

Abstract
Lawsones and indenopyrazoles are the prevalent structural motifs and building blocks in pharmaceuticals and bioactive molecules, but their synthesis has always remained challenging as no comprehensive protocol has been outlined to date. Herein, a metal-free, ring-expansion reaction of indantrione with diazomethanes, generated in situ from the N-tosylhydrazones, has been developed for the synthesis of lawsone and indenopyrazole derivatives in acetonitrile and alcohol solvents, respectively. It provides these valuable lawsone and pyrazole skeletons in good yields and high levels of diastereoselectivity from simple and readily available starting materials. DFT calculations were used to explore the mechanism in different solutions. The synthetic application example also showed the prospects of this method for the preparation of valuable compounds.
Subject terms: Synthetic chemistry methodology, Synthetic chemistry methodology, Homogeneous catalysis
Lawsones and indenopyrazoles harbor an interesting benzo-fused cyclic ketone scaffold as an essential structural motif in diverse bioactive molecules, however, their synthesis remains challenging. Here, the authors report an efficient synthesis of lawsones and indenopyrazolones via the solvent-controlled regioselective ring-expansion of indantriones involving a 1,2-carbonyl shift.
Introduction
The generation of privileged natural product scaffolds is a successful strategy for obtaining libraries of bioactive compounds1–4. Scaffold generation focuses on the application of methodologies capable of generating structures that cover a larger part of the chemical and biological space5–7. Since scaffold generation is pivotal during the early stage of drug discovery, issues like chemical efficiency and easy diversification are crucial in chemical reaction development. Using diazo compounds to expand the size of cyclocarbonyl compounds by one carbon unit is a classical homologation method for the generation of structurally complex and diverse scaffolds8–11. In the last century, the ring expansion of isatins and other cyclic ketones with multiple diazo variants (e.g., diazomethane and α-diazoesters) was reported12–15. Several catalytic ring-expansion reactions also have been performed by the Kingsbury16, Maruoka17, and Feng groups18. However, the ring-expansion reaction of benzo-fused cyclic dione substrate faces regioselectivity because of 1,2-aryl migration and 1,2-carbonyl migration reaction pathways (Fig. 1a). To the best of our knowledge, the regioselective ring-expansion reaction of indantrione has not been reported. In a continuation of our research with diazo compounds, we report herein the regioselective ring expansion of indantrione with α-aryldiazomethane involving 1,2-carbonyl migration (Fig. 2b).
Fig. 1. Regiochemistry of the ring expansion of cyclic ketones.
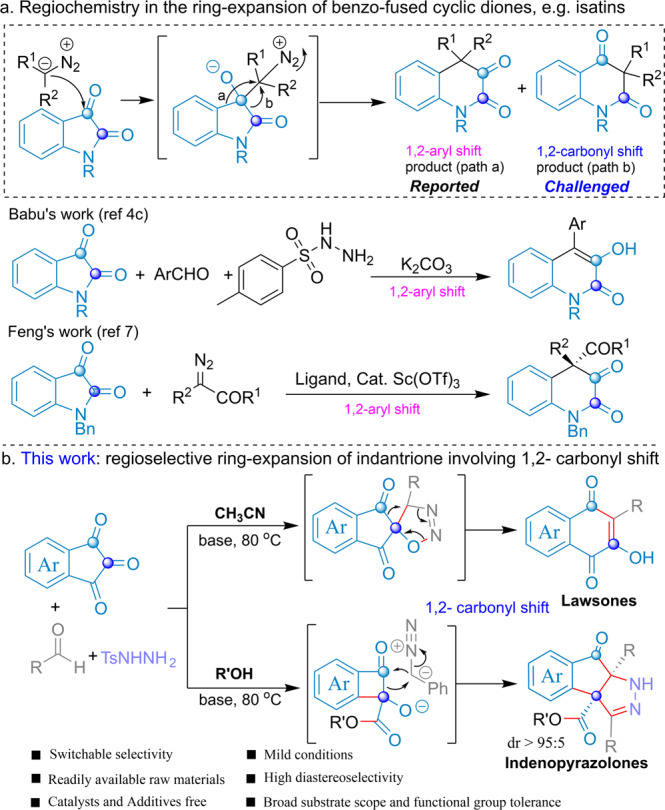
a The selective ring expansion of isatins. b This work, the selective ring-expansion of indantrione.
Fig. 2. Representative bioactive compounds.
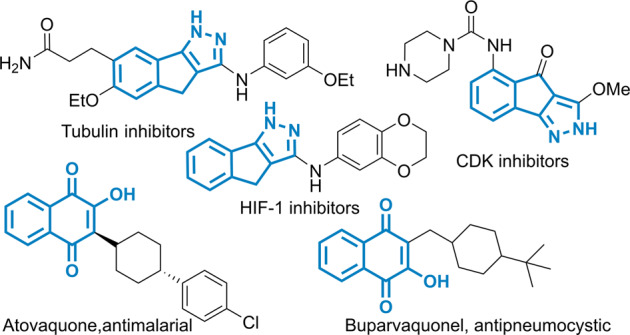
Such as Lawsone derivatives with tubulin, CDK, and HIF-1 inhibitions and indenopyrazole derivatives with antimalarial and antipneumocystic activities.
Lawsones and indenopyrazole derivatives constitute important classes of polycyclic systems embedded in a plethora of natural products and synthetic compounds showing antimalarial (Atovaquone)19, antipneumocystic (Buparvaquone)20 bioactivities, and protease inhibitory activities such as cyclin-dependent kinase (CDK)21,22, hypoxia-inducible factor (HIF)-123,24, and tubulin25–27 (Fig. 2). In addition, lawsones are also the essential synthons28 for the synergistic nucleophilicity and electrophilicity to overcome various of synthetic challenges29–31. Despite being appealing structural motifs, highly efficient approaches to prepare lawsones and indenopyrazoles are limited32–41. The reported protocols are generally multi-step reactions, which require high loadings of metal catalysts and properly functionalized substrates that are not readily available. The solvent-controlled ring-expansion reaction of indantrione and α-aryldiazomethane would be a straightforward method for preparing these lawsones and indenopyrazole compounds. Herein, a metal-free, ring-expansion reaction of indantrione with diazomethanes, generated in situ from the N-tosylhydrazones, has been developed for the synthesis of lawsone and indenopyrazole derivatives in acetonitrile and alcohol solvents, respectively.
Results and discussion
Optimization of the reaction conditions
We screened an array of solvents and bases (Table 1) for their reactivity and chemoselectivity in the ring-expansion reaction of indantrione 1a, benzaldehyde 2a, and p-methylbenzenesulfonohydrazide 3. This survey led to the following optimized reaction conditions: (1) 1,2,3-Indantrione 1a (1.0 mmol) and Cs2CO3 (2.0 equiv.) were added to α-aryldiazomethane and stirred at 80 °C in MeCN for 3 h. The desired lawsone 4a product was obtained in 90% yield (Table 1, entry 1); (2) 1,2,3-Indantrione 1a (1.0 mmol) and Cs2CO3 (3.0 equiv.) were added to the α-aryldiazomethane and stirred at 80 °C in EtOH for 10 h. The desired indenopyrazolone 5a product was obtained in 83% yield with a dr value >95:5 (Table 1, entry 5). A small amount of lawson product 4a was generated in a 15% yield. This result shows that solvent-controlled regioselective ring-expansion reaction had a 1,2-carbonyl migratory tendency with two different products.
Table 1.
Reaction optimizationa,b,c.
| Entry | Solvent | Base | Time (h) | Yield (%) | |
|---|---|---|---|---|---|
| 4a | 5(dr) | ||||
| 1 | CH3CN | Cs2CO3 | 10 | 90 | nd |
| 2 | 1,4-dioxane | Cs2CO3 | 10 | 31 | nd |
| 3 | toluene | Cs2CO3 | 10 | nd | nd |
| 4 | THF | Cs2CO3 | 10 | 47 | nd |
| 5 | EtOH | Cs2CO3 | 10 | 15 | 83 (>95:5) |
| 6 | MeOH | Cs2CO3 | 10 | 8 | 87 (>95:5) |
| 7 | n-propanol | Cs2CO3 | 10 | 27 | 65 (>95:5) |
| 8 | H2O | Cs2CO3 | 10 | 5 | nd |
| 9 | EtOAc | Cs2CO3 | 10 | 67 | nd |
| 10 | DMSO | Cs2CO3 | 10 | 51 | nd |
| 11 | CH3CN | DBU | 3 | 85 | nd |
| 12 | CH3CN | K2CO3 | 3 | 83 | nd |
| 13 | CH3CN | n-BuOK | 3 | 15 | nd |
| 14 | CH3CN | Na2CO3 | 3 | 77 | nd |
| 15 | CH3CN | Et3N | 3 | nd | nd |
| 16 | CH3CN | NaOH | 3 | 35 | nd |
| 17 | EtOH | DBU | 10 | 3 | 80 (>95:5) |
| 18 | EtOH | K2CO3 | 10 | 5 | 74 (>95:5) |
| 19 | EtOH | t-BuOK | 10 | 35 | 62 (>95:5) |
| 20 | EtOH | Na2CO3 | 10 | nd | 77 (>95:5) |
| 21 | EtOH | Et3N | 10 | nd | 36 (>95:5) |
| 22 | EtOH | NaOH | 10 | 46 | 51 (>95:5) |
nd not detected.
aReagents and conditions: (1) In a 25 mL reaction tube, benzaldehyde 2a (1.2 mmol), p-toluenesulfonyl hydrazide 3 (1.2 mmol), solvent (10 mL), under ambient atmosphere (1 atm), at room temperature for 0.5–1 h. (2) indantrione 1a (1.0 mmol), base (2.0 mmol), at 80 °C for 10 h.
bThe dr value was determined by 1H-NMR analysis of crude products.
cIsolated yield based on 1a.
Substrate scope of indantriones and aldehydes for producing lawsones
We then explored the scope of this reaction with a range of indantriones 1 and aldehydes 2 (Fig. 3) (see Supplementary Notes 1, 2) for producing lawsones. As anticipated, the ring-expansion reaction of indantriones generally tolerated a broad range of substituted aryl aldehydes bearing either electron-donating or electron-withdrawing substituents, such as methyl, fluoro, chloro, bromo, nitro, nitrile, trifluoromethyl, and naphthyl groups. The corresponding hydroxynaphtho-quinones were afforded in high to excellent yield. The electron-donating groups slightly reduced the reaction yield compared with electron-withdrawing groups (e.g., 4b, p-F, 96% and 4j, p-CF3, 93% yields vs. 4e, p-CH3, 90% and 4h, p-OMe, 85% yields). The position of substituents did not significantly affect the reaction yield (e.g., 4b, para-F; 4m, meta-F; 4u, ortho-F; with yields of 96%, 95%, and 90%, respectively). Polysubstituted aryl aldehydes were also suitable for this reaction (e.g., 4aa and 4ab with 85% and 84% yields, respectively). Borate ester-substituted aryl aldehydes (4t, 91% yield), heteroaryl aldehydes (4ad and 4ag, 93% and 81% yields, respectively), and some large sterically hindered aryl aldehydes (4af and 4ah, 85% and 80% yields, respectively) also reacted smoothly. The ring-expansion reaction also produced equally satisfying results using alkyl aldehydes, e.g., prenylaldehyde (4ai, 74% yield), butyraldehyde (4aj, 71% yield), and 2-ethylcaproaldehyde (4ak, 50% yield) as substrates. The corresponding ring-expansion products were also obtained under the standard conditions for substituted indantriones and benzaldehyde (4al 88%, and 4am 85% yields). To verify the relative configurations of the hydroxynaphtho-quinones, 4h, and 4ae were selected as representative compounds and characterized by X-ray crystallography (see Supplementary Data 1, 2, and see Supplementary Note 3, Figs. S1 and S2) (CCDC 2149716 (4h), 2149715 (4ae), 2149717 (5a), 2149718 (5f), 2149719 (5q), 2149720 (5r) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif).
Fig. 3. Substrate scope of indantriones and aldehydes for producing lawsones.

a General conditions: (1) In a 25 mL reaction tube, aldehyde 2 (1.2 mmol), p-toluenesulfonyl hydrazide 3 (1.2 mmol), CH3CN solvent (10 mL), under ambient atmosphere (1 atm), stirred for 0.5–1 h. (2) indantrione 1a (1.0 mmol), Cs2CO3 (2.0 mmol), at 80 °C for 3 h. b Isolated yield based on 1a.
Substrate scope of indantriones and aldehydes for producing indenopyrazoles
Similarly, we investigated the scope of this reaction using a range of indantriones 1 and aldehydes 2 (Fig. 4) (see Supplementary Notes 1, 2) to produce indenopyrazolones. Under the standard reaction conditions, aryl aldehydes with various substituents, such as methyl, fluoro, chloro, bromo, or methoxy groups, smoothly reacted to afford the indenopyrazolone derivatives in high to excellent yields and high stereoselectivities (dr, ≥95:5). Electron-donating groups also slightly reduced the reaction yield compared with electron-withdrawing groups (e.g., 5b, p-CH3, 83% and 5f, p-OCH3, 79% yield vs. 5c, p-F, 91% and 5d, p-Cl, 88% yields). The position and number of substituents did not significantly affect the reaction yield (e.g., 5c, para-F; 5i, meta-F; 5 m ortho-F; with yields of 91%, 90%, and 81%, respectively). The reaction also produced equally satisfying results using heteroaryl aldehydes (e.g., 5q, 85% yield) and sterically bulky aryl aldehydes (e.g., 5t, 81% yield). In different alcohol solutions, the corresponding esterified indenopyrazolone derivatives were obtained, such as 5o–5ab, with medium to excellent yields and high stereoselectivities (dr, ≥95:5). The corresponding products were also obtained under standard conditions for substituted indantriones and benzaldehyde (5ad 85%, and 5ae 87% yields). Unfortunately, alkyl-substituted aldehydes were found to be incompatible with this transformation and did not yield any product. 5a, 5f, 5q, and 5r were characterized by X-ray crystallography (see Supplementary Data 3–6, and see Supplementary Note 3, Figs. S3–S6) (CCDC 2149716 (4h), 2149715 (4ae), 2149717 (5a), 2149718 (5f), 2149719 (5q), 2149720 (5r) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif).
Fig. 4. Substrate scope of indantriones and aldehydes for producing indenopyrazoles.

a General conditions: (1) In a 25 mL reaction tube, aldehyde 2 (2.1 mmol), p-toluenesulfonyl hydrazide 3 (2.1 mmol), R’OH solvent (20 mL), under ambient atmosphere (1 atm), stirred for 0.5–1 h. (2) indantrione 1a (1.0 mmol), Cs2CO3 (3.0 mmol), at 80 °C for 10 h. b The dr value was determined by 1H-NMR analysis of crude products. c Isolated yield based on 1a.
Mechanistic studies
To elucidate the reaction mechanism of the regioselective ring-expansion of indantrione and α-aryldiazomethanes, various control experiments were conducted (Fig. 5). The results indicated that: (1) the addition of TEMPO or BHT did not inhibit the reaction under standard conditions, and no radical-TEMPO/BHT coupling products were detected (Fig. 5, eq. 1); (2) isotope-labeling experiments in deuterated methanol showed that the ester group of the indenopyrazolone product was formed by bonding the C1 atom of indantrione with deuterated methyl (Fig. 5, eq. 2); (3) using lawsone 4a and α-aryldiazomethane as the starting material in alcohol solution, indenopyrazolone product 5a was not obtained. This shows that the reaction mechanism of the two products proceeded via different paths (Fig. 5, eq. 3); (4) by using the mixed solvent of acetonitrile and ethanol under standard conditions, the yield of the indenopyrazolone product increased gradually upon increasing the proportion of ethanol. On the contrary, the yield of lawsones increased upon increasing the acetonitrile proportion (Fig. 5, eq. 4). This shows that the solvent had an obvious selectivity for the two different products; (5) Using ninhydrin 1a’ as the starting material, the indenopyrazolone product was also obtained in an alcohol solvent under standard conditions (Fig. 5, eq. 5). The lawsone product was obtained in acetonitrile solvent (Fig. 5, eq. 6).
Fig. 5. Control experiments.

Free radical capture experiment (eq. 1). Deuterium hydrogen exchange experiment (eq. 2). Standard reaction of intermediate 4 in ethanol solvent (eq. 3). Mixed solvent proportion control experiment (eq. 4). Reaction of ninhydrin hydrate under standard conditions (eqs. 5 and 6).
To better understand the regioselectivity of the ring-expansion process and the cascade reaction mechanism, based on the experimental results, we performed theoretical calculations (see Supplementary Data 8) on the possible reaction pathways of this solvent-controlled regioselective ring-expansion reaction of indantrione and α-aryldiazomethane42–44. Initially, aldehyde 2 and p-toluenesulfonyl hydrazide 3 condensed in solution to form aryl alkylenediamine a (Fig. 6). Then, the aryl alkenylamine removed the benzosulfonyl group to obtain α-aryldiazomethane dipoles b and b’. Subsequently, indantriones underwent two different ring-expansion reactions in different solvents. In acetonitrile, the ring-expansion reaction of indantrione proceeded via the competitive pathways A and B. In reaction pathway B, the carboanion of the α-aryldiazomethane dipole b nucleophile attacked the carbonyl C1 atom of indantrione. Then, intermediate INT-4 formed through the cycloaddition of transition state TS-3 (ΔG = 28.2 kcal mol−1). Next, intermediate INT-4 underwent intramolecular ring expansion and denitrogenation. After transition state TS-4 (ΔG = 30.6 kcal mol−1), intermediate INT-5 formed, which underwent enol tautomerism to obtain 4a’ (Fig. 6, pathway B). Similarly, in reaction pathway A, α-aryldiazomethane dipole b attacked carbonyl C2 of indantrione via transition state TS-1 (ΔG = 22.0 kcal mol−1) to form intermediate INT-2. Then, the intermediate underwent intramolecular ring expansion and denitrogenation. After transition state TS-2 (ΔG = 22.1 kcal mol−1), intermediate INT-3 formed, and finally, tautomerism occurred to form stable product 4a. Notably, our calculations showed that the energy barrier of the initial step TS-3 (ΔG = 22.0 kcal mol−1) of pathway B was higher than TS-1 (ΔG = 15.8 kcal mol−1) of pathway A. The energy barrier difference of the two-step transition state was about 6.2 kcal mol−1 (Fig. 6). The results indicated that nucleophilic addition between indantrione and α-aryldiazomethane was more favorable at the C2 site than at the C1 site. The reaction was more likely to produce reaction product 4a. This is consistent with the experimental results. Based on the calculation results, the proposed mechanism is shown in Fig. 7, path a.
Fig. 6. Calculated reaction pathways and free energy profiles for the regioselectivity of the ring-expansion process in acetonitrile solvent.

DFT calculations were performed with B3LYP-D3(BJ)-SMD(acetonitrile)/6-311++G(d,p)//B3LYP-D3(BJ)/6-31G(d) (for details see SI, Table S7).
Fig. 7. Proposed mechanism.

a Reaction formation mechanism of Lawson derivatives. b Reaction formation mechanism of indenopyrazole derivatives.
In alcohol solution, the alcohol oxygen anion first nucleophilically attacked the carbonyl C1 atom of indantriones to form intermediate INT-6 (Fig. 8). Then, the intermediate underwent a 1,2-aryl migration rearrangement to obtain the four-membered-ring intermediate, INT-7 via TS-5 (ΔG = 20.1 kcal mol−1). α-Aryldiazomethane b was then nucleophilically added to the cyclocarbonyl carbon atom of intermediate INT-7 to obtain INT-9 via TS-6 (ΔG = 41.2 kcal mol−1). Next, under the catalysis of Cs2HCO3+, INT-9 underwent transition state TS-7 (ΔG = −52.8 kcal mol−1) and TS-8 (ΔG = −57.49 kcal mol−1) to undergo dehydration to form intermediate INT-11. Subsequently, α-aryldiazomethane dipole b attacked the conjugated double bond of intermediate INT-11 via TS-9 (ΔG = −55.5 kcal mol−1) to obtain INT-13. This process was diastereoselective. Finally, intermediate INT-13 underwent heterocyclic condensation and isomerization to obtain product 5a (Fig. 8). Based on the calculation results, the proposed mechanism is shown in Fig. 7, path b.
Fig. 8. Calculated reaction pathways and free energy profiles for the regioselectivity of the ring-expansion process in ethanol solvent.

DFT calculations were performed with B3LYP-D3(BJ)-SMD(ethanol)/[other:6-311++G(d,p);Cs: Lanl2DZ] //B3LYP-D3(BJ)/[other: 6-31G (d); Cs: Lanl2DZ] (for details see SI, Table S8).
Synthetic application
To test the robustness of this solvent-controlled regioselective ring-expansion reaction, a series of multigram-scale experiments were performed (Fig. 9) (see Supplementary Method 1). When 10 mmol of 1a’ and 21 mmol of 2a in EtOH or MeOH solvent were subjected to the standard reaction conditions, product 5a or 5o was isolated in 76% and 81% yield, respectively. Additionally, when 10 mmol of 1a and 12 mmol of 2a in MeCN solvent were subjected to the standard reaction conditions, product 4a was isolated in an 89% yield. The synthetic versatility of lawsones and indenopyrazolones was then explored in an array of derivatizations (see Supplementary Method 2). The carbonyl group of compound 5a was reduced to a hydroxyl group 6a under lithium aluminum hydride reduction, but the ester group was unaffected. Compound 6b was obtained by the selective methoxy stripping of compound 5n under the action of boron tribromide. Lawsone product 4a can be used as a reaction synthon to construct a variety of products with diverse structures that can react with phenylalkynes to construct benzannulated bicyclo[3.3.0]octanes compound 6d with biological activity45. Compound 4a also reacted with diphenylacetylene to obtain alkylidene phthalide 6c, which is a key intermediate for the synthesis of bioactive natural products46. The synthesized bioactive natural product 4a was directly acetylated to obtain hydroxyacetyl product 6e. Notably, compound 4w easily formed carbazoloquinone derivative 6f under metal catalysis, which is of great interest as a privileged structure for anticancer drug molecules47.
Fig. 9. Synthetic application.

Multigram-scale experiments and synthetic versatility of lawsones and indenopyrazolones. General conditions: see SI, Supplementary Method 2.
Conclusions
In summary, we have described the first solvent-controlled regioselective ring-expansion reaction of indantrione and α-aryldiazomethanes. This reaction preferentially provides the 1,2-carbonyl migration product. In acetonitrile solvent, the reaction products were lawsone derivatives. In alcohol solution, the reaction products were stereoselective indenopyrazolone derivatives. A series of substrates underwent the reaction smoothly, providing highly-functionalized lawsone derivatives and indenopyrazolone derivatives in high yields (up to 95%) and with high levels of diastereoselectivity (up to 95:5 dr). A range of functional groups was also tolerated under the mild reaction conditions. The mechanism control experiment was used to determine the possible reaction mechanism pathways. The synthetic application example also demonstrated the prospect of this method for preparing valuable compounds.
Methods
General procedure for preparing lawsones
A mixture of aldehydes (2, 1.2 mmol) and p-toluenesulfonyl hydrazide (1.2 mmol) in acetonitrile (20 ml) was stirred at room temperature for 0.5 h until complete consumption of starting materials (monitored by TLC). Then, indantrione (1, 1 mmol) and Cs2CO3 (2 equiv) were added to the crude product and stirred at 80 °C for 3 h. After the reaction was finished, the solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate 8:1) to afford the desired product 4. The products were further identified by FTIR spectroscopy, NMR spectroscopy, and HRMS, see Supplementary Data 7.
General procedure for preparing Indenopyrazoles
A mixture of aldehydes (2, 2.1 mmol) and p-toluenesulfonyl hydrazide (2.1 mmol) in alcohol (30 ml) was stirred at room temperature for 0.5–1 h until complete consumption of starting materials (monitored by TLC). Then, indantrione (1, 1 mmol) and Cs2CO3 (3 equiv) were added to the crude product and stirred at 80 °C for 10 h. After the reaction was finished, the solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate 25:1) to afford the desired product 5. The products were further identified by FTIR spectroscopy, NMR spectroscopy, and HRMS (see Supplementary Data 7).
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
The authors gratefully acknowledge for financial support of the Program for the NSFC (Nos. 22267021, 22067020), Yunnan Fundamental Research Projects (202101AS070034, 202001BB050009), and the Program for Excellent Young Talents, Yunnan University.
Author contributions
Y.J. conceived and directed the project. BW.H., PY.J., L.J., and X.Y. performed the experiments. WX.Y. performed the theoretical calculations. J.L., YC.J., and Y.J. analyzed the results, and Y.J. wrote the manuscript.
Peer review
Peer review information
Communications Chemistry thanks Fernando da Silva and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and the Supplementary Information as well as from the authors upon reasonable request. The X-ray crystallographic coordinates for structures 4h, 4ae, 5a, 5f, 5q, and 5r, reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under CCDC 2149716 (4h, Supplementary Data 1), 2149715 (4ae, Supplementary Data 2), 2149717 (5a, Supplementary Data 3), 2149718 (5f, Supplementary Data 4), 2149719 (5q, Supplementary Data 5) and 2149720 (5r, Supplementary Data 6), respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The compound characterizations are available in Supplementary Data 7. DFT calculations are available in Supplementary Data 8. The computed output file is in the open database (https://zenodo.org/) link (DOI:10.5281/zenodo.7296393).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Bingwei Hu, Wenxin Yan, Peiyun Jiang.
Contributor Information
Yinchun Jiao, Email: yinchunjiao@hnust.edu.cn.
Yi Jin, Email: jinyi@ynu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-022-00807-z.
References
- 1.Rizzo S, Waldmann H. Development of a natural-product-derived chemical toolbox for modulation of protein function. Chem. Rev. 2014;114:4621. doi: 10.1021/cr400442v. [DOI] [PubMed] [Google Scholar]
- 2.Nandy JP, et al. Advances in solution-and solid-phase synthesis toward the generation of natural product-like libraries. Chem. Rev. 2009;109:1999–2060. doi: 10.1021/cr800188v. [DOI] [PubMed] [Google Scholar]
- 3.Andrei SA, et al. Rationally designed semisynthetic natural product analogues for stabilization of 14-3-3 protein–protein interactions. Angew. Chem. Int. Ed. 2018;57:13470–13474. doi: 10.1002/anie.201806584. [DOI] [PubMed] [Google Scholar]
- 4.Itoh H, et al. Development of a high-throughput strategy for discovery of potent analogues of antibiotic lysocin E. Nat. Commun. 2019;10:1–11. doi: 10.1038/s41467-019-10754-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat. Chem. 2016;8:531–541. doi: 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- 6.Grigalunas M, Brakmann S, Waldmann H. Chemical evolution of natural product structure. J. Am. Chem. Soc. 2022;144:3314–3329. doi: 10.1021/jacs.1c11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bon RS, Waldmann H. Bioactivity-guided navigation of chemical space. Acc. Chem. Res. 2010;43:1103. doi: 10.1021/ar100014h. [DOI] [PubMed] [Google Scholar]
- 8.Ford A, et al. Modern organic synthesis with α-diazocarbonyl compounds. Chem. Rev. 2015;115:9981–10080. doi: 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]
- 9.Ye T, McKervey MA. Organic synthesis with. alpha.-diazo carbonyl compounds. Chem. Rev. 1994;94:1091–1160. doi: 10.1021/cr00028a010. [DOI] [Google Scholar]
- 10.Candeias NR, Paterna R, Gois PM. Homologation reaction of ketones with diazo compounds. Chem. Rev. 2016;116:2937–2981. doi: 10.1021/acs.chemrev.5b00381. [DOI] [PubMed] [Google Scholar]
- 11.Tan F, et al. Catalytic Asymmetric Homologation of Ketones with α-Alkyl α-Diazo Esters. J. Am. Chem. Soc. 2021;143:2394–2402. doi: 10.1021/jacs.0c12683. [DOI] [PubMed] [Google Scholar]
- 12.Mosettig E, Burger A. Ring enlargement with diazomethane in the hydroaromatic series1. J. Am. Chem. Soc. 1930;52:3456–3463. doi: 10.1021/ja01371a070. [DOI] [Google Scholar]
- 13.Gutsche CD. The reaction of diazomethane and its derivatives with aldehydes and ketones. Org. React. 2004;8:364–430. [Google Scholar]
- 14.Tangella Y, et al. Regioselective ring expansion of isatins with in situ generated α-aryldiazomethanes: direct access to viridicatin alkaloids. Org. lett. 2018;20:3639–3642. doi: 10.1021/acs.orglett.8b01417. [DOI] [PubMed] [Google Scholar]
- 15.Skubi KL, et al. Enantioselective excited-state photoreactions controlled by a chiral hydrogen-bonding iridium sensitizer. J. Am. Chem. Soc. 2017;139:17186–17192. doi: 10.1021/jacs.7b10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rendina VL, Moebius DC, Kingsbury JS. An enantioselective synthesis of 2-aryl cycloalkanones by Sc-catalyzed carbon insertion. Org. lett. 2011;13:2004–2007. doi: 10.1021/ol200402m. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto T, Naganawa Y, Maruoka K. Desymmetrizing asymmetric ring expansion of cyclohexanones with α-diazoacetates catalyzed by chiral aluminum Lewis acid. J. Am. Chem. Soc. 2017;133:8834–8837. doi: 10.1021/ja202070j. [DOI] [PubMed] [Google Scholar]
- 18.Li W, et al. A catalytic asymmetric ring-expansion reaction of isatins and α-alkyl-α-diazoesters: highly efficient synthesis of functionalized 2‐quinolone derivatives. Angew. Chem. Int. Ed. 2012;124:8772–8775. doi: 10.1002/ange.201204594. [DOI] [PubMed] [Google Scholar]
- 19.Paton DG, et al. Exposing Anopheles mosquitoes to antimalarials blocks Plasmodium parasite transmission. Nature. 2019;567:239–243. doi: 10.1038/s41586-019-0973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mäntylä A, et al. Synthesis, in vitro evaluation, and antileishmanial activity of water-soluble prodrugs of buparvaquone. J. Med. Chem. 2004;47:188–195. doi: 10.1021/jm030868a. [DOI] [PubMed] [Google Scholar]
- 21.Nugiel DA, et al. Synthesis and evaluation of indenopyrazoles as cyclin-dependent kinase inhibitors. 2. Probing the indeno ring substituent pattern. J. Med. Chem. 2002;45:5224–5232. doi: 10.1021/jm020171+. [DOI] [PubMed] [Google Scholar]
- 22.Yue EW, et al. Synthesis and evaluation of indenopyrazoles as cyclin-dependent kinase inhibitors. 3. Structure–activity relationships at C3. J. Med. Chem. 2002;45:5233–5248. doi: 10.1021/jm0201722. [DOI] [PubMed] [Google Scholar]
- 23.Fuse S, et al. Design, synthesis, and evaluation of indeno[2,1-c]pyrazolones for use as inhibitors against hypoxia-inducible factor (HIF)-1 transcriptional activity. Bioorg. Med. Chem. 2020;28:115207. doi: 10.1016/j.bmc.2019.115207. [DOI] [PubMed] [Google Scholar]
- 24.Minegishi H, Fukashiro S, Ban HS, Nakamura H. Discovery of Indenopyrazoles as a New Class of Hypoxia Inducible Factor (HIF)-1 Inhibitors. ACS Med. Chem. Lett. 2013;4:297–301. doi: 10.1021/ml3004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan I, et al. Design, synthesis and biological evaluation of 1, 4-dihydro indeno[1,2-c] pyrazole linked oxindole analogues as potential anticancer agents targeting tubulin and inducing p53 dependent apoptosis. Eur. J. Med. Chem. 2018;144:104–115. doi: 10.1016/j.ejmech.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y-N, et al. Design, synthesis, and biological evaluation of 1-methyl-1,4-dihydroindeno[1,2-c]pyrazole analogues as potential anticancer agents targeting tubulin colchicine binding site. J. Med. Chem. 2016;59:5341–5355. doi: 10.1021/acs.jmedchem.6b00071. [DOI] [PubMed] [Google Scholar]
- 27.Minegishi H, et al. Methyl 3-((6-Methoxy-1,4-dihydroindeno[1,2-c]pyrazol-3-yl)amino)benzoate (GN39482) as a tubulin polymerization inhibitor identified by MorphoBase and ChemProteoBase profiling methods. J. Med. Chem. 2015;58:4230–4241. doi: 10.1021/acs.jmedchem.5b00035. [DOI] [PubMed] [Google Scholar]
- 28.Sadeghpour M, Olyaei A, Adl A. Recent progress on the synthesis of henna-based dibenzoxanthenes. N. J. Chem. 2021;45:13669–13691. doi: 10.1039/D1NJ02475B. [DOI] [Google Scholar]
- 29.Rueping M, Sugiono E, Merino E. Asymmetric iminium ion catalysis: an efficient enantioselective synthesis of pyranonaphthoquinones and β-lapachones. Angew. Chem. Int. Ed. 2008;47:3046–3049. doi: 10.1002/anie.200705110. [DOI] [PubMed] [Google Scholar]
- 30.Ramachary DB, Anif Pasha M, Thirupathi G. Organocatalytic asymmetric formal [3+2] cycloaddition as a versatile platform to access methanobenzo [7] annulenes. Angew. Chem. Int. Ed. 2017;56:12930–12934. doi: 10.1002/anie.201706557. [DOI] [PubMed] [Google Scholar]
- 31.Kaya U, et al. Enantioselective synthesis of 4 H-pyranonaphthoquinones via sequential squaramide and silver catalysis. Chem. Commun. 2016;52:1669–1672. doi: 10.1039/C5CC09592A. [DOI] [PubMed] [Google Scholar]
- 32.Louvis ADR, et al. Synthesis, characterization and biological activities of 3-aryl-1, 4-naphthoquinones–green palladium-catalysed Suzuki cross coupling. N. J. Chem. 2016;40:7643–7656. doi: 10.1039/C6NJ00872K. [DOI] [Google Scholar]
- 33.Yu D, Chen XL, Ai BR, Zhang XM, Wang JY. Tetrabutylammonium iodide catalyzed hydroxylation of naphthoquinone derivatives with tert-butyl hydroperoxide as an oxidant. Tetrahedron Lett. 2018;59:3620–3623. doi: 10.1016/j.tetlet.2018.08.052. [DOI] [Google Scholar]
- 34.Allan KM, Hong BD, Stoltz BM. Expedient synthesis of 3-hydroxyisoquinolines and 2-hydroxy-1, 4-naphthoquinones via one-pot aryne acyl-alkylation/condensation. Org. Biomol. Chem. 2009;7:4960–4964. doi: 10.1039/b913336d. [DOI] [PubMed] [Google Scholar]
- 35.Schank K, La Vecchia L, Lick C. Chemie freier, cyclischer vicinaler Tricarbonyl-Verbindungen (1, 2, 3-Trione’), Teil 1, Reaktionsweise von Diazomethan und seinen Derivaten mit 5, 5-Dimethylcyclohexan-1, 2, 3-trion (=Oxo-dimedon’) und verwandten Cyclohexan-1, 2, 3-trionen. Helv. Chim. Acta. 2001;84:2071–2088. doi: 10.1002/1522-2675(20010711)84:7<2071::AID-HLCA2071>3.0.CO;2-D. [DOI] [Google Scholar]
- 36.Ramu G, Kodiripaka BG, Chaitanya KR, Babu BN. A facile and metal-free domino reaction of TsDAM and 2-alkenylarylaldehyde: An easy access to 8-hydroxy-2, 8-dihydro indeno [2, 1-c] pyrazoles. Org. Biomol. Chem. 2021;19:4118–4125. doi: 10.1039/D1OB00262G. [DOI] [PubMed] [Google Scholar]
- 37.Chaturvedi AK, Kant R, Rastogi N. Access to the phosphorylindenopyrazole scaffold via a metal-free domino reaction of diazoalkylphosphonates with 3-bromophthalides. J. Org. Chem. 2016;81:11291–11296. doi: 10.1021/acs.joc.6b02267. [DOI] [PubMed] [Google Scholar]
- 38.Davies DL, Ellul CE, Macgregor SA, McMullin CL, Singh K. Experimental and DFT studies explain solvent control of C–H activation and product selectivity in the Rh (III)-catalyzed formation of neutral and cationic heterocycles. J. Am. Chem. Soc. 2015;137:9659–9669. doi: 10.1021/jacs.5b04858. [DOI] [PubMed] [Google Scholar]
- 39.Deng Y, et al. Solvent tunes the selectivity of hydrogenation reaction over α-MoC catalyst. J. Am. Chem. Soc. 2018;140:14481–14489. doi: 10.1021/jacs.8b09310. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, et al. Solvent-enabled radical selectivities: controlled syntheses of sulfoxides and sulfides. Angew. Chem. Int. Ed. 2017;55:1094–1097. doi: 10.1002/anie.201508729. [DOI] [PubMed] [Google Scholar]
- 41.Liu C, Zhu C, Cai Y, Jiang H. Solvent-switched oxidation selectivities with O2: controlled synthesis of α-difluoro (thio) methylated alcohols and ketones. Angew. Chem. Int. Ed. 2021;60:12038–12045. doi: 10.1002/anie.202017271. [DOI] [PubMed] [Google Scholar]
- 42.Deng S, Qu CX, Jiao YC, Liu WQ, Shi F. Insights into 2-indoly lmethanol-involved cycloadditions: origins of regioselectivity and enantioselectivity. J. Org. Chem. 2020;85:11641–11653. doi: 10.1021/acs.joc.0c01123. [DOI] [PubMed] [Google Scholar]
- 43.Li T-Z, et al. Regio- and enantioselective (3+3) cycloaddition of nitrones with 2-indolylmethanols enabled by cooperative organocatalysis. Angew. Chem. Int. Ed. 2021;60:2355–2363. doi: 10.1002/anie.202011267. [DOI] [PubMed] [Google Scholar]
- 44.Wu P, et al. Design and synthesis of axially chiral aryl-pyrroloindoles via the strategy of organocatalytic asymmetric (2 + 3) cyclization. Fund. Res. 2022 doi: 10.1016/j.fmre.2022.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar AS, Thirupathi G, Reddy GS, Ramachary DB. Stereoselective insertion of benzynes into lawsones: synthesis of biologically important benzannulated bicyclo [3.3. 0] octanes. Chemistry. 2019;25:1177–1183. doi: 10.1002/chem.201805798. [DOI] [PubMed] [Google Scholar]
- 46.Borthakur S, Baruah S, Sarma B, Gogoi S. Pd (II)-catalyzed synthesis of alkylidene phthalides via a decarbonylative annulation reaction. Org. Lett. 2019;21:2768–2771. doi: 10.1021/acs.orglett.9b00726. [DOI] [PubMed] [Google Scholar]
- 47.Barcia JC, et al. A new approach to the synthesis of benzo [b] naphtho [2, 3-b] furan-6, 11-diones and 2-benzyl-3-hydroxynaphthalene-1, 4-diones. SynOpen. 2017;1:0156–0165. doi: 10.1055/s-0036-1591729. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and the Supplementary Information as well as from the authors upon reasonable request. The X-ray crystallographic coordinates for structures 4h, 4ae, 5a, 5f, 5q, and 5r, reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under CCDC 2149716 (4h, Supplementary Data 1), 2149715 (4ae, Supplementary Data 2), 2149717 (5a, Supplementary Data 3), 2149718 (5f, Supplementary Data 4), 2149719 (5q, Supplementary Data 5) and 2149720 (5r, Supplementary Data 6), respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The compound characterizations are available in Supplementary Data 7. DFT calculations are available in Supplementary Data 8. The computed output file is in the open database (https://zenodo.org/) link (DOI:10.5281/zenodo.7296393).