

Abstract
The widespread utilization of mupirocin to treat methicillin-resistant Staphylococcus aureus (MRSA)-caused infectious diseases has led to the emergence of mupirocin-resistant Staphylococcus aureus (MuRSA), posing a serious global medical threat. In order to counteract MuRSA, we develop a d-α-tocopherol polyethylene glycol 1000 succinate (TPGS) modified mupirocin and silver complex (TPGS/Mup-Ag) to combat MuRSA. The surfactivity of TPGS endows Mup-Ag with a homogeneous and small particle size (∼16 nm), which significantly enhances bacterial internalization. Silver ions are released from the mupirocin-Ag complex (Mup-Ag) to exert a synergistic antibacterial activity with mupirocin. Results manifest that our strategy reduces the concentration of mupirocin that induces 50% bacterial death from about 1000 μmol/mL to about 16 μmol/mL. In vitro bacterial infection model suggests that TPGS/Mup-Ag can not only eliminate both intracellular and inhibit bacterial adhesion, but also living cells are not affected. Results of in vivo experiments demonstrate that TPGS/Mup-Ag can effectively inhibit the progression of skin infection and accelerate wound healing, as well as alleviate systemic inflammation in both the subcutaneous infection model and the wound infection model. Furthermore, this study may contribute to the development of therapeutic agents for antibiotic-resistant bacteria and offer ideas for silver-based bactericides.
Keywords: Drug resistance, Staphylococcus aureus, Mupirocin, Skin and tissue infection, Silver
Graphical abstract
1. Introduction
Over the past few decades, antibiotics isolated from natural products and chemically synthesized antibiotics have become key tools in the treatment of bacterial infections [1]. Nevertheless, antibiotic resistance has developed because of the indiscriminate and excessive use of antibiotics. Consequently, the emergence of drug-resistant bacteria has gradually become a major challenge to human health [2]. Staphylococcus aureus (S. aureus) is the second most common cause of nonsocomial bloodstream infection worldwide [3,4]. and is responsible for a great majority of skin infections, including superficial infections such as impetigo and infected abrasions, as well as invasive infections such as cellulitis, subcutaneous abscesses, and wound infections [5,6]. After the clinical identification of methicillin-resistant S. aureus (MRSA) in 1960 [7], mupirocin (pseudomonic acid A, Mup) was the only approved antibiotic for the decolonization of MRSA in patients and healthcare personnel [8]. Unfortunately, with the wide use of mupirocin in clinical practice, a declining sensitivity and increasing resistance of S. aureus to mupirocin gradually emerged and has been reported as mupirocin-resistant S. aureus (MuRSA) in waves around the world, posing a significant threat to public health [7,[9], [10], [11]]. What is worse, the prevalence of high-level mupirocin resistance among Staphylococcus aureus (8.5%, 95% CI 6.3–10.7%) was almost the same as for MRSA (8.1%, 95% CI 6.8–9.4%) [12]. Therefore, there is an urgent need for the development of novel strategies that possess potent bactericidal efficacy to combat MuRSA and reverse the dilemma of bacterial drug resistance against mupirocin.
Having been extensively researched in the field of drug delivery such as tumor therapy and infection treatment, nanoparticles hold the potential to solve the problem of antibiotic resistance [[13], [14], [15], [16], [17]]. Among these, silver nanoparticles (AgNPs) have received much attention for their wide antimicrobial spectrum and rather low bactericidal concentration [[18], [19], [20]]. The attachment and aggregation of AgNPs on biofilms can alter the morphology and permeability of the cell membrane [21,22], increase membrane rigidity by converting it from order to disorder state [23,24], and disintegrate essential components on cell membrane such as proteins and fatty acids. 23 Besides, AgNPs uptake by bacteria can interfere with intracellular lipids, proteins, and notably, DNA, aggravating the disruption process of bacteria [25,26]. On the other hand, AgNPs constantly release silver ions, which, due to the electrostatic attraction, possess a high affinity to biomolecules distributed on membrane surfaces [27], resulting in membrane proteins transformation and the destruction of cellular energy supply systems [28]. However, the toxicity of AgNPs cannot be ignored. Low concentrations of AgNPs can cause cellular damage featuring reactive oxygen species (ROS) generation [29], inflammation [30], and apoptosis [31]. Since silver was found to be non-toxic in minute concentrations and is less prone to developing bacterial resistance compared with antibiotics [20], reducing the dosage of silver may be a promising strategy for designing effective antibacterial agents.
Recently, the strategy of combining antibiotics with AgNPs has moved into our focus. Vazquez-Muñoz et al. discovered a synergistic antimicrobial effect of AgNPs combined with kanamycin on S. aureus, E. coli, and S. Typhimurium with fractional inhibitory concentration indexes (FICI) of 0.35, 0.46, and 0.47, respectively. Prominently, AgNPs at low concentrations alone with kanamycin at 50% of the minimum inhibitory concentration (MIC) could alter the bacterial membrane potential and cause ultrastructural damage, which facilitated kanamycin exerting a bactericidal effect [32]. Kaur et al. reported that amikacin- and vancomycin-conjugated AgNPs possessed significantly enhanced bactericidal capacity on E. coli and S. aureus, as the zone of inhibition (ZOI) of these two strains shrank by 55% and 60% respectively [33]. Therefore, the silver-antibiotic conjugate strategy provides sanguine scope for resolving drug resistance of mupirocin and effectively eliminating MuRSA.
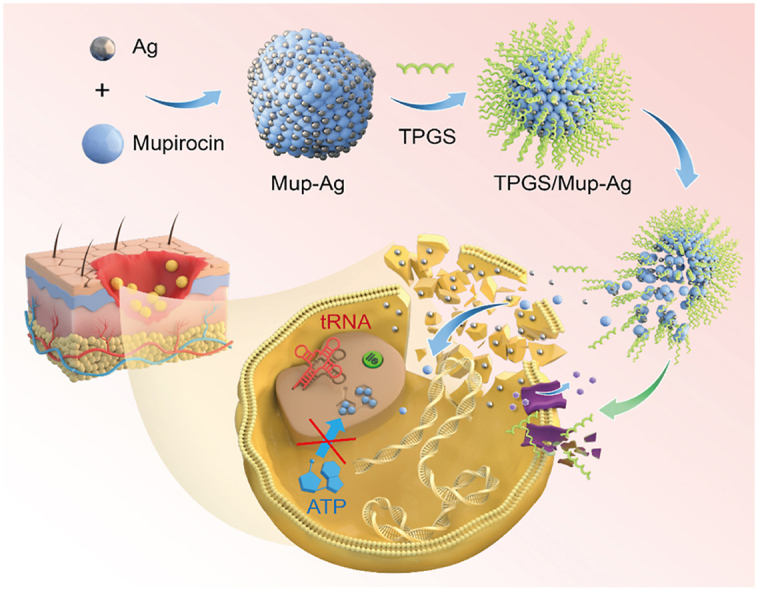
In this article, leveraging the precipitation reaction between mupirocin and sliver, we first report a Mup-Ag complex for the safe and efficient decolonization of MuRSA. Under physiological conditions, because silver ions released from the Mup-Ag complex produce a synergistic bactericidal effect with mupirocin, the dosage of silver was greatly reduced, avoiding the toxicity of silver ions at a high concentration on normal cells and tissues. D-alpha tocopheryl polyethylene glycol succinate (TPGS), a US Food and Drug Administration (FDA) approved pharmaceutical adjuvant, has been shown to enhance the antibacterial capacity of antibiotics against drug-resistant bacteria by affecting the bacteria cell membrane and inhibiting efflux pump activity. Significantly, TPGS ameliorates the dispersibility of Mup-Ag due to its surfactivity, endowing Mup-Ag with homogeneous and small particle size and thus allowing for easier transit through the bacterial membrane. We demonstrate that the TPGS-modified Mup-Ag complex (TPGS/Mup-Ag) can function as an efficient bactericidal agent against MuRSA without exerting a toxic effect on normal cells. Finally, we examine the anti-infective activity of TPGS/Mup-Ag in mice subcutaneous infection model and wound infection model caused by MuRSA.
2. Materials and methods
2.1. Materials, cell and bacteria culture, and animals
Silver nitrate, mupirocin, and sodium hydroxide were obtained from Aladdin Bio-Chem Technology Co. Limited (Shanghai, China). d-α-tocopherol polyethylene glycol 1000 succinate (TPGS) was obtained from Sigma Aldrich Co., Limited (USA). Crystal Violet Staining Solution was obtained from Servicebio Technology Co., Limited (Wuhan, China). Calcein acetoxymethyl (Calcein-AM) and propidium iodide (PI) solutions were obtained from Beyotime Biotechnology Co. Limited (Shanghai, China). LB medium and MH medium were obtained from Solarbio Science & Technology Co. Limited (Beijing, China). All other chemicals were of analytical or chromatographic grade.
Human immortalized keratinocytes (HaCaT) were obtained from Cell Resource Center of China Science Academy. HaCaT was cultured in Dulbecco's modified Eagle medium (Gibco BRL, USA) containing 10% fetal bovine serum (BI, Biologic Industries, China) and penicillin/streptomycin (100 U/mL, 100 U/mL) at 37 °C in a humidified atmosphere containing 5% CO2. Cells were digested regularly using trypsin/EDTA.
Mupirocin-resistant Staphylococcus aureus (MuRSA) and mupirocin-sensitive Staphylococcus aureus (MuSSA) were collected from Department of Dermatology, Zhejiang Provincial People's Hospital. The isolates were cultivated on nutrient agar plates, then harvested and cultured in LB medium or MH medium in an aerophilic environment at 37 °C, 180 rpm grown to an OD600 of 0.2–0.3 that corresponds to the exponential phase.
Female BABL/c mice (20 ± 2 g) were obtained from the Shanghai Silaike Laboratory Animal Limited Liability Company. All mice were free of pathogens and fed with enough food and water. All experiments were performed in compliance with guidelines set by Experimental Animal Center, Zhejiang Provincial People's Hospital (approval number: A20220009). All animal procedures were conducted under national regulations and approved by the local animal experimentation ethical committee.
2.2. Synthesis and characterization of Mup-Ag complex
90 mg mupirocin and 6 mL deionized water were mixed, followed by adding 0.36 mL 0.5 M NaOH and stirring for 20 min. 0.5 mL deionized water containing 30 mg AgNO3 was dropwise dropped into the aforementioned solution under vigorous stirring for 1 h at room temperature. Purification of the prepared Mup-Ag was achieved by repeated ultracentrifugation (14,000 rpm, 10 min) and washing with an aqueous solution of methanol (methanol/water = 9/1, v/v). The aqueous solvent was removed by freeze-drying and then Mup-Ag complex was obtained. The whole process was shielded from light. The chemical structure of Mup-Ag was confirmed by 1H NMR spectra and X-ray diffraction (XRD). A drop of Mup-Ag suspension at a concentration of 0.1 mg/mL was dropped onto a copper grid coated with carbon film and a transmission electron microscope (TEM) (JEOL JEM-1230, Japan) was employed to observe the morphology of Mup-Ag. The elements in Mup-Ag were characterized by scanning electron microscopy (SEM) equipped with energy-dispersive X-ray spectroscopy (SEM-EDS).
2.3. Preparation of and characterization of TPGS/Mup-Ag complex
4 mg Mup-Ag and 196 mg TPGS were dissolved in 1 mL deionized water under ice bath sonication (intermittent operation for 2 s on and 3 s off, repeated 20 times). The purification was achieved by dialysis against deionized water (MWCO = 3500 Da) for 4 h, followed by lyophilization. TPGS/Mup-Ag suspension at a concentration of 0.5 mg/mL was used for TEM imaging and the size distribution of TPGS/Mup-Ag was measured by DLS (Zetasizer, Malvern Co., UK) [34]. The concentration of Ag+ was detected by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) (PerkinElmer NexION 300X) using 1 mg Mup-Ag and 10 mg TPGS/Mup-Ag. Drug loading (DL) and encapsulation efficiency (EE) of Mup were measured by high-performance liquid chromatography (HPLC) according to the following formulas:
The dialysis membrane diffusion technique was utilized to investigate the drug release behavior. 5 mg TPGS/Mup-Ag was loaded into a dialysis bag and immersed in 30 mL of PBS (pH 6.8). An incubator shaker was used to maintain 34 °C and shake horizontally at 60 rpm. The release medium was withdrawn and replaced with free PBS at predefined time points.
2.4. Cell culture and cytotoxicity assay
MTT assay was employed to evaluate the cytotoxicity of TPGS/Mup-Ag on HaCaT cells. Briefly, HaCaT cells were seeded into 96-well plates with a density of 5 × 104 cells/mL and then incubated overnight. Various concentrations of AgNO3, Mup, Mup-Ag, TPGS/Mup, and TPGS/Mup-Ag were added before a 24-h incubation. Afterward, 20 μL of MTT solution (5 mg/mL) was added to each well for further incubation. After 4 h, the medium was discarded, and 100 μL of DMSO was added to each well to dissolve the purple formazan crystals. The absorbance at 570 nm was detected by a microplate reader (Bio-Rad, model 680, USA).
2.5. In vitro antibacterial capacity
2.5.1. MIC and MBC determination and growth curve
The bacteria used in this study were those harvested in the mid-exponential growth phase (absorbance at 620 nm was 0.2–0.3), (hereinafter inclusive). The minimal inhibitory concentrations (MICs) value of AgNO3, Mup, Mup-Ag, TPGS/Mup, and TPGS/Mup-Ag was identified as the lowest concentration in which the bacterial growth was completely inhibited after 20 h incubation at 37 °C by the broth microdilution method. For minimal bactericidal concentration (MBC) tests, 50 μL of mixtures containing formulations and bacterial inoculum from the MIC tests were incubated in agar plates for 24 h at 37 °C. MBC value was identified as the lowest concentration of formulations under which the plates did not show any bacterial growth. Then, the bacteria were incubated with different preparations. At predetermined time points, the absorbance of these cultures at a wavelength of 620 nm was detected. Experiments were repeated three times.
2.5.2. Observation of the morphological destruction of bacteria
MuRSA and MuSSA were treated for 4 h with different preparations: PBS, AgNO3 (0.14 μg/mL), Mup (0.41 μg/mL), Mup-Ag (0.50 μg/mL), TPGS/Mup (24.80 μg/mL), and TPGS/Mup-Ag (30.00 μg/mL), hereinafter inclusive. Isolates were collected by centrifugation (5000 rpm, 6 min). After rinsing with PBS three times, the bacteria samples were fixed and dehydrated with 30, 50, 70, 80, 90, and 100% ethanol for 15 min in sequence. The samples were finally sputter-coated with gold and observed using SEM and TEM.
2.5.3. Determination of biofilm formation by crystal violet assay
MuRSA and MuSSA suspensions (150 μL, 5 × 105 CFU/mL) were placed in 96-well plates and incubated at 37 °C overnight. After adding different preparations, the incubation was continued at 37 °C for another 8 h to allow further bacterial biofilm formation. After removing the medium and rinsing with PBS gently, the bacterial biofilms were fixed with 95 formalin for 15 min, followed by staining with 0.1% crystal violet solution for 10 min. The wells were washed with PBS three times to remove the redundant dye, followed by air drying for 1 h. Finally, crystal-violet-stained biofilms were dissolved in 200 μL of 33% acetic acid and assessed upon OD590 by a microplate reader (Bio-Rad, model 680, USA) [35].
2.5.4. Live/dead bacteria staining
After MuRSA and MuSSA were incubated with different preparations for 4 h, two isolates were collected and rinsed with PBS three times. Then bacteria were cultured in the presence of calcein-AM (2 μM) and PI (4.5 μM) for 30 min in the dark. Then the bacteria were rinsed and resuspended in PBS containing 50% glycerol. A confocal fluorescence microscope (LSM 880 with fast AiryScan, Zeiss) was employed to observe the survival of the bacteria.
2.6. Mechanism of anti-drug resistant
2.6.1. EB accumulation
After incubating with TPGS at different concentrations (20 μM, 66 μM, and 100 μM) and CCCP at 100 μM for 1 h, bacteria were further incubated with ethidium bromide (EB) (2 μg/mL) for another 3 h to enable EB accumulation. The EB-incubated bacteria without the pretreatment of CCCP or TPGS was used as the negative control group. After incubation, the bacteria were collected by centrifugation (5000 rpm, 10 min) and observed using a confocal fluorescence microscope and semi-quantitatively analyzed by ImageJ. For the quantification of EB accumulation, the fluorescence intensity of EB was measured using a spectrofluorometer (λex = 530 nm, λem = 600 nm). The increased percentage of EB accumulation after TPGS or CCCP treatment was calculated by comparing it with the fluorescence intensity of the negative control group.
2.6.2. Real-time RT-PCR
Oligonucleotide primers used for the real-time reverse transcription-PCR (real-time RT-PCR) were designed and purchased from Servicebio Technology Co., Limited (China). MuRSA was incubated with different preparations for 20 h at 37 °C. After centrifugation (8000 rpm, 4 °C) for 5 min, DNase-treated RNA templates were extracted from the pelleted bacterial cultures by RNAiso Plus, followed by RNA concentration quantification by a nanodrop spectrophotometer (Thermo Scientific, MA, USA). cDNA was synthesized using a PrimeScript RT-PCR kit (G3330, Servicebio Tech., China). Real-time PCR was performed using an SYBR Premix Ex Taq kit (G3320, Servicebio Tech., China) on an Applied Biosystems StepOnePlus Real-Time PCR system. The usage of primers is presented in Table S2. The relative quantity of mRNA corresponding to the NorA was determined, and expression analysis was performed subsequently using the 2−ΔΔCq method.
2.7. Bacterial infection model in vitro
2.7.1. Co-culture of bacteria and cells
HaCaT cells were placed into the 12-well plates with a density of 5 × 104 cells per well for overnight incubation. Afterward, HaCaT cells were infected with MuRSA for 4 h with a multiplicity of infection (MOI) of 20. Different preparations were added to each well and incubation for 12 h. After treatment, the cells were fixed in 2.5% glutaraldehyde solution, rinsed by PBS, fixed by osmium, dehydrated with ethanol, and embedded with an embedding agent. The coexistence of HaCaT and MuRSA was observed by TEM. In the meantime, HaCaT cells were cultured in the presence of calcein-AM (2 μM) and PI (4.5 μM) for 30 min in the dark, followed by suspending in PBS containing 50% glycerol and observation by a confocal fluorescence microscope.
2.7.2. Investigation of intracellular bacteria
Prepared bacteria isolates were added to the HaCaT cells at an MOI of 10 and incubated for 1 h at 37 °C. Afterward, the cells were rinsed and the medium was replaced with a fresh medium containing 50 μg/mL gentamicin to kill extracellular MuRSA for 1 h. Then, each group was added with different preparations and incubated for another 12 h. Infected cells were washed three times and then cultured with PBS containing 0.02% Triton X-100 to lyse the cells and release intracellular invaded bacteria. The invasion rate was calculated as follows:
2.7.3. Bacterial adhesion
After HaCaT cells were infected by MuRSA (MOI = 10) and incubated for 1 h, HaCaT cells were rinsed six times to remove unadhered bacteria. Cells were lysed by PBS containing 0.02% Triton X-100 and adhered bacteria were quantified by serial dilution plating. The adhesion rate was calculated as follows:
2.8. Animal modeling
The mice subcutaneous infection model was established according to the previously reported method [36]. Briefly, the backs of female BABL/c mice were shaved and removed by depilatory cream. 100 μL MuRSA (2 × 106 CFU) at the mid-logarithmic growth phase was subcutaneously injected into the back skin of mice. The needle was at 30° to the skin and penetrated the skin by 4 mm. For the mice wound infection model, mice were anesthetized with 2% Pentobarbital Sodium (40 mg/kg) and the back skin was shaved and cleaned. The whole skin layer was cut with a #11 scalpel (length: 0.8 cm, interval: 1.5 mm), followed by inoculating with MuRSA (20 μL, 2 × 106 CFU) using a micropipette [37]. The treatment protocol and dosage are as follows: PBS group: 0.5 mL twice a day; Ag group: 0.5 mL silver acetate solution (CAg+ = 0.01 mg/mL) twice a day; Mup group: 0.5 mL mupirocin PBS solution (CMup = 0.1 mg/mL) twice a day; Mup-Ag group: 0.5 mL Mup-Ag suspension (CAg+ = 0.01 mg/mL, CMup = 0.1 mg/mL) twice a day; TPGS/Mup group: 0.5 mL TPGS/Mup solution (CTPGS = 5 mg/mL, CMup = 0.1 mg/mL) twice a day; TPGS/Mup-Ag group: 0.5 mL TPGS/Mup-Ag solution (CTPGS = 5 mg/mL, CAg+ = 0.01 mg/mL, CMup = 0.1 mg/mL) twice a day. After modeling, the mice were weighed daily. The wound size was measured using ImageJ and was presented as a percentage of the original wound area.
2.9. In vivo antibacterial effect
Mice were sacrificed with 10% chloral hydrate on the 9th day of treatment and the back skin was harvested. 20 μL homogenates of skin tissue were mixed with 180 μL sterile PBS before being diluted to a series of concentrations. The suspensions were spread on solid media and cultured at 37 °C for 24 h. The colony count method was employed to detect the antibacterial effect in vivo. Meanwhile, the blood of mice was collected for routine analysis using an automatic hematology analyzer (TEK8500 VET, TECOM).
2.10. Immunohistochemistry and immunofluorescence
The immunohistochemistry and immunofluorescence staining of wounds were carried out on subcutaneous infection mice and wound infection mice after being treated for 11 days and 9 days, respectively.
2.11. Statistical analysis
All data are presented as mean ± standard error. The student's t-test was used for the comparison between two groups. One-way ANOVA and the LSD t-test were carried out for statistical analysis, using GraphPad Prism software version 7 (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
3. Results and discussion
3.1. Preparation and characterization of TPGS/Mup-Ag
Mup-Ag complex was synthesized by the precipitation reaction between the carboxylate radical of mupirocin and silver ion (Ag+) (Fig. 1A). The 1H NMR spectrum of mupirocin (Fig. 1B) exhibited the peaks characterizing –CH3 (0.88 ppm, 1.20 ppm, and 2.07 ppm), –CH2- in the hydrocarbon chain (1.20–1.55 ppm), and –CH2- in cyclic hydrocarbon chain (∼3.60 ppm). Peaks characteristic of –COOCH2- (3.97 ppm), –CH2- in oxirane (2.45–2.60 ppm), and –CH2COOH (2.18 ppm) could also be observed. After the chelation between the carboxyl group and Ag+, the peak representing the proton on carboxyl α position moved to the high field (2.18 ppm–2.12 ppm, –CH–COOAg) due to the electron-donor induction effect of Ag+, which is consistent with other observations in the literature [38,39]. The differential scanning calorimetry (DSC, Fig. 1C) of Mup-Ag exhibited two exothermic peaks which represent the melting point and clearing point (177 °C and 230 °C), while Mup only showed an endothermic peak at 77 °C. Further, a TEM was employed to observe the morphology of the Mup-Ag complex. As shown in Fig. 1D, Mup-Ag possessed an irregular shape with a size of over several hundred nanometers. Scanning electron microscopy with energy dispersive spectrometry (SEM-EDS) was used to verify the element distribution of Mup-Ag. As shown in Fig. 1E and G, peaks representing C, O, and Ag could be observed on the surface of the Mup-Ag complex, suggesting that the Mup-Ag complex was successfully synthesized. The particle size of the TPGS micelle was measured to be 9.83 ± 0.78 nm (Fig. 1H). After the modification of TPGS, the particle size of the TPGS/Mup-Ag turned out to be 15.99 ± 1.24 nm and still showed a narrow size distribution (Fig. 1I). High-performance liquid chromatography (HPLC) and inductively coupled plasma mass spectrometry (ICP-MS) were used to determine the drug loading (DL) and encapsulation efficiency (EE) of Mup- Ag. As a result, when the dosage of Mup-Ag was 2%, the EE was measured to be 79.35 ± 0.23% and DL was 1.63 ± 0.11%. The drug release behaviors of different formulations were investigated under simulated skin physiological conditions (pH 6.8) at 34 °C. As displayed in Figure S1, TPGS/Mup showed a sustained drug release behavior for about 12 h. Since Mup dissociates from silver ions in physiological environment, the drug release of TPGS/Mup-Ag extended to about 15 h and the release rate of Mup at 24 h was 77.65%. Cell viability of the TPGS/Mup-Ag group was above 80% at 31.25 μM compared with 31.36% in AgNO3 and 31.79 in the Mup-Ag group, indicating that the TPGS/Mup-Ag significantly improved the biocompatibility of Ag (Figure S2).
Fig. 1.

Characterization of the Mup-Ag complex and TPGS/Mup-Ag. (A) Schematic illustration showing Mup-Ag and TPGS/Mup-Ag preparation. (B) 1H NMR spectra of Mup (bottom) and Mup-Ag (top). (C) DSC thermograms for Mup (bottom) and Mup-Ag (top) with a heating rate of 10 °C/min (D) A TEM image of Mup-Ag. (E) Low magnification SEM image of Mup-Ag. (F) High magnification SEM image of Mup-Ag. (G) Energy dispersive X-ray (EDX) analysis showing the elementary composition of Mup-Ag. (H) Size distribution and polydispersity index (PDI) of TPGS micelle and a TEM image. (I) Size distribution and polydispersity index (PDI) of TPGS/Mup-Ag micelle and a TEM image.
3.2. Antibacterial capacity of TPGS/Mup-Ag in vitro
The in vitro antibacterial capacity of TPGS/Mup-Ag was investigated on mupirocin-resistant Staphylococcus aureus (MuRSA) and mupirocin-sensitive Staphylococcus aureus (MuSSA). As shown in Fig. 2A, the minimal inhibitory concentrations (MICs) of Mup towards MuRSA and MuSSA were 1000 μg/mL and 125 μg/mL respectively, which were consistent with other observations [40]. TPGS/Mup-Ag exhibited the lowest MICs at 15.63 μg/mL towards MuRSA and 7.82 μg/mL towards MuSSA, which could be related to the synergistic effect between Ag+, Mup, and TPGS. Similarly, as displayed in Fig. 2B, TPGS/Mup-Ag showed a minimal bactericidal concentration (MBC) of 31.25 μg/mL towards MuRSA and 15.63 μg/mL toward MuSSA, which were significantly lower than any other groups. The concrete dosage of each component in the preparations, we calculated the absolute concentrations of mupirocin and silver corresponding to the MIC and MBC values of different preparations. As shown in Table S1, the following table, when the concentration of TPGS/Mup-Ag reached its MIC to MuRSA, the CAg+ was found to be 3.37 μg/mL. By contrast, the MIC of Ag+ was 6.74 μg/mL. Similarly, when the concentration of TPGS/Mup-Ag reached its MBC to MuRSA, the CAg+ was found to be 6.74 μg/mL while the MBC of Ag+ was 13.75 μg/mL. All these results suggested that when silver formed a complex with mupirocin, the concentration of silver ion with the same bactericidal effect was significantly reduced. We further investigated the effect of the preparations on the growth of MuRSA and MuSSA (Fig. 2C). The OD600 value of MuRSA remarkably increased at 4 h of incubation with Mup, indicating that Mup was unable to inhibit the growth of MuRSA. However, the visible growth of bacteria was completely inhibited by Mup-Ag, TPGS/Mup, and TPGS/Mup-Ag. In the case of MuSSA, Mup still showed an unsatisfied inhibition of bacteria. As for both these two isolates, TPGS/Mup-Ag exhibited the best antibacterial capacity compared with other preparations. Figure S3 showed that the TPGS/Mup-Ag reduces the 50%-bactericidal concentration of mupirocin from about 1000 μmol/mL to about 16 μmol/mL, indicating that the prepared preparation could substantially enhance the sensitivity of MuRSA to mupirocin and reverse the mupirocin resistance of MuRSA.
Fig. 2.

Evaluation of the antibacterial capacity of TPGS/Mup-Ag towards MuRSA and MuSSA in vitro. (A) MIC and (B) MBC susceptibility semiquantitative profiles of TPGS/Mup-Ag against MuRSA and MuSSA obtained by the microplate broth dilution method (n = 3). (C) Growth curves of MuRSA and MuSSA incubated with different preparations (n = 3). (D) Representative TEM images and (E) SEM images of MuRSA and MuSSA after incubating with different preparations for 6 h. (F) Fluorescence staining images of MuRSA and MuSSA after incubating with different preparations for 4 h. Live and dead bacteria were stained with calcein-AM (green fluorescence) and dead bacteria were stained with PI (red fluorescence). (G, H) Crystal violet staining and determination of the relative biofilm formation rate of MuRSA and MuSSA using a microplate reader upon OD590. Data are presented as the mean ± SD: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To visualize the antibacterial capacity of TPGS/Mup-Ag, TEM was used to observe bacteria morphology. As displayed in Fig. 2D, bacteria without any treatment showed a typical ball structure with an intact and distinct wall. The bacteria morphology changed obviously after being treated with different bactericidal substances. The wall of bacteria that incubated with Ag and Mup-Ag transformed and fractured, while Mup led to the appearance of gaps inside bacteria. TPGS/Mup-Ag caused the greatest damage compared with other preparations. We also used SEM to study the morphological changes of the two bacterial strains upon treatments (Fig. 2E). The remarkably thick cell wall of S. aureus could be clearly observed, which may contribute to its drug resistance [41,42]. Upon TPGS/Mup-Ag treatment, the surface of bacteria cracked, and contents leaked out. Adhesion between bacteria occurred, followed by the disappearance of the basic form of bacteria. In Fig. 2F, live bacteria were stained with calcein acetoxymethyl (calcein-AM) which showed green fluorescence whereas dead bacteria were stained with propidium iodide (PI) and showed red fluorescence [43]. After being incubated with different preparations for 4 h, MuRSA and MuSSA showed death to different extents, as reflected by different numbers of red fluorescence signals. Significantly, after being treated with TPGS/Mup-Ag, nearly all bacteria were stained red, suggesting that TPGS/Mup possessed the best bactericidal effect. By calculating the ratio of the red fluorescence signal and green fluorescence signal in each image with ImageJ (Figure S4), we found that after TPGS/Mup treatment, the death of MuRSA and MuSSA was consistent, indicating that TPGS increased the susceptibility of MuRSA to mupirocin to the same level as MuSSA. Bacterial biofilms are one of the factors of infectious diseases and have been proven to contribute to bacterial antibiotic tolerance [44,45]. Biofilms can be viewed as a protector that allows bacteria to survive antibiotics and a desiccated environment [44,46]. Therefore, using the crystal violet staining, we assessed the relative biofilm biomass upon different treatments to reveal the inhibitory effect of TPGS/Mup-Ag on biofilm formation (Fig. 2G and H). Among all preparations, TPGS/Mup-Ag showed the greatest effect of biofilm destruction, as reflected by 7.28 ± 3.53% and 2.69 ± 0.53% (MuRSA and MuSSA) of the OD value of the control groups.
3.3. Bacterial infection model in vitro
During the initial stage of skin infection, staphylococcus aureus enters keratinocytes and persists in the cells to avoid glia-mediated immune responses and clearance responses [47]. Hence, it is pivotal to further investigate the antibacterial capacity of TPGS/Mup-Ag in the presence of keratinocytes. The in vitro bacterial infection model was established by the co-culture of MuRSA and human immortal keratinocyte cell line (HaCaT). As displayed in Fig. 3A–C, the 4-h infection of MuRSA led to a remarkable death of HaCaT, as reflected by the obvious red fluorescence signal in the PBS group. The cytotoxicity of silver also resulted in considerable cell death. Nevertheless, the number of dead cells decreased to different extents after different treatments. The PI+ cell rate of the TPGS/Mup group is significantly lower than that of the Mup group, indicating that TPGS could enhance the bactericidal effect of Mup and subsequently reduce cell death (12.16 ± 0.38% vs 5.70 ± 0.56%, Mup vs TPGS/Mup, p < 0.001). The slightest cell death was observed after TPGS/Mup-Ag treatment, as only 5.37% of HaCaT was calculated as death. Next, we used TEM to visually explore the coexistence of MuRSA and HaCaT. As shown in Fig. 3B, abundant MuRSA could be observed inside HaCaT cells whose morphology was severely damaged and whose cell membrane was torn. Treatment with Ag could effectively reduce the amount of MuRSA, but cellular vacuoles and destruction disclosed the cytotoxicity of silver. TPGS/Mup-Ag showed the best bactericidal effect as there were hardly any bacteria around HaCaT cells and the cell morphology was no distinct difference was found from normal cells. At present, although not much effort has been made to study the intracellular bactericidal effect of a drug delivery system [48], the intracellular survival and proliferation of bacteria are one of the major reasons for infection persistence and recurrence, even after successful antimicrobial treatment [49]. Therefore, we determined the intracellular survival rate of MuRSA by removing the extracellular bacteria. As shown in Fig. 3D, the number of bacteria that invaded HaCaT cells was about 260% of the initial amount, which could be attributed to bacterial proliferation inside cells. Because the MuRSA was resistant to Mup, 56.27% of the bacteria still remained inside HaCaT cells. The lowest level of intracellular bacteria was observed in the TPGS/Mup-Ag group, as reflected by 5.25% of the initial bacteria calculated. Apart from invasion, the colonization of MuRSA also contributes to the pathogenesis of bacterial infection [50]. Accordingly, we further investigate the effect of TPGS/Mup-Ag on MRSA adhesion to HaCaT cells. As shown in Figs. 3E and 11.67% of the MuRSA was found to adhere to the surface of HaCaT cells. The number of bacteria that adhered on the surface of HaCaT after Mup was significantly higher than that of TPGS/Mup (4.15 ± 0.30% vs 2.65 ± 0.36%, Mup vs TPGS/Mup, p < 0.05), and the number of bacteria that adhered on the surface of HaCaT after Mup-Ag was higher than that of TPGS/Mup-Ag (1.52 ± 0.22% vs 0.43 ± 0.07%, Mup-Ag vs TPGS/Mup-Ag, p < 0.05), indicating that TPGS effectively enhanced the antibacterial ability of Mup as well as Mup-Ag. This is consistent with the results reported in the literature [51,52].
Fig. 3.

In vitro antibacterial capacity in bacterial infection model. (A) Fluorescence images of HaCaT cells infected by MuRSA for 4 h, followed by treating with different preparations for 12 h. Live and dead cells were respectively stained with calcein-AM (green fluorescence) and dead cells were stained with PI (red fluorescence). (B) Representative TEM images of MuRSA-infected HaCaT cells (MOI = 20) after being treated with different preparations for 12 h. (C) The ratio of PI-positive cells to total cells in (A). (D) Survival rate of intracellular MuRSA cells after being treated with different preparations for 12 h (n = 3). (E) Rate of MuRSA adhesion to HaCaT after exposure to different preparations for 12 h (n = 3). Data are presented as the mean ± SD: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
3.4. Mechanism of overcoming drug resistance
To understand the mechanism of the enhanced anti-bacterial effect of TPGS/Mup-Ag, we prepared ICG-Ag complex and TPGS/ICG-Ag for bacterial internalization assessments. The particle sizes of these two preparations had no significant difference compared with TPGS/Mup-Ag and Mup-Ag (p = 0.611) (Figure S5). In Fig. 4A, both the TPGS/ICG-Ag and ICG-Ag showed a gradual increase in fluorescence signal inside the bacteria with a prolonged incubation time. TPGS/ICG-Ag displayed a stronger bacterial internalization behavior than ICG-Ag at both 2 h and 6 h. The fluorescence intensities in MuRSA after being incubated with TPGS/ICG-Ag were 1.27-and 1.37-fold greater than those of the Mup-Ag group at 2 h and 6 h, respectively (Fig. 4B). The stronger internalization could be attributed to the dispersion effect of TPGS reducing the particle size of the complex, which facilitates bacterial phagocytosis [51]. Then, the effect of TPGS treatment on the activity of bacterial efflux pumps was investigated by measuring the expression level of efflux pump-related genes NorA by real-time RT-PCR (Fig. 4C). Previous studies have reported that NorA is the main kind of major facilitator superfamily (MFS) efflux pump which provides MRSA with resistance to numerous antimicrobials [53,54]. The bacterial culture of MuRSA harvested in the mid-exponential growth phase was incubated with Ag, Mup, Mup-Ag, TPGS/Mup, and TPGS/Mup-Ag for 20 h at 37 °C. As shown, the expression level of NorA in MuRSA was 6.26-fold compared with that in MuSSA, confirming that the overexpression of NorA was involved in the drug resistance in MuRSA. After treating with TPGS and TPGS/Mup-Ag, NorA expression levels decreased by 5.12-fold and 5.29-fold respectively, while no significant difference was found between untreated bacteria and Mup-Ag treated bacteria, suggesting that the presence of TPGS played an important role in inhibiting efflux pump genes. Although the expression level of NorA was higher than that of MuSSA, the significantly down-regulated gene expression level still highlighted that TPGS and TPGS/Mup-Ag were able to largely inhibit the activity of efflux pumps and pose a great potential to overcome bacterial resistance. To visualize the effectiveness of TPGS on the activity of the efflux pump, we used EB as an efflux pump substrate as the accumulation of EB was inversely proportional to the activity of the efflux pump [55]. Meanwhile, carbonyl cyanide m-chlorophenylhydrazone (CCCP) was employed in the positive control group as a proton pump inhibitor. As shown in Fig. 4D, the fluorescence intensity of EB in the control group was relatively low, and it increased with the rising concentration of added TPGS. As shown in Fig. 4E, the increased percentage of accumulated EB rose from 18.84% to 55.80% when MuRSA was exposed in TPGS from 20 to 100 μM. CCCP is well-known for its capacity to inhibit efflux pumps and lead to increased EB accumulation. Despite the slightly lower EB level compared with the CCCP group at the identical concentration (100 μM), MuRSA that was treated with TPGS still showed a large increase in EB accumulation. Therefore, TPGS could be speculated to have an inhibitory effect on MuRSA efflux pumps of MuRSA, which was consistent with the results obtained from the above-mentioned RT-PCR measurement. Although the mechanism of the inhibition of bacterial efflux pump caused by TPGS has not been fully elucidated, previous studies suggested that TPGS, as a surfactant, can modulate efflux pump activity in mammalian cells by changing the liquidity of cytomembrane, inhibiting ATPase, and other possible mechanisms [56,57]. In our study, efflux pumps of bacteria functioned depending on the energy supplement from membrane proton force and ATPase, and the mechanism of TPGS modulating the efflux pump activity of mammalian cells could be used to explain the inhibited efflux pump after TPGS treatment.
Fig. 4.

Mechanism of overcoming drug resistance. (A) Confocal fluorescent images of MuRSA after 2 h and 6 h of incubation with TPGS/Mup-Ag and Mup-Ag. (B) Quantitative analysis of fluorescence in (A) with imageJ. (C) Expression levels of NorA mRNA transcripts of MuRSA treated with different preparations for 20 h assessed by RT-PCR. Untreated MuRSA was used as a control. (D) Confocal fluorescent images of MuRSA after exposure to EB for 4 h, following pretreatment with TPGS and CCCP (n = 3). (E) Quantitative analysis of the increase in EB accumulation compared with the negative control groups (n = 3). Data are presented as the mean ± SD: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
3.5. In vivo anti-infective efficacy on subcutaneous infection model
In this part, we examined the in vivo anti-infective activity of TPGS/Mup-Ag using a MuRSA-induced subcutaneous infection model on mice (Fig. 5A). Mice were separated into 6 groups: PBS, Ag, Mup, Mup-Ag, TPGS/Mup, and TPGS/Mup-Ag. As shown in Fig. 5B, one day after the bacterial solution was injected subcutaneously, punilaoile stules appeared on the infected skin, followed by a visible abscess. After receiving different treatments for more than 11 continuous days, scar sizes gradually decreased to varying degrees as time prolonged. By the 11th day of treatment, the wound size of untreated mice was about 48.57% of that of the day after modeling. Because of the insensitivity of Mup, an unsatisfied therapeutic effect could be observed, as the wound size remained at 41.73% of the original size. Among all groups, the best effect of promoting wound healing was achieved by the treatment of TPGS/Mup-Ag, in which the wound area shrank to 18.67 ± 5.72% of the original size (Fig. 5C). Next, the levels of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) were detected in wound tissues were analyzed to determine skin inflammation. As shown in Fig. 5D and F, both the two factors increased sharply after infection (39.42 ± 3.69 pg/mg prot in IL-6 and 72.19 ± 6.64 pg/mg prot in TNF-α). Nevertheless, IL-6 and TNF-α both showed the most significant decline when treated with TPGS/Mup-Ag, with 6.16 ± 1.46 pg/mg prot in IL-6 and 15.23 ± 3.26 pg/mg prot in TNF-α. Afterward, lymphocyte (LYMPH) count, white blood cell (WBC) count, and neutrophil (NEU) percentage were measured to reveal the inflammation levels in mice. Upon different treatments, remarkable reductions in levels of the three hematology indexes (lymphocyte count, LYMPH; white blood cell count, WBC; neutrophil percentage, NEU) indicated that our preparations effectively alleviated the infection (Fig. 5G–I). In bacterial colony analysis, Mup-treated wounds still showed a comparatively higher number of bacteria and TPGS/Mup-Ag treated mice possessed the fewest bacterial colonies compared with other groups due to the anti-bacterial effect of Mup combined with Ag, as well as the inhibition of TPGS on NorA efflux pump (Fig. 5J and K). The recovery of the infected skin tissues was further evaluated by histological analysis using hematoxylin and eosin (H&E) and Masson staining (Fig. 5I and J), so as the neutrophil by MPO staining. After bacterial infection and subsequent subcutaneous abscess, obvious thickened epidermal and extensive infiltration of inflammatory cells could be observed, as reflected by the increased layers of keratinocyte, and dispersed green fluorescence signal respectively. Usually, when bacterial infection occurs, a large number of neutrophils migrate from the bloodstream to the lesion [58,59]. By contrast, mice treated with TPGS/Mup-Ag showed a remarkable decrease in infiltrated inflammatory cells and H&E staining also suggested that skin structure recovered to an almost normal state.
Fig. 5.

In vivo antibacterial capacity on mice subcutaneous infection model. (A) Experimental design and treatment protocols (created with BioRender.com). (B) Photographs of the MuRSA-infected abscesses of mice that were treated with different preparations for 11 consecutive days. (C) Curves showing wound areas after various treatments (wound area on the day after infection was used as 100%). (D) Curves show the body weight throughout the treatment process. (E) IL-6 and (F) TNF-α levels in wound tissue on the ninth day (n = 6). (G) Lymphocyte count, (H) red blood cell count, and (I) neutrophil percentage in mice blood at the ninth day (n = 6). (J) Photographs of bacterial colonies obtained from infected tissues of mice on the ninth day and bacteria colonies were counted by Image J (K) (n = 6). (L–M) Representative images of wound tissue after H&E and Masson staining. (N) Fluorescent images of wound tissue after staining with anti-MPO antibody, in which the green and blue indicate neutrophil infiltration and nucleus respectively. Data are presented as the mean ± SD: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
3.6. In vivo anti-infective efficacy on wound infection model
Among various skin pathogens, Staphylococcus aureus is the most frequently isolated one from skin wounds and can cause a wide range of clinical infections [60]. Therefore, mouse wound infection models established that can replicate various features of infected human wounds were adopted according to a widely reported method (Fig. 6A and B) [[61], [62], [63]]. As shown in Fig. 6A, mice showed three clear wounds on the back with a length of 0.8 cm and an interval of 1.5 mm. After inoculating MuRSA on the wound, the same treatment was also applied to evaluate the anti-infective activity of TPGS/Mup-Ag. On day 9, wounds in the PBS group showed a severe suppurative scar (74.34 ± 6.39% of the initial wound size), indicating that MuRSA-infected wounds had a slow self-healing. Compared with the Mup group, wounds in TPGS/Mup showed an obviously faster rate (68.53 ± 8.07 vs 27.01 ± 6.75, Mup vs TPGS/Mup, p < 0.001). This may be because TPGS enhanced the sensitivity of mupirocin. The difference between the Mup-Ag group and TPGS/Mup-Ag group could also be attributed to this reason (Fig. 6C). In bacterial colony analysis (Fig. 6D), the fewest bacterial colonies were detected in TPGS/Mup-Ag group, reinforcing the excellent anti-infective capacity of MuRSA. Further, H&E and Masson showed that the epidermis and dermal structure of scalpel-caused wounds gradually recovered after treatment (Fig. 6E and F). F4/80 staining images further confirmed the anti-infective capacity of our preparations, as hardly any red fluorescent signal could be seen in the TPGS/Mup-Ag group (Fig. 6G). Also, TNF-α and IL-6 as inflammatory indicators were detected in wound tissues (Fig. 6H and I). The two factors showed a considerable rose after infection (43.91 ± 9.23 pg/mg prot in TNF-α and 33.17 ± 3.96 pg/mg prot in IL-6). Notably, TNF-α and IL-6 both showed the most significant decline when treated with TPGS/Mup-Ag, with 10.18 ± 2.29 pg/mg prot in TNF-α and 4.70 ± 2.41 pg/mg prot in IL-6, indicating a superior therapeutic effect of TPGS/Mup-Ag over other preparations.
Fig. 6.

In vivo antibacterial capacity in mice wound infection model. (B) Experimental design and treatment protocols (created with BioRender.com). (A) Photographs of the MuRSA-infected wounds of mice that were treated with different preparations for nine consecutive days. (C) Curves showing wound areas after various treatments (wound area on the day after infection was used as 100%) (n = 6). (D) Photographs of bacterial colonies obtained from infected tissues of mice on the ninth day. (E–F) Representative images of wound tissue after H&E and Masson staining. (G) Fluorescent images of wound tissue after staining with anti-F4/80 antibody, in which the red and blue fluorescent signals indicate macrophage and nucleus respectively. (H) TNF-α and (I) IL-6 levels in wound tissue on the ninth day (n = 6). Data are presented as the mean ± SD: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
4. Conclusion
In summary, we developed a TPGS-based mupirocin and silver complex for the treatment of skin infections caused by mupirocin-resistant S. aureus. Because the modification of TPGS endowed Mup-Ag complex with a particle size of less than 20 nm, bacterial endocytosis towards TPGS/Mup-Ag was significantly enhanced, leading to decreased MIC and MBC and prominent bacterial death. TEM and SEM images further visualized that TPGS/Mup-Ag could effectively destroy the bacterial walls, followed by the disappearance of normal bacterial morphology. Besides, TPGS/Mup-Ag effectively eliminated both intracellular and extracellular bacteria and inhibit bacterial adhesion, without affecting living cells. Activity of the efflux pump in MuRSA was considerably inhibited by TPGS, along with the expression level of efflux pump-related genes NorA, thereby the Mup concentration inside MuRSA was significantly enhanced. Finally, in mice with subcutaneous infection and wound infection, TPGS/Mup-Ag treated mice showed a significantly smaller wound size and recovered levels of inflammatory cytokines. TPGS/Mup-Ag accelerated wound healing, controlled skin infection, and alleviated systemic inflammation levels. We demonstrated that TPGS/Mup-Ag overcame the drug resistance of MuRSA to mupirocin and could be a promising candidate for skin and tissue infection diseases caused by mupirocin-resistant S. aureus. Also, this work may offer new ideas for the development of silver-based antibacterial preparations.
Credit author statement
Xiao-Hua Tao, Yong-Zhong Du: conceptualization and supervision. Ming-Chen Sun: Methodology, Validation, investigation, writing - original draft, writing-reviewing and Editing. Ying-Fang Chen: Methodology and validation. Di Liu, Xiao-Ling Xu, Yu-Chan You: Investigation and supervision. Wei Lu, Yun-Juan Shi, Ming-Yang Ren, and Yi-Bin Fan: Methodology.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by Zhejiang Province Public Welfare Technology Research Project (LGF20H110003) and Zhejiang Province Health Technology Plan (2022KY049).
Footnotes
Supplementary material to this article can be found online at https://doi.org/10.1016/j.mtbio.2022.100534.
Contributor Information
Yong-Zhong Du, Email: duyongzhong@zju.edu.cn.
Xiao-Hua Tao, Email: txhtxh@sina.com.
Appendix A. Supplementary data
In vitro drug release, cytotoxicity on HaCaT cells, MuRSA and MuSSA viability under a series of concentrations of various preparations, quantitative analysis of fluorescence signal in Fig. 2f, and primers used for the RT-PCR assay.
Appendix B. Supplementary material
The following are the Supplementary material to this article:
Data availability
Data will be made available on request.
References
- 1.Levy S.B., Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 2004;10(12 Suppl):S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Aminov R.I. The role of antibiotics and antibiotic resistance in nature. Environ. Microbiol. 2009;11(12):2970–2988. doi: 10.1111/j.1462-2920.2009.01972.x. [DOI] [PubMed] [Google Scholar]
- 3.Tong S.Y., Davis J.S., Eichenberger E., Holland T.L., Fowler V.G., Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015;28(3):603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wertheim H.F., Melles D.C., Vos M.C., van Leeuwen W., van Belkum A., Verbrugh H.A., Nouwen J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005;5(12):751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 5.Saeed K., Marsh P., Ahmad N. Cryptic resistance in Staphylococcus aureus: a risk for the treatment of skin infection? Curr. Opin. Infect. Dis. 2014;27(2):130–136. doi: 10.1097/QCO.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 6.McCaig L.F., McDonald L.C., Mandal S., Jernigan D.B. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg. Infect. Dis. 2006;12(11):1715–1723. doi: 10.3201/eid1211.060190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eriksen K.R. ["Celbenin"-resistant staphylococci] Ugeskr Laeger. 1961;123:384–386. [PubMed] [Google Scholar]
- 8.Caffrey A.R., Quilliam B.J., LaPlante K.L. Risk factors associated with mupirocin resistance in meticillin-resistant Staphylococcus aureus. J. Hosp. Infect. 2010;76(3):206–210. doi: 10.1016/j.jhin.2010.06.023. [DOI] [PubMed] [Google Scholar]
- 9.Mupirocin-resistant Staphylococcus aureus. Lancet. 1987;2(8555):387–388. [PubMed] [Google Scholar]
- 10.Hayden M.K., Lolans K., Haffenreffer K., Avery T.R., Kleinman K., Li H., Kaganov R.E., Lankiewicz J., Moody J., Septimus E., Weinstein R.A., Hickok J., Jernigan J., Perlin J.B., Platt R., Huang S.S. Chlorhexidine and mupirocin susceptibility of methicillin-resistant Staphylococcus aureus isolates in the REDUCE-MRSA trial. J. Clin. Microbiol. 2016;54(11):2735–2742. doi: 10.1128/JCM.01444-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gostev V., Kruglov A., Kalinogorskaya O., Dmitrenko O., Khokhlova O., Yamamoto T., Lobzin Y., Ryabchenko I., Sidorenko S. Molecular epidemiology and antibiotic resistance of methicillin-resistant Staphylococcus aureus circulating in the Russian Federation. Infect. Genet. Evol. 2017;53:189–194. doi: 10.1016/j.meegid.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Dadashi M., Hajikhani B., Darban-Sarokhalil D., van Belkum A., Goudarzi M. Mupirocin resistance in Staphylococcus aureus: a systematic review and meta-analysis. J Glob Antimicrob Re. 2020;20:238–247. doi: 10.1016/j.jgar.2019.07.032. [DOI] [PubMed] [Google Scholar]
- 13.Xu X.L., Zhang N.N., Shu G.F., Liu D., Qi J., Jin F.Y., Ji J.S., Du Y.Z. A luminol-based self-illuminating nanocage as a reactive oxygen species amplifier to enhance deep tumor penetration and synergistic therapy. ACS Nano. 2021;15(12):19394–19408. doi: 10.1021/acsnano.1c05891. [DOI] [PubMed] [Google Scholar]
- 14.Crofts T.S., Gasparrini A.J., Dantas G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017;15(7):422–434. doi: 10.1038/nrmicro.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell M.J., Billingsley M.M., Haley R.M., Wechsler M.E., Peppas N.A., Langer R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021;20(2):101–124. doi: 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui Y., Yi X., Hou F., Wibowo D., Zhang F., Zhao D., Gao H., Zhao C.X. Role of nanoparticle mechanical properties in cancer drug delivery. ACS Nano. 2019;13(7):7410–7424. doi: 10.1021/acsnano.9b03924. [DOI] [PubMed] [Google Scholar]
- 17.Rizzello L., Cingolani R., Pompa P.P. Nanotechnology tools for antibacterial materials. Nanomedicine. 2013;8(5):807–821. doi: 10.2217/nnm.13.63. [DOI] [PubMed] [Google Scholar]
- 18.Sharma V.K., Yngard R.A., Lin Y. Silver nanoparticles: green synthesis and their antimicrobial activities. Adv. Colloid Interface Sci. 2009;145(1–2):83–96. doi: 10.1016/j.cis.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Mishra V., Nayak P., Singh M., Tambuwala M.M., Aljabali A.A., Chellappan D.K., Dua K. Pharmaceutical aspects of green synthesized silver nanoparticles: a boon to cancer treatment. Anti Cancer Agents Med. Chem. 2021;21(12):1490–1509. doi: 10.2174/1871520620666200918111024. [DOI] [PubMed] [Google Scholar]
- 20.Mathur P., Jha S., Ramteke S., Jain N.K. Pharmaceutical aspects of silver nanoparticles. Artif. Cell Nanomed. Biotechnol. 2018;46(sup1):115–126. doi: 10.1080/21691401.2017.1414825. [DOI] [PubMed] [Google Scholar]
- 21.Sondi I., Salopek-Sondi B. Silver nanoparticles as antimicrobial agent: a case study on E. coli as a model for Gram-negative bacteria. J. Colloid Interface Sci. 2004;275(1):177–182. doi: 10.1016/j.jcis.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi S., Sakamoto K. Effect of hydrolysis on mechanical properties of tricalcium phosphate/poly-L: -lactide composites. J. Mater. Sci. Mater. Med. 2009;20(1):379–386. doi: 10.1007/s10856-008-3583-2. [DOI] [PubMed] [Google Scholar]
- 23.Ivask A., Elbadawy A., Kaweeteerawat C., Boren D., Fischer H., Ji Z., Chang C.H., Liu R., Tolaymat T., Telesca D., Zink J.I., Cohen Y., Holden P.A., Godwin H.A. Toxicity mechanisms in Escherichia coli vary for silver nanoparticles and differ from ionic silver. ACS Nano. 2014;8(1):374–386. doi: 10.1021/nn4044047. [DOI] [PubMed] [Google Scholar]
- 24.Lok C.N., Ho C.M., Chen R., He Q.Y., Yu W.Y., Sun H., Tam P.K., Chiu J.F., Che C.M. Proteomic analysis of the mode of antibacterial action of silver nanoparticles. J. Proteome Res. 2006;5(4):916–924. doi: 10.1021/pr0504079. [DOI] [PubMed] [Google Scholar]
- 25.Rinna A., Magdolenova Z., Hudecova A., Kruszewski M., Refsnes M., Dusinska M. Effect of silver nanoparticles on mitogen-activated protein kinases activation: role of reactive oxygen species and implication in DNA damage. Mutagenesis. 2015;30(1):59–66. doi: 10.1093/mutage/geu057. [DOI] [PubMed] [Google Scholar]
- 26.Mijakovic I., Petranovic D., Macek B., Cepo T., Mann M., Davies J., Jensen P.R., Vujaklija D. Bacterial single-stranded DNA-binding proteins are phosphorylated on tyrosine. Nucleic Acids Res. 2006;34(5):1588–1596. doi: 10.1093/nar/gkj514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt K.B., Bard A.J. Interaction of silver(I) ions with the respiratory chain of Escherichia coli: an electrochemical and scanning electrochemical microscopy study of the antimicrobial mechanism of micromolar Ag+ Biochemistry. 2005;44(39):13214–13223. doi: 10.1021/bi0508542. [DOI] [PubMed] [Google Scholar]
- 28.Yamanaka M., Hara K., Kudo J. Bactericidal actions of a silver ion solution on Escherichia coli, studied by energy-filtering transmission electron microscopy and proteomic analysis. Appl. Environ. Microbiol. 2005;71(11):7589–7593. doi: 10.1128/AEM.71.11.7589-7593.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn J.M., Eom H.J., Yang X., Meyer J.N., Choi J. Comparative toxicity of silver nanoparticles on oxidative stress and DNA damage in the nematode, Caenorhabditis elegans. Chemosphere. 2014;108:343–352. doi: 10.1016/j.chemosphere.2014.01.078. [DOI] [PubMed] [Google Scholar]
- 30.Xu L., Dan M., Shao A., Cheng X., Zhang C., Yokel R.A., Takemura T., Hanagata N., Niwa M., Watanabe D. Silver nanoparticles induce tight junction disruption and astrocyte neurotoxicity in a rat blood-brain barrier primary triple coculture model. Int. J. Nanomed. 2015;10:6105–6118. doi: 10.2147/IJN.S85265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun C., Yin N., Wen R., Liu W., Jia Y., Hu L., Zhou Q., Jiang G. Silver nanoparticles induced neurotoxicity through oxidative stress in rat cerebral astrocytes is distinct from the effects of silver ions. Neurotoxicology. 2016;52:210–221. doi: 10.1016/j.neuro.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Bruna T., Maldonado-Bravo F., Jara P., Caro N. Silver nanoparticles and their antibacterial applications. Int. J. Mol. Sci. 2021;22(13) doi: 10.3390/ijms22137202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaur A., Kumar R. Enhanced bactericidal efficacy of polymer stabilized silver nanoparticles in conjugation with different classes of antibiotics. RSC Adv. 2019;9(2):1095–1105. doi: 10.1039/C8RA07980C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W., Zhang Y., Kumari J., Engelkamp H., Kouwer P.H.J. Magnetic stiffening in 3D cell culture matrices. Nano Lett. 2021;21(16):6740–6747. doi: 10.1021/acs.nanolett.1c00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rewak-Soroczynska J., Sobierajska P., Targonska S., Piecuch A., Grosman L., Rachuna J., Wasik S., Arabski M., Ogorek R., Wiglusz R.J. New approach to antifungal activity of fluconazole incorporated into the porous 6-Anhydro-alpha-l-Galacto-beta-d-Galactan structures modified with nanohydroxyapatite for chronic-wound treatments-in vitro evaluation. Int. J. Mol. Sci. 2021;22(6) doi: 10.3390/ijms22063112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youn C., Archer N.K., Miller L.S. Research techniques made simple: mouse bacterial skin infection models for immunity Research. J. Invest. Dermatol. 2020;140(8):1488–1497. doi: 10.1016/j.jid.2020.04.012. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archer N.K., Wang Y., Ortines R.V., Liu H., Nolan S.J., Liu Q., Alphonse M.P., Dikeman D.A., Mazhar M., Miller R.J., Anderson L.S., Francis K.P., Simon S.I., Miller L.S. Preclinical models and methodologies for monitoring Staphylococcus aureus infections using noninvasive optical imaging. Methods Mol. Biol. 2020;2069:197–228. doi: 10.1007/978-1-4939-9849-4_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubbiani R., Kitanovic I., Alborzinia H., Can S., Kitanovic A., Onambele L.A., Stefanopoulou M., Geldmacher Y., Sheldrick W.S., Wolber G., Prokop A., Wolfl S., Ottt I. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010;53(24):8608–8618. doi: 10.1021/jm100801e. [DOI] [PubMed] [Google Scholar]
- 39.Busetto L., Cassani M.C., Femoni C., Macchioni A., Mazzoni R., Zuccaccia D. Synthesis, molecular structures and solution NMR studies of N-heterocyclic carbene-amine silver complexes. J. Organomet. Chem. 2008;693(15):2579–2591. doi: 10.1016/j.jorganchem.2008.04.030. [DOI] [Google Scholar]
- 40.Sritharadol R., Hamada M., Kimura S., Ishii Y., Srichana T., Tateda K. Mupirocin at subinhibitory concentrations induces biofilm formation in Staphylococcus aureus. Microb. Drug Resist. 2018;24(9):1249–1258. doi: 10.1089/mdr.2017.0290. [DOI] [PubMed] [Google Scholar]
- 41.Hanaki H., Kuwahara-Arai K., Boyle-Vavra S., Daum R.S., Labischinski H., Hiramatsu K. Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-resistant Staphylococcus aureus clinical strains Mu3 and Mu50. J. Antimicrob. Chemother. 1998;42(2):199–209. doi: 10.1093/jac/42.2.199. [DOI] [PubMed] [Google Scholar]
- 42.Kuroda M., Kuwahara-Arai K., Hiramatsu K. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem. Biophys. Res. Commun. 2000;269(2):485–490. doi: 10.1006/bbrc.2000.2277. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y.L., He M., Miron R.J., Chen A.Y., Zhao Y.B., Zhang Y.F. Temperature/pH-Sensitive nanoantibiotics and their sequential assembly for optimal collaborations between antibacterial and immunoregulation. ACS Appl. Mater. Interfaces. 2017;9(37):31589–31599. doi: 10.1021/acsami.7b10384. [DOI] [PubMed] [Google Scholar]
- 44.Flemming H.C., Wingender J., Szewzyk U., Steinberg P., Rice S.A., Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 2016;14(9):563–575. doi: 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- 45.Karygianni L., Ren Z., Koo H., Thurnheer T. Biofilm matrixome: extracellular components in structured microbial communities. Trends Microbiol. 2020;28(8):668–681. doi: 10.1016/j.tim.2020.03.016. [DOI] [PubMed] [Google Scholar]
- 46.Xiu W., Gan S., Wen Q., Qiu Q., Dai S., Dong H., Li Q., Yuwen L., Weng L., Teng Z., Mou Y., Wang L. Biofilm microenvironment-responsive nanotheranostics for dual-mode imaging and hypoxia-relief-enhanced photodynamic therapy of bacterial infections. Research. 2020 doi: 10.34133/2020/9426453. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bitschar K., Wolz C., Krismer B., Peschel A., Schittek B. Keratinocytes as sensors and central players in the immune defense against Staphylococcus aureus in the skin. J. Dermatol. Sci. 2017;87(3):215–220. doi: 10.1016/j.jdermsci.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Kaur S., Harjai K., Chhibber S. Bacteriophage-aided intracellular killing of engulfed methicillin-resistant Staphylococcus aureus (MRSA) by murine macrophages. Appl. Microbiol. Biotechnol. 2014;98(10):4653–4661. doi: 10.1007/s00253-014-5643-5. [DOI] [PubMed] [Google Scholar]
- 49.Kubica M., Guzik K., Koziel J., Zarebski M., Richter W., Gajkowska B., Golda A., Maciag-Gudowska A., Brix K., Shaw L., Foster T., Potempa J. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One. 2008;3(1):e1409. doi: 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Srisuwan S., Voravuthikunchai S.P. Rhodomyrtus tomentosa leaf extract inhibits methicillin-resistant Staphylococcus aureus adhesion, invasion, and intracellular survival in human HaCaT keratinocytes. Microb. Drug Resist. 2017;23(8):1002–1012. doi: 10.1089/mdr.2016.0284. [DOI] [PubMed] [Google Scholar]
- 51.Kang X.Q., Shu G.F., Jiang S.P., Xu X.L., Qi J., Jin F.Y., Liu D., Xiao Y.H., Lu X.Y., Du Y.Z. Effective targeted therapy for drug-resistant infection by ICAM-1 antibody-conjugated TPGS modified beta-Ga2O3:Cr(3+) nanoparticles. Theranostics. 2019;9(10):2739–2753. doi: 10.7150/thno.33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang X.Q., Qiao Y., Lu X.Y., Jiang S.P., Li W.S., Wang X.J., Xu X.L., Qi J., Xiao Y.H., Du Y.Z. Tocopherol polyethylene glycol succinate-modified hollow silver nanoparticles for combating bacteria-resistance. Biomater. Sci. 2019;7(6):2520–2532. doi: 10.1039/c9bm00343f. [DOI] [PubMed] [Google Scholar]
- 53.Du D., Wang-Kan X., Neuberger A., van Veen H.W., Pos K.M., Piddock L.J.V., Luisi B.F. Author Correction: multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol. 2018;16(9):577. doi: 10.1038/s41579-018-0060-x. [DOI] [PubMed] [Google Scholar]
- 54.Handzlik J., Matys A., Kiec-Kononowicz K. Recent advances in multi-drug resistance (MDR) efflux pump inhibitors of gram-positive bacteria S. aureus. Antibiotics. 2013;2(1):28–45. doi: 10.3390/antibiotics2010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peleg A.Y., Adams J., Paterson D.L. Tigecycline efflux as a mechanism for nonsusceptibility in acinetobacter baumannii. Antimicrob. Agents Chemother. 2007;51(6):2065–2069. doi: 10.1128/AAC.01198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Assanhou A.G., Li W.Y., Zhang L., Xue L.J., Kong L.Y., Sun H.B., Mo R., Zhang C. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials. 2015;73:284–295. doi: 10.1016/j.biomaterials.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 57.Zhao S., Tan S., Guo Y., Huang J., Chu M., Liu H., Zhang Z. pH-sensitive docetaxel-loaded D-α-tocopheryl polyethylene glycol succinate-poly(β-amino ester) copolymer nanoparticles for overcoming multidrug resistance. Biomacromolecules. 2013;14(8):2636–2646. doi: 10.1021/bm4005113. [DOI] [PubMed] [Google Scholar]
- 58.Li J., Liu X., Tan L., Cui Z., Yang X., Liang Y., Li Z., Zhu S., Zheng Y., Yeung K.W.K., Wang X., Wu S. Zinc-doped Prussian blue enhances photothermal clearance of Staphylococcus aureus and promotes tissue repair in infected wounds. Nat. Commun. 2019;10(1):4490. doi: 10.1038/s41467-019-12429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fan X., Yang F., Huang J., Yang Y., Nie C., Zhao W., Ma L., Cheng C., Zhao C., Haag R. Metal-organic-framework-Derived 2D carbon nanosheets for localized multiple bacterial eradication and augmented anti-infective therapy. Nano Lett. 2019;19(9):5885–5896. doi: 10.1021/acs.nanolett.9b01400. [DOI] [PubMed] [Google Scholar]
- 60.Dunyach-Remy C., Ngba Essebe C., Sotto A., Lavigne J.P. Staphylococcus aureus toxins and diabetic foot ulcers: role in pathogenesis and interest in diagnosis. Toxins. 2016;8(7) doi: 10.3390/toxins8070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo Y., Ramos R.I., Cho J.S., Donegan N.P., Cheung A.L., Miller L.S. In vivo bioluminescence imaging to evaluate systemic and topical antibiotics against community-acquired methicillin-resistant Staphylococcus aureus-infected skin wounds in mice. Antimicrob. Agents Chemother. 2013;57(2):855–863. doi: 10.1128/AAC.01003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ortines R.V., Liu H., Cheng L.I., Cohen T.S., Lawlor H., Gami A., Wang Y., Dillen C.A., Archer N.K., Miller R.J., Ashbaugh A.G., Pinsker B.L., Marchitto M.C., Tkaczyk C., Stover C.K., Sellman B.R., Miller L.S. Neutralizing alpha-toxin accelerates healing of Staphylococcus aureus-infected wounds in nondiabetic and diabetic mice. Antimicrob. Agents Chemother. 2018;62(3) doi: 10.1128/AAC.02288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zolfaghari P.S., Packer S., Singer M., Nair S.P., Bennett J., Street C., Wilson M. In vivo killing of Staphylococcus aureus using a light-activated antimicrobial agent. BMC Microbiol. 2009;9:27. doi: 10.1186/1471-2180-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.