

Abstract
The social and public health burdens of ischemic stroke have been increasing worldwide. In addition to focal brain damage, acute ischemic stroke (AIS) provokes systemic abnormalities across peripheral organs. AIS profoundly alters the autonomic nervous system, hypothalamic-pituitary-adrenal axis, and immune system, which further yield deleterious organ-specific consequences. Poststroke systemic pathological alterations in turn considerably contribute to the progression of ischemic brain injury, which accounts for the substantial impact of systemic complications on stroke outcomes. This review provides a comprehensive and updated pathophysiological model elucidating the systemic effects of AIS. To address their clinical significance and inform stroke management, we also outline the resulting systemic complications at particular stages of AIS and highlight the mechanisms. Future therapeutic strategies should attempt to integrate the treatment of primary brain lesions with interventions for secondary systemic complications, and should be tailored to patient individualized characteristics to optimize stroke outcomes.
Keywords: Acute ischemic stroke, Autonomic dysfunction, Hypothalamic-pituitary-adrenal axis activation, Systemic immune alterations, Systemic complications, Stroke management
1. Introduction
Stroke is the second most common cause of death and disability-adjusted life years worldwide. The global burden of stroke remains high and is expected to continue to increase due to population growth and ageing. Acute ischemic stroke (AIS) is known to account for 84.4% of prevalent strokes (Johnson et al., 2019).
Stroke is an abrupt cerebrovascular event that generally occurs in the course of chronic systemic disease processes. Stroke development is typically accompanied by a series of comorbidities involving multiple organ systems, mostly including, but not confined to, the cardiovascular system. Age and sex are two crucial nonmodifiable factors with considerable impact on the incidence and disease outcomes of stroke. Ageing, together with the interplay with sex, confers a higher risk and poorer outcomes of stroke, given age and sex differences typically shape the spectrum of stroke risk factors, pre-stroke comorbidities, and poststroke complications (Ahnstedt and McCullough, 2019). Approximately 80% of patients hospitalized for a first-time stroke and transient ischemic attack, have two or more chronic comorbidities, including but not limited to hypertension, atrial fibrillation, diabetes mellitus, ischemic heart disease, and chronic kidney disease (CKD); and this proportion increases to nearly 90% in elderly patients (Yousufuddin et al., 2017). With this in mind, it is not unexpected that the organism’s organ systems involving both the CNS and periphery are already subject to varying degrees of structural and functional alterations preceding acute stroke in a large proportion of patients (especially in older subjects). Additionally, both the brain and peripheral organs are challenged owing to sustained exposure to chronic low-grade systemic inflammation that occurs with normal ageing or pathological contexts such as autoimmune disorders and chronic infections (Ferrucci and Fabbri, 2018). All these “silent” pathologies before stroke onset set the stage for the emergence of stroke-induced systemic abnormalities.
Following AIS onset, excitotoxicity, oxidative stress, and neuroinflammatory episodes are immediately elicited within the brain, with the latter being central to secondary brain injury. Beyond the drastic pathological changes within the ischemic brain, AIS induces extensive extra-brain pathophysiological alterations, including autonomic dysfunction, activation of the hypothalamic-pituitary-adrenal (HPA) axis, and systemic immune dysregulation (Iadecola et al., 2020; Meisel et al., 2005), which, working in concert, provokes or aggravates structural changes and functional impairment of multiple peripheral organs, alone or in most cases, in conjunction with pre-existing comorbidities (Fig. 1). The resulting peripheral disturbances in turn exacerbate brain injury by affecting crucial aspects of stroke progression, for instance, neuroinflammation, thereby forming a positive feedback loop. Clinically, these abnormalities are pervasively reflected as a range of multisystem complications, which commonly emerge at particular stages and complicate the clinical course of stroke (Kumar et al., 2010). Altogether, it is more accurate to view stroke as a systemic disease with farreaching effects on peripheral organs.
Fig. 1.

Systemic effects of stroke. Stroke induces a relative sympathetic predominance, activation of the hypothalamic-pituitary-adrenal (HPA) axis, and systemic immune dysregulation. In conjunction with brain-released damage-associated molecular patterns (DAMPs), heightened sympathetic and glucocorticoid signaling elicits dynamic immune changes that involve peripheral immune compartments. Driven by sympathetic-mediated gut permeability, bacterial components and metabolites translocate to the periphery, functioning as pathogen-associated molecular patterns (PAMPs) that exacerbate systemic inflammation and facilitate infections. Together, these pathophysiological alterations contribute to structural changes and functional impairment of multiple peripheral organs, thereby causing multi-system complications. In turn, massive inflammatory mediators derived from the periphery, including the classical immune organs, liver, and gut, reach the damaged brain and substantially aggravate brain injury. ACTH, adrenocorticotropic hormone; CAs, catecholamines; GCs, glucocorticoids; iNKT, invariant natural killer T; CKD, chronic kidney disease; Neu, neutrophils; Mo/Mø (monocytes/macrophages); LC, lymphocytes. Figure adapted from REF (Meisel et al., 2005).
Whether these seemingly unrelated complications simply coincide with or are attributable to stroke are not well documented. Deciphering the mechanistic underpinnings of these complications is of serious clinical significance. Given the limited effective treatments for rescuing the ischemic brain, novel therapeutic approaches that target major elements driving the crosstalk between the CNS and periphery must be developed to block the pathophysiological cascades. Meanwhile, stroke therapeutic paradigms should integrate the treatment of primary brain damage with interventions for secondary systemic complications, rather than treating these disorders as two distinct entities. In this review, we reinforce the notion that stroke is a systemic disease by synthesizing current experimental and clinical advances concerning the underlying mechanisms. Here, we focus on the acute and subacute phase of AIS, the definition of which adopted here is: the first 6 h after stroke onset for hyperacute phase; 6–24 h for acute phase; 24 h to 6 weeks for subacute phase (Bahn et al., 1996). This review also highlights the clinical features of the resulting systemic complications, including incidences, temporal courses, risk factors, and clinical prognosis. Raising knowledge of these will assist in timely identification, accurate diagnosis, and prompt interventions for systemic complications, which represents a feasible opportunity for clinicians to improve stroke outcomes.
2. Pathophysiological alterations after AIS
Following the disruption of cerebral blood flow, ischemic neural cells release damage-associated molecular patterns (DAMPs) that fuel drastic inflammation in the focal ischemic region within minutes, characterized by resident glial activation, massive secretion of inflammatory mediators, and infiltration of peripheral immune cells through the breached blood-brain barrier (BBB). Furthermore, inflammatory responses elicited in situ propagate throughout the brain, referred to as global brain inflammation (Shi et al., 2019).
Central autonomic networks and the hypothalamus are stimulated by either direct ischemic injuries or locally increased proinflammatory cytokines. Consequently, autonomic derangement and the HPA axis activation occur; the latter originates from the release of corticotropin-releasing factor and downstream adrenocorticotropic hormone (ACTH) from the alerted hypothalamus (Fig. 1) (Courties et al., 2019; Meisel et al., 2005). Both primary and secondary lymphoid organs are extensively innervated by sympathetic nerves, with distinct adrenoreceptor (AR) subtypes, as well as glucocorticoid receptors, present on resident immune cells. Accordingly, systemic immune dysregulation ensues. Generally, the occurrence of these pathophysiological processes largely depends on the extent of brain damage, as well as the lesion location (as described in details below). It is known that most experimental stroke studies just include male subjects or focus on young males. However, it has been gaining emerging evidence that both age and sex play important roles in stroke-induced central and peripheral pathophysiological changes. Ageing significantly exaggerates neuroinflammatory responses to stroke, as specifically indicated by enhanced neutrophil pathogenicity, thereby causing adverse functional outcomes (Ritzel et al., 2018; Roy-O’Reilly et al., 2020). Additionally, increased accumulation of T lymphocytes in the ischemic brain has been demonstrated in aged versus young mice, in young males versus females (Manwani et al., 2013; Ritzel et al., 2016), and in aged males versus females (Ahnstedt et al., 2020).
2.1. Autonomic dysfunction
Poststroke autonomic dysfunction involves both the sympathetic and parasympathetic nervous system, which eventually culminates in a relative sympathetic predominance (Krause et al., 2017; Mo et al., 2019). In clinical settings, current methods to evaluate autonomic function include measurement of heart rate variability, baroreflex sensitivity, and circulating/urinary catecholamines (including norepinephrine and epinephrine) or its metabolites (including metanephrine and normetanephrine), either of which, however, only partially reflects sympathetic activity. Abnormal heart rate variability and baroreflex sensitivity primarily signify cardiovascular autonomic disturbances. Catecholamine levels only represent the systemic arm of sympathetic nervous system, which is derived from the adrenal medulla and “spillover” from sympathetic nerve terminals, while the local arm that exerts targeted innervation to almost every organ within the body cannot be assessed using noninvasive diagnostic procedures.
One study reported that nearly 80% of AIS patients displayed autonomic dysfunction that persisted up to 6 months after stroke (Xiong et al., 2014). Patients with greater stroke severity and larger infarcts involving the central autonomic network (particularly the insular cortex) are prone to autonomic dysfunction (Hilz et al., 2011; Korpelainen et al., 1996; Meyer et al., 2004; Nayani et al., 2016; Ozdemir and Hachinski, 2008; Sander et al., 2001; Sykora et al., 2009; Tokgozoglu et al., 1999; Yperzeele et al., 2015). As a crucial region for maintaining sympathovagal balance, the insular cortex is frequently affected following cerebral ischemia within the middle cerebral artery (MCA) territory (Fink et al., 2005). Insular stroke has been widely identified as an important promoter for autonomic dysfunction and cardiovascular abnormalities after AIS. A consensus on the lateralization of insular involvement has not been reached, though most studies incline towards the right insular stroke in autonomic disturbances (Meyer et al., 2004; Nayani et al., 2016; Ozdemir and Hachinski, 2008; Sander et al., 2001; Scheitz et al., 2018; Sykora et al., 2009; Tokgozoglu et al., 1999). In contrast to the simplified laterality hypothesis that the right and left insular cortex primarily regulate sympathetic and parasympathetic tone, respectively, recent evidence suggests a functional heterogeneity of different insular subregions among which the dorsal anterior region mainly controls the parasympathetic outflow (Krause et al., 2017). In an observational study of 384 AIS patients, Walter and colleagues have also demonstrated a robust association between insular lesion and poststroke sympathetic overactivation, as indicated by increased plasma levels of metanephrine and normetanephrine that predominantly represent the non-neuronal sources derived from adrenomedullary glands (Walter et al., 2013). Catecholamine elevation has been repeatedly reported in rodents undergoing extensive cerebral ischemia (Mracsko et al., 2014; Singh et al., 2016; Wang et al., 2014) or infarction involving the insular cortex (Smith et al., 1986), and in a proportion of AIS patients, particularly those with insular lesions and/or large infarct volumes (Liesz et al., 2013; Meyer et al., 2004; Sander et al., 2001; Walter et al., 2013).
Driven by either locally intensified sympathetic innervation or increased circulating catecholamine levels, the consequences of post-stroke autonomic dysfunction involve not only the well-known cardiac complications but also abnormalities of multiple peripheral organs, such as systemic immune alterations, gastrointestinal dysfunction, and glucose metabolic changes (Mo et al., 2019). Independent of the initial stroke severity, autonomic dysfunction is recognized to be associated with adverse clinical outcomes in AIS patients (Nayani et al., 2016; Xiong et al., 2018).
2.2. HPA axis activation
Consequent to the direct central drive of ischemic lesion, cerebral ischemia induces a rapid activation of the HPA system in the first hours in a fraction of patients after AIS onset (Fassbender et al., 1994). Interestingly, there is a dissociation between circulating cortisol and ACTH, that is, a temporary rise of ACTH only in the hyperacute phase (at 4 h after ischemia onset) but a prolonged increase of cortisol that persists for several days, which has been recognized in both animal models and patients of ischemic stroke (Fassbender et al., 1994; Johansson et al., 2000; Mracsko et al., 2014). This implies other mechanisms underlying sustained glucocorticoid production. Proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6, are known to stimulate the HPA axis at different levels. In this line, it has long been reported that increased levels of plasma IL-6 corelate with elevated plasma cortisol in AIS patients, thus leading to the supposed concept that elevated proinflammatory cytokines represent another significant contributor to hypercortisolemia beyond the hyperacute phase of stroke (Johansson et al., 2000). Moreover, increased gene expression of these cytokines in the hypothalamus has recently been validated in rodent models of the MCA occlusion (MCAO), and is associated with increased corticotropin-releasing factor expression (Courties et al., 2019).
According to one recent review, most clinical studies reported that elevated serum cortisol levels corelated with greater stroke severity (Barugh et al., 2014). In accordance with the previous observation that patients only with severe stroke present with an elevation in plasma cortisol (Liesz et al., 2013), extensive cerebral infarction in animal models were reported to give rise to hypercortisolemia, while moderatesized cortical infarcts had no effect on cortisol levels (Mracsko et al., 2014). Elevated circulating cortisol levels may be related to unfavorable outcomes and increased mortality. However, whether this association is independent of baseline stroke severity has not been determined with certainty (Barugh et al., 2014).
2.3. Dynamic alterations in systemic immunity
Known as a major contributor to stroke-associated infections, stroke-induced immunosuppression is the prevailing conceptual model for poststroke systemic immune dysfunction, which, of note, represents only one prominent aspect of systemic immune alterations following stroke. Alongside the drastic neuroinflammation localized to the brain, DAMPs leaking into the circulation, in concert with disturbances of the autonomic nervous system and HPA axis, instigate complex and multiphasic immune perturbations in the periphery, including the immediate immune activation and subsequent immunosuppression (Fig. 2), with the former being relatively underappreciated in previous studies. Presumably, it is not a specific time point but a transition period (12–24 h after stroke onset) during which an immunocompromised state is boosted and ultimately exceeds the initial systemic immune activation that subsides but does not necessarily disappear. Infarct size is a major determinant of the extent of systemic immune disturbances, as repeatedly confirmed in animal models (Liesz et al., 2009; Mracsko et al., 2014) and AIS patients (Audebert et al., 2004; Buck et al., 2008; Hug et al., 2009; Liesz et al., 2013; Urra et al., 2017). Notably, stroke not only perturbs immune homeostasis in the classical peripheral immune organs (that is, the bone marrow, thymus, spleen, lymph nodes, and gut-associated lymphoid tissues), but also interferes with immune cells residing in other organs, such as in the liver and lung (see below).
Fig. 2.
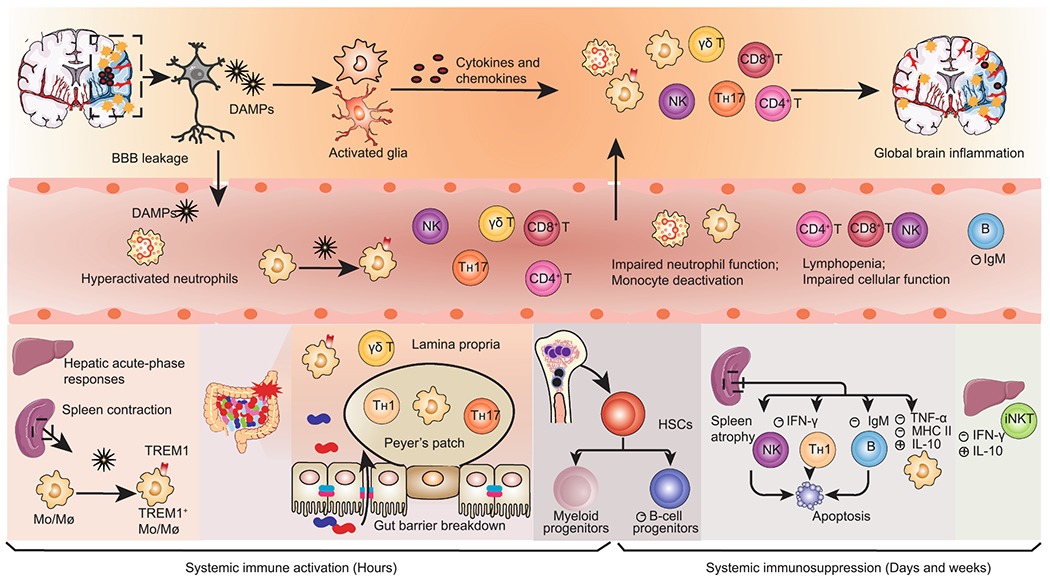
Dynamic alterations in systemic immunity after AIS. Ischemic neural cells release damage-associated molecular patterns (DAMPs) that trigger local inflammatory cascades, including glial activation and infiltration of peripheral immune cells. Simultaneously, DAMPs reach the circulation and elicit systemic immune activation, remarked by activation of peripheral innate immune cells and massive secretion of proinflammatory mediators in the liver, spleen, gut, and circulation. Splenic Mo/Mø (monocytes/macrophages) are rapidly deployed into the blood. Peripheral Mo/Mø display elevated TREM1+ expression in response to both brain-derived DAMPs and gut-derived bacterial components arising from gut leakage. Systemic immune activation is rapidly superseded by a long-lasting immunosuppression that encompasses spleen atrophy, lymphopenia and impaired function of both the innate and adaptive immunity. The loss of spleen marginal zone B cells causes a declined secretion of immunoglobulin M (IgM). The bone marrow displays enhanced myelopoiesis and deficient B lymphopoiesis, with the former contributing to early circulating neutrophilia and monocytosis and the latter favoring later immunosuppression. AIS, acute ischemic stroke; BBB, blood-brain barrier; CRP, C-reactive protein; TREM1, triggering receptor expressed on myeloid cells 1; HSCs, hematopoietic stem cells; iNKT, invariant natural killer T.
2.3.1. Systemic immune activation
Immediately (within hours) after stroke, systemic inflammatory responses are elicited in response to signals from stroke-damaged brains, which encompass activation of innate immune cells and massive secretion of proinflammatory mediators in the periphery (Chapman et al., 2009; Liesz et al., 2015; Offner et al., 2006a; Vahidy et al., 2016). Supporting the initial immune scenario, a proportion of AIS patients without underlying infections present with an elevated body temperature, increased serum C-reactive protein (CRP) and cytokine levels, and transient leukocytosis soon after stroke onset, indicating noninfectious systemic inflammation (Audebert et al., 2004). Elevated plasma IL-6 levels within 24 h from stroke onset correlate significantly with lesion volume and stroke severity in AIS patients (Basic Kes et al., 2008), and remain a reliable predictor of poor recovery after adjustment of stroke severity, age, and preceding infections (Whiteley et al., 2012). Over the past decade, advances in the understanding of the mechanisms driving stroke-induced systemic immune activation have emerged, which primarily involve sympathetic hyperactivation and brain-released DAMPs, as well as gut-derived pathogen-associated molecular patterns (PAMPs) (Liu et al., 2019).
Circulating neutrophils and monocytes are the first responders to penetrate the ischemic brain and substantiate postischemic neuroinflammation (Iadecola et al., 2020). A numerical increase of neutrophils and monocytes has been consistently observed in human patients (Buck et al., 2008; Cai et al., 2020; Hug et al., 2009; Roth et al., 2018; Vogelgesang et al., 2008; Weisenburger-Lile et al., 2019) and rodents (Cai et al., 2020; Kim et al., 2014; Wang et al., 2014) soon after cerebral ischemia. Circulating hyperactivated neutrophils is suggested to be induced within 6 h after clinical onset of stroke (Weisenburger-Lile et al., 2019). Experimental research has suggested that the primary mechanisms mediating neutrophilia and monocytosis involve the mobilization from the spleen (Kim et al., 2014) and bone marrow displaying enhanced myelopoiesis (Courties et al., 2015; Denes et al., 2011). Increased sympathetic innervation is responsible for the myelopoiesis bias via activating β3-ARs on hematopoietic niche cells (Courties et al., 2015; Roth et al., 2018). Additionally, monocytes that have egressed from the bone marrow are further diverted to the spleen, making the spleen an expanded and sustained source of monocyte production (Roth et al., 2018).
Converging experimental evidence has documented the immediate deployment of splenic proinflammatory monocytes into the circulation in the acute phase after stroke (<24 h), concurrent with a significant reduction in spleen size and weight, namely, spleen contraction (Kim et al., 2014; Seifert et al., 2012). Clinically, spleen contraction has been observed in a proportion of patients within 24–48 h after stroke onset, followed by a re-expansion along with improved clinical recovery (Chiu et al., 2016; Sahota et al., 2013; Vahidy et al., 2016). Moreover, spleen contraction within the first 24 h of clinical stroke onset is suggested to be correlated with increased serum cytokine levels (Vahidy et al., 2016), as well as early systemic inflammatory response syndrome in African-American patients but not in patients older than 75 years (Zha et al., 2018). Splenocytes exhibit elevated secretion of proinflammatory mediators, such as IL-6, TNF-α, and C-C motif chemokine ligand 2 (CCL2) after stroke (Liesz et al., 2015; Offner et al., 2006a). Currently, it remains a challenge to characterize the ongoing splenic cellular responses in stroke patients. Elucidating the mechanisms of acute splenic responses and contribution to early peripheral immune activation should be considered a priority since it represents one promising avenue to develop rational immunomodulating approaches for treating stroke.
Importantly, it has been recognized for a long time that focal brain injury triggers hepatic acute-phase responses within hours after the CNS insult. Early elevation of serum and hepatic C-X-C motif chemokine ligand 1 (CXCL1) and CCL2 coincides with an increase in circulating neutrophils and monocytes and enhanced neutrophil and monocyte recruitment to the liver, and of interest, later to the brain (Campbell et al., 2003, 2005). The peak levels of CXCL1 in the liver, circulation, and lung, along with upregulation of hepatic CXCL1 gene expression, occur within hours and precede that in the ischemic brain (Chapman et al., 2009). One separate study also reported the activation of hepatic proinflammatory pathways following cerebral ischemia, accompanied by activation of hepatic adrenergic signaling. In this study, administration of propranolol, a nonselective β-AR inhibitor, abrogated these inflammatory processes, which provides a mechanistic explanation that sympathetic activation mediates hepatic inflammatory changes (Wang et al., 2014). Likewise, a sympathetic-mediated adipose inflammation following experimental stroke was reported by the same group in another study (Wang et al., 2011). One experimental research using novel SPECT imaging approaches observed a local lung inflammation, as well as a transient gut inflammation, as early as 2 h after cerebral ischemia (Szigeti et al., 2015). Overall, the emerge of inflammatory chemokines in the periphery is unlikely to be a result of leak from the injured brain.
Recently, several independent lines of experimental evidence have addressed the critical role of DAMPs in triggering systemic inflammatory activation in the absence of exogenous pathogens following stroke. High-mobility group box 1 (HMGB1), a prototypic DAMP with potent immunogenicity, is passively released from necrotic neuronal cells, reaches the circulation, and provokes massive cytokine secretion and monocytic activation in the spleen via acting on the receptor of advanced glycation end-products (RAGE) (Liesz et al., 2015; Roth et al., 2018). Plasma HMGB1 levels are elevated within hours after extensive cerebral ischemia in rodent models (Kim et al., 2018; Liesz et al., 2015; Roth et al., 2018) and human patients (Liesz et al., 2015; Roth et al., 2018; Schulze et al., 2013). One clinical study has demonstrated a correlation between elevated plasma HMGB1 levels and increased circulating hyperactivated neutrophil subsets within 6 h after AIS onset (Weisenburger-Lile et al., 2019). According to one pioneering experimental finding, coupled with brain-released DAMPs, translocated gut-derived PAMPs from sympathetic-dependent gut permeability induce pronounced expression of triggering receptor expressed on myeloid cells 1 (TREM1), a potent proinflammatory modulator, on monocytes/macrophages in the intestine, blood, and spleen, which occurs as early as 4.5 h after MCAO (Liu et al., 2019). Overall, it appears that sterile and nonsterile inflammation occur simultaneously in response to stroke in the hyperacute phase, both of which are derived endogenously, and drive critical components of systemic immune activation.
It is increasingly recognized that peripherally derived cytokines, chemokines, and “primed” inflammatory cells (primarily myeloid cells) aggravate ischemic brain injury and injurious neuroinflammation via compromising BBB integrity and substantiating brain trafficking (Campbell et al., 2003, 2005; Kim et al., 2014; Liu et al., 2019; Seifert et al., 2012). Meanwhile, peripheral systemic inflammatory responses partially involve, albeit not decisively in some cases, a range of abnormalities involving multiple peripheral organs, such as gut barrier leakage, acute lung injury, cardiac neurogenic arrhythmia, and insulin resistance in the liver and adipose tissue (see Section 3).
2.3.2. Systemic immunosuppression
As the initial systemic immune activation rapidly subsides, a sustained immunocompromised state supervenes, which is initiated as early as 12 h from stroke onset, becomes most significant on day 3, and lasts for several weeks (Prass et al., 2003; Vogelgesang et al., 2008) It is conceivable that the immunocompromised state is not simply a passive process consequent to the exhaustion of the initial immune activation; instead, it is orchestrated by the interplay of multiple aspects of neurogenic signaling that actively suppress the immune function, involving both the adaptive and innate immunity. Converging experimental studies have consistently elucidated the profound role of post-stroke sympathetic/HPA axis activation and the resulting stress mediators (i.e. catecholamines and cortisol) in compromising immune function (Courties et al., 2019; Liu et al., 2017; McCulloch et al., 2017; Prass et al., 2003; Wong et al., 2011). Nevertheless, their conclusive roles have not been convincingly established based on current clinical evidence, mostly due to the considerable difficulty in determining their genuine contribution independent of stroke severity.
Systemic immunodeficiency in the subacute phase of stroke is the best addressed component of systemic immune alterations, including prolonged lymphopenia, defective lymphocytic function, and monocyte deactivation (Haeusler et al., 2008; Hug et al., 2009; Liu et al., 2017; McCulloch et al., 2017; Offner et al., 2006b; Prass et al., 2003; Vogelgesang et al., 2008). Extensive lymphopenia results from enhanced apoptosis and contributes to spleen atrophy (Liu et al., 2017; McCulloch et al., 2017; Offner et al., 2006b) and thymus atrophy (Offner et al., 2006b). Dysfunction of T helper 1 (TH1) and natural killer (NK) cells is signified by a deficient production of interferon-γ (IFN-γ) that is crucial for antibacterial defense (Haeusler et al., 2008; Liu et al., 2017; Offner et al., 2006b; Prass et al., 2003). Monocytes exhibit markedly reduced expression of major histocompatibility complex class II (MHC II) molecules, a decreased ratio of secreted TNF-α/IL-10, and the consequent insufficient antigen-presentation, as evidenced by both experimental (Prass et al., 2003) and clinical studies (Haeusler et al., 2008; Hug et al., 2009; Vogelgesang et al., 2008). All the immunosuppressive signatures have long been acknowledged to result from the synergetic effects of catecholamines and glucocorticoids, with the former generally believed to play dominant roles (Liu et al., 2017; Prass et al., 2003). Several experimental and clinical studies otherwise called into question the role of catecholamines but implied the role of glucocorticoids in poststroke lymphopenia (Liesz et al., 2013; Mracsko et al., 2014; Zierath et al., 2018). Besides its immune-stimulatory effects in the hyperacute phase, the HMGB1-RAGE pathway has also been implicated in the later immunosuppression, the blockade of which prevents the expansion of splenic immuno-incompetent monocytes and partially ameliorates lymphopenia after severe experimental stroke (Liesz et al., 2015).
One experimental study has identified B lymphopoiesis defects in the bone marrow as a critical driver of poststroke immunosuppression, which is predominantly instigated by increased glucocorticoid release (Courties et al., 2019). Despite the antiinflammatory signature and relatively increased proportion of regulatory T (Treg) cells in the spleen and blood after stroke (Offner et al., 2006b), the effect of Treg cells on systemic immunity is still a matter of debate. One experimental study provided a direct evidence supporting the association between immunosuppression and Treg cells generated in the bone marrow where Treg proportion and mobilization were both increased, mediated by heightened β2- and β3-adrenergic signaling (Wang et al., 2015). Importantly, β2-adrenergic signaling has been reported to trigger a drastic loss of innate-like B cells in the splenic marginal zone and the subsequent decrease in circulating immunoglobulin M levels, thereby facilitating lung infection in stroke mice (McCulloch et al., 2017). Similarly, hepatic invariant NKT (iNKT) cells display a β-AR-dependent immunocompromised phenotype that potentiates infections in stroke rodents (Wong et al., 2011), which was later reported in the blood of AIS patients (Wong et al., 2017). Additionally, impaired respiratory burst in circulating neutrophils and monocytes was suggested to correlate with poststroke infections in AIS patients (Ruhnau et al., 2014). One experimental study ascribed the immunocompromised phenotype of neutrophils, as indicated by their reduced activation, chemotaxis, and phagocytosis, to enhanced catecholamine binding to neutrophil β2-ARs following stroke (Nicholls et al., 2018).
2.3.3. Immune alterations in the chronic phase
So far, there remains a paucity of data to clarify the long-term changes in systemic immunity after stroke. Notably, stroke induces a secondary and complicated autoreactive immune responses against brain-released antigens both inside and outside the brain, which becomes prominent weeks after stroke (Iadecola et al., 2020). Within the injured brain, it remains controversial whether autoimmune response against brain antigens is beneficial by promoting neurological recovery or detrimental by potentiating delayed neuropsychiatric sequelae after the index stroke. In regard to its systemic effects, persistent immuno-reactivity to brain antigens might keep the disturbed system immunity alerted and remain superimposed on the chronic low-grade inflammation that contributes to stroke development, which could persistently affect the organism, for instance, by predisposing to recurrent cardiocerebrovascular events. Clinically, cardiocerebrovascular events and infectious diseases represent two primary causes of hospital readmissions throughout 5-year and 10-year periods after the index stroke (Rohweder et al., 2017; Sennfält et al., 2020). A detailed discussion of these underappreciated alterations in the chronic phase of stroke is beyond the focus of this review.
3. Clinical significance
Widespread disturbances of the autonomic nervous system, HPA axis, and immune system, all working collaboratively, provoke pathological alterations across peripheral organs, alone or in conjunction with pre-stroke comorbid conditions. Elucidation of the potential mechanisms enables the implementation of preventive and therapeutic options to counteract these systemic pathophysiological alterations.
Systemic complications have long been documented in the literature with substantial heterogeneity in the reported incidences, risk factors, and clinical prognosis. In contrast to early deaths within the first week that primarily arise from direct neurological sequelae of the initial stroke, deaths thereafter are mainly attributed to systemic complications, the proportion of which is higher in AIS patients than in patients with hemorrhagic stroke; among these, pneumonia and severe cardiac events are the two leading causes of death (Prosser et al., 2007; Silver et al., 1984). Generally, severe stroke and advanced age are two crucial risk factors that render stroke patients susceptible to systemic complications. The role of sex differences in the occurrence of certain complications, such as stroke-associated pneumonia (SAP) and Takotsubo cardiomyopathy, has been widely recognized, though the exact mechanisms have not been well clarified yet.
A range of biomarkers, such as heart rate variability, plasma glucocorticoids and catecholamines, and various immunological parameters, have been identified to be associated with specific profiles of complications after stroke. Although their additional predictive value over easily accessible clinical features, such as stroke severity, lesion site, and age, is rather marginal in clinical practice, prioritizing practical biomarker candidates helps to inform medical decision-making to personalize and improve stroke care. Validation of practical biomarker candidates proposed by experimental and clinical studies should be further prioritized.
3.1. Infections
Infections, mainly SAP and urinary tract infection (UTI), remain one of the most common and serious systemic complications following stroke (Ingeman et al., 2011). Despite improved care in stroke units, SAP is still a leading cause of death that peaks between the second and fourth weeks after AIS (Prosser et al., 2007). More than half of SAP cases arise within the first 3 days after stroke onset (Kishore et al., 2015). Owing to the substantial heterogeneity between studies, ranging from the study population to clinical settings and definitions of SAP, the reported incidence of SAP varied widely from 3.9% to 56.6%, with a higher incidence recorded in neurological intensive care units (Hannawi et al., 2013). Incident pneumonia affects approximately 15% of AIS patients (Kishore et al., 2015). The universally acknowledged risk factors for predicting SAP include stroke severity indicated by National Institutes of Health Stroke Scale (NIHSS) on admission, advanced age, dysphagia, atrial fibrillation, male sex, and admission hyperglycemia. SAP remarkably worsens stroke prognosis by causing a prolonged length of stay, poor functional outcomes, and higher short- and long-term mortality after AIS (Finlayson et al., 2011; Yu et al., 2016).
Together with the clues that dysphagia and aspiration alone are not sufficient to explain the high susceptibility to SAP (Meisel et al., 2005), immunosuppression has been strongly implicated in the pathogenesis of SAP by converging experimental and clinical evidence (Haeusler et al., 2008; Prass et al., 2003; Vogelgesang et al., 2008; Wong et al., 2011). In this line, a high neutrophil-to-lymphocyte ratio (Nam et al., 2018) and decreased monocytic MHC II expression (Hoffmann et al., 2017) have been suggested as a predictor of SAP in AIS patients, with the latter suggested to be a precondition for the independent association of dysphagia with SAP. Noteworthy, age-related decline in T cell function and the fact that females show greater capacity of antigen responses and a less prominent loss of naive T cells compared to males during ageing are likely to account for the increased susceptibility of males to SAP (Takahashi and Iwasaki, 2021). Locally in the pulmonary immune compartment, reduced phagocytic capability and MHC II expression in alveolar macrophages have been observed in stroke models (Hoyer et al., 2019; Samary et al., 2018). The precise role of vagal cholinergic antiinflammatory signaling that acts on the α7 nicotinergic acetylcholine receptor (α7nAChR) on alveolar macrophages and epithelial cells in pulmonary antibacterial defense remains elusive. Although this pathway was once suggested to facilitate the development of spontaneous pneumonia in stroke mice (Engel et al., 2015), depletion of the α7nAChR was insufficient to reverse pulmonary impaired antibacterial defense and failed to prevent aspiration-induced pneumonia after MCAO (Jagdmann et al., 2020).
Though multiple clinical observations described the correlation of poststroke infections with plasma/urinary levels of catecholamines or its metabolites, an independent association has not been determined with certainty (Chamorro et al., 2007; Harms et al., 2011; Klehmet et al., 2009; Liesz et al., 2013; Urra et al., 2009a, 2009b; Vogelgesang et al., 2010; Zierath et al., 2018). Heart rate variability indices have recently been identified as a predictor for poststroke infections by Hoyer and colleagues (Bramer et al., 2019; Günther et al., 2012). Clinical evidence concerning its relation with serum/urinary cortisol levels is similarly contradictory (Harms et al., 2011; Klehmet et al., 2009; Liesz et al., 2013; Urra et al., 2009a, 2009b; Vogelgesang et al., 2010).
Strikingly, culturable enteropathogenic bacteria have been identified in the lung of stroke animals (Crapser et al., 2016; Stanley et al., 2016; Tascilar et al., 2010; Wen et al., 2019), suggesting the gut microbiota as an alternative source of pathogens driving SAP. Although a similar finding was once reported in the sputum and blood of a small cohort of patients experiencing poststroke infections (Stanley et al., 2016), conclusive evidence from current clinical data is still limited to support the role of the gut microbiota in SAP, which is likely due to the low rate of positive culturable bacteria in the blood and sputum specimens of SAP patients. Moreover, it remains to be established what proportion of SAP is driven by bacterial translocation versus dysphagia and aspiration under immunosuppressive conditions. In conclusion, SAP is not simply a bacterial infectious disease, but a respiratory syndrome resulting from complex interactions among bacterial, chemical, and immunocompromised factors, which accounts for the failure of prophylactic antibiotics treatment to prevent pneumonia and improve functional outcomes in stroke patients (Kalra et al., 2015; Westendorp et al., 2015). Poststroke infections further exacerbate systemic and central inflammation triggered by stroke, thereby worsening stroke outcomes (Dénes et al., 2011; McColl et al., 2007). In this paradigm, elevated serum HMGB1 concentrations are reported to be further upregulated and underlie the augmented inflammation in animal models of poststroke infections, supporting the concept that sterile and nonsterile inflammation do not exist in isolation but strengthen each other after stroke (Kim et al., 2018).
With a comparable incidence to SAP, UTI occurs in 10–19% of stroke patients, mostly after 48 h of hospitalization (Smith et al., 2019; Westendorp et al., 2011). Older age, female sex, and urinary catheterization are frequently reported risk factors for UTI (Smith et al., 2019). Interestingly, few studies have identified stroke severity as an independent predictor of UTI. Unlike SAP, UTI has less impact on stroke outcomes and is suggested to be not independently associated with poststroke mortality (Westendorp et al., 2011). Indwelling catheters, a routine procedure to relieve retention of urine, is postulated to be the main explanation for poststroke UTI (Smith et al., 2019). Although immunosuppression is essential for SAP pathogenesis, no definitive evidence suggesting its specific relevance to UTI is available (Urra et al., 2017), and prophylactic antibiotics have been shown to mitigate the risk of UTI after AIS (Kalra et al., 2015; Vermeij et al., 2018). In summary, UTI is distinct from other types of poststroke infections, and thus should not be grouped with others in clinical research.
3.2. Cardiac complications
Analogous to the frequency and fatality of SAP, cardiac complications represent another leading cause of death after the first week of AIS (Prosser et al., 2007). Poststroke cardiac complications manifest as symptoms and electrocardiographic abnormalities indicating myocardial injury, and/or arrhythmias, which have been classified under the term “stroke-heart syndrome” (Scheitz et al., 2018). Considering the large burden of pre-existing cardiovascular disease in stroke patients, it is sometimes difficult to establish whether cardiac abnormalities coincide with or are the consequence of AIS. Though pre-existing structural and coronary heart disease may precipitate stroke-heart syndrome, cardiac complications can occur in the absence of overt cardiac disease (Scheitz et al., 2018). Nevertheless, attribution of cardiac alterations to stroke itself, must be approached with caution to avoid misdiagnosis and delayed treatment of primary life-threatening ischemic cardiac events.
During the first 3 months after stroke, approximately 20% of AIS patients present with serious cardiac adverse events, which mostly occur between 48 and 72 h after stroke and account for one-fifth of stroke-related mortality (Prosser et al., 2007; Silver et al., 1984). Risk factors include pre-existing congestive heart failure, diabetes mellitus, renal dysfunction, high-risk electrocardiographic changes, severe stroke, and stroke involving the right insular cortex (Prosser et al., 2007; Scheitz et al., 2018). Arrhythmia is detected in approximately 30% of stroke patients and atrial fibrillation is the most common type. The incidence of arrhythmia peaks within the first 24 h after admission and decreases markedly thereafter (Kallmunzer et al., 2012). Newly diagnosed atrial fibrillation is observed in one quarter of patients with AIS (Sposato et al., 2015), and was recently suggested to confer comparable risks of recurrent stroke and death within one year after stroke to known atrial fibrillation (Hsieh et al., 2018). Elevation of serum high-sensitivity cardiac troponin (cTn) levels is noted in a large proportion of AIS patients (Scheitz et al., 2015), particularly in those with infarcts involving the right insula (Ay et al., 2006), or specifically, its dorsal subregion (Krause et al., 2017). Notably, half of AIS patients with similar baseline cTn levels to patients with non-ST-elevation acute coronary syndrome have no angiographic evidence of coronary artery disease (Mochmann et al., 2016). Only dynamic changes in cTn signify acute myocardial damage resulting from coronary or noncoronary causes (e.g. neurogenic mechanisms), with the latter usually exhibiting modestly elevated cTn levels (Mochmann et al., 2016; Scheitz et al., 2015), but a cutoff to differentiate between these two conditions has not been established. Takotsubo cardiomyopathy, a reversible type of neurogenic myocardial injury without culprit stenosis of the involved coronary artery, is relatively uncommon but probably underappreciated in AIS patients; it shows an obvious female bias and mainly affects patients with moderate-to-severe stroke and right insular stroke. Given its rapid recovery, Takotsubo cardiomyopathy was previously considered a benign condition, which, however, was recently suggested to be correlated with increased in-hospital mortality and worse functional outcomes on discharge after stroke (Jung et al., 2016), and contribute to a proportion of major adverse cardiocerebrovascular events in the long term (Templin et al., 2015).
The significant association of poststroke autonomic imbalance with cardiovascular abnormalities has been appreciated for decades in extensive experimental and clinical research (Meyer et al., 2004; Nayani et al., 2016; Ozdemir and Hachinski, 2008; Sander et al., 2001; Tokgozoglu et al., 1999). Sympathovagal imbalance, interacting with concomitant cardiovascular disease if present, contributes to abnormalities in cardiac function and structure in the context of catecholamine surges from either myocardial sympathetic terminals or the circulation (Fig. 3). Arrhythmia, on the one hand, arises from neurogenic causes, that is, aberrant cardiac automaticity and conductivity driven by augmented myocardial sympathetic drive. Interestingly, recent experimental studies have revealed the contribution of cardiac resident macrophages (Hoyer et al., 2019) and left atrial myocardial inflammatory infiltration (Balint et al., 2019) to rhythm dysregulation after stroke. On the other hand, cardiogenic arrhythmia resulting from pre-existing cardiac comorbidities and electrolyte disturbances is not uncommon in stroke patients. It remains unclear whether neurogenic mechanisms or pre-existing cardiac disease are the major contributing factors to newly diagnosed atrial fibrillation (Hsieh et al., 2018).
Fig. 3.
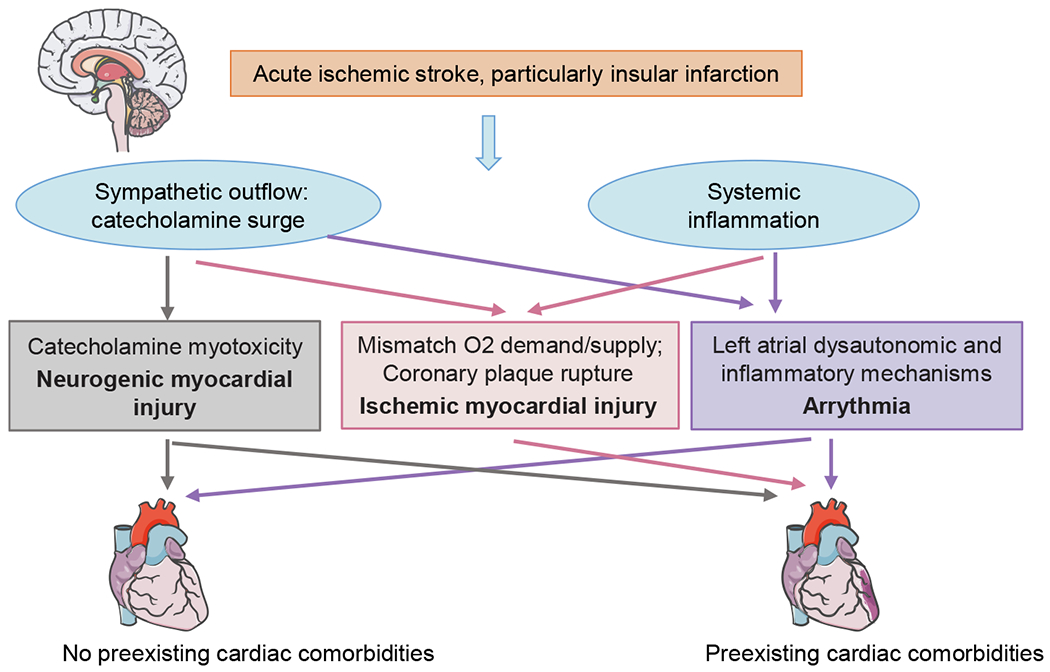
The mechanisms of cardiac abnormalities after acute ischemic stroke. In conjunction with systemic inflammation, cardiac autonomic imbalance, mostly reflected as sympathetic predominance, interacts with concomitant cardiovascular diseases (if any), and results in acute myocardial injury and arrhythmia, particularly after insular infarction.
Poststroke acute myocardial injury is classified as neurogenic and ischemic. The former is characterized by contraction band necrosis caused by massive catecholamines acting on myocytes; the latter, interacting with coexisting coronary atherostenosis, is provoked by either a mismatch between oxygen demand and oxygen delivery secondary to sympathetic hyperactivity and/or other comorbidities (such as arrhythmia and severe infections), or occasionally coronary plaque rupture (Anders et al., 2013; Scheitz et al., 2015). AIS renders atherosclerotic plaques unstable by triggering enhanced vascular inflammation and atherogenic monocyte recruitment via the synergistic effects of sympathetic deployment of monocytes and subsequent monocytic and endothelial activation by brain-released HMGB1 (Roth et al., 2018), which may reasonably account for the substantially increased risk of incident major adverse cardiovascular events 30 days after the first-ever AIS (Sposato et al., 2020). The persistent impact of poststroke sympathetic hyperactivation (Bieber et al., 2017), as well as enhanced macrophage trafficking and proinflammatory cytokine levels in the heart (Yan et al., 2018), has been suggested to contribute to chronic cardiac dysfunction in rodent models.
3.3. Acute lung injury
Acute lung injury (ALI) or acute respiratory distress syndrome, a more severe form of ALI, usually develops after severe brain injury, such as traumatic brain injury and subarachnoid hemorrhage. Indeed, both animal and human studies that document ALI after AIS are scarce. An animal model with large infarct volumes was previously utilized to construct neurogenic pulmonary edema in the setting of ischemic stroke (Toung et al., 2005). One retrospective study reported that 3.4% of stroke patients developed aspiration-related acute respiratory distress syndrome (Zhao et al., 2015). This condition was reported to be associated with high in-hospital mortality (Bai et al., 2017). Although less frequently observed in patients with large ischemic stroke, pulmonary edema manifests primarily in patients with poor cardiovascular reserve (Toung et al., 2005). Once direct myocardial injury occurs, the baseline deficient cardiopulmonary reserve could be further aggravated in a relevant proportion of AIS patients, thereby potentiating pulmonary edema. Supplemental oxygen is routinely recommended to correct hypoxemia and maintain oxygen saturation >94% in AIS patient (Powers et al., 2019).
The main mechanisms of ALI include neurogenic pulmonary edema, diffuse alveolar-capillary leakage, and enhanced pulmonary inflammation. Regardless of systemic hemodynamic changes, neurogenic pulmonary edema results from pulmonary vasoconstriction and endothelial damage, both of which have been suggested to be direct consequences of increased pulmonary adrenergic innervation from the injured medulla stimulated by a sudden elevation in intracranial pressure following severe brain injury, termed “pulmonary venule adrenergic hypersensitivity”. The direct adrenergic effects are proposed to be mediated by acting on α-ARs of pulmonary vascular beds, as evidenced by the finding that α-adrenergic blockade prevented the development of pulmonary edema in animals experiencing severe CNS injury (Winklewski et al., 2014). On the other hand, the local inflammatory milieu in the lung participates in alveolar-capillary injury. One recent study illustrated lung inflammation after experimental cerebral ischemia, although histologically evident ALI did not formally occur (Austin et al., 2019). Increased levels of proinflammatory mediators (such as IL-6, CXCL1, and TNF-α) and accumulation of macrophages in the pulmonary immune compartment have been documented in stroke animals (Austin et al., 2019; Chapman et al., 2009; Samary et al., 2018). The underlying mechanisms of lung inflammation following brain injury remain unknown. Robust evidence is lacking to underpin the hypothesis of increased β-adrenergic effects on alveolar macrophages (Winklewski et al., 2014). Additionally, SAP is expected to further aggravate lung inflammation. Overall, although ALI does not typically emerge after AIS, the lung is still vulnerable to subsequent insults, for example, infections and mechanical ventilation, in the context of lung inflammation and an impaired alveolar-capillary barrier.
3.4. Gastrointestinal complications
Gastrointestinal complications, including dysphagia, gut dysmotility (specifically, delayed gastric emptying, constipation, and fecal incontinence), and gastrointestinal bleeding, are present in up to 50% of AIS patient (Camara-Lemarroy et al., 2014). As two common symptoms in AIS patients, constipation and fecal incontinence are major contributors to reduced quality of life. Gastrointestinal bleeding occurs in 1.2–8.5% of AIS patients, mostly in the first week from stroke onset, and is more prevalent among Asians (Camara-Lemarroy et al., 2014; Chou et al., 2017; Fu, 2019; Ogata et al., 2014; Rumalla and Mittal, 2016). Risk factors include pre-existing peptic ulcer, malignancies, advanced age, stroke severity, and posterior circulation ischemia (Camara-Lemarroy et al., 2014; Ogata et al., 2014; Rumalla and Mittal, 2016). Gastrointestinal bleeding might result from a combination of multiple factors, including stress-induced gastric acid secretion, administration of antiplatelet drugs, gastric mucosal sympathetic vasoconstriction, and systemic inflammation (Camara-Lemarroy et al., 2014). The presence of gastrointestinal bleeding is associated with increased short- and longterm mortality and unfavorable outcomes after AIS (Chou et al., 2017; Donnell et al., 2008; Fu, 2019; Ogata et al., 2014; Rumalla and Mittal, 2016).
Convincing experimental studies have revealed that stroke leads to a functionally (Singh et al., 2016) or morphologically (Crapser et al., 2016; Liu et al., 2019; Stanley et al., 2016; Wen et al., 2019) defective intestinal barrier and gut dysbiosis (Houlden et al., 2016; Singh et al., 2016; Spychala et al., 2018; Stanley et al., 2016); the former occurs as early as 3–4.5 h after MCAO (Liu et al., 2019; Stanley et al., 2016; Wen et al., 2019). The extent of these abnormalities is suggested to be positively correlated with stroke severity (Crapser et al., 2016; Singh et al., 2016). Alterations in the intestinal bacterial composition have also been demonstrated in AIS patients (Li et al., 2019b; Yamashiro et al., 2017). These concealed gut abnormalities remain asymptomatic, underdiagnosed, and thus not treated, yet with significant impact on stroke progression and prognosis. Gut-derived metabolites, such as trimethylamine N-oxide (TMAO), may represent promising serological biomarkers for the early identification of these hidden gut abnormalities, since plasma TMAO levels are suggested to be higher in AIS patients compared to healthy controls and are associated with greater stroke severity (Wu et al., 2020).
The brain and gut have intimate interactions via complex neuroimmune-endocrine pathways and gut microbial metabolites, thus forming the bidirectional brain-gut axis (Cryan et al., 2020). The exact mechanisms underlying the “top-down” signaling through which AIS provokes gastrointestinal disturbances remain incompletely understood. Locally increased sympathetic outflow has been identified as one crucial signaling mediating poststroke gut permeability and dysbiosis (Houlden et al., 2016; Liu et al., 2019; Stanley et al., 2016). Under these pathological conditions, bacteria or bacterial metabolites translocate into the periphery (that is, the blood, liver, spleen, and, lung), which has been postulated to be an important cause of poststroke infections, particularly pneumonia (Crapser et al., 2016; Stanley et al., 2016; Tascilar et al., 2010; Wen et al., 2019). Indeed, the ratio of adrenergic to cholinergic neurons is significantly reduced due to a loss of cholinergic neurons in the submucosal plexus of the ileum, thereby causing a relative increase in sympathetic output. Administration of β-AR blockers was suggested to dampen stroke-induced gut leakage and the ensuing systemic dissemination of gut bacteria in stroke animals (Stanley et al., 2016).
Local intestinal immune homeostasis is tightly controlled by commensal microbiota. Recent experimental studies have substantially advanced our knowledge of the “bottom-up” communication by elucidating the role of the microbiota-gut-brain axis in poststroke systemic and cerebral inflammation. Driven by gut leakage and dysbiosis, TREM1+ monocytes/macrophages and proinflammatory T cell phenotypes (TH1, TH17, and IL-17+ γδT) are induced in the intestinal Peyer’s patches or the lamina propria, which penetrate the brain and exacerbate ischemic brain injury (Benakis et al., 2016; Liu et al., 2019; Singh et al., 2016). TREM1 upregulation further aggravates the gut permeability and subsequent bacterial translocation, thus forming a vicious positive-feedback loop (Liu et al., 2019). Moreover, systemic dissemination of gut-derived bacterial components and metabolites stimulates the immune system and aggravates systemic inflammation (Crapser et al., 2016). A dysbiotic gut microbiota was also reported to be associated with systemic inflammation in AIS patients, as indicated by increased leukocyte counts and high-sensitivity CRP levels (Yamashiro et al., 2017). Additionally, TMAO-associated expansion of circulating proinflammatory monocytes has been suggested to be a potential mechanism underlying the enhanced likelihood of recurrent cardiocerebrovascular events in AIS patients (Haghikia et al., 2018).
Ageing itself is a robust negative regulator of the gut microbiome, which, in combination with stroke-induced dysbiosis, significantly exacerbates systemic inflammation and ischemic brain damage (Spychala et al., 2018). Furthermore, poststroke intestinal permeability and gut-derived bacterial translocation are exacerbated in aged stroke mice, thereby leading to more severe infections (Crapser et al., 2016; Wen et al., 2019), which presumably accounts for a proportion of the robust association between advanced age and poststroke infections. One recent experimental study has first demonstrated a more pronounced increase in gut permeability and a more prolonged alteration in microbiota diversity in aged males compared to aged females despite their similar extent of ischemic brain injury (Ahnstedt et al., 2020). Promisingly, restoring gut homeostasis by youthful microbiota transplantation is suggested to improve behavioral and cognitive outcomes in aged stroke mice (Lee et al., 2020; Spychala et al., 2018). As all the aforementioned putative mechanisms to date originated from experimental studies, clinical data are warranted to evaluate the effects of gut dysbiosis on stroke outcome and test the efficacy of microbiota transplantation.
3.5. Acute kidney injury
Poststroke acute kidney injury (AKI) and pre-stroke CKD are intricately interrelated. CKD is a clear risk factor for AIS and unfavorably impacts stroke recovery. Furthermore, concomitant CKD is an important factor contributing to AKI after stroke, which in turn substantially exacerbates the progression of itself (Chelluboina and Vemuganti, 2019). The reported prevalence of poststroke AKI varied within a wide range with differences in diagnostic criteria and study populations (e.g. including patients with or without CKD). One recent meta-analysis has reported that AKI is prevalent in ~13% of AIS patients, which is lower than in patients with intracerebral hemorrhage (Zorrilla-Vaca et al., 2018). The most widely acknowledged risk factor for poststroke AKI is renal insufficiency on admission. Other related factors include advanced age, presence of cardiac comorbidities (ischemic heart disease and heart failure), stroke severity, and anemia (Arnold et al., 2018, 2019). AKI is a powerful independent predictor of stroke prognosis, including both short- and long-term mortality, and long-term incident cardiovascular events, the rates of which are positively correlated with AKI severity (Arnold et al., 2018, 2019). Intuitively, one might expect that underlying CKD mediates the negative impact of AKI on stroke outcomes because CKD itself is independently associated with increased mortality after stroke (Tsagalis et al., 2009). However, among AIS patients without coexisting CKD, AKI still correlates with higher rates of in-hospital mortality and disability at discharge (Saeed et al., 2014). Therefore, AKI may worsen stroke prognosis independent of its influence on the progression of concurrent CKD.
Considering that a fraction of stroke patients without CKD also develop AKI, it is reasonable to posit that stroke itself can trigger AKI (Saeed et al., 2014). Consequent to poststroke sympathetic hyperactivity, systemic and local hemodynamic variation diminishes the renal blood flow that supplies the glomerulus and tubule, thus causing a reduced glomerular filtration rate and acute tubular injury (Saeed et al., 2014). Although a relationship between contrast exposure and AKI was not reported (Khatri et al., 2014), further studies are needed to clarify whether contrast-related nephrotoxicity is involved in the development of AKI. In addition, increased levels of circulating inflammatory mediators after AIS may also contribute to AKI, which requires further investigation. AKI can further promote injuries to distant organs, particularly the brain, lung, heart, gut, and liver, likely mediated by uremic toxins, amplified systemic inflammatory responses, and oxidative stress (Husain-Syed et al., 2020).
3.6. Metabolic changes
Experimental cerebral ischemia is reported to trigger significant alterations in hepatic metabolic pathways, such as energy/glucose metabolism and amino acid metabolism (Wesley et al., 2019), as well as hepatic ketogenesis (Koch et al., 2017). Hyperadrenergic signaling provokes hepatic ketogenesis (Koch et al., 2017) and activation of hepatic proinflammatory pathways, with the latter playing critical roles in hepatic insulin resistance and upregulated expression of hepatic gluconeogenic enzymes, thereby contributing to poststroke hyperglycemia (Wang et al., 2014). Accordingly, β-AR antagonist administration mitigates poststroke hepatic ketogenesis in mice fed fat-rich diets (Koch et al., 2017), hepatic inflammatory responses, and subsequent glucose dysregulation (Lin et al., 2020; Wang et al., 2014).
Hyperglycemia occurs in 30–40% of AIS patients (Luitse et al., 2012), even in patients without a known history of diabetes mellitus, a large subgroup of whom ultimately exhibit impaired glucose metabolism or diabetes mellitus during follow-up (Kruyt et al., 2010). Regardless of the diabetes history, poststroke early-phase hyperglycemia is followed by a decrease in blood glucose levels within 24 h after stroke onset, but a further rise during 48–88 h (Kruyt et al., 2010). Persistent hyperglycemia during the first 24 h is associated with poststroke infection, unfavorable functional outcomes, and increased 30-/90-day mortality, particularly among nonlacunar patients and patients without a preceding diagnosis of diabetes mellitus (Fang et al., 2013; Mi et al., 2018; Yong and Kaste, 2008; Zonneveld et al., 2017). Clinical findings demonstrate that hyperglycemia is a key player in antagonizing the therapeutic benefits of thrombolysis treatment and precipitating hemorrhagic transformation. The plausible mechanistical explanations include impaired recanalization, increased ischemia/reperfusion injury, and BBB disruption under hyperglycemic conditions (Hafez et al., 2014; Kruyt et al., 2010). Hyperglycemia combined with leukocytosis on admission has been suggested to exhibit better predictive values for in-hospital mortality and pneumonia after AIS than hyperglycemia alone (You et al., 2019). Compared with the standard control treatment, however, intensive glycaemic control to maintain blood glucose levels between 80 and 130 mg/dL fails to improve 90-day functional outcomes (Johnston et al., 2019).
Both increased glucose production and insulin resistance underlie poststroke hyperglycemia. Increased levels of glucocorticoids and catecholamines stimulate glucose production through augmented glycogenolysis and gluconeogenesis. Meanwhile, insulin resistance arises from increased hepatic and adipose proinflammatory cytokine levels owing to massive sympathetic discharge (Wang et al., 2011, 2014). One clinical study also supported the role of sympathetic predominance in driving hyperglycemia given the observed independent association between hyperglycemia and decreased baroreflex sensitivity in non-diabetes AIS patients (Sykora et al., 2012). The beneficial effects of β-AR blockade on improving poststroke hyperglycemia have been delineated in animal models (Lin et al., 2020; Luger et al., 2018; Wang et al., 2011, 2014). Similar results were demonstrated in one retrospective study on AIS patients without a known diagnosis of diabetes mellitus and receiving β-AR blockers before stroke onset (Dziedzic et al., 2012).
Abnormal thyroid hormone metabolism is another common form of metabolic derangements after AIS, characterized by a reduction in serum triiodothyronine (T3) levels with normal thyroid-stimulating hormone concentrations, namely, non-thyroidal illness syndrome, affecting more than half of AIS patients according one small-scale epidemiological study (Zhang and Meyer, 2010). Mounting clinical studies have suggest a correlation of lower serum T3 levels with stroke severity and poor functional outcomes (Jiang et al., 2017; Lamba et al., 2018; Wang et al., 2017; Zhang and Meyer, 2010). Interestingly, this association was recently reported to be age-dependent and become nonsignificant in AIS patients below 65 years (Li et al., 2019a). Two separate studies reported an independent association with higher risk of hemorrhagic transformation in AIS patients receiving intravenous thrombolysis (Liu et al., 2016; Qiu et al., 2017). Moreover, lower serum T3 levels even within normal ranges have been reported to be independently associated with unfavorable outcomes in a relatively large cohort of AIS patients (Xu et al., 2016). Overall, the practical utility of serum T3 levels in predicting AIS prognosis merits further well-designed investigations.
The mechanism mediating non-thyroidal illness syndrome in clinically euthyroid AIS patients remains elusive. Early studies have recognized the role of IL-6 in its development (Abo-Zenah et al., 2008; Boelen et al., 1993; Yamazaki et al., 1996). Several lines of clinical studies have demonstrated that serum CRP levels is negatively associated with T3 concentrations after AIS (Bunevicius et al., 2014; Ma et al., 2016; Xu et al., 2016). Together, it is generally postulated that poststroke systemic immune activation and consequent release of proinflammatory mediators are the key elements in driving non-thyroidal illness syndrome via impairing peripheral triiodothyronine conversion and central inhibition of the hypothalamic-pituitary-thyroid axis (Bunevicius et al., 2014; Ma et al., 2016; Zhang and Meyer, 2010).
3.7. Venous thromboembolism
The incidence of venous thromboembolism, including deep vein thrombosis and pulmonary embolism, is 10 per 1000 person-years in AIS patients, which is 20-fold higher during the first month and 11-fold higher from 1 to 3 months after stroke than in the general population, as reported in one large prospective population-based cohort study (Rinde et al., 2016). Hemorrhagic stroke appears to confer a higher risk of venous thromboembolism than AIS (Ji et al., 2019; Stecker et al., 2014). Deep vein thrombosis develops rapidly after stroke onset, the incidence of which peaks within the first week (Ji et al., 2019). Although pulmonary embolism afflicts only 1% of AIS patients (Pongmoragot et al., 2013), clinicians need to stay alert to this life-threatening complication, which accounts for a fraction of in-hospital deaths that mostly manifest during the 2nd-4th weeks (Silver et al., 1984). Pulmonary embolism is recognized to be independently associated with higher rates of death at 30 days and 1 year, and disability at discharge in AIS patients. Risk factors for pulmonary embolism encompass age, stroke severity, history of cancer, prior venous thromboembolism disease, and incident deep vein thrombosis (Pongmoragot et al., 2013).
Regardless of the shared risk factors between venous thromboembolism and AIS, including advanced age, obesity, and atrial fibrillation, stroke-entailing conditions (such as immobilization and secondary infections) primarily contribute to the short-term development of venous thromboembolism triggered by stroke (Morelli et al., 2019; Rinde et al., 2016). Systemic inflammation may also contribute to the increased likelihood of venous thromboembolism after AIS, as its occurrence was suggested to be associated with increased serum CRP levels (Bembenek et al., 2011). Intermittent pneumatic compression is recommended to prevent deep vein thrombosis and the ensuing pulmonary embolism in immobile stroke patients, while the benefit of prophylactic anticoagulant therapy is still under rigorous debate (Powers et al., 2019).
4. Therapeutic implications and perspectives
Currently, reperfusion therapies remain the only effective evidence-based treatment for AIS. The universal implementation of recanalization therapies, however, is hampered by the narrow therapeutic window, strict criteria for patient selection, limited accessibility to sophisticated imaging and thrombectomy techniques, and the likelihood of futile recanalization and hemorrhagic transformation. Given its powerful neuroprotective benefits revealed in a vast body of preclinical literature, therapeutic hypothermia has emerged as an extremely attractive stroke therapy, the conclusive efficacy of which is being investigated with strong enthusiasm in current random clinical trials (Baron, 2018). Despite the elusive efficacy of several immunomodulatory drugs, such as fingolimod, minocycline, and natalizumab, immunomodulation has shown potential as adjunct therapeutic strategies with a wider interventional time frame for treating stroke (Iadecola et al., 2020). Given the crucial roles of endogenous brain-derived DAMPs and gut-derived PAMPs in poststroke systemic immune dysregulation, neutralization of these highly immunogenic molecules or blockade of their downstream receptors may represent a novel immunomodulatory strategy. Modulating the gut microbiota by fecal substitute transplant, even when initiated 3 days after MCAO, has been suggested to improve stroke recovery via alleviating peripheral and central inflammation, especially in aged rodents (Lee et al., 2020; Singh et al., 2016; Spychala et al., 2018). Notwithstanding this attractive line of research, more efforts are warranted to explore whether, and to what extent, therapeutic effects translate to clinical practice.
While extensive interventions are targeted to limit either primary or secondary brain injury, systemic complications that compromise stroke recovery must be prevented or corrected, which might be more relevant for the long-term morbidity of stroke patients. In view of the deleterious consequences of systemic pathophysiological alterations following stroke, tempering these disturbed stress pathways might ameliorate systemic complications. Current guidelines have not addressed whether and how poststroke autonomic disturbances should be managed. Extensive experimental studies have reported the benefits of pharmacological blockade of adrenergic signaling in alleviating a range of poststroke systemic abnormalities, including immunosuppression and the ensuing infections (Liu et al., 2017; McCulloch et al., 2017; Prass et al., 2006; Prass et al., 2003; Romer et al., 2015; Wang et al., 2015; Wong et al., 2011), gut permeability (Stanley et al., 2016), and hyperglycemia (Lin et al., 2020; Luger et al., 2018; Wang et al., 2011, 2014) (Table 1). Nonetheless, clinical evidence for administration of β-blockers, which is mainly based on cross-sectional studies, is rather inconclusive, with most focusing on neurological outcomes, mortality or poststroke infections as endpoint measures (Table 2). These conflicting data make it difficult to determine the safety and efficacy of the off-label administration of β-blockers in treating stroke.
Table 1.
Pharmacological interventions targeting neurohumoral stress pathways after experimental stroke.
| Animal model | Drugs | Results | Study |
|---|---|---|---|
| Mouse, 60 min tMCAO | Propranolol† | Decreased gut permeability and bacterial load in the lung, bronchoalveolar lavage fluid, liver and spleen at 24 h after MCAO | (Stanley et al., 2016) |
| Mouse, 60 min tMCAO | 6-OHDA¶; propranolol; RU486* | Either propranolol or RU486 reversed lymphopenia at 12 h after MCAO; yet only propranolol and 6-OHDA normalized T-cell IFN-γ production and prevented spontaneous lung infection | (Prass et al., 2003) |
| Mouse, 60 min tMCAO | Propranolol | Mitigation of pneumonia and bacteremia induced by S. pneumoniae aspirated exogenously | (Prass et al., 2006) |
| Mouse, 60 min tMCAO | Propranolol; RU486 | Reduced spleen atrophy, attenuated T- and NK-lymphopenia, normalized NK cell function, reduced infection induced by exogenous bacterial inoculation and improved functional outcomes mediated by the coordinated effects of propranolol and RU486 | (Liu et al., 2017) |
| Mouse, 40 min tMCAO | Propranolol | Preserved counts of splenic marginal zone B cells, increased plasma IgM concentrations and reduced lung infection, with insignificantly reduced infarct volumes | (McCulloch et al., 2017) |
| Mouse, 30 min tMCAO | 6-OHDA; propranolol | Restored hepatic iNKT cell function and reduced bacterial load in the lung, liver and spleen at 24 h after MCAO | (Wong et al., 2011) |
| Mouse, 45 min tMCAO | 6-OHDA; RU486; α-blocker phentolamine; propranolol; β2-blocker butoxamine; β3-blocker SR59230A | 6-OHDA and propranolol, but not RU486 and phentolamine, abrogated the expansion and mobilization of bone marrow Treg cells on days 1, 3, and 7 after stroke, which was specifically attributed to the blockade of β2- and β3-ARs, respectively, and blockade of β3-ARs lowered the bacterial load in the lung 7 d after MCAO | (Wang et al., 2015) |
| Mouse, 60 min tMCAO | Propranolol and RU486 | Reduced lung infection 3 d after MCAO, decreased infarct volume within 7 d after MCAO and increased 30-d survival | (Romer et al., 2015) |
| Rats, pMCAO | Propranolol | Reduced activation of hepatic proinflammatory pathways, attenuated hepatic insulin resistance, decreased expression of gluconeogenic enzymes and mitigated hyperglycemia at 24 h after MCAO | (Wang et al., 2014) |
| Rats, pMCAO | Propranolol | Suppressed secretion of proinflammatory cytokines in the adipose tissue and mitigation of impaired glucose tolerance at 24 h after MCAO | (Wang et al., 2011) |
| Mouse, 60 min tMCAO | Propranolol | Improved glucose tolerance at 24 h after MCAO | (Luger et al., 2018) |
| Mouse, 90 min tMCAO | Propranolol | Reduced hepatic ketogenesis in mice fed fat-rich diets at 24 h after MCAO | (Koch et al., 2017) |
tMCAO, transient middle cerebral artery occlusion; pMCAO, permanent middle cerebral artery occlusion; IgM, immunoglobulin M; iNKT, invariant natural killer T; Treg, regulatory T cells;
Propranolol is a non-selective β-adrenoreceptor blocker;
6-hydroxydopamine HBr (6-OHDA) ablates peripheral sympathetic innervation;
RU486 is a glucocorticoid receptor antagonist.
Table 2.
Clinical medication of β-blockers in ischemic stroke patients.
| Stroke subtype | Design | Sample size and group | Outcome measures | Findings | Study |
|---|---|---|---|---|---|
| AIS within 24 h of onset | Retrospective | 833, 10.6% on β-blockers during hospitalization | 30-day mortality | The inpatient use of β-blockers was associated with a reduced risk of early death from cardiovascular causes. | (Dziedzic et al., 2007) |
| Ischemic stroke within 14 days | Prospective | 111, 19.8% on β-blockers at stroke onset | Stroke severity on presentation, sympathovagal tone evaluated by HRV, ESR, and thrombin and HbA1C levels | The use of β-blockers at stroke onset was associated with less severe stroke, lower sympathetic tone, ESR, and thrombin and HbA1C levels. | (Laowattana and Oppenheimer, 2007) |
| AIS within 6 h of onset | Retrospective analysis of two previous large RCTs | 1375, 19.2% on β-blockers prior to stroke | Baseline stroke severity and 3-month functional outcome | No association with either outcome measure. | (Westendorp et al., 2016) |
| AIS within 24 h of onset and without pre-stroke diagnosis of diabetes mellitus | Retrospective | 603, 10.4% on β-blockers prior to stroke | Glucose level on admission and fasting glucose on day 1 after admission | The use of β-blockers before stroke was associated with lower plasma glucose on admission and 1 day thereafter. | (Dziedzic et al., 2012) |
| AIS | Retrospective | 2804, 15.5% receiving both pre-stroke and inpatient β-blockers | In in-hospital mortality | The continuation use of β-blockers both prior to stroke and during the first 3 days of hospitalization was associated with decreased in in-hospital mortality, while null association was found in patients receiving β-blockers only at home or only during hospitalization. | (Phelan et al., 2015) |
| First-time stroke, including 83.7% ischemic stroke, 11.8% ICH, and 4.5% SAH | Prospective | 100,043, 23.1% on β-blockers prior to stroke | 30-day mortality | Pre-stroke use of β-blockers was not associated with 30-day mortality in all stroke subtypes | (Sundbøll et al., 2015) |
| 88.3% ischemic stroke and 11.7% hemorrhagic stroke | Retrospective | 625, 48.2% receiving β-blockers† | Pneumonia, UTI, and death | β-blocker therapy was associated with a decrease in the risk of UTI, but a higher 30-day mortality, while no difference was found on the risk of pneumonia. | (Maier et al., 2015) |
| AIS | Retrospective | 5212, 22.2% receiving β-blockers before stroke onset and 4.7% newly on β-blockers within 3 days after stroke onset | Pneumonia, functional outcome and mortality at 3 months | Both pre-stroke and on-stroke β-blocker medication were associated with a reduced risk of pneumonia; the latter was also associated with reduced mortality; neither were associated with functional outcome. | (Sykora et al., 2015) |
| Acute ischemic (84%) and hemorrhagic stroke (11%) within 24 h after onset | Retrospective analysis of the PASS RCTs | 2538, 35% receiving β-blockers before stroke onset | Infections, including pneumonia, UTI, and other infections, functional outcome and mortality at 3 months | Pre-stroke use of β-blockers was associated with increased risks of infections and pneumonia, but not associated with 3-month mortality or functional outcome. | (Westendorp et al., 2016) |
| AIS | Prospective | 1431, 33.8% on β-blockers within the first 3 days of admission, 19.9% on selective β-blockers, while 18.4% on non-selective β-blockers | Infections, including pneumonia, UTI, and bacteremia, discharge functional outcome, and in-hospital death | No association was found between β-blocker therapy and in-hospital death or discharge functional outcome. Non-selective, but not selective β-blocker use, was associated with an increased risk of UTI. | (Starr et al., 2017) |
| Hypertensive patients with acute ischemic (90.9%) or hemorrhagic (9.1%) stroke | Retrospective | 1126, 54.6% on β-blockers before stroke onset | 3-month functional outcome and mortality | No association with either outcome measure. | (Eizenberg et al., 2018) |
| Severe AIS receiving endovasculartherapy¶ | Retrospective | 306, 51.6% receiving both pre-stroke and inpatient β-blockers | Pneumonia, UTI, sepsis, and mortality | No association with either outcome measure, although a non-significant trend of increased UTI frequency was observed in a subgroup of patients with insular/anterio-medial cortex infarcts | (Maier et al., 2018) |
AIS, acute ischemic stroke; HRV. heart rate variability; ESR, erythrocyte sedimentation rate; HbA1C, hemoglobin A1C; ICH, intracerebral hemorrhage; SAH, subarachnoid hemorrhage; RCTs, randomized clinical trials; UTI, urinary tract infection; PASS, the Preventive Antibiotics in Stroke Study.
β-blockers medication was defined as the continuous use both prior to stroke and during hospitalization;
AIS patients with advanced age (72 ± 13 years) and severe stroke at baseline (a median National Institutes of Health Stroke Score of 16).
Several putative reasons might account for this translation failure. Most experimental studies mimicked clinical AIS using transient MCAO models with extensive infarcts that trigger strong neuroendocrine responses; and the increases in either local sympathetic outflow or circulating stress mediators were verified, thus providing direct indications for corresponding drug administration. Another important translational gap is that experimental studies mostly leverage pretreatment strategy. The timing of intervention is a crucial determinant of the therapeutic efficacy given the rapid development of systemic abnormalities. Indeed, delayed administration of propranolol at 24 h after MCAO was suggested to fail to prevent immunosuppression (Prass et al., 2003). Another obvious but important issue to raise is that adrenergic signaling exerts such widespread biological effects that its blockade may culminate in unwelcome events, particularly in patients with potential contradictions. Considering the conflicting clinical data of pharmacological autonomic manipulation, it is of paramount importance to determine who is eligible for this medication and when it should be applied to maximize the benefits and minimize adverse side effects. Future sufficiently powered prospective studies with well-designed patient stratification are desperately needed to investigate the safety and efficacy of relevant pharmacological agents and lay the foundation for their potential application in clinical trials.
To date, specialized stroke unit care remains the most effective therapeutic modality for stroke. Considering the small proportion of patients eligible for reperfusion therapies, the overall benefit of stroke unit care is presumably suggested to arise from the intensive management of secondary complications and the early initiation of rehabilitation. To achieve optimal stroke recovery, future therapeutic regimens should coordinate multitarget pharmacological and non-pharmacological approaches and integrate the treatment of primary brain damage with interventions for secondary systemic complications.
5. Conclusions
While brain damage starts locally in the ischemic brain parenchyma, AIS provokes systemic disturbances across peripheral organs. Different spectra of systemic complications develop, depending on the size and location of cerebral infarcts, as well as patient demographics and preexisting comorbidities. Disturbances of the autonomic function, HPA axis, and systemic immunity are fundamental to the occurrence of systemic complications that are associated with poor clinical outcomes throughout the course of stroke. This review informs clinicians and researchers in the systemic manifestation of stroke, and emphasizes the notion that the damaged brain and secondary peripheral organ dysfunction should not be addressed in isolation in the management of stroke. Based on the context of patient characteristics, including age, sex, the initial ischemic events, pre-stroke comorbidities, and, if possible, measurements of serum catecholamine and glucocorticoid levels and autonomic function, therapeutic paradigms should holistically consider abnormalities occurring across peripheral organs, quickly recognize systemic complications, and ultimately tailor prompt treatment to optimize stroke outcomes.
Acknowledgements
We thank members of Hao clinical team and McCullough laboratory for debates about the systemic effects of stroke and the mechanisms and consequent complications. We also thank Prof. FDS (Tianjin Medical University General Hospital, Beijing Tiantan Hospital) and Prof. QL (Tianjin Medical University General Hospital) for providing suggestions on this manuscript.
Funding
This review was supported by the National Natural Science Foundation of China (81825008 and 82090043 to JH) and the National Institute of Health (R01NS103592 to LDM).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abo-Zenah HA, et al. , 2008. Relating circulating thyroid hormone concentrations to serum interleukins-6 and -10 in association with non-thyroidal illnesses including chronic renal insufficiency. BMC Endocr. Disord 8, 1. 10.1186/1472-6823-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnstedt H, McCullough LD, 2019. The impact of sex and age on T cell immunity and ischemic stroke outcomes. Cell. Immunol 345, 103960 10.1016/j.cellimm.2019.103960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnstedt H, et al. , 2020. Sex differences in T cell immune responses, gut permeability and outcome after ischemic stroke in aged mice. Brain Behav. Immun 87, 556–567. 10.1016/j.bbi.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders B, et al. , 2013. What does elevated high-sensitive troponin I in stroke patients mean: concomitant acute myocardial infarction or a marker for high-risk patients? Cerebrovasc. Dis 36, 211–217. 10.1159/000353875. [DOI] [PubMed] [Google Scholar]
- Arnold J, et al. , 2018. Incidence and impact on outcomes of acute kidney injury after a stroke: a systematic review and meta-analysis. BMC Nephrol. 19, 283. 10.1186/s12882-018-1085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J, et al. , 2019. Acute kidney injury calculated using admission serum creatinine underestimates 30-day and 1-year mortality after acute stroke. Clin. Kidney J 13, 46–54. 10.1093/ckj/sfz049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert HJ, et al. , 2004. Systemic inflammatory response depends on initial stroke severity but is attenuated by successful thrombolysis. Stroke 35, 2128–2133. 10.1161/01.Str.0000137607.61697.77. [DOI] [PubMed] [Google Scholar]
- Austin V, et al. , 2019. Ischaemic stroke in mice induces lung inflammation but not acute lung injury. Sci. Rep 9, 3622. 10.1038/s41598-019-40392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay H, et al. , 2006. Neuroanatomic correlates of stroke-related myocardial injury. Neurology 66, 1325–1329. 10.1212/01.wnl.0000206077.13705.6d. [DOI] [PubMed] [Google Scholar]
- Bahn MM, Oser AB, Cross DT 3rd, 1996. CT and MRI of stroke. J. Magn. Reson. Imaging 6, 833–845. 10.1002/jmri.1880060518. [DOI] [PubMed] [Google Scholar]
- Bai W, et al. , 2017. Blood glutamate levels are closely related to acute lung injury and prognosis after stroke. Front. Neurol 8, 755. 10.3389/fneur.2017.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balint B, et al. , 2019. Left atrial microvascular endothelial dysfunction, myocardial inflammation and fibrosis after selective insular cortex ischemic stroke. Int. J. Cardiol 292, 148–155. 10.1016/j.ijcard.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Baron JC, 2018. Protecting the ischaemic penumbra as an adjunct to thrombectomy for acute stroke. Nat. Rev. Neurol 14, 325–337. 10.1038/s41582-018-0002-2. [DOI] [PubMed] [Google Scholar]
- Barugh AJ, et al. , 2014. Cortisol levels and the severity and outcomes of acute stroke: a systematic review. J. Neurol 261, 533–545. 10.1007/s00415-013-7231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basic Kes V, et al. , 2008. Pro-inflammatory and anti-inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clin. Biochem 41, 1330–1334. 10.1016/j.clinbiochem.2008.08.080. [DOI] [PubMed] [Google Scholar]
- Bembenek J, et al. , 2011. Early stroke-related deep venous thrombosis: risk factors and influence on outcome. J. Thromb. Thrombolysis 32, 96–102. 10.1007/sl1239-010-0548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C, et al. , 2016. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med 22, 516–523. 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieber M, et al. , 2017. Stroke-induced chronic systolic dysfunction driven by sympathetic overactivity. Ann. Neurol 82, 729–743. 10.1002/ana.25073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelen A, Platvoet-Ter Schiphorst MC, Wiersinga WM, 1993. Association between serum interleukin-6 and serum 3,5,3’-triiodothyronine in nonthyroidal illness. J. Clin. Endocrinol. Metab 77, 1695–1699. 10.1210/jeem.77.6.8263160. [DOI] [PubMed] [Google Scholar]
- Bramer D, et al. , 2019. Very low frequency heart rate variability predicts the development of post-stroke infections. Transl. Stroke Res 10, 607–619. 10.1007/sl2975-018-0684-1. [DOI] [PubMed] [Google Scholar]
- Buck BH, et al. , 2008. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 39, 355–360. 10.1161/strokeaha.107.490128. [DOI] [PubMed] [Google Scholar]
- Bunevicius A, et al. , 2014. Ischemic stroke functional outcomes are independently associated with C-reactive protein concentrations and cognitive outcomes with triiodothyronine concentrations: a pilot study. Endocrine 45, 213–220. 10.1007/s12020-013-9958-2. [DOI] [PubMed] [Google Scholar]
- Cai W, et al. , 2020. Functional dynamics of neutrophils after ischemic stroke. Transl. Stroke Res 11, 108–121. 10.1007/sl2975-019-00694-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara-Lemarroy CR, Ibarra-Yruegas BE, Gongora-Rivera F, 2014. Gastrointestinal complications after ischemic stroke. J. Neurol. Sci 346, 20–25. 10.1016/j.jns.2014.08.027. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, et al. , 2003. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J.: Off. Publ. Federation Am. Soc. Exp. Biol 17, 1168–1170. 10.1096/fj.02-0757fje. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, et al. , 2005. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am. J. Pathol 166, 1487–1497. 10.1016/S0002-9440d0)62365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro A, et al. , 2007. Catecholamines, infection, and death in acute ischemic stroke. J. Neurol. Sci 252, 29–35. 10.1016/j.jns.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Chapman KZ, et al. , 2009. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J. Cereb. Blood Flow Metab 29, 1764–1768. 10.1038/jcbfin.2009.113. [DOI] [PubMed] [Google Scholar]
- Chelluboina B, Vemuganti R, 2019. Chronic kidney disease in the pathogenesis of acute ischemic stroke. J. Cereb. Blood Flow Metab 39, 1893–1905. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu NL, et al. , 2016. The volume of the spleen and its correlates after acute stroke. J. Stroke Cerebrovasc. Dis 25, 2958–2961. 10.1016/j.jstrokecerebrovasdis.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YF, Weng WC, Huang WY, 2017. Association between gastrointestinal bleeding and 3-year mortality in patients with acute, first-ever ischemic stroke. J. Clin. Neurosci 44, 289–293. 10.1016/j.jocn.2017.06.068. [DOI] [PubMed] [Google Scholar]
- Courties G, et al. , 2019. Glucocorticoids regulate bone marrow B lymphopoiesis after stroke. Circ. Res 124, 1372–1385. 10.1161/circresaha.118.314518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courties G, et al. , 2015. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res 116, 407–417. 10.1161/circresaha.116.305207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapser J, et al. , 2016. Ischemic stroke induces gut permeability and enhances bacterial translocation leading to sepsis in aged mice. Aging (Albany NY) 8, 1049–1063. 10.18632/aging.100952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, et al. , 2020. The gut microbiome in neurological disorders. Lancet Neurol. 19, 179–194. 10.1016/Sl474-4422(19)30356-4. [DOI] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Kovács KJ, 2011. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood- brain barrier damage and brain oedema independently of infarct size. J. Neuroinflammation 8, 164. 10.1186/1742-2094-8-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, et al. , 2011. Experimental stroke-induced changes in the bone marrow reveal complex regulation of leukocyte responses. J. Cereb. Blood Flow Metab 31, 1036–1050. 10.1038/jcbfm.2010.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnell MJ, et al. , 2008. Gastrointestinal bleeding after acute ischemic stroke. Neurology 71, 650. 10.1212/01.wnl.0000319689.48946.25. [DOI] [PubMed] [Google Scholar]
- Dziedzic T, et al. , 2012. Beta-blockers use and risk of hyperglycemia in acute stroke patients. Atherosclerosis 223, 209–211. 10.1016/j.atherosclerosis.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Dziedzic T, et al. , 2007. Beta-blockers reduce the risk of early death in ischemic stroke. J. Neurol. Sci 252, 53–56. 10.1016/j.jns.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Eizenberg Y, et al. , 2018. Pre admission treatment with Beta-blockers in hypertensive patients with acute stroke and 3-month outcome-Data from a national stroke registry. J. Clin. Hypertens. (Greenwich) 20, 568–572. 10.1111/jch.13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel O, et al. , 2015. Cholinergic pathway suppresses pulmonary innate immunity facilitating pneumonia after stroke. Stroke 46,3232–3240. 10.1161/strokeaha.115.008989. [DOI] [PubMed] [Google Scholar]
- Fang Y, et al. , 2013. Hyperglycaemia in acute lacunar stroke: a Chinese hospital-based study. Diab. Vase. Dis. Res 10, 216–221. 10.1177/1479164112459663. [DOI] [PubMed] [Google Scholar]
- Fassbender K, et al. , 1994. Pattern of activation of the hypothalamic-pituitary-adrenal axis in acute stroke. Relation to acute confusional state, extent of brain damage, and clinical outcome. Stroke 25, 1105–1108. 10.1161/01.str.25.6.1105. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Fabbri E, 2018. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol 15, 505–522. 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JN, et al. , 2005. Insular cortex infarction in acute middle cerebral artery territory stroke: predictor of stroke severity and vascular lesion. Arch. Neurol 62, 1081–1085. 10.1001/archneur.62.7.1081. [DOI] [PubMed] [Google Scholar]
- Finlayson O, et al. , 2011. Risk factors, inpatient care, and outcomes of pneumonia after ischemic stroke. Neurology 77, 1338–1345. 10.1212/WNL.0b013e31823152bl. [DOI] [PubMed] [Google Scholar]
- Fu J, 2019. Factors affecting the occurrence of gastrointestinal bleeding in acute ischemic stroke patients. Medicine (Baltimore) 98, e16312. 10.1097/md.0000000000016312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther A, et al. , 2012. Heart rate variability - a potential early marker of sub-acute post-stroke infections. Acta Neurol. Scand 126, 189–196. 10.1111/j.1600-0404.2011.01626.x. [DOI] [PubMed] [Google Scholar]
- Haeusler KG, et al. , 2008. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc. Dis 25, 50–58. 10.1159/000111499. [DOI] [PubMed] [Google Scholar]
- Hafez S, et al. , 2014. Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Transl Stroke Res. 5, 442–453. 10.1007/s12975-014-0336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghikia A, et al. , 2018. Gut microbiota-dependent trimethylamine N-oxide predicts risk of cardiovascular events in patients with stroke and is related to proinflammatory monocytes. Arterioscler. Thromb. Vase. Biol 38, 2225–2235. 10.1161/atvbaha.118.311023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannawi Y, et al. , 2013. Stroke-associated pneumonia: major advances and obstacles. Cerebrovasc. Dis 35, 430–443. 10.1159/000350199. [DOI] [PubMed] [Google Scholar]
- Harms H, et al. , 2011. Influence of stroke localization on autonomic activation, immunodepression, and post-stroke infection. Cerebrovasc. Dis 32, 552–560. 10.1159/000331922. [DOI] [PubMed] [Google Scholar]
- Hilz MJ, et al. , 2011. High NIHSS values predict impairment of cardiovascular autonomic control. Stroke 42, 1528–1533. 10.1161/strokeaha.110.607721. [DOI] [PubMed] [Google Scholar]
- Hoffmann S, et al. , 2017. Stroke-induced immunodepression and dysphagia independently predict stroke-associated pneumonia - the PREDICT study. J. Cereb. Blood Flow Metab 37, 3671–3682. 10.1177/0271678xl6671964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden A, et al. , 2016. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav. Immun 57, 10–20. 10.1016/j.bbi.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer FF, et al. , 2019. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity 51, 899–914.e897. 10.1016/j.immuni.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CY, et al. , 2018. Characteristics and outcomes of ischemic stroke in patients with known atrial fibrillation or atrial fibrillation diagnosed after stroke. Int. J. Cardiol 261, 68–72. 10.1016/j.ijcard.2017.ll.047. [DOI] [PubMed] [Google Scholar]
- Hug A, et al. , 2009. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke 40, 3226–3232. 10.1161/strokeaha.109.557967. [DOI] [PubMed] [Google Scholar]
- Husain-Syed F, Rosner MH, Ronco C, 2020. Distant organ dysfunction in acute kidney injury. Acta Physiol. (Oxf) 228, e13357. 10.1111/apha.13357. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Buckwalter MS, Anrather J, 2020. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J. Clin. Invest 130, 2777–2788. 10.1172/jcil35530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingeman A, et al. , 2011. In-hospital medical complications, length of stay, and mortality among stroke unit patients. Stroke 42, 3214–3218. 10.1161/strokeaha.110.610881. [DOI] [PubMed] [Google Scholar]
- Jagdmann S, et al. , 2020. Impact of key nicotinic AChR subunits on post-stroke pneumococcal pneumonia. Vaccines (Basel) 8. 10.3390/vaccines8020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R, et al. , 2019. Higher risk of deep vein thrombosis after hemorrhagic stroke than after acute ischemic stroke. J. Vase. Nurs 37, 18–27. 10.1016/j.jvn.2018.10.006. [DOI] [PubMed] [Google Scholar]
- Jiang X, et al. , 2017. Prognostic value of thyroid hormones in acute ischemic stroke - a meta analysis. Sci. Rep 7, 16256. 10.1038/s41598-017-16564-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson A, et al. , 2000. Cortisol axis abnormalities early after stroke–relationships to cytokines and leptin. J. Intern. Med 247, 179–187. 10.1046/j.1365-2796.2000.00600.x. [DOI] [PubMed] [Google Scholar]
- Johnson CO, et al. , 2019. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 439–458. 10.1016/s1474-4422(19)30034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston KC, et al. , 2019. Intensive vs standard treatment of hyperglycemia and functional outcome in patients with acute ischemic stroke: the SHINE randomized clinical trial. JAMA 322, 326–335. 10.1001/jama.2019.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JM, et al. , 2016. Takotsubo-like myocardial dysfunction in ischemic stroke: a hospital-based registry and systematic literature review. Stroke 47, 2729–2736. 10.1161/strokeaha.116.014304. [DOI] [PubMed] [Google Scholar]
- Kallmunzer B, et al. , 2012. Serious cardiac arrhythmias after stroke: incidence, time course, and predictors–a systematic, prospective analysis. Stroke 43, 2892–2897. 10.1161/strokeaha.112.664318. [DOI] [PubMed] [Google Scholar]
- Kalra L, et al. , 2015. Prophylactic antibiotics after acute stroke for reducing pneumonia in patients with dysphagia (STROKE-INF): a prospective, cluster-randomised, open-label, masked endpoint, controlled clinical trial. Lancet 386, 1835–1844. 10.1016/s0140-6736(15)00126-9. [DOI] [PubMed] [Google Scholar]
- Khatri M, et al. , 2014. Acute kidney injury is associated with increased hospital mortality after stroke. J. Stroke Cerebrovasc. Dis 23, 25–30. 10.1016/j.jstrokecerebrovasdis.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, et al. , 2014. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J. Cereb. Blood Flow Metab 34, 1411–1419. 10.1038/jcbfm.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ID, et al. , 2018. Alarmin HMGB1 induces systemic and brain inflammatory exacerbation in post-stroke infection rat model. Cell Death Dis. 9, 426. 10.1038/s41419-018-0438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore AK, et al. , 2015. How is pneumonia diagnosed in clinical stroke research? A systematic review and meta-analysis. Stroke 46, 1202–1209. 10.1161/strokeaha.114.007843. [DOI] [PubMed] [Google Scholar]
- Klehmet J, et al. , 2009. Stroke-induced immunodepression and post-stroke infections: lessons from the preventive antibacterial therapy in stroke trial. Neuroscience 158, 1184–1193. 10.1016/j.neuroscience.2008.07.044. [DOI] [PubMed] [Google Scholar]
- Koch K, et al. , 2017. Hepatic ketogenesis induced by middle cerebral artery occlusion in mice. J. Am. Heart Assoc 6 10.1161/jaha.117.005556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpelainen JT, et al. , 1996. Abnormal heart rate variability as a manifestation of autonomic dysfunction in hemispheric brain infarction. Stroke 27, 2059–2063. 10.1161/01.str.27.ll.2059. [DOI] [PubMed] [Google Scholar]
- Krause T, et al. , 2017. Stroke in right dorsal anterior insular cortex Is related to myocardial injury. Ann. Neurol 81, 502–511. 10.1002/ana.24906. [DOI] [PubMed] [Google Scholar]
- Kruyt ND, et al. , 2010. Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat. Rev. Neurol 6, 145–155. 10.1038/nrneurol.2009.231. [DOI] [PubMed] [Google Scholar]
- Kumar S, Selim MH, Caplan LR, 2010. Medical complications after stroke. Lancet Neurol. 9, 105–118. 10.1016/s1474-4422(09)70266-2. [DOI] [PubMed] [Google Scholar]
- Lamba N, et al. , 2018. A prognostic role for Low tri-iodothyronine syndrome in acute stroke patients: a systematic review and meta-analysis. Clin. Neurol. Neurosurg 169, 55–63. 10.1016/j.dineuro.2018.03.025. [DOI] [PubMed] [Google Scholar]
- Laowattana S, Oppenheimer SM, 2007. Protective effects of beta-blockers in cerebrovascular disease. Neurology 68, 509–514. 10.1212/01.wnl.0000253186.23949.fd. [DOI] [PubMed] [Google Scholar]
- Lee J, et al. , 2020. Gut microbiota-derived short-chain fatty acids promote poststroke recovery in aged mice. Circ. Res 127, 453–465. 10.1161/circresaha.119.316448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LQ, et al. , 2019a. The prognostic value of total T3 after acute cerebral infarction is age-dependent: a retrospective study on 768 patients. BMC Neurol. 19, 54. 10.1186/s12883-019-1264-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, et al. , 2019b. Change of intestinal microbiota in cerebral ischemic stroke patients. BMC Microbiol. 19, 191. 10.1186/s12866-019-1552-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, et al. , 2015. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci 35, 583. 10.1523/JNEUROSCI.2439-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, et al. , 2009. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke 40, 2849–2858. 10.1161/strokeaha.109.549618. [DOI] [PubMed] [Google Scholar]
- Liesz A, et al. , 2013. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS ONE 8, e74839. 10.1371/journal.pone.0074839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, et al. , 2020. Effects of β-adrenergic blockade on metabolic and inflammatory responses in a rat model of ischemic stroke. Cells 9. 10.3390/cells9061373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, et al. , 2016. Low free triiodothyronine levels are related to symptomatic intracranial hemorrhage and poor functional outcomes after intravenous thrombolysis in acute ischemic stroke patients. Neurol. Res 38, 429–433. 10.1080/01616412.2016.1178480. [DOI] [PubMed] [Google Scholar]
- Liu Q, et al. , 2017. Brain ischemia suppresses immunity in the periphery and brain via different neurogenic innervations. Immunity 46, 474–487. 10.1016/j.immuni.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Liu Q, et al. , 2019. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat. Immunol 20, 1023–1034. 10.1038/s41590-019-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger S, et al. , 2018. Beta adrenoceptor blockade ameliorates impaired glucose tolerance and alterations of the cerebral ceramide metabolism in an experimental model of ischemic stroke, 1756286418769830 Ther. Adv. Neurol. Disord 11. 10.1177/1756286418769830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitse MJ, et al. , 2012. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 11, 261–271. 10.1016/s1474-4422(12)70005-4. [DOI] [PubMed] [Google Scholar]
- Ma L, et al. , 2016. Low triiodothyronine: a new facet of inflammation in acute ischemic stroke. Clin. Chim. Acta 458, 63–67. 10.1016/j.cca.2016.04.023. [DOI] [PubMed] [Google Scholar]
- Maier IL, et al. , 2018. Influence of beta-blocker therapy on the risk of infections and death in patients at high risk for stroke induced immunodepression. PLoS ONE 13, e0196174. 10.1371/journal.pone.0196174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier IL, et al. , 2015. Effect of beta-blocker therapy on the risk of infections and death after acute stroke–a historical cohort study. PLoS ONE 10, e0116836. 10.1371/journal.pone.0116836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwani B, et al. , 2013. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp. Neurol 249, 120–131. 10.1016/j.expneurol.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM, 2007. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J. Neurosci 27, 4403–4412. 10.1523/jneurosci.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch L, Smith CJ, McColl BW, 2017. Adrenergic-mediated loss of splenic marginal zone B cells contributes to infection susceptibility after stroke. Nat. Commun 8, 15051. 10.1038/ncommsl5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel C, et al. , 2005. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci 6, 775–786. 10.1038/nrnl765. [DOI] [PubMed] [Google Scholar]
- Meyer S, et al. , 2004. Lateralization in autonomic dysfunction in ischemic stroke involving the insular cortex. NeuroReport 15, 357–361. 10.1097/00001756-200402090-00029. [DOI] [PubMed] [Google Scholar]
- Mi D, et al. , 2018. Correlation of hyperglycemia with mortality after acute ischemic stroke, 1756285617731686 Ther. Adv. Neurol. Disord 11. 10.1177/1756285617731686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo J, et al. , 2019. Autonomic disturbances in acute cerebrovascular disease. Neurosci. Bull 35, 133–144. 10.1007/s12264-018-0299-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochmann HC, et al. , 2016. Coronary angiographic findings in acute ischemic stroke patients with elevated cardiac troponin: the troponin elevation in acute ischemic stroke (TRELAS) study. Circulation 133, 1264–1271. 10.1161/circulationaha.l15.018547. [DOI] [PubMed] [Google Scholar]
- Morelli VM, et al. , 2019. The role of stroke as a trigger for incident venous thromboembolism: results from a population-based case-crossover study. TH Open 3, e50–e57. 10.1055/s-0039-1681020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mracsko E, et al. , 2014. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav. Immun 41, 200–209. 10.1016/j.bbi.2014.05.015. [DOI] [PubMed] [Google Scholar]
- Nam KW, et al. , 2018. High neutrophil-to-lymphocyte ratio predicts stroke-associated pneumonia. Stroke 49, 1886–1892. 10.1161/strokeaha.118.021228. [DOI] [PubMed] [Google Scholar]
- Nayani S, et al. , 2016. Autonomic dysfunction in first ever ischemic stroke: prevalence, predictors and short term neurovascular outcome. Clin. Neurol. Neurosurg 150, 54–58. 10.1016/j.clineuro.2016.08.022. [DOI] [PubMed] [Google Scholar]
- Nicholls AJ, et al. , 2018. Activation of the sympathetic nervous system modulates neutrophil function. J. Leukoc. Biol 103, 295–309. 10.1002/jlb.3ma0517-194rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, et al. , 2006a. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab 26, 654–665. 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H, et al. , 2006b. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J. Immunol 176, 6523–6531. 10.4049/jimmunol.176.ll.6523. [DOI] [PubMed] [Google Scholar]
- Ogata T, et al. , 2014. Gastrointestinal bleeding in acute ischemic stroke: recent trends from the fukuoka stroke registry. Cerebrovasc. Dis. Extra 4, 156–164. 10.1159/000365245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir O, Hachinski V, 2008. Brain lateralization and sudden death: its role in the neurogenic heart syndrome. J. Neurol. Sci 268, 6–11. 10.1016/j.jns.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Phelan C, et al. , 2015. Effect of β-adrenergic antagonists on in-hospital mortality after ischemic stroke. J Stroke Cerebrovasc Dis 24, 1998–2004. 10.1016/j.jstrokecerebrovasdis.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongmoragot J, et al. , 2013. Pulmonary embolism in ischemic stroke: clinical presentation, risk factors, and outcome. J. Am. Heart Assoc 2, e000372 10.1161/jaha.113.000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers WJ, et al. , 2019. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 50, e344–e418. 10.1161/str.0000000000000211. [DOI] [PubMed] [Google Scholar]
- Prass K, et al. , 2006. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke 37, 2607–2612. 10.1161/01.STR.0000240409.68739.2b. [DOI] [PubMed] [Google Scholar]
- Prass K, et al. , 2003. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J. Exp. Med 198, 725–736. 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser J, et al. , 2007. Predictors of early cardiac morbidity and mortality after ischemic stroke. Stroke 38, 2295–2302. 10.1161/strokeaha.106.471813. [DOI] [PubMed] [Google Scholar]
- Qiu M, Fang M, Liu X, 2017. Low free triiodothyronine levels predict symptomatic intracranial hemorrhage and worse short-term outcome of thrombolysis in patients with acute ischemia stroke. Medicine (Baltimore) 96, e8539. 10.1097/md.0000000000008539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinde LB, et al. , 2016. Ischemic stroke and risk of venous thromboembolism in the general population: the tromso study. J. Am. Heart Assoc 5 10.1161/jaha.116.004311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, et al. , 2016. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J. Immunol 196, 3318–3330. 10.4049/jimmunol.1502021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, et al. , 2018. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 136, 89–110. 10.1007/s00401-018-1859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohweder G, et al. , 2017. Hospital readmission within 10 years post stroke: frequency, type and timing. BMC Neurol. 17, 116. 10.1186/s12883-017-0897-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer C, et al. , 2015. Blocking stroke-induced immunodeficiency increases CNS antigen-specific autoreactivity but does not worsen functional outcome after experimental stroke. J. Neurosci 35, 7777–7794. 10.1523/jneurosci.1532-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, et al. , 2018. Brain-released alarmins and stress response synergize in accelerating atherosclerosis progression after stroke. Sci. Transl. Med 10 10.1126/scitranslmed.aao1313. [DOI] [PubMed] [Google Scholar]
- Roy-O’Reilly MA, et al. , 2020. Aging exacerbates neutrophil pathogenicity in ischemic stroke. Aging (Albany NY) 12, 436–461. 10.18632/aging.102632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhnau J, et al. , 2014. Stroke alters respiratory burst in neutrophils and monocytes. Stroke 45, 794–800. 10.1161/strokeaha.113.003342. [DOI] [PubMed] [Google Scholar]
- Rumalla K, Mittal MK, 2016. Gastrointestinal bleeding in acute ischemic stroke: a population-based analysis of hospitalizations in the United States. J. Stroke Cerebrovasc. Dis 25, 1728–1735. 10.1016/j.jstrokecerebrovasdis.2016.03.044. [DOI] [PubMed] [Google Scholar]
- Saeed F, et al. , 2014. Acute renal failure is associated with higher death and disability in patients with acute ischemic stroke: analysis of nationwide inpatient sample. Stroke 45, 1478–1480. 10.1161/strokeaha.114.004672. [DOI] [PubMed] [Google Scholar]
- Sahota P, et al. , 2013. Changes in spleen size in patients with acute ischemic stroke: a pilot observational study. Int. J. Stroke 8, 60–67. 10.1111/ijs.12022. [DOI] [PubMed] [Google Scholar]
- Samary CS, et al. , 2018. Focal ischemic stroke leads to lung injury and reduces alveolar macrophage phagocytic capability in rats. Crit. Care 22, 249. 10.1186/s13054-018-2164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander D, et al. , 2001. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology 57, 833–838. 10.1212/wnl.57.5.833. [DOI] [PubMed] [Google Scholar]
- Scheitz JF, et al. , 2018. Stroke-heart syndrome: clinical presentation and underlying mechanisms. Lancet Neurol. 17, 1109–1120. 10.1016/s1474-4422(18)30336-3. [DOI] [PubMed] [Google Scholar]
- Scheitz JF, et al. , 2015. Application and interpretation of high-sensitivity cardiac troponin assays in patients with acute ischemic stroke. Stroke 46, 1132–1140. 10.1161/strokeaha.114.007858. [DOI] [PubMed] [Google Scholar]
- Schulze J, et al. , 2013. Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke 44, 246–248. 10.1161/strokeaha.112.676072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert HA, et al. , 2012. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J. Neuroimmune Pharmacol 7, 1017–1024. 10.1007/s11481-012-9406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennfält S, et al. , 2020. Patterns in hospital readmissions after ischaemic stroke - an observational study from the Swedish stroke register (Riksstroke). Eur. Stroke J 5, 286–296. 10.1177/2396987320925205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K, et al. , 2019. Global brain inflammation in stroke. Lancet Neurol. 18, 1058–1066. 10.1016/S1474-4422(19)30078-X. [DOI] [PubMed] [Google Scholar]
- Silver FL, et al. , 1984. Early mortality following stroke: a prospective review. Stroke 15, 492–496. 10.1161/01.str.15.3.492. [DOI] [PubMed] [Google Scholar]
- Singh V, et al. , 2016. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J. Neurosci 36, 7428. 10.1523/JNEUROSCI.1114-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Almallouhi E, Feng W, 2019. Urinary tract infection after stroke: a narrative review. J. Neurol. Sci 403, 146–152. 10.1016/j.jns.2019.06.005. [DOI] [PubMed] [Google Scholar]
- Smith KE, et al. , 1986. Changes in plasma catecholamine levels after insula damage in experimental stroke. Brain Res. 375, 182–185. 10.1016/0006-8993(86)90973-x. [DOI] [PubMed] [Google Scholar]
- Sposato LA, et al. , 2015. Diagnosis of atrial fibrillation after stroke and transient ischaemic attack: a systematic review and meta-analysis. Lancet Neurol. 14, 377–387. 10.1016/s1474-4422(15)70027-x. [DOI] [PubMed] [Google Scholar]
- Sposato LA, et al. , 2020. First-ever ischemic stroke and incident major adverse cardiovascular events in 93 627 older women and men. Stroke 51, 387–394. 10.1161/strokeaha.119.028066. [DOI] [PubMed] [Google Scholar]
- Spychala MS, et al. , 2018. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann. Neurol 84, 23–36. 10.1002/ana.25250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley D, et al. , 2016. Translocation and dissemination of commensal bacteria in poststroke infection. Nat. Med 22, 1277–1284. 10.1038/nm.4194. [DOI] [PubMed] [Google Scholar]
- Starr JB, Tirschwell DL, Becker KJ, 2017. Increased infections with β-blocker use in ischemic stroke, a β(2)-receptor mediated process? Neurol. Sci 38, 967–974. 10.1007/s10072-017-2877-x. [DOI] [PubMed] [Google Scholar]
- Stecker M, et al. , 2014. Risk factors for DVT/PE in patients with stroke and intracranial hemorrhage. Open Neurol. J 8, 1–6. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundbøll J, et al. , 2015. Impact of preadmission treatment with calcium channel blockers or beta blockers on short-term mortality after stroke: a nationwide cohort study. BMC Neurol. 15, 24. 10.1186/s12883-015-0279-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora M, et al. , 2012. Association of non-diabetic hyperglycemia with autonomic shift in acute ischaemic stroke. Eur. J. Neurol 19, 84–90. 10.1111/j.1468-1331.2011.03438.x. [DOI] [PubMed] [Google Scholar]
- Sykora M, et al. , 2009. Impaired baroreceptor reflex sensitivity in acute stroke is associated with insular involvement, but not with carotid atherosclerosis. Stroke 40, 737–742. 10.1161/strokeaha.108.519967. [DOI] [PubMed] [Google Scholar]
- Sykora M, Siarnik P, Diedler J, 2015. β-blockers, pneumonia, and outcome after ischemic stroke: evidence from virtual international stroke trials archive. Stroke 46, 1269–1274. 10.1161/strokeaha.114.008260. [DOI] [PubMed] [Google Scholar]
- Szigeti K, et al. , 2015. A novel SPECT-based approach reveals early mechanisms of central and peripheral inflammation after cerebral ischemia. J. Cereb. Blood Flow Metab 35, 1921–1929. 10.1038/jcbfm.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Iwasaki A, 2021. Sex differences in immune responses. Science 371, 347. 10.1126/science.abe7199. [DOI] [PubMed] [Google Scholar]
- Tascilar N, et al. , 2010. Bacterial translocation in experimental stroke: what happens to the gut barrier? Bratisl. Lek. Listy 111, 194–199. [PubMed] [Google Scholar]
- Templin C, et al. , 2015. Clinical features and outcomes of takotsubo (stress) cardiomyopathy. N. Engl. J. Med 373, 929–938. 10.1056/NEJMoa1406761. [DOI] [PubMed] [Google Scholar]
- Tokgozoglu SL, et al. , 1999. Effects of stroke localization on cardiac autonomic balance and sudden death. Stroke 30, 1307–1311. 10.1161/01.str.30.7.1307. [DOI] [PubMed] [Google Scholar]
- Toung TJ, et al. , 2005. Increases in lung and brain water following experimental stroke: effect of mannitol and hypertonic saline, discussion 259-260 Crit. Care Med. 33, 203–208. 10.1097/01.ccm.0000150659.15558.23. [DOI] [PubMed] [Google Scholar]
- Tsagalis G, et al. , 2009. Renal dysfunction in acute stroke: an independent predictor of long-term all combined vascular events and overall mortality. Nephrol. Dial. Transplant 24, 194–200. 10.1093/ndt/gfn471. [DOI] [PubMed] [Google Scholar]
- Urra X, et al. , 2009a. Monocytes are major players in the prognosis and risk of infection after acute stroke. Stroke 40, 1262–1268. 10.1161/strokeaha.108.532085. [DOI] [PubMed] [Google Scholar]
- Urra X, et al. , 2009b. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 158, 1174–1183. 10.1016/j.neuroscience.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Urra X, et al. , 2017. Neuroanatomical correlates of stroke-associated infection and stroke-induced immunodepression. Brain Behav. Immun 60, 142–150. 10.1016/j.bbi.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Vahidy FS, et al. , 2016. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J. Cereb. Blood Flow Metab 36, 1012–1021. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij JD, et al. , 2018. Antibiotic therapy for preventing infections in people with acute stroke. Cochrane Database Syst. Rev 1, Cd008530. 10.1002/14651858.CD008530.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelgesang A, et al. , 2008. Analysis of lymphocyte subsets in patients with stroke and their influence on infection after stroke. Stroke 39, 237–241. 10.1161/strokeaha.107.493635. [DOI] [PubMed] [Google Scholar]
- Vogelgesang A, et al. , 2010. Functional status of peripheral blood T-cells in ischemic stroke patients. PLoS ONE 5, e8718. 10.1371/journal.pone.0008718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter U, et al. , 2013. Insular stroke is associated with acute sympathetic hyperactivation and immunodepression. Eur. J. Neurol 20, 153–159. 10.1111/j.1468-1331.2012.03818.x. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. , 2015. Cerebral ischemia increases bone marrow CD4+CD25+FoxP3+ regulatory T cells in mice via signals from sympathetic nervous system. Brain Behav. Immun 43, 172–183. 10.1016/j.bbi.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 2017. Low T(3) levels as a predictor marker predict the prognosis of patients with acute ischemic stroke. Int. J. Neurosci 127, 559–566. 10.1080/00207454.2016.1211649. [DOI] [PubMed] [Google Scholar]
- Wang YY, et al. , 2011. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am. J. Physiol. Endocrinol. Metab 300, E155–E163. 10.1152/ajpendo.00301.2010. [DOI] [PubMed] [Google Scholar]
- Wang YY, et al. , 2014. Activation of hepatic inflammatory pathways by catecholamines is associated with hepatic insulin resistance in male ischemic stroke rats. Endocrinology 155, 1235–1246. 10.1210/en.2013-1593. [DOI] [PubMed] [Google Scholar]
- Weisenburger-Lile D, et al. , 2019. Harmful neutrophil subsets in patients with ischemic stroke: association with disease severity. Neurol. Neuroimmunol. Neuroinflamm 6, e571 10.1212/nxi.0000000000000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen SW, et al. , 2019. Advanced age promotes colonic dysfunction and gut-derived lung infection after stroke. Aging Cell 18, e12980. 10.1111/acel.12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley UV, et al. , 2019. Local and systemic metabolic alterations in brain, plasma, and liver of rats in response to aging and ischemic stroke, as detected by nuclear magnetic resonance (NMR) spectroscopy. Neurochem. Int 127, 113–124. 10.1016/j.neuint.2019.01.025. [DOI] [PubMed] [Google Scholar]
- Westendorp WF, et al. , 2011. Post-stroke infection: a systematic review and metaanalysis. BMC Neurol. 11, 110. 10.1186/1471-2377-ll110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp WF, et al. , 2016. Pre-stroke use of beta-blockers does not lower post-stroke infection rate: an exploratory analysis of the preventive antibiotics in stroke study. Cerebrovasc. Dis 42, 506–511. 10.1159/000450926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp WF, et al. , 2015. The Preventive Antibiotics in Stroke Study (PASS): a pragmatic randomised open-label masked endpoint clinical trial. Lancet 385, 1519–1526. 10.1016/s0140-6736(14)62456-9. [DOI] [PubMed] [Google Scholar]
- Whiteley W, et al. , 2012. The use of blood biomarkers to predict poor outcome after acute transient ischemic attack or ischemic stroke. Stroke 43, 86–91. 10.1161/strokeaha.Ill.634089. [DOI] [PubMed] [Google Scholar]
- Winklewski PJ, Radkowski M, Demkow U, 2014. Cross-talk between the inflammatory response, sympathetic activation and pulmonary infection in the ischemic stroke. J. Neuroinflammat 11, 213. 10.1186/s12974-014-0213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CH, et al. , 2011. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science 334, 101–105. 10.1126/science.1210301. [DOI] [PubMed] [Google Scholar]
- Wong CH, et al. , 2017. Prolonged activation of invariant natural killer T cells and TH2-skewed immunity in stroke patients. Front. Neurol 8, 6. 10.3389/neur.2017.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, et al. , 2020. Relationship between elevated plasma trimethylamine N-oxide levels and increased stroke injury. Neurology 94, e667–e677. 10.1212/wnl.0000000000008862. [DOI] [PubMed] [Google Scholar]
- Xiong L, et al. , 2014. Autonomic dysfunction in different subtypes of post-acute ischemic stroke. J. Neurol. Sci 337, 141–146. 10.1016/j.jns.2013.11.036. [DOI] [PubMed] [Google Scholar]
- Xiong L, et al. , 2018. Autonomic dysfunction predicts clinical outcomes after acute ischemic stroke: a prospective observational study. Stroke 49, 215–218. 10.1161/strokeaha.117.019312. [DOI] [PubMed] [Google Scholar]
- Xu XY, Li WY, Hu XY, 2016. Alteration of thyroid-related hormones within normal ranges and early functional outcomes in patients with acute ischemic stroke. Int. J. Endocrinol 2016, 3470490. 10.1155/2016/3470490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro K, et al. , 2017. Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS ONE 12, e0171521. 10.1371/journal.pone.0171521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki K, et al. , 1996. Interleukin-6 (IL-6) inhibits thyroid function in the presence of soluble IL-6 receptor in cultured human thyroid follicles. Endocrinology 137, 4857–4863. 10.1210/endo.137.ll.8895357. [DOI] [PubMed] [Google Scholar]
- Yan T, et al. , 2018. Inflammatory responses mediate brain-heart interaction after ischemic stroke in adult mice. J. Cereb. Blood Flow Metab . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong M, Kaste M, 2008. Dynamic of hyperglycemia as a predictor of stroke outcome in the ECASS-II trial. Stroke 39, 2749–2755. 10.1161/strokeaha.108.514307. [DOI] [PubMed] [Google Scholar]
- You S, et al. , 2019. Combined utility of white blood cell count and blood glucose for predicting in-hospital outcomes in acute ischemic stroke. J. Neuroinflammation 16, 37. 10.1186/s12974-019-1422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousufuddin M, et al. , 2017. Impact of multiple chronic conditions in patients hospitalized with stroke and transient ischemic attack. J. Stroke Cerebrovasc. Dis 26, 1239–1248. 10.1016/j.jstrokecerebrovasdis.2017.01.015. [DOI] [PubMed] [Google Scholar]
- Yperzeele L, et al. , 2015. Heart rate variability and baroreceptor sensitivity in acute stroke: a systematic review. Int. J. Stroke 10, 796–800. 10.1111/ijs.12573. [DOI] [PubMed] [Google Scholar]
- Yu YJ, et al. , 2016. Association between pneumonia in acute stroke stage and 3-year mortality in patients with acute first-ever ischemic stroke. J. Clin. Neurosci 33, 124–128. 10.1016/j.jocn.2016.02.039. [DOI] [PubMed] [Google Scholar]
- Zha A, et al. , 2018. Association between splenic contraction and the systemic inflammatory response after acute ischemic stroke varies with age and race. Transl. Stroke Res 9, 484–492. 10.1007/s12975-017-0596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Meyer MA, 2010. Clinical analysis on alteration of thyroid hormones in the serum of patients with acute ischemic stroke. Stroke Res. Treat 2010. 10.4061/2010/290678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J-N, Liu Y, Li H-C, 2015. Aspiration-related acute respiratory distress syndrome in acute stroke patient. PLoS ONE 10, e0118682. 10.1371/journal.pone.Ol18682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierath D, et al. , 2018. Cortisol is more important than metanephrines in driving changes in leukocyte counts after stroke. J Stroke Cerebrovasc. Dis 27, 555–562. 10.1016/j.jstrokecerebrovasdis.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonneveld TP, et al. , 2017. Hyperglycemia predicts poststroke infections in acute ischemic stroke. Neurology 88, 1415–1421. 10.1212/wnl.0000000000003811. [DOI] [PubMed] [Google Scholar]
- Zorrilla-Vaca A, et al. , 2018. Acute kidney injury following acute ischemic stroke and intracerebral hemorrhage: a meta-analysis of prevalence rate and mortality risk. Cerebrovasc. Dis 45, 1–9. 10.1159/000479338. [DOI] [PubMed] [Google Scholar]