. Dig. Dis 33, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Cover C, Bajt ML, 2006. Role of caspases in acetaminophen-induced liver injury. Life Sci. 78, 1670–1676. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ, 1993. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am. J. Physiol 264, G801–G809. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A, 2012a. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev 44, 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Ramachandran A, 2020. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem. Toxicol 138, 111240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Williams CD, Ramachandran A, Bajt ML, 2012b. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int. 32, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LP, Farrar HC, Darville TL, Sullivan JE, Givens TG, Kearns GL, Wasserman GS, Simpson PM, Hinson JA, Pediatric Pharmacology Research Unit Network, National Institute of Child Health and Human Development, 2001. Elevation of serum interleukin 8 levels in acetaminophen overdose in children and adolescents. In: Clin. Pharmacol. Ther, 70, pp. 280–286. [DOI] [PubMed] [Google Scholar]
- James LP, McCullough SS, Knight TR, Jaeschke H, Hinson JA, 2003a. Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic. Res 37, 1289–1297. [DOI] [PubMed] [Google Scholar]
- James LP, McCullough SS, Lamps LW, Hinson JA, 2003b. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol. Sci 75, 458–467. [DOI] [PubMed] [Google Scholar]
- James LP, Simpson PM, Farrar HC, Kearns GL, Wasserman GS, Blumer JL, Reed MD, Sullivan JE, Hinson JA, 2005. Cytokines and toxicity in acetaminophen overdose. J. Clin. Pharm 45, 1165–1171. [DOI] [PubMed] [Google Scholar]
- Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB, 1973. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther 187, 195–202. [PubMed] [Google Scholar]
- Kataoka H, Kono H, Patel Z, Kimura Y, Rock KL, 2014. Evaluation of the contribution of multiple DAMPs and DAMP receptors in cell death-induced sterile inflammatory responses. PLoS One 9, e104741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubes P, Mehal WZ, 2012. Sterile inflammation in the liver. Gastroenterology 143, 1158–72. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Sunil VR, Gardner CR, Laskin JD, 2011. Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol 51, 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson JA, Farhood A, Hopper RD, Bajt ML, Jaeschke H, 2000. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol. Sci 54, 509–516. [DOI] [PubMed] [Google Scholar]
- Lawson JA, Fisher MA, Simmons CA, Farhood A, Jaeschke H, 1999. Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol. Appl. Pharmacol 156, 179–186. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Han D, Gunawan B, Kaplowitz N, 2006. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 43, 1220–1230. [DOI] [PubMed] [Google Scholar]
- Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Melgaço JG, Oliveira AG, Pinto MA, Lima CX, De Paula AM, Cara DC, Leite MF, Teixeira MM, Menezes GB, 2012. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56, 1971–1982. [DOI] [PubMed] [Google Scholar]
- Martin-Murphy BV, Holt MP, Ju C, 2010. The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett 192, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima K, Yang D, Oppenheim JJ, 2022. Interleukin-8: an evolving chemokine. Cytokine 153, 155828. [DOI] [PubMed] [Google Scholar]
- McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H, 2012. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Invest 122, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H, Acute Liver Failure Study Group, 2014. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology 60, 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H, 2011. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 53, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB, 1973. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther 187, 185–194. [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A, 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol 19, 683–765. [DOI] [PubMed] [Google Scholar]
- Nguyen NT, Umbaugh DS, Sanchez-Guerrero G, Ramachandran A, Jaeschke H, 2022. Kupffer cells regulate liver recovery through induction of chemokine receptor CXCR2 on hepatocytes after acetaminophen overdose in mice. Arch. Toxicol 96, 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H, 2019. Acetaminophen hepatotoxicity. Semin. Liver Dis 39, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Jaeschke H, 2020. A mitochondrial journey through acetaminophen hepatotoxicity. Food Chem. Toxicol 140, 111282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumack BH, Bateman DN, 2012. Acetaminophen and acetylcysteine dose and duration: past, present and future. Clin. Toxicol. (Philos.) 50, 91–98. [DOI] [PubMed] [Google Scholar]
- Schmidt LE, Dalhoff K, 2005. Alpha-fetoprotein is a predictor of outcome in acetaminophen-induced liver injury. Hepatology 41, 26–31. [DOI] [PubMed] [Google Scholar]
- Singhal R, Ganey PE, Roth RA, 2012. Complement activation in acetaminophen-induced liver injury in mice. J. Pharmacol. Exp. Ther 341, 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stravitz RT, Lee WM, 2019. Acute liver failure. Lancet 394, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, Hughes MJ, Francis B, Wojtacha D, Man TY, Dear JW, Devey LR, Mowat AM, Pollard JW, Park BK, Jenkins SJ, Simpson KJ, Hume DA, Wigmore SJ, Forbes SJ, 2015. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology 149, 1896–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weemhoff JL, Woolbright BL, Jenkins RE, McGill MR, Sharpe MR, Olson JC, Antoine DJ, Curry SC, Jaeschke H, 2017. Plasma biomarkers to study mechanisms of liver injury in patients with hypoxic hepatitis. Liver Int. 37, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, Kanneganti TD, Park BK, Jaeschke H, 2011. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol. Appl. Pharmacol 252, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, Bajt ML, Farhood A, Jaeschke H, 2010a. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int. 30, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, Jaeschke H, 2014. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol. Appl. Pharmacol 275, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, Farhood A, Jaeschke H, 2010b. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol. Appl. Pharmacol 247, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright BL, Jaeschke H, 2017. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol 66, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Cook SF, Sharpe MR, Winefield RD, Wilkins DG, Rollins DE, Jaeschke H, 2015. Time course of acetaminophen-protein adducts and acetaminophen metabolites in circulation of overdose patients and in HepaRG cells. Xenobiotica 45, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H, 2014. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol. Appl. Pharmacol 279, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, Fu L, Tian C, Yang J, He F, Tang L, 2019. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun 10, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Matsushima K, Oppenheim JJ, Leonard EJ, 1987. Neutrophil chemotactic factor produced by lipopolysaccharide (LPS)-stimulated human blood mononuclear leukocytes: partial characterization and separation from interleukin 1 (IL 1). J. Immunol 139, 788–793. [PubMed] [Google Scholar]
- Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, Wang W, Zhang S, Iwakura Y, Meng G, Fu YX, Hou B, Tang H, 2018. Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell. Mol. Immunol 15, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]