

Abstract
An increasing number of invasive fruit fly pests are colonizing new grounds. With this study, we aimed to uncover the invasion pathways of the oriental fruit fly, Bactrocera dorsalis into the islands of the Indian Ocean. By using genome‐wide SNP data and a multipronged approach consisting of PCA, ancestry analysis, phylogenetic inference, and kinship networks, we were able to resolve two independent invasion pathways. A western invasion pathway involved the stepping‐stone migration of B. dorsalis from the east African coast into the Comoros, along Mayotte and into Madagascar with a decreasing genetic diversity. The Mascarene islands (Reunion and Mauritius), on the contrary, were colonized directly from Asia and formed a distinct cluster. The low nucleotide diversity suggests that only a few genotypes invaded the Mascarenes. The presence of many long runs of homozygosity (ROH) in the introduced populations is indicative of population bottlenecks, with evidence of a more severe bottleneck for populations along the western migration pathway than on the Mascarene islands. More strict phytosanitary regulations are recommended in order to prevent the further spread of B. dorsalis.
Keywords: Bactrocera dorsalis, Indian Ocean, invasive species, pest species, phylogeography
1. INTRODUCTION
In response to globalization, there has been an increased concern surrounding invasive phytophagous insect species, which is motivated by the accumulating economic losses in the fruit trade (Suckling et al., 2013; Welsh et al., 2021). The oriental fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae), is a major pest species natively found in wet tropical regions of Asia (Clarke et al., 2019; Zeng et al., 2019). The host range of B. dorsalis consists of a large variety of plants in both the species' native (Tan & Serit, 1994; Zeng et al., 2019) and invasive range (Hassani et al., 2016; Moquet et al., 2021; Rwomushana et al., 2008). With increasing invasions worldwide, many efforts have been undertaken to eradicate or suppress the species locally (Seewooruthun, Soonnoo, & Alleck, 2000; Vargas et al., 2010; Zeng et al., 2019), including the Sterile Insect Technique (SIT), a method relying on the release of sterile male flies that compete with resident fertile males in order to suppress the target pest population (Dyck et al., 2021). Additionally, B. dorsalis is effectively displacing native fruit fly species as has been recently shown by Moquet et al. (2021), drawing the attention of researchers.
Up to now, the genetic population structure of B. dorsalis has been studied using microsatellite markers (Kim et al., 2021; Shi et al., 2012) and mtDNA (i.e., COI gene; Ayyasamy & Kamala Jayanthi, 2020; Garzón‐Orduña et al., 2019; Li et al., 2012; San Jose et al., 2018) revealing a rich genetic diversity of B. dorsalis in its Asian range. Moreover, results of Garzón‐Orduña et al. (2019) indicate that researchers are only scratching the surface of B. dorsalis mitochondrial haplotype diversity. Spatial structuring of B. dorsalis populations is generally faint with high levels of admixture over large distances, complicating origin tracing of intercepted flies using molecular techniques (Choudhary et al., 2016; Kim et al., 2021; Shi et al., 2012). However, the weak spatial structure of B. dorsalis could also be related to the markers used so far, which may not allow high‐resolution characterization of population structure.
Within the last century B. dorsalis has invaded the USA in Hawaii (Vargas et al., 2010), California (Barr et al., 2014), and Florida (Alvarez et al., 2016; Steck et al., 2019; Zeng et al., 2019), was introduced to Africa and Oceania (Vargas et al., 2015), and expanded its range in China (Garzón‐Orduña et al., 2019; Shi et al., 2012). While recent range expansions in India (Choudhary et al., 2016) and several other Asian countries (Zeng et al., 2019) have also been suggested, these statements likely originate from the confounding of “first detection” with “first taxonomic record” where the latter does not imply that the species was previously absent and incomplete knowledge of B. dorsalis taxonomic history (Clarke et al., 2019). Furthermore, B. dorsalis was first documented in (East) Africa in the Coast Province of Kenya, during routine field surveys by Lux et al. (2003) where it was first inaccurately described as B. invadens (Ekesi et al., 2006; Schutze et al., 2015; Zhang et al., 2016). From there, it has rapidly expanded westwards and southwards along the Indian Ocean coast and has reached South Africa in 2010 (Drew et al., 2005; Zeng et al., 2019). During the last decade, B. dorsalis managed to be re‐established in Florida in 2015 (Steck et al., 2019) and caused several outbreaks in California during 2006–2012 and 2021 (Barr et al., 2014; USDA APHIS, 2021) after initial eradication. First detections of B. dorsalis in Europe occurred in Italy (Nugnes et al., 2018) and France (https://draaf.paca.agriculture.gouv.fr/) in recent years.
Range expansions of B. dorsalis are fairly well documented, but little research has been done on the genetic nature of migration pathways of B. dorsalis on a regional scale (Garzón‐Orduña et al., 2019; Zeng et al., 2019). Especially information on human‐mediated range expansions, which can be complex in nature (e.g., colonization of oceanic islands) is generally lacking and could provide new information beneficial to sanitary protocols such as improved quarantine systems and better detection and screening methods (Kim et al., 2021; Suckling et al., 2013; Welsh et al., 2021). One example of human‐mediated range expansion is the spread of B. dorsalis throughout islands in the Indian Ocean. Here, B. dorsalis was first detected in 2005 on Grande Comore, an island part of the Union des Comores (De Meyer et al., 2012). The two other major islands of the country, Anjouan and Mohéli, are assumed to have been colonized later based on monitoring of B. dorsalis abundances on the islands (Hassani et al., 2016). Colonization of the Comoros was rapidly followed by an occurrence in the French Overseas Department Mayotte in 2007 (De Meyer et al., 2012), and eventually, B. dorsalis reached Madagascar in 2010 (Raoelijaona et al., 2012). The first records of B. dorsalis on Mauritius, situated to the east of Madagascar, date back to 1996, where it was declared eradicated after a nationwide eradication program in 1999 (Seewooruthun, Permalloo, et al., 2000; Seewooruthun, Soonnoo, & Alleck, 2000). In 2013, the species was recorded again, but its re‐occurrence was soon followed by a second eradication (Sookar & Deguine, 2016). In 2015, B. dorsalis was trapped for the third time in Mauritius, and no large‐scale eradication programs have been pursued since then (Sookar et al., 2020).
Knowledge of the invasion pathway and genetic structure of B. dorsalis in the Indian Ocean is lacking and could be beneficial for policymakers (Szyniszewska, 2016). Moreover, the invasion of the islands in the Indian Ocean by B. dorsalis could be a prime example of founder effects as a byproduct of island hopping (Mayr, 1942; Sendell‐Price et al., 2021; Tinghitella et al., 2011). Colonized populations may undergo a step‐wise reduction in genetic diversity relative to their sources and rapid differentiation from source populations (Sendell‐Price et al., 2021; Tinghitella et al., 2011).
Given the different dates for historical recordings on several islands in the western Indian Ocean and possible discrepancies between the detection timeline and actual range expansion, we aim to explore the colonization pathway of this invasive species and its genetic relationship with the population in its native Asian range and invaded range on mainland Africa. Furthermore, we expect to pick up an erosion of genetic diversity within the invasive range, especially on oceanic islands, and a buildup of homozygous genomic regions within the invasive range compared with the genetic source. In order to test the above, we performed whole genome (re‐)sequencing of B. dorsalis samples collected in the Indian Ocean, adjacent continental Africa, and Asia. A multi‐pronged analytic approach was employed, including an allele frequency‐based tree building algorithm and network theory on SNP markers.
2. MATERIALS AND METHODS
2.1. Sampling, sequence filtering, and genotyping
For this study, we used 237 B. dorsalis specimens obtained from 21 populations in Asia (seven populations, 66 specimens), Africa (three populations, 30 specimens), and several islands in the Indian Ocean (11 populations and 141 specimens): Grande Comoro, Anjouan, Mohéli (Comoros W, E, and C, respectively), Mayotte, Madagascar, and the Mascarene islands (Mauritius and Réunion) using methyl eugenol traps. More than one population was included for Mayotte, Madagascar, and Réunion (two, three, and two, respectively; Table S1). Additionally, seven specimens of one Bactrocera zonata population collected in Tel Aviv, Israel, were included to serve as an outgroup for phylogeographic tree reconstruction. Samples were collected between 2016 and 2021, with the exception of those from Burundi and Malawi, which were collected in 2011. See Table S1 for further details on coordinates, number of samples, and collection year per location.
After morphological species identification (https://fruitflykeys.africamuseum.be/), DNA extraction was performed using the Qiagen Blood & Tissue extraction kit following the manufacturer's recommendations. The Asian and African samples were extracted with a different kit: the Promega Wizard SV Genomic DNA Purification System. Elution was carried out with 120 μl buffer AE. DNA concentration was evaluated using a Qubit fluorometer (Thermo Fisher Scientific). Samples that did not meet the minimum concentration guideline of 10 ng/μl were reconcentrated using a Concentrator Plus (Eppendorf). DNA samples were then submitted for whole genome sequencing (150 bp paired‐end) on the NovaSeq 6000 platform with an average input size of 350 bp and a minimum output of 6 Gb (rendering a 12.7× theoretical coverage of the B. dorsalis reference genome).
Assessment of the general quality of the raw reads was performed using “fastQC” (https://github.com/s‐andrews/FastQC). Reads were then quality trimmed with “fastp” (Chen et al., 2018), discarding read pairs of which more than 2% of the bases failed to meet a quality threshold of 10. Trimmed reads were aligned to the B. dorsalis reference genome (GenBank assembly accession: GCA_020283865.1) using the bwa‐mem command from the burrows‐wheeler aligner tool (Li & Durbin, 2009). Variant calling was performed using the GATK Haplotypecaller algorithm implemented in “Elprep” (Herzeel et al., 2021). “ElPrep” is a tool designed as an in‐memory and multithreaded toolset to fully take advantage of the processing power available with modern servers and consists of a 5‐step variant calling best practices pipeline. After individual variant calling, gVCF files were combined using the GATK CombineVCFs command (McKenna et al., 2010). Joint variant calling was performed separately for each of the six chromosomes using GATK GenotypeGVCFs, and chromosome VCF files were merged afterward. Joint calling allows for the rescue of low‐coverage genotypes in one sample when another sample provides evidence supporting the existence of the genotype. Biallelic variants were hard filtered using GATK VariantFiltration using the recommended settings (quality‐by‐depth ratio (QD) < 2.0 || read mapping quality (MQ) < 40.0 || probability of strand bias (FS) > 60.0 || symmetric odds ratio (SOR) > 3.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0). Filtering on QD normalizes QUAL by the number of reads supporting every SNP and is preferred above filtering quality and depth in individual steps. Samples with missingness larger than 20% were omitted from further analysis. Furthermore, SNPs with more than two alleles, a minimum depth and minor allele count lower than three and a missingness higher than 10% were omitted from further analysis using “VCFtools” (Danecek et al., 2011). Finally, to account for the effects of linkage between SNPs in the datasets, we pruned the VCF file based on a pairwise correlation threshold of 0.1 and a window size of 10,000 bases using the package “SNPRelate” (Zheng et al., 2012) implemented in R (version 4.0.3).
2.2. Population genetic parameters
Multiple estimates of genetic diversity were calculated using the same filters as stated above, but no minor allele filtering or linkage pruning was applied in order to best approximate the real genomic diversity. We used “STACKS populations” (Catchen et al., 2013) to estimate observed heterozygosity (H O), expected heterozygosity (H E), nucleotide diversity (π), and the number of private alleles (A PR) at the population level. A measure of genetic differentiation (F ST) between population pairs was calculated using the same software. Lastly, we attributed populations to a higher level grouping (i.e., Asia, Africa, the Comoros islands, Mayotte, Madagascar, and the Mascarene islands) based on admixture results and geographical borders and repeated the estimation for all genetic parameters at this level.
2.3. Runs of homozygosity
To infer the demographic history and more specifically, historical bottlenecks, we estimated the total number (NROH) and total length (SROH) of long runs of homozygosity (ROH) using “bcftools” (Danecek et al., 2021). Different demographic scenarios cause different ROH signatures (Ceballos et al., 2018), hence, ROH analyses are increasingly being used in population genomic studies (e.g., in the range‐restricted Pyrenean Desman, Escoda & Castresana, 2021; Swedish wels catfish, Jensen et al., 2021 and in killer whales, Foote et al., 2021). Since ROH are sensitive to minor allele frequency (MAF) filtering and linkage pruning, such filters were not applied before calculating the total number of ROH and the total summed length of ROH (Meyermans et al., 2020). To test for significant differences between the six geographical groups, a parametric linear model was carried out using SROH as the predictor variable. A p‐value was calculated for every pairwise comparison between geographical groups, and a Tukey's range test was applied to correct for multiple testing using the package “mixlm” (https://github.com/khliland/mixlm) implemented in R.
2.4. Admixture analysis and PCA
To infer admixture between samples across the whole dataset, ancestry coefficients for K number of ancestral populations were calculated using the sNMF function (Frichot et al., 2014) of the “LEA” package (Frichot & François, 2015) implemented in R. K varied from 2 to 10 while performing 10 replicate runs for each K. For each value of K, the best run was selected based on the cross‐entropy criterion. Ancestral proportions (Q values) were then visualized for K = 2 up to the value of K with the lowest cross‐entropy level + 1 to ensure that further subdivision is not meaningful using the R package “ggplot2” (Wickham, 2017).
In order to investigate and visualize the partitioning of genetic variation, an analysis of principal components (PCA) was performed using the snpgdsPCA function of the R package “SNPRelate” (Zheng et al., 2012). To test for significant differences between geographical groups, a permutational ANOVA (permanova) was carried out using the first seven eigenvectors as predictor variables and employing 10,000 permutations using the pairwise.adonis2 function of the “pairwiseAdonis” package (https://github.com/pmartinezarbizu/pairwiseAdonis). A p‐value was calculated for every pairwise comparison, and the Benjamini–Hochberg method (Benjamini & Hochberg, 1995) was applied to correct for multiple testing.
2.5. PoMo phylogenetic tree
In order to infer a population‐level phylogenetic tree, a Polymorphisms‐aware phylogenetic Model (PoMo) was constructed (De Maio et al., 2015). This approach is well‐suited for tree estimation in scenarios of incomplete lineage sorting and accounts for ancestral variation while being computationally more efficient than other methods.
First, a VCF file was constructed, including all B. dorsalis individuals, and the B. zonata individuals as the outgroup. The outgroup individuals were variant called in a separate cohort and filtered in an identical manner. Missing data filters were not applied for the outgroup.
Secondly, bcftools consensus (Li, 2011) was used to create a whole genome alignment in fasta format using the B. dorsalis reference genome (GenBank assembly accession: GCA_020283865.1). The alignment was then converted to a “counts file” containing the allele counts per population for each site with the publicly available python library “cflib” (https://github.com/pomo‐dev/cflib). Sites with missing data were subsequently omitted from the count's file. To account for the assumption of independent sites, 1 million sites across the whole genome were randomly selected of which 8680 were variant sites. Based on these data, we employed a generalized time‐reversible polymorphism‐aware evolutionary model with eight rate heterogeneity categories (GTR + G8 + P; Schrempf et al., 2019) to reconstruct a maximum‐likelihood phylogeny in “IQ‐tree” (Nguyen et al., 2015). This substitution model features an expanded nucleotide state alphabet so that taxa can have polymorphic states, and thus extends traditional models that are based on nucleotide states that are fixed within a taxon (Schrempf et al., 2016). One‐thousand ultrafast bootstrap trees were constructed to calculate node support (Hoang et al., 2018). The resulting tree reconstruction was visualized with ITOL (Letunic & Bork, 2021).
2.6. Kinship networks
As a flexible and highly scalable approach for analyzing individual‐based genetic relationships, a kinship network was constructed. The advantage of using an individual‐based metric over a population‐based one such as F ST is that no a priori assumptions are made about (meta‐)population structure and thus allowing the data to directly inform the fine‐grain genetic structure (Jones & Manseau, 2022). Kinship is ideally suited to infer the impact of recent events on relationships between individuals (such as captive breeding programs; Escoda et al., 2019) but is also applicable to wild populations without prior knowledge of demography, shared ancestry, or pedigree (Staples et al., 2014).
Kinship was estimated using the snpgdsIBDMLE function within the R package “SNPRelate” (Zheng et al., 2012) applying the Expectation–Maximization (EM) algorithm for exploring the maximum value of the log‐likelihood function (Milligan, 2003; Zheng et al., 2012). Two networks were created. The first network includes all edges with edge weight in proportion to the respective kinship value. A second network was constructed by extracting the backbone structure of the network, simplifying the network by retaining the most important edges, using the disparity filter algorithm by Serrano et al. (2009) implemented in the R package “backbone” (available at: https://CRAN.R‐project.org/package=backbone). By imposing a significance level α = 0.05 or 0.10, the links that carry weights that are not different than under the null hypothesis can be pruned from the network. This method is preferred above a global weight threshold pruning method and reveals the multilayered and often complex nature of the network (Jones & Manseau, 2022). Pruning at a small value of α reveals those edges that constitute a central part of the network backbone while pruning at a higher value of α can reveal the more shallow substructures within the network. Nodes without edges are not shown. Node degree is calculated as the number of edges surrounding the respective node.
3. RESULTS
On average, 56,655,392 reads were produced per sample with an average number of mapped reads equal to 48,252,941 (85.17% mapping success). After SNP calling and application of GATK hard filtering, 140,132,105 SNPs remained. Five samples were omitted from further analysis based on missingness larger than 20% (three samples from Reunion, one sample from Burundi, and one sample from Sri Lanka). Consecutive filtering at minimum depth and minor allele count higher than or equal to three, site missingness lower than 10% and linkage pruning rendered 909,776 biallelic SNPs for further PCA and kinship network analysis.
3.1. Population genetic parameters
Samples from Asia exhibited the highest nucleotide diversity (π = 0.0183) and the highest number of private alleles (1,633,403) compared with all other regions (Table 1). The number of private alleles for every Asian population separately was high and relatively evenly distributed (average = 215,444, SD = 41,516). Within the Asian group, Telangana (India E, Table S1) showed the highest nucleotide diversity (π = 0.0192, Table 2). The African region and the Comoros had the second highest nucleotide diversity (π = 0.0157 for both) followed by Madagascar (π = 0.0148), Mayotte (π = 0.0147), and the Mascarenes (π = 0.0123; Table 1). Within the African group, Kenya showed the highest diversity (π = 0.0162), whereas Malawi (π = 0.0151) and Burundi (π = 0.0149) were ranked below the Comoros. Group‐wise heterozygosity (H O) varied between 0.0165 (Asia) and 0.0113 (Mascarenes). Standard errors were equal to or smaller than 0.00005 for all parameters.
TABLE 1.
Genetic parameters for the group level.
| Group | #Individuals | H E | H O | π | A PR |
|---|---|---|---|---|---|
| Asia | 65 | 0.0182 | 0.0165 | 0.0183 | 1,633,403 |
| Africa | 29 | 0.0154 | 0.0136 | 0.0157 | 32,875 |
| Comoros | 45 | 0.0155 | 0.0136 | 0.0157 | 44,288 |
| Mayotte | 20 | 0.0143 | 0.0145 | 0.0147 | 13,108 |
| Madagascar | 38 | 0.0146 | 0.0134 | 0.0148 | 37,854 |
| Mascarenes | 35 | 0.0122 | 0.0113 | 0.0123 | 164,730 |
TABLE 2.
Genetic parameters on the level of populations.
| Population | #Individuals | H E | H O | π | A PR |
|---|---|---|---|---|---|
| China | 10 | 0.0174 | 0.0167 | 0.0183 | 249,212 |
| India E | 6 | 0.0176 | 0.0173 | 0.0192 | 155,451 |
| India S | 10 | 0.0159 | 0.0167 | 0.0168 | 175,864 |
| India N | 10 | 0.0173 | 0.0161 | 0.0182 | 240,583 |
| Sri Lanka | 9 | 0.0160 | 0.0154 | 0.0169 | 173,927 |
| Malaysia | 10 | 0.0172 | 0.0166 | 0.0181 | 252,907 |
| Thailand | 10 | 0.0177 | 0.0170 | 0.0186 | 260,164 |
| Kenya | 10 | 0.0154 | 0.0157 | 0.0162 | 11,117 |
| Malawi | 10 | 0.0143 | 0.0130 | 0.0151 | 8538 |
| Burundi | 9 | 0.0140 | 0.0117 | 0.0149 | 7021 |
| Comoros E | 15 | 0.0154 | 0.0144 | 0.0160 | 16,892 |
| Comoros W | 15 | 0.0147 | 0.0131 | 0.0152 | 12,184 |
| Comoros C | 15 | 0.0146 | 0.0132 | 0.0151 | 10,913 |
| Mayotte W | 10 | 0.0135 | 0.0148 | 0.0142 | 6270 |
| Mayotte E | 10 | 0.0137 | 0.0143 | 0.0144 | 6548 |
| Madagascar E | 15 | 0.0140 | 0.0135 | 0.0145 | 13,155 |
| Madagascar N | 15 | 0.0142 | 0.0132 | 0.0147 | 14,239 |
| Madagascar S | 8 | 0.0129 | 0.0132 | 0.0138 | 7972 |
| Réunion W | 15 | 0.0120 | 0.0116 | 0.0124 | 42,483 |
| Réunion E | 5 | 0.0109 | 0.0114 | 0.0122 | 15,497 |
| Mauritius | 15 | 0.0115 | 0.0111 | 0.0119 | 35,972 |
Genetic differentiation (F ST) was 0.0089, 0.0090, 0.0092, 0.0093, and 0.0070 between Asia and Africa, Comoros, Mayotte, Madagascar, and the Mascarenes, respectively (Table 3). Genetic differentiation was high between pairwise comparisons of the Mascarenes and Mayotte (0.0416), Africa (0.0345), Comoros (0.0311), and Madagascar (0.0349) (Table 3). Pairwise differentiation at the population level can be found in Table S2.
TABLE 3.
Pairwise F ST values between regional groups.
| Region | Asia | Africa | Comoros | Mayotte | Madagascar | Mascarenes |
|---|---|---|---|---|---|---|
| Asia | 0.0089 | 0.0090 | 0.0091 | 0.0093 | 0.0070 | |
| Africa | 0.0111 | 0.0216 | 0.0187 | 0.0345 | ||
| Comoros | 0.0145 | 0.0141 | 0.0311 | |||
| Mayotte | 0.0141 | 0.0416 | ||||
| Madagascar | 0.0349 |
3.2. Runs of homozygosity
Asia was the region with the highest number of ROH (2200 ± 122) while the number of ROH (NROH) was similar and lower for the rest of the regions (Africa: 1366 ± 52.8, Comoros: 1614 ± 78.2, Mayotte: 1729 ± 60.9, Madagascar: 1699 ± 42.7, Mascarenes: 1639 ± 53.1, Figure 1). The summed length of ROH (SROH) was significantly lower for Asia (12,451,121 ± 847,024) when compared to the rest of the regions (average across regions: 76,443,349 ± 2,423,295, Tukey contrasts test: p < 0.001), suggesting less inbreeding (Ceballos et al., 2018, Figure 1). Significant differences for SROH were found for all comparisons between regions (p < 0.001) except between Comoros–Mayotte (p = 0.067), Africa–Comoros (p = 0.14), and Madagascar–Mayotte (p = 0.79). See Table S3 for the full results of the post hoc comparisons.
FIGURE 1.
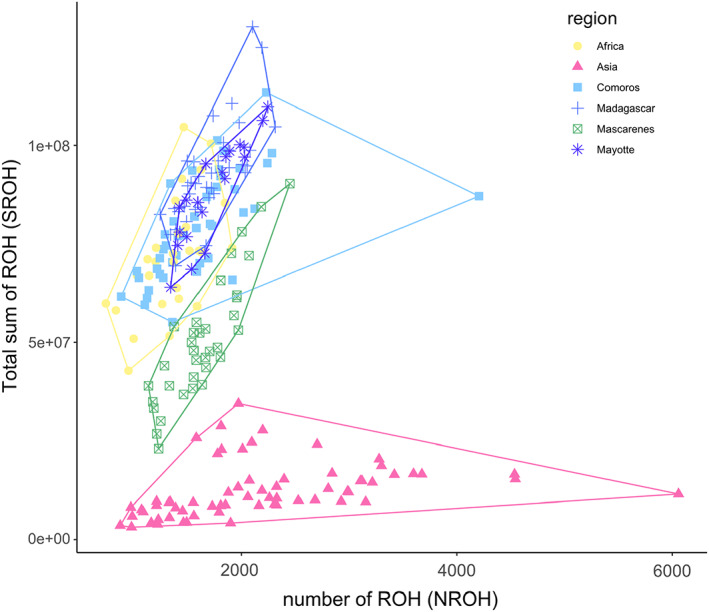
Number of runs of homozygosity (NROH) and total sum of lengths of runs of homozygosity (SROH) for Bactrocera dorsalis in six main geographical regions. Higher NROH coupled with a higher SROH is indicative of stronger bottlenecks while a higher SROH indicates more profound inbreeding (Ceballos et al., 2018). A linear regression line is plotted.
3.3. Admixture analysis, PCA, and PoMo tree
The sNMF‐based estimates of shared ancestry point toward the presence of four distinct genetic clusters with K = 5 as the optimal K according to the level of cross‐entropy (Figure 2). The first cluster contains all Asian samples, excluding five samples from Tamil Nadu (India S, Table S1), which form a cluster on their own. A third cluster suggests a clinal pattern from the African mainland toward Madagascar and contains individuals from Africa, all three Comoros islands, Mayotte and Madagascar. A fourth cluster encompasses all individuals from Mauritius and Réunion (Figure 2). Results from the PCA corroborate the presence of four genetic clusters with each of the clusters coinciding with results from the admixture analysis considering K = 4 or larger (Figure 3). Interestingly, five out of the 10 samples from Tamil Nadu (India S, Table S1) are forming a distinct cluster, agreeing with results from the admixture analysis (Figure 2). Individuals from Africa were more closely related to individuals sampled in the Comoros, corroborating the clinal pattern seen in the admixture type analysis. The first and second principal components explained 3.58% and 2.57% of total variations, respectively. The permanova showed significant structuring (p adj < 0.05) for all but one pairwise comparison between the six major geographical groups. There was no observed significant difference between Mayotte and the Comoros (p = 0.105).
FIGURE 2.
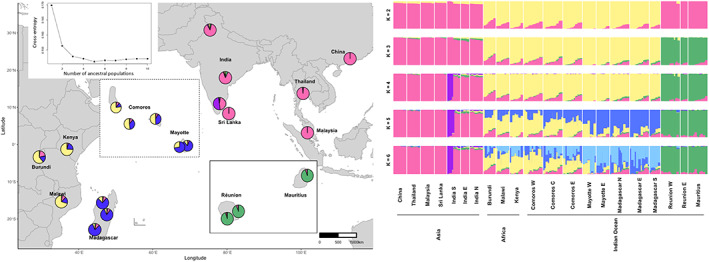
Ancestry coefficients resulting from the sNMF analysis for Bactrocera dorsalis in the study region showing the presence of four distinct clusters as represented on a geographical map for K = 5 (a) and in a barplot (b). Cross‐entropy for every value of K is displayed in the top left corner. A first cluster contains all Asian B. dorsalis apart from five samples collected in Tamil Nadu (India S, Table S1), which form a cluster on their own. A third cluster exhibits a clinical pattern and contains individuals from Africa, all three of the Comoros islands, Mayotte, and Madagascar while a fourth cluster encompasses all individuals from the Mascarenes (Mauritius and Reunion).
FIGURE 3.
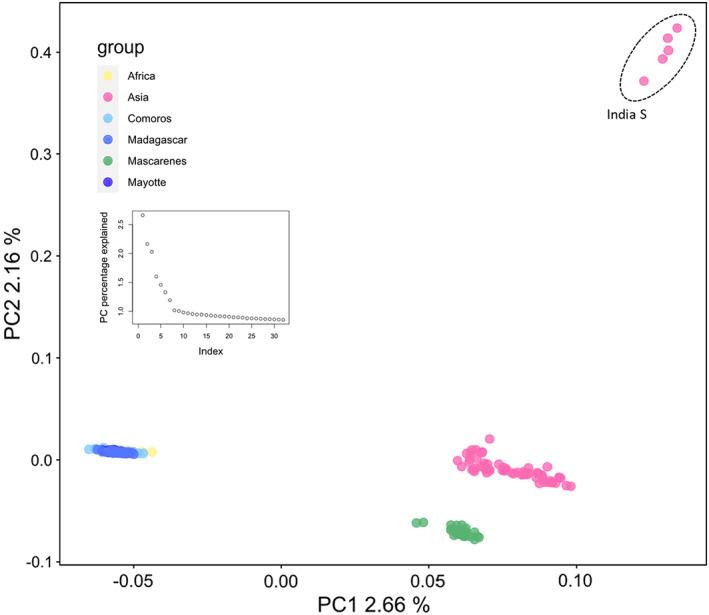
Analysis of principal components (PCA). Results of PCA are indicating that genetic variation can be partitioned into four well‐defined clusters. A first cluster contained all Asian Bactrocera dorsalis exclusively. A second cluster contained individuals from Africa, all three of the Comoros islands, Mayotte, and Madagascar, and a third cluster encompassed all individuals from the Mascarenes (Mauritius and Reunion). An additional fourth cluster comprised five out of the 10 individuals sampled in Tamil Nadu, India (India S, Table S1).
Overall, the topology of the PoMo tree was strongly supported based on bootstrap values (Figure 4). The phylogeography of B. dorsalis in the Indian Ocean encompassed two major clades. One clade contained samples from Mauritius‐Réunion (eastern clade) and was paraphyletic to a clade containing the African mainland populations together with the rest of the island populations (western clade). The deep divergence between the western and the eastern clade suggested different origins. Within the western clade, a hierarchical structuring can be observed that coincides with an eastwards direction of invasion from the African mainland (Kenya, Burundi) toward Mayotte and Madagascar. Comoros W was the most basal island within the Comoros and is the most related to the African mainland populations. The rest of the Comoros islands together with Mayotte and Madagascar are paraphyletic to Comoros W (Figure 4).
FIGURE 4.

PoMo phylogenetic tree for Bactrocera dorsalis in the Indian Ocean with the inclusion of Asian and African populations and Bactrocera zonata as an outgroup. Different colors represent the six geographical regions (from top to bottom: Asia, the Mascarenes, Africa, the Comoros, Mayotte, and Madagascar). Only bootstrap values below 100 are shown.
3.4. Kinship networks
Estimated kinship between all dyads was 0.013 on average with a standard deviation of 0.022. A first (kinship >0.25) and second (kinship >0.125) degree of relationship occurred eight and 80 times, respectively, in a total of 26,796 combinations. First‐degree relationships occurred four times in India S, two times in Mauritius, once in Mayotte E and once in Mayotte W. Second‐degree relationships were also exclusively detected between individuals belonging to the same population and could primarily be found in the Western group (28 in Madagascar, 20 in Mayotte and two in the Comoros), followed by the Mascarenes (11 in Mauritius and nine in Réunion) and Asia (10 in India S). The average degree (number of edges per node) within the pruned network (p ≤ 0.05) was 4.93 with a standard deviation of 3.40.
Three distinct clusters could be observed when all edges between individuals are conserved (Figure 5a). Samples from the Mascarene islands (Eastern cluster) seem to exhibit a closer proximity to the Asian cluster than samples from Africa, the Comoros, Mayotte, and Madagascar (Western cluster) indicating a higher level of kinship (Figure 5a).
FIGURE 5.
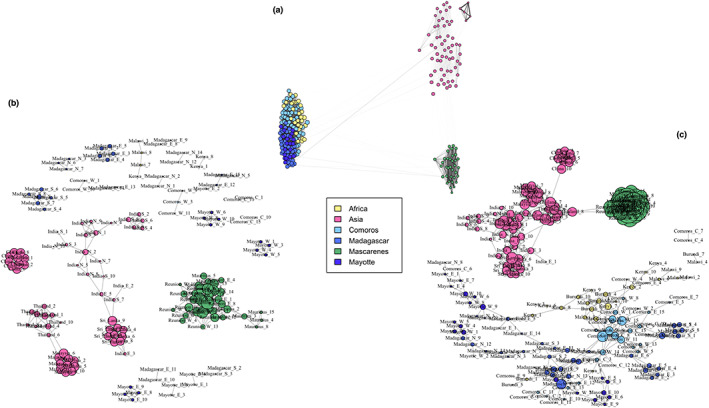
Kinship networks of Bactrocera dorsalis in the Indian Ocean in the force‐directed Fruchtermann–Reingold layout. In the first network (a), all edges are represented, and edge opacity and thickness are proportional to the weight of the respective edge. In the second (b) and third network (c), a backbone pruning method was applied, so that only edges with p‐value ≤0.05 and 0.10 are conserved, respectively.
Within the pruned (p ≤ 0.05) network, extensive geographic structuring could be observed within the Asian group with cluster outlines largely coinciding with country borders for Sri Lanka, China, Thailand, and Malaysia (Figure 5b). No edges can be observed between the Asian populations and Africa or populations within the Indian Ocean. The Mascarene islands form a distinct and compact cluster with four Mauritian individuals displaying a more peripheral position (Figure 5b). When applying less stringent pruning (p ≤ 0.10), a more connected network resulted (600 instead of 387 edges). All Asian populations formed one group sharing two edges with the Mascarenes. Africa, Comoros, Mayotte, and Madagascar constituted another group within the network (Figure 5c). Interestingly, it is the most Western island (Grande Comoro, Table S1) that serves as the primary connector with Africa as exemplified by the large degree of Comoros_W_15 (6) and Comoros_W_3 (5) and other Comoros islands only play a minor role in this. The network structure is less dense within Mayotte and Madagascar and clustering can be observed only to a minor extent.
4. DISCUSSION
In light of tracing the origin of invasive fruit fly incursions, researchers have increasingly focused on resolving the population structure of various tephritid species (Deschepper et al., 2021; Dupuis et al., 2018; Kim et al., 2021; Narde et al., 2005; Popa‐Báez et al., 2021; Ruiz‐Arce et al., 2020; Virgilio et al., 2010). For example, by amplifying the N5N4 mitochondrial region in Ceratitis capitata, Ruiz‐Arce et al. (2020) were able to discriminate between six worldwide geographical regions but struggled to provide a more detailed population genetic structure. A comparable approach was used by Kim et al. (2021) and Qin et al. (2018), who used COI and NAD6 haplotype diversity, respectively, in combination with microsatellite markers to infer regional clustering patterns of B. dorsalis in a large part of its Asian range. Here, they found evidence for founder effects but did not achieve proof for extensive genetic structure, details that could inform invasion pathways. To our knowledge, our study is the first to use SNP markers resulting from whole‐genome resequencing to examine tephritid population structuring and have high enough resolution to characterize invasion routes. Within the Asian range, we were able to discriminate between populations using a combination of population‐wide allele frequency information in the PoMo analysis and kinship metrics to add granularity at the individual level (Figures 4 and 5). We found populations in Telangana, India (Table S1), and Sri Lanka to be a sister group to populations in Punjab and Tamil Nadu, India (Table S1), China, Thailand, and Malaysia. These results corroborate unpublished work by Y. Zhang, S. Liu, M. De Meyer, M. Virgilio, S. Feng, Y. Qin, S. Singh, S. L. Wee, F. Jiang, S. Guo, X. Zhou, H. Li, P. Deschepper, H. Delatte, A. van Sauers‐Muller, T. S. Syamsudin, A. P. Kawi, M. Kasina, K. Badji, F. Said, L. Liu, Z. Zhao, Z. Li (in preparation) who focus on the worldwide phylogeography of B. dorsalis. Since there may be ongoing admixture between the sampled locations, a bifurcating population tree is likely not entirely representative of the individual‐level genetic relationships but rather renders a general perspective on the phylogeny of B. dorsalis (De Maio et al., 2015).
The colonization of islands in the Indian Ocean by B. dorsalis is a relatively recent phenomenon (Zeng et al., 2019) and is characterized by two independent invasion pathways, as pointed out by distinct partitioning of individuals in all of the analyses performed here (sNMF ancestry estimation, Figure 2; PCA, Figure 3; PoMo tree, Figure 4). The Comoros, Mayotte, and Madagascar, together with the African populations, belonged to one pathway, while the Mascarenes were contained within a separate cluster (Figures 2, 3, 4). Source populations for both colonization pathways are likely different but share genetic variation to some extent. However, fully shared genetically diverse origins cannot be excluded, with drift effects and bottlenecks driving the divergent genetic structure. In the latter case, we would expect to see a larger degree of shared ancestry (Figure 2) in the case of a single origin for both pathways, certainly because the sampling took place only 4–6 years after the invasion of the Mascarenes, rendering little time for drift effects to alter the genetic composition in such a drastic way as observed here. Furthermore, the ML tree reconstruction showed a strong support for two independent invasion routes within the Indian Ocean with a basal position of the Mascarenes (Figure 5).
It has to be noted that every population in this study is characterized by a different time frame between the moment of sampling and the time of colonization, which could bias the amount of genetic differentiation that can be observed between the source and introduced population. However, drift especially acts strongly in the early stages after a population bottleneck and wears off over time (Gilchrist et al., 2012). The fast lifecycle of B. dorsalis could imply that drift rapidly changes population genetic structure and differentiation after colonization took place and time and thus only a limited time is needed to pick up signatures of a genetic bottleneck after the invasion.
4.1. Western invasion pathway
A western invasion pathway is characterized by step‐wise island hopping starting from the east African coast and ending in Madagascar. Grande Comore (Comoros W) was most likely the first Indian Ocean island to be colonized as suggested by the backbone structure of the kinship network (Figure 5c) and is consistent with the timing of the first observations of B. dorsalis on the Comoros (De Meyer et al., 2012). Additionally, the Comorian populations were reconstructed as a paraphyletic group, with Comoros W (Grande Comore, Table S1) as the group most closely related to an African population, namely the Malawi population (Figure 4). International trade data corroborate the link between the Comoros W and the African mainland. The main port of entry and the international airport are situated in Moroni, Grande Comore (Comoros W), where fresh goods are imported from different parts of the world including neighboring countries such as Madagascar, Tanzania, Kenya, or Mozambique (2.6%, 0.8%, 0.6%, and 0.2% of total goods import, respectively) (2020 data from https://OEC.world). Of all Indian Ocean countries, Comoros exhibits the strongest similarity of genetic ancestry with the African region (Figure 2) and has a similar genetic diversity (Table 1), further pointing to Comoros as the western point of entry into the Indian Ocean. From there on, B. dorsalis invaded Mayotte and Madagascar resulting in an increase of genetic differentiation to populations of the African mainland (Tables 1 and 2). Additionally, a decrease in the amount of the yellow cluster in the admixture plots is suggestive of genetic drift in the form of founder effects (Figure 2a; Tables 1 and 2). Similarly, Sendell‐Price et al. (2021) found evidence for founder‐induced loss of genetic diversity in an island‐hopping field cricket (Teleogryllus oceanicus) and increased genetic differentiation when moving further away from the source. The significantly higher SROH for Mayotte and Madagascar compared with Africa and Comoros further corroborates an eastward direction of invasion coupled with increased inbreeding (Figure 1). Interestingly, the network structure of B. dorsalis in Madagascar largely coincides with population delimitation (Figure 5a,c), suggesting that multiple introduction events are at the basis of the Malagasy populations. Alternatively, B. dorsalis may have spread through Madagascar while experiencing bottlenecks, which could explain the markedly lower nucleotide diversity in Madagascar S compared with the other Malagasy populations (π = 0.0138 vs. 0.0145 and 0.0147).
4.2. Eastern invasion pathway
Réunion and Mauritius are always clustered together (Figures 2, 3, 4, 5) and are significantly related (Figure 5b). In contrast to islands along the western invasion pathway, the Mascarene islands were colonized directly from Asia and our results do not suggest a relationship with the western pathway. The weak links between both pathways observed in Figure 5a disappear when backbone pruning is applied and thus suggests the insignificance of these links (Figure 5b,c). Pinpointing an Asian source for B. dorsalis on the Mascarene islands is challenging and would require more exhaustive sampling, including all origin countries of fresh fruit imports. For example, trade data reveal that the three largest providers of fresh fruits and vegetables for Mauritius are China, the United Arab Emirates, and India each constituting 17.6%, 11.5%, and 9.9% of the import (2020 data from https://OEC.world). It is known from the literature that B. dorsalis has reached Mauritius on several occasions, followed by eradication programs after which the species was considered eradicated from the island (Seewooruthun, Permalloo, et al., 2000; Seewooruthun, Soonnoo, & Alleck, 2000; Sookar & Deguine, 2016). Our data agree with recolonization of the Mascarene islands from a genetically diverse source. If B. dorsalis had been in a dormant phase with low numbers, one might expect to see a heavy impact of a bottleneck, which is not what was observed here. While the phylogenetic reconstruction might point toward colonization of Mauritius from Réunion, the more derived position of the Mauritian population in the tree topology might also be explained by the later sampling date of Mauritius compared with Réunion (2021 and 2019, respectively). Furthermore, a surveillance network with methyl eugenol traps has been installed on Réunion island since 2013, and traps were checked every 2 weeks; the first B. dorsalis individual was detected in May 2017 on Réunion (Moquet et al., 2021), whereas it was detected in 2015 on Mauritius (Sookar et al., 2020). The ROH analysis revealed that populations on the Mascarene islands are less impacted by a bottleneck than populations on other Indian Ocean islands. However, genetic diversity is lower on the Mascarenes when compared to other island populations. The lower diversity in combination with smaller SROH could indicate that only a limited number of different genotypes were introduced on the Mascarene islands (Figure 1). Hence, the low genetic diversity is a product of the recency of the founder effect rather than long‐term drift on the islands themselves. This is in contrast to Madagascar and Mayotte where the lower genetic diversity compared with the African mainland is likely related to a series of bottlenecks upon introduction on the islands, accompanied by prolonged periods of drift and inbreeding as indicated by the higher SROH (Figure 1). Additionally, the high number of private alleles, both on a regional and at a population level (Table 1), corroborates the lack of drift on the Mascarenes though caution is advised when interpreting the number of private alleles since this statistic is influenced by a priori assignment of samples to groups.
Ultimately, we think that this study could provide policymakers with valuable information to prevent the re‐establishment of B. dorsalis after successful eradication or prevent new invasions. Long‐distance dispersal in the case of isolated areas such as oceanic islands is usually linked with human activities. The main driver for the invasion of agricultural pests is still considered to trade (see Hulme, 2021 for a review). Nevertheless, other major pathways such as luggage of airline passengers (Liebhold et al., 2006) and international shipment of goods (both private and through internet commerce, Lenda et al., 2014) are considered of increasing importance. In popular tourist destinations with a large proportion of the local population having foreign roots and family ties, as is the case in the majority of the western Indian Ocean islands, these pathways are even more pertinent. Characterizing invasion pathways can, therefore, assist policymakers in developing appropriate measures and policies to curb further introductions. It will allow them to target particular trade routes and visitors for more scrutinous phytosanitary inspections, which can even be modeled and adapted by taking into account aspects such as traffic peaks, travel, seasons, and temporal abundance of potential pests in countries of origin (Szyniszewska, 2016). Models for a particular horticultural pest such as the one developed in this research can then be extrapolated to other organisms that are similarly associated with the same crops. Moreover, looking at infestation rates and host range of the invasive B dorsalis populations from the Mascarenes (Moquet et al., 2021; Sookar et al., 2020) compared with the ones from Madagascar or Comoros (Hassani et al., 2016; Rasolofoarivao et al., 2022) it seems that the Mascarenes populations have slightly different ecological parameters with a broader host range and higher infestation rates, highlighting the importance of preventive measures for pest control.
5. CONCLUSION
Our study is the first to investigate the population structure and invasion history of B. dorsalis in the Indian Ocean. By using whole genome SNP data, we resolved two independent invasion pathways. A western invasion pathway involves stepping‐stone migration of B. dorsalis from the east African coast into Comoros, along Mayotte, and into Madagascar with a decreasing genetic diversity. Our findings are in line with the first detection of B. dorsalis on the African mainland in 2003, followed by a relatively quick invasion of Comoros. The Mascarene islands were colonized directly from Asia and formed a distinct cluster. The low nucleotide diversity suggests that only a few genotypes invaded the Mascarenes. The presence of many long runs of homozygosity (ROH) in the introduced populations indicates population bottlenecks, with a more severe bottleneck effect for populations along the western migration pathway than on the Mascarene islands.
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest.
BENEFIT‐SHARING STATEMENT
All geographic samples were collected by the different co‐authors in compliance with their national jurisdiction or sourced from collections obtained prior to the Nagoya Protocol ratification.
Supporting information
Tables S1–S3
ACKNOWLEDGEMENTS
The authors acknowledge the Plant Protection Platform (3P, IBISA) where DNA extractions were conducted and David Nestel for providing us with samples of B. zonata. Funding for this research was provided by the EU‐FF‐IPM H2020 FF‐IPM project (Grant ID: 818184), and the National Natural Science Foundation of China (Grant ID: 31972341). The computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, FWO, and the Flemish Government—department EWI.
Deschepper, P. , Vanbergen, S. , Zhang, Y. , Li, Z. , Hassani, I. M. , Patel, N. A. , Rasolofoarivao, H. , Singh, S. , Wee, S. L. , De Meyer, M. , Virgilio, M. , & Delatte, H. (2023). Bactrocera dorsalis in the Indian Ocean: A tale of two invasions. Evolutionary Applications, 16, 48–61. 10.1111/eva.13507
Contributor Information
Pablo Deschepper, Email: pablo.deschepper@africamuseum.be.
Sam Vanbergen, Email: sam.vanbergen@africamuseum.be.
DATA AVAILABILITY STATEMENT
With the aim of full access and benefit‐sharing within the framework of the Nagoya Protocol, a zenodo data archive was created containing a filtered VCF file, a metadata file, and a count file. The VCF file contains raw SNP information with only the GATK filters applied. The data can be accessed using the following doi: 10.5281/zenodo.6602072. Raw paired‐end read data are stored as a BioProject (PRJNA893460). Admittance to related genetic resources and databases is herewith provided. Research and development results are fully shared by all co‐authors involved, and joint ownership of relevant intellectual property rights is assured.
REFERENCES
- Alvarez, S. , Evans, E. , & Hodges, A. (2016). Estimated costs and regional economic impacts of the oriental fruit fly (Bactrocera dorsalis) outbreak in Miami‐Dade county, Florida . UF/IFAS Extension, FE988.
- Ayyasamy, A. , & Kamala Jayanthi, P. D. (2020). Population structure inferred from COI data analyses for Bactrocera dorsalis (Diptera: Tephritidae) in the Karnataka region of India. Oriental Insects, 54(1), 41–57. 10.1080/00305316.2019.1579115 [DOI] [Google Scholar]
- Barr, N. B. , Ledezma, L. A. , Leblanc, L. , San Jose, M. , Rubinoff, D. , Geib, S. M. , Fujita, B. , Bartels, D. W. , Garza, D. , Kerr, P. , Hauser, M. , & Gaimari, S. (2014). Genetic diversity of Bactrocera dorsalis (Diptera: Tephritidae) on the Hawaiian Islands: Implications for an introduction pathway into California. Journal of Economic Entomology, 107(5), 1946–1958. 10.1603/EC13482 [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22, 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos, F. C. , Joshi, P. K. , Clark, D. W. , Ramsay, M. , & Wilson, J. F. (2018). Runs of homozygosity: Windows into population history and trait architecture. Nature Reviews Genetics, 19, 220–234. 10.1038/nrg.2017.109 [DOI] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. , & Gu, J. (2018). Fastp: An ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics, 34, i884–i890. 10.1093/bioinformatics/bty560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary, J. S. , Naaz, N. , Prabhakar, C. S. , & Lemtur, M. (2016). Genetic analysis of oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae) populations based on mitochondrial cox1 and nad1 gene sequences from India and other Asian countries. Genetica, 144(5), 611–623. 10.1007/s10709-016-9929-7 [DOI] [PubMed] [Google Scholar]
- Clarke, A. R. , Li, Z. , Qin, Y. , Zhao, Z.‐H. , Liu, L. , & Schutze, M. K. (2019). Bactrocera dorsalis (Hendel) (Diptera: Tephritidae) is not invasive through Asia: It's been there all along. Journal of Applied Entomology, 143(8), 797–801. 10.1111/jen.12649 [DOI] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , Durbin, R. , & 1000 Genomes Project Analysis Group . (2011). The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Bonfield, J. K. , Liddle, J. , Marshall, J. , Ohan, V. , Pollard, M. O. , Whitwham, A. , Keane, T. , McCarthy, S. A. , Davies, R. M. , & Li, H. (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2), giab008. 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio, N. , Schrempf, D. , & Kosiol, C. (2015). PoMo: An allele frequency‐based approach for species tree estimation. Systematic Biology, 64(6), 1018–1031. 10.1093/sysbio/syv048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer, M. , Quilici, S. , Franck, A. , Attie, M. , Chadhouliati, A. C. , Issimaila, M. A. , Youssoufa, M. A. , Abdoul‐Karime, A. L. , Barbet, A. , & White, I. M. (2012). Records of frugivorous fruit flies (Diptera: Tephritidae: Dacini) from the Comoro archipelago. African Invertebrates, 53(1), 1–9. 10.10520/EJC119227 [DOI] [Google Scholar]
- Deschepper, P. , Todd, T. N. , Virgilio, M. , De Meyer, M. , Barr, N. B. , & Ruiz‐Arce, R. (2021). Looking at the big picture: Worldwide population structure and range expansion of the cosmopolitan pest Ceratitis capitata (Diptera, Tephritidae). Biological Invasions, 23(11), 3529–3543. 10.1007/s10530-021-02595-4 [DOI] [Google Scholar]
- Drew, R. A. I. , Tsuruta, K. , & White, I. M. (2005). A new species of pest fruit fly (Diptera: Tephritidae: Dacinae) from Sri Lanka and Africa. African Entomology, 13(1), 149–154. 10.10520/EJC32620 [DOI] [Google Scholar]
- Dupuis, J. R. , Sim, S. B. , San Jose, M. , Leblanc, L. , Hoassain, M. A. , Rubinoff, D. , & Geib, S. M. (2018). Population genomics and comparisons of selective signatures in two invasions of melon fly, Bactrocera cucurbitae (Diptera: Tephritidae). Biological Invasions, 20(5), 1211–1228. 10.1007/s10530-017-1621-z [DOI] [Google Scholar]
- Dyck, V. A. , Hendrichs, J. , & Robinson, A. (2021). Sterile insect technique: Principles and practice in area‐wide integrated pest management (2nd ed.). CRC Press. 10.1201/9781003035572 [DOI] [Google Scholar]
- Ekesi, S. , Nderitu, P. W. , & Rwomushana, I. (2006). Field infestation, life history and demographic parameters of the fruit fly Bactrocera invadens (Diptera: Tephritidae) in Africa. Bulletin of Entomological Research, 96(4), 379–386. 10.1079/BER2006442 [DOI] [PubMed] [Google Scholar]
- Escoda, L. , & Castresana, J. (2021). The genome of the Pyrenean desman and the effects of bottlenecks and inbreeding on the genomic landscape of an endangered species. Evolutionary Applications, 14, 1898–1913. 10.1111/eva.13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoda, L. , Fernández‐González, Á. , & Castresana, J. (2019). Quantitative analysis of connectivity in populations of a semi‐aquatic mammal using kinship categories and network assortativity. Molecular Ecology Resources, 19, 310–326. 10.1111/1755-0998.12967 [DOI] [PubMed] [Google Scholar]
- Foote, A. D. , Hooper, R. , Alexander, A. , Baird, R. W. , Baker, C. S. , Ballance, L. , Barlow, J. , Brownlow, A. , Collins, T. , Constantine, R. , Dalla Rosa, L. , Davison, N. J. , Durban, J. W. , Esteban, R. , Excoffier, L. , Martin, S. L. F. , Forney, K. A. , Gerrodette, T. , Gilbert, M. T. P. , … Morin, P. A. (2021). Runs of homozygosity in killer whale genomes provide a global record of demographic histories. Molecular Ecology, 30, 6162–6177. 10.1111/mec.16137 [DOI] [PubMed] [Google Scholar]
- Frichot, E. , & François, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6(8), 925–929. 10.1111/2041-210X.12382 [DOI] [Google Scholar]
- Frichot, E. , Mathieu, F. , Trouillon, T. , Bouchard, G. , & François, O. (2014). Fast and efficient estimation of individual ancestry coefficients. Genetics, 196(4), 973–983. 10.1534/genetics.113.160572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzón‐Orduña, I. J. , Geib, S. M. , & Barr, N. B. (2019). The genetic diversity of Bactrocera dorsalis (Diptera: Tephritidae) in China and neighboring countries: A review from published studies. Journal of Economic Entomology, 112(4), 2001–2006. 10.1093/jee/toz073 [DOI] [PubMed] [Google Scholar]
- Gilchrist, A. S. , Cameron, E. C. , Sved, J. A. , & Meats, A. W. (2012). Genetic consequences of domestication and mass rearing of pest fruit fly Bactrocera tryoni (Diptera: Tephritidae). Journal of Economic Entomology, 105, 1051–1056. 10.1603/EC11421 [DOI] [PubMed] [Google Scholar]
- Hassani, I. M. , Raveloson‐Ravaomanarivo, L. H. , Delatte, H. , Chiroleu, F. , Allibert, A. , Nouhou, S. , Quilici, S. , & Duyck, P. F. (2016). Invasion by Bactrocera dorsalis and niche partitioning among tephritid species in Comoros. Bulletin of Entomological Research, 106(6), 749–758. 10.1017/S0007485316000456 [DOI] [PubMed] [Google Scholar]
- Herzeel, C. , Costanza, P. , Decap, D. , Fostier, J. , Wuyts, R. , & Verachtert, W. (2021). Multithreaded variant calling in elPrep 5. PLoS One, 16, e0244471. 10.1371/journal.pone.0244471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, D. T. , Chernomor, O. , von Haeseler, A. , Minh, B. Q. , & Vinh, L. S. (2018). UFBoot2: Improving the ultrafast bootstrap approximation. Molecular Biology and Evolution, 35(2), 518–522. 10.1093/molbev/msx281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme, P. E. (2021). Unwelcome exchange: International trade as a direct and indirect driver of biologic al invasions worldwide. One Earth, 4, 666–679. 10.1016/j.oneear.2021.04.015 [DOI] [Google Scholar]
- Jensen, A. , Lillie, M. , Bergström, K. , Larsson, P. , & Höglund, J. (2021). Whole genome sequencing reveals high differentiation, low levels of genetic diversity and short runs of homozygosity among Swedish wels catfish. Heredity, 127, 79–91. 10.1038/s41437-021-00438-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T. B. , & Manseau, M. (2022). Genetic networks in ecology: A guide to population, relatedness, and pedigree networks and their applications in conservation biology. Biological Conservation, 267, 109466. 10.1016/j.biocon.2022.109466 [DOI] [Google Scholar]
- Kim, H. , Kim, S. , Kim, S. , Lee, Y. , Lee, H. S. , Lee, S. J. , Choi, D. S. , Jeon, J. , & Lee, J. H. (2021). Population genetics for inferring introduction sources of the oriental fruit fly, Bactrocera dorsalis: A test for quarantine use in Korea. Insects, 12(10), 851. 10.3390/insects12100851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenda, L. , Skórka, P. , Knops, J. M. H. , Moroń, D. , Sutherland, W. J. , Kuszewska, K. , & Woyciechowski, M. (2014). Effect of the internet commerce on dispersal modes of invasive alien species. PLoS One, 9, e99786. 10.1371/journal.pone.0099786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Research, 49(W1), W293–W296. 10.1093/nar/gkab301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27, 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wu, Y. , Chen, H. , Wu, J. , & Li, Z. (2012). Population structure and colonization of Bactrocera dorsalis (Diptera: Tephritidae) in China, inferred from mtDNA COI sequences. Journal of Applied Entomology, 136(4), 241–251. 10.1111/j.1439-0418.2011.01636.x [DOI] [Google Scholar]
- Liebhold, A. M. , Work, T. T. , McCullough, D. G. , & Cavey, J. F. (2006). Airline baggage as a pathway for alien insect species invading the United States. American Entomologist, 52, 48–54. 10.1093/ae/52.1.48 [DOI] [Google Scholar]
- Lux, S. A. , Copeland, R. S. , White, I. M. , Manrakhan, A. , & Billah, M. K. (2003). A new invasive fruit fly species from the Bactrocera dorsalis (Hendel) group detected in East Africa. International Journal of Tropical Insect Science, 23(4), 355–361. 10.1017/S174275840001242X [DOI] [Google Scholar]
- Mayr, E. (1942). Systematics and the origin of species. Columbia University Press. [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyermans, R. , Gorssen, W. , Buys, N. , & Janssens, S. (2020). How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genomics, 21(1), 94. 10.1186/s12864-020-6463-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan, B. G. (2003). Maximum‐likelihood estimation of relatedness. Genetics, 163, 1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moquet, L. , Payet, J. , Glenac, S. , & Delatte, H. (2021). Niche shift of tephritid species after the oriental fruit fly (Bactrocera dorsalis) invasion in La Réunion. Diversity and Distributions, 27(1), 109–129. 10.1111/ddi.13172 [DOI] [Google Scholar]
- Narde, F. , Carapelli, A. , Dalla, R. , Roderick, G. K. , & Frati, F. (2005). Population structure and colonization history of the olive fly, Bactrocera oleae (Diptera, Tephritidae). Molecular Ecology, 14(9), 2729–2738. 10.1111/j.1365-294x.2005.02610.x [DOI] [PubMed] [Google Scholar]
- Nguyen, L. T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2015). IQ‐TREE: A fast and effective stochastic algorithm for etimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugnes, F. , Russo, E. , Viggiani, G. , & Bernardo, U. (2018). First record of an invasive fruit fly belonging to the Bactrocera dorsalis Complex (Diptera: Tephritidae) in Europe. Insects, 9(4), 182. 10.3390/insects9040182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa‐Báez, Á. D. , Lee, S. F. , Yeap, H. L. , Westmore, G. , Crisp, P. , Li, D. , Catullo, R. , Cameron, E. C. , Edwards, O. R. , Taylor, P. W. , & Oakeshott, J. G. (2021). Tracing the origins of recent Queensland fruit fly incursions into South Australia, Tasmania and New Zealand. Biological Invasions, 23(4), 1117–1130. 10.1007/s10530-020-02422-2 [DOI] [Google Scholar]
- Qin, Y. , Krosch, M. N. , Schutze, M. K. , Zhang, Y. , Wang, X. X. , Prabhakar, C. S. , Susanto, A. , Hee, A. K. W. , Ekesi, S. , Badji, K. , Khan, M. , Wu, J. J. , Wang, Q. L. , Yan, G. , Zhu, L. H. , Zhao, Z. H. , Liu, L. J. , Clarke, A. R. , & Li, Z. H. (2018). Population structure of a global agricultural invasive pest, Bactrocera dorsalis (Diptera: Tephritidae). Evolutionary Applications, 11(10), 1990–2003. 10.1111/eva.12701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoelijaona, J. , Raoelijaona, A. , Ratovonomenjanahary, T. , Brunet, C. , De Meyer, M. , Vayssières, J. , & Quilici, S. (2012). Situation of Bactrocera invadens (Diptera: Tephritidae) in Madagascar. In Proceedings of the 2nd international team meeting, Kolymbari (Greece). [Google Scholar]
- Rasolofoarivao, H. , Ravaomanarivo, L. H. R. , & Delatte, H. (2022). Host plant ranges of fruit flies (Diptera: Tephritidae) in Madagascar. Bulletin of Entomological Research, 112(1), 1–12. 10.1017/S0007485321000511 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Arce, R. , Todd, T. N. , Deleon, R. , Barr, N. B. , Virgilio, M. , De Meyer, M. , & McPheron, B. A. (2020). Worldwide phylogeography of Ceratitis capitata (Diptera: Tephritidae) using mitochondrial DNA. Journal of Economic Entomology, 113, 1455–1470. 10.1093/jee/toaa024 [DOI] [PubMed] [Google Scholar]
- Rwomushana, I. , Ekesi, S. , Gordon, I. , & Ogol, C. K. P. O. (2008). Host plants and host plant preference studies for Bactrocera invadens (Diptera: Tephritidae) in Kenya, a new invasive fruit fly species in Africa. Annals of the Entomological Society of America, 101(2), 331–340. 10.1603/0013-8746(2008)101[331:HPAHPP]2.0.CO;2 [DOI] [Google Scholar]
- San Jose, M. , Doorenweerd, M. , Leblanc, L. , Barr, N. , Geib, S. , & Rubinoff, D. (2018). Tracking the origin of fly invasions: Using mitochondrial haplotype diversity to identify potential source populations in two genetically intertwined fruit fly species (Bactrocera carambolae and Bactrocera dorsalis [Diptera: Tephritidae]). Journal of Economic Entomology, 111, 2914–2926. 10.1093/jee/toy272 [DOI] [PubMed] [Google Scholar]
- Schrempf, D. , Minh, B. Q. , De Maio, N. , von Haeseler, A. , & Kosiol, C. (2016). Reversible polymorphism‐aware phylogenetic models and their application to tree inference. Journal of Theoretical Biology, 407, 362–370. 10.1016/j.jtbi.2016.07.042 [DOI] [PubMed] [Google Scholar]
- Schrempf, D. , Minh, B. Q. , von Haeseler, A. , & Kosiol, C. (2019). Polymorphism‐aware species trees with advanced mutation models, bootstrap, and rate heterogeneity. Molecular Biology and Evolution, 36(6), 1294–1301. 10.1093/molbev/msz043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutze, M. K. , Aketarawong, N. , Amornsak, W. , Armstrong, K. F. , Augustinos, A. A. , Barr, N. , Bo, W. , Bourtzis, K. , Boykin, L. M. , Caceres, C. , & Cameron, S. L. (2015). Synonymization of key pest species within the Bactrocera dorsalis species complex (Diptera: Tephritidae): Taxonomic changes based on a review of 20 years of integrative morphological, molecular, cytogenetic, behavioural and chemo‐ecological data. Systematic Entomology, 40(2), 456–471. 10.1111/syen.12113 [DOI] [Google Scholar]
- Seewooruthun, S. I. , Permalloo, S. , Sookar, P. , & Soonnoo, A. R. (2000). The oriental fruit fly, Bactrocera dorsalis eradicated from Mauritius . Proceedings of the Indian Ocean Commission, Regional Fruit Fly Symposium, Flic En Flac, Mauritius, 5th–9th June, 2000, 207–210.
- Seewooruthun, S. I. , Soonnoo, A. R. , & Alleck, M. (2000). Eradication of an exotic fruit fly from Mauritius . Area‐wide control of fruit flies and other insect pests. Joint Proceedings of the International Conference on Area‐Wide Control of Insect Pests, 28 May–2 June, 1998 and the Fifth International Symposium on Fruit Flies of Economic Importance, Penang, Malay.
- Sendell‐Price, A. T. , Ruegg, K. C. , Robertson, B. C. , & Clegg, S. M. (2021). An Island‐hopping bird reveals how founder events shape genome‐wide divergence. Molecular Ecology, 30(11), 2495–2510. 10.1111/mec.15898 [DOI] [PubMed] [Google Scholar]
- Serrano, M. Á. , Boguñá, M. , & Vespignani, A. (2009). Extracting the multiscale backbone of complex weighted networks. Proceedings of the National Academy of Sciences of the United States of America, 106, 6483–6488. 10.1073/pnas.0808904106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, W. , Kerdelhué, C. , & Ye, H. (2012). Genetic structure and inferences on potential source areas for Bactrocera dorsalis (Hendel) based on mitochondrial and microsatellite markers. PLoS One, 7(5), e37083. 10.1371/journal.pone.0037083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookar, P. , & Deguine, J. P. (2016). Integrated management of fruit flies: Case studies from the Indian Ocean Islands. Springer International Publishing; http://agritrop.cirad.fr/584982/1/ID584982.pdf [Google Scholar]
- Sookar, P. , Patel, N. , & Ramkalawon, P. (2020). Bactrocera dorsalis, an invasive fruit fly species . Book of abstracts 4th International TEAM meeting (La Grande Motte, FR, 2020‐10‐05/09), p49. https://www.alphavisa.com/team/2020/documents/Abstract‐book_TEAM‐2020.pdf
- Staples, J. , Qiao, D. , Cho, M. H. , Silverman, E. K. , Nickerson, D. A. , & Below, J. E. (2014). PRIMUS: Rapid reconstruction of pedigrees from genome‐wide estimates of identity by descent. American Journal of Human Genetics, 95, 553–564. 10.1016/j.ajhg.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck, G. J. , Fox, A. J. , Carrillo, D. , Dean, D. , Roda, A. , Epsky, N. D. , & Smith, T. R. (2019). Oriental fruit fly eradication in Florida 2015–2016, program implementation, unique aspects, and lessons learned. American Entomologist, 65(2), 108–121. 10.1093/ae/tmz023 [DOI] [Google Scholar]
- Suckling, D. M. , Stringer, L. D. , Stephens, A. E. A. , Woods, B. , Williams, D. G. , Baker, G. , & El‐Sayed, A. M. (2013). From integrated pest management to integrated pest eradication: Technologies and future needs. Pest Management Science, 70(2), 179–189. 10.1002/ps.3670 [DOI] [PubMed] [Google Scholar]
- Szyniszewska, A. M. (2016). Analysis of seasonal risk for importation of the Mediterranean fruit fly, Ceratitis capitata (Diptera: Tephritidae), via air passenger traffic arriving in Florida and California. Journal of Economic Entomology, 109, 2317–2328. 10.1093/jee/tow196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, K. H. , & Serit, M. (1994). Adult population dynamics of Bactrocera dorsalis (Diptera: Tephritidae) in relation to host phenology and weather in two villages of Penang Island, Malaysia. Environmental Entomology, 23(2), 267–275. 10.1093/ee/23.2.267 [DOI] [Google Scholar]
- Tinghitella, R. M. , Zuk, M. , Beveridge, M. , & Simmons, L. W. (2011). Island hopping introduces Polynesian field crickets to novel environments, genetic bottlenecks and rapid evolution. Journal of Evolutionary Biology, 24(6), 1199–1211. 10.1111/j.1420-9101.2011.02255.x [DOI] [PubMed] [Google Scholar]
- USDA APHIS . (2021). APHIS establishes an oriental fruit fly (Bactrocera dorsalis) quarantine in Santa Clara county, California . https://www.aphis.usda.gov/aphis/newsroom/stakeholder‐info/stakeholder‐messages/plant‐health‐news/da‐2021‐31
- Vargas, R. I. , Piñero, J. C. , & Leblanc, L. (2015). An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific region. Insects, 6, 297–318. 10.3390/insects6020297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas, R. I. , Piñero, J. C. , Mau, R. F. , Jang, E. B. , Klungness, L. M. , McInnis, D. , Harris, E. B. , McQuate, G. , Bautista, R. C. , & Wong, L. (2010). Area‐wide suppression of the Mediterranean fruit fly, Ceratitis capitata, and the oriental fruit fly, Bactrocera dorsalis, in Kamuela, Hawaii. Journal of Insect Science, 10(1), 135. 10.1673/031.010.13501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgilio, M. , Delatte, H. , Backeljau, T. , & De Meyer, M. (2010). Macrogeographic population structuring in the cosmopolitan agricultural pest Bactrocera cucurbitae (Diptera: Tephritidae). Molecular Ecology, 19(13), 2713–2724. 10.1111/j.1365-294X.2010.04662.x [DOI] [PubMed] [Google Scholar]
- Welsh, M. J. , Turner, J. A. , Epanchin‐Niell, R. S. , Monge, J. J. , Soliman, T. , Robinson, A. P. , Kean, J. M. , Phillips, C. , Stringer, L. D. , Vereijssen, J. , Liebhold, A. M. , Kompas, T. , Ormsby, M. , & Brockerhoff, E. G. (2021). Approaches for estimating benefits and costs of interventions in plant biosecurity across invasion phases. Ecological Applications, 31(5), e02319. 10.1002/eap.2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2017). ggplot2 – Elegant graphics for data analysis (3rd ed.). Springer. [Google Scholar]
- Zeng, Y. , Reddy, G. V. P. , Li, Z. , Qin, Y. , Wang, Y. , Pan, X. , Jiang, F. , Gao, F. , & Zhao, Z. H. (2019). Global distribution and invasion pattern of oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). Journal of Applied Entomology, 143(3), 165–176. 10.1111/jen.12582 [DOI] [Google Scholar]
- Zhang, L. J. , Jiang, L. H. , Wei, C. Y. , Liu, R. S. , Liu, X. L. , Li, J. G. , & Xue, H. J. (2016). The status of Bactrocera invadens Drew, Tsuruta & White (Diptera: Tephritidae) inferred from complete mitochondrial genome analysis. Mitochondrial DNA Part B, 1(1), 680–681. 10.1080/23802359.2016.1219638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S. M. , Laurie, C. , & Weir, B. S. (2012). A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–3328. 10.1093/bioinformatics/bts606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Data Availability Statement
With the aim of full access and benefit‐sharing within the framework of the Nagoya Protocol, a zenodo data archive was created containing a filtered VCF file, a metadata file, and a count file. The VCF file contains raw SNP information with only the GATK filters applied. The data can be accessed using the following doi: 10.5281/zenodo.6602072. Raw paired‐end read data are stored as a BioProject (PRJNA893460). Admittance to related genetic resources and databases is herewith provided. Research and development results are fully shared by all co‐authors involved, and joint ownership of relevant intellectual property rights is assured.