

Abstract
Neutrophil recruitment to the inflamed endothelium is a multistep process and is of utmost importance in the development of the hallmark vaso-occlusive crisis in sickle cell disease (SCD). However, there lacks a standardized, clinically feasible approach for assessing neutrophil recruitment to the inflamed endothelium for individualized risk stratification and therapeutic response prediction in SCD. Here, we describe a microfluidic device functionalized with E-selectin, a critical endothelial receptor for the neutrophil recruitment process, as a strategy to assess neutrophil binding under physiologic flow in normoxia and clinically relevant hypoxia in SCD. We show that hypoxia significantly enhances neutrophil binding to E-selectin and promotes the formation of neutrophil-platelet aggregates. Moreover, we identified two distinct patient populations: a more severe clinical phenotype with elevated lactate dehydrogenase levels and absolute reticulocyte count but lowered fetal hemoglobin levels associated with constitutively less neutrophil binding to E-selectin. Mechanistically, we demonstrate that the extent of neutrophil activation correlates with membrane L-selectin shedding, resulting in the loss of ligand interaction sites with E-selectin. We also show that inhibition of E-selectin significantly reduces leukocyte recruitment to activated endothelial cells. Our findings add mechanistic insight into neutrophil-endothelial interactions under hypoxia and provide a clinically feasible means for assessing neutrophil binding to E-selectin using clinical whole blood samples, which can help guide therapeutic decisions for SCD patients.
Keywords: Lab-on-a-chip, E-selectin, L-selectin, neutrophils, platelets, sickle cell disease, hypoxia, sickle cell disease, neutrophil rolling, cell recruitment
1. Introduction
Leukocyte recruitment to the inflamed endothelium is a sequential, multistep process that involves leukocyte and endothelial cell adhesion molecules that support leukocyte tethering and rolling, firm adhesion, and transmigration under shear stress. Leukocytes initially attach and roll over the activated endothelium through interactions between selectins (P- and E-selectin) and their ligands (McEver 2015). Activated integrins on leukocytes then interact with adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule (VCAM-1) on the activated endothelium, thereby regulating leukocyte firm attachment and migration (Zarbock and Ley 2008). The first leukocyte subtype recruited to sites of inflammation is innate cells, primarily neutrophils (Williams et al. 2011). The adhesion molecule L-selectin is expressed on the neutrophil membrane and plays a pivotal role in neutrophil rolling by directly binding to the endothelial E-selectin (Ivetic 2018; Zöllner et al. 1997). Notably, L-selectin is rapidly shed from the cell surface during neutrophil recruitment and activation. Several studies describe this process as a concerted mechanism to reduce adhesion receptor density to allow downstream integrin-mediated firm attachment (Hafezi-Moghadam and Ley 1999; Ivetic et al. 2019).
In sickle cell disease (SCD), long-standing vascular inflammation and endothelial dysfunction leading to chronic overexpression of adhesive molecules, precipitating an inflammatory and pro-thrombotic milieu that favors leukocyte interactions with the vessel wall. In an early in vivo study, adherent leukocytes triggered vaso-occlusion through increased heterotypic interactions with circulating sickle red blood cells (RBCs) (Turhan et al. 2002). In this study, transgenic sickle mice deficient in both E- and P-selectin exhibited severe leukocyte recruitment defects and were protected from tumor necrosis factor (TNF)-α-induced vaso-occlusion. The same research group showed that RBC and platelet capture by adherent leukocytes was significantly reduced in E-selectin knockout mice but not in P-selectin knockouts in a humanized mouse model of SCD, demonstrating the role of E-selectin in SCD pathophysiology (Hidalgo et al. 2009).
Moreover, adults with homozygous SCD often suffer from vasculopathy that arises from significant, episodic hypoxia (Caboot and Allen 2014; Kato et al. 2009; Machado and Gladwin 2005). The conventional paradigm is that hypoxia contributes to pathogenic vaso-occlusive events in SCD, primarily through impaired RBC deformability (Inanc et al. 2021; Man et al. 2022; Man et al. 2020b; Man et al. 2020c; Man et al. 2021), increased RBC adhesion to endothelial-derived adhesion molecules including E-selectin (Kong et al. 2004; Kucukal et al. 2020b; Papageorgiou et al. 2018; Zund et al. 1996), elevated blood viscosity (Connes et al. 2016; Kucukal et al. 2020a), and activation of the coagulation system (Kucukal et al. 2021; Nasimuzzaman and Malik 2019). However, this pathophysiologic model does not account for the potential contribution of hypoxia to neutrophil adhesiveness or to the formation of heterotypic cell-cell interactions. Notably, a recent study revealed that enhanced neutrophil-platelet interactions in murine models of SCD promoted vaso-occlusion in the pulmonary arterioles (Bennewitz et al. 2017). Mounting evidence also shows that hypoxia is a critical regulator of key neutrophil proinflammatory functions, including increased cell survival, motility, aggregation, and glycolytic enzyme activities (Cramer et al. 2003; Egners et al. 2016; Walmsley et al. 2005).
In the current study, we mimicked neutrophil recruitment to the inflamed endothelium. We developed an E-selectin functionalized microfluidic device to assess neutrophil binding to E-selectin in healthy individuals and subjects with SCD under normoxic and clinically relevant hypoxic conditions. Microchannels coated with E-selectin were coupled with a micro gas exchanger to mimic microvascular damage. E-selectin is expressed by activated endothelial cells, and local hypoxia is induced by inadequate oxygen delivery. We found that hypoxia enhanced neutrophil binding to E-selectin and significantly increased the formation of neutrophil aggregates in samples of subjects with homozygous SCD (HbSS) compared to healthy individuals (HbAA). Importantly, in subjects with SCD, we uncovered two subpopulations with distinct profiles of neutrophil binding to E-selectin constitutively. We demonstrate that the extent of neutrophil activation and membrane L-selectin shedding drive this differential binding of neutrophils to E-selectin and correlate with the clinical severity of SCD. Our findings add mechanistic insight into neutrophil-endothelial interactions under hypoxia and provide a clinically feasible means for assessing neutrophil binding to E-selectin using clinical whole blood samples, which can help guide therapeutic decisions for SCD patients.
2. Materials and methods
Reagents and materials used in this study and additional methods are available in the supplementary material.
2.1. Blood samples
Deidentified venous blood samples were drawn into ethylenediaminetetraacetic acid (EDTA)-containing vacutainers unless stated otherwise at University Hospitals Cleveland Medical Center (UHCMC) in Cleveland, Ohio, according to an institutional review board approved protocol (IRB 05-14-07C). All patient samples were obtained during non-crisis visits to UHCMC Adult Sickle Cell Disease Clinic. Informed consent was obtained from all study participants (patients and healthy individuals). Clinical variables of participants with SCD were obtained from the electronic medical records system. All blood samples were stored at 4 °C before being tested, and all microfluidic experiments were performed within 8 hours of venipuncture.
2.2. Microfluidic device and flow adhesion experiments
As previously described (Alapan et al. 2016; Alapan et al. 2014; Man et al. 2020a), a double-sided adhesive (DSA) film defining the microchannel geometries was laser micro-machined and sandwiched between a polymethyl methacrylate (PMMA) cap and a microscope glass slide. Holes of 0.61 mm diameter were spaced 41 mm from each other and drilled on the PMMA cap to form the inlet and outlet ports. The microchannel height was approximately 50 μm determined by the DSA thickness, mimicking the size scale of post-capillary venules. N-γ-maleimidobutyryl-oxysuccinimide ester (GMBS) was used to immobilize E-selectin onto the microchannel bottom surface covalently. Briefly, assembled microchannels were rinsed with phosphate-buffered saline (PBS, 1X) and 100% ethanol and incubated twice for 15 min at room temperature with GMBS working solution (prepared by dissolving 25 mg of GMBS in 0.25 mL of dimethyl sulfoxide and further diluting with ethanol at 0.28 % v/v). The microchannels were washed twice with ethanol and PBS and incubated with an E-selectin working solution (25 μg/mL in PBS) for 1.5 hours at room temperature. To prevent nonspecific adhesion, 2% bovine serum albumin (BSA) was added into the microchannels, which were maintained at 4°C overnight.
Prior to experiments, microchannels were slowly brought to room temperature, and each microchannel was rinsed with Hank’s buffer solution. Whole blood samples in EDTA were re-calcified with Hank’s buffer at 1:1 v/v in the ambient environment and perfused into the microchannels at 1 dyne/cm2. This shear rate corresponds to the typical shear stress values observed in post-capillary venules. Non-adherent cells were washed off with Hank’s buffer under the same shear stress for 15 min, and phase-contrast images were acquired with an inverted microscope (Olympus IX83). Bound neutrophils and neutrophil aggregates were manually quantified within a 32 mm2 window in Adobe Photoshop (Adobe Inc., San Jose, CA). Events of neutrophil aggregation are defined as a minimum of two neutrophils morphologically in contact with each other. In the acquired images, when two or more neutrophils formed an aggregation event, it was manually quantified as one neutrophil aggregate. The entire experiment takes approximately 30 min, including microscopic imaging. The microfluidic device is single-use and was disposed of following each experiment.
2.3. Micro-gas exchanger for hypoxia induction
To study neutrophil adhesion to E-selectin under clinically relevant hypoxia, we fabricated a micro-gas exchanger to deoxygenate blood, as previously described (Kim et al. 2017). Briefly, a gas-permeable inner tubing was placed within a gas-impermeable outer tubing, and 95% N2 and 5% CO2-controlled gas was allowed to flow in the spacing, such that oxygen diffused from the blood through the permeable tubing wall and mixed with the controlled gas, resulting in an oxygen saturation level of 83% at the inlet of the microchannel. The micro gas exchanger was connected to the microchannels for hypoxia induction, and adhesion experiments were conducted as described above.
2.4. Endothelial cell experiments
In some experiments, microfluidic devices were coated with 0.2 mg/mL fibronectin at 37°C for 1 hour, after which human pulmonary microvascular endothelial cells (HPMECs) at 1×106 cells/mL were seeded in the microchannels and incubated to allow attachment and spreading (Kucukal et al. 2018). Cells were cultured at 37°C and 5% CO2 under a 100 μL/min flow in a culture medium for 48–72 hours until a sufficiently confluent (>90%) monolayer over the microchannel surface was achieved (Noomuna et al. 2020). Confluent HPMEC monolayers were incubated with 25 ng/mL TNF-α in a culture medium at 37°C for 6 hours. Controls were incubated with a culture medium under the same experimental conditions. The medium was replaced every two hours during incubation. In some microchannels, HPMECs were treated with 50 μg/mL of an adhesion-blocking antibody against E-selectin in the last 30 min of TNF-α activation. Heparinized whole blood samples were used for flow adhesion experiments as described above.
2.5. Statistical analysis
Data are reported as mean ± standard deviation (SD). Image fluorescence intensity was quantified by ImageJ software. Statistical analyses were performed with Minitab 19 (Minitab, State College, PA). Data were first analyzed for normality, followed by appropriate tests to compare different groups: paired t-test for normally distributed paired data, one-way ANOVA for normally distributed unpaired data, and nonparametric Mann-Whitney U test for non-normally distributed data. Statistical significance was set at a 95% confidence level for all tests (P < 0.05).
3. Results
3.1. A microfluidic device for assessment of neutrophil binding to E-selectin using clinical whole blood samples
We fabricated a microfluidic device using a simple lamination technique. We coated the microchannel surface with E-selectin through a GMBS cross-linking mechanism to mimic the microvasculopathy in SCD, in which E-selectin is abnormally upregulated on the inflamed vascular endothelium (Fig. 1A). The microchannel is 25mm*4mm*50μm (length*width*height), which enables a large surface area for adhesion event observation and a thickness similar to a post-capillary venule in human vasculature. We further coupled the microfluidic device with a micro-gas exchanger to mimic the inadequate oxygen supply in SCD. We analyzed the effects of hypoxia on neutrophil recruitment (Fig. 1B). Fifteen microliters (μL) of diluted whole blood were injected into the microchannel at 1 dyne/cm2. The microchannel was rinsed with Hank’s buffer at 1 dyne/cm2. Following rinsing, we observed the accumulation of a homogeneous cell population of roughly 12-μm diameter in size; fluorescent labeling of cells for neutrophil elastase confirmed that adhered cells were indeed of neutrophil origin (Fig. 1C insets). Therefore, we established that the inflamed endothelium-mimicking microfluidic device enables assessment of neutrophil binding to E-selectin using clinical whole blood samples.
Figure 1. Overview of the inflamed endothelium-mimicking microfluidic device for assessment of neutrophil binding to E-selectin under physiologic flow in normoxia and clinically relevant hypoxia in SCD.
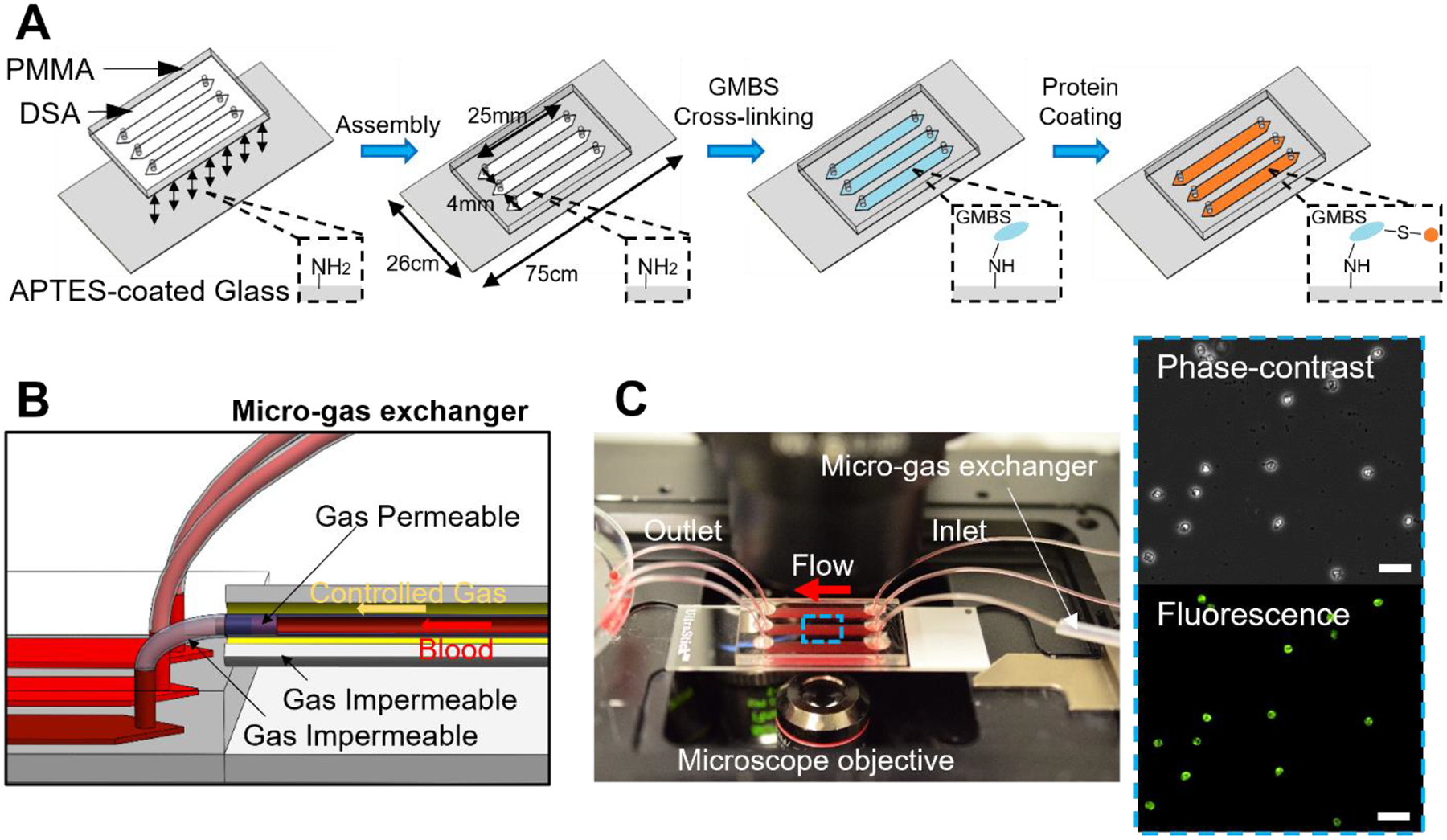
(A) Schematics of the fabrication and protein coating process of the microfluidic device. (B) Schematics of the microfluidic device integrated with the micro-gas exchanger for hypoxia induction. (C) Macroscopic view of the microfluidic device mounted on an inverted microscope for blood testing. Insets: Adherent cells were fixed, permeabilized, and fluorescently labeled with anti-human neutrophil elastase antibody. Scale: 50 μm.
3.2. Hypoxia has a more profound effect on neutrophil binding to E-selectin in SCD
We next asked if neutrophil recruitment to the inflamed endothelium is influenced by clinically relevant hypoxia. We tested blood samples from 11 subjects with HbSS and 5 with HbAA under normoxic or hypoxic conditions using the inflamed endothelium-mimicking microfluidic device. Representative neutrophil distribution within the microchannels in HbAA and HbSS samples under variable oxygen tension are shown in Fig. S1. We found that hypoxia significantly increased the number of neutrophils binding to E-selectin in HbAA samples compared to normoxic conditions (Fig. 2A, mean ± SD = 1611 ± 631 vs. 481 ± 224, P = 0.004). Similarly, the number of neutrophils binding to E-selectin under hypoxia was significantly greater than normoxia in samples of HbSS subjects (Fig. 2A, mean ± SD = 5246 ± 4633 vs. 2057 ± 1856, P = 0.018). Notably, the number of neutrophils binding to E-selectin was significantly greater in samples from subjects with HbSS than in HbAA under both normoxic and hypoxic states (Fig. 2A, P = 0.005 & P = 0.031). These data indicate that subjects with HbSS have constitutively greater neutrophil binding to E-selectin than HbAA, which is significantly enhanced under hypoxic conditions.
Figure 2. Hypoxia enhances neutrophil binding on E-selectin and neutrophil-platelet aggregation.
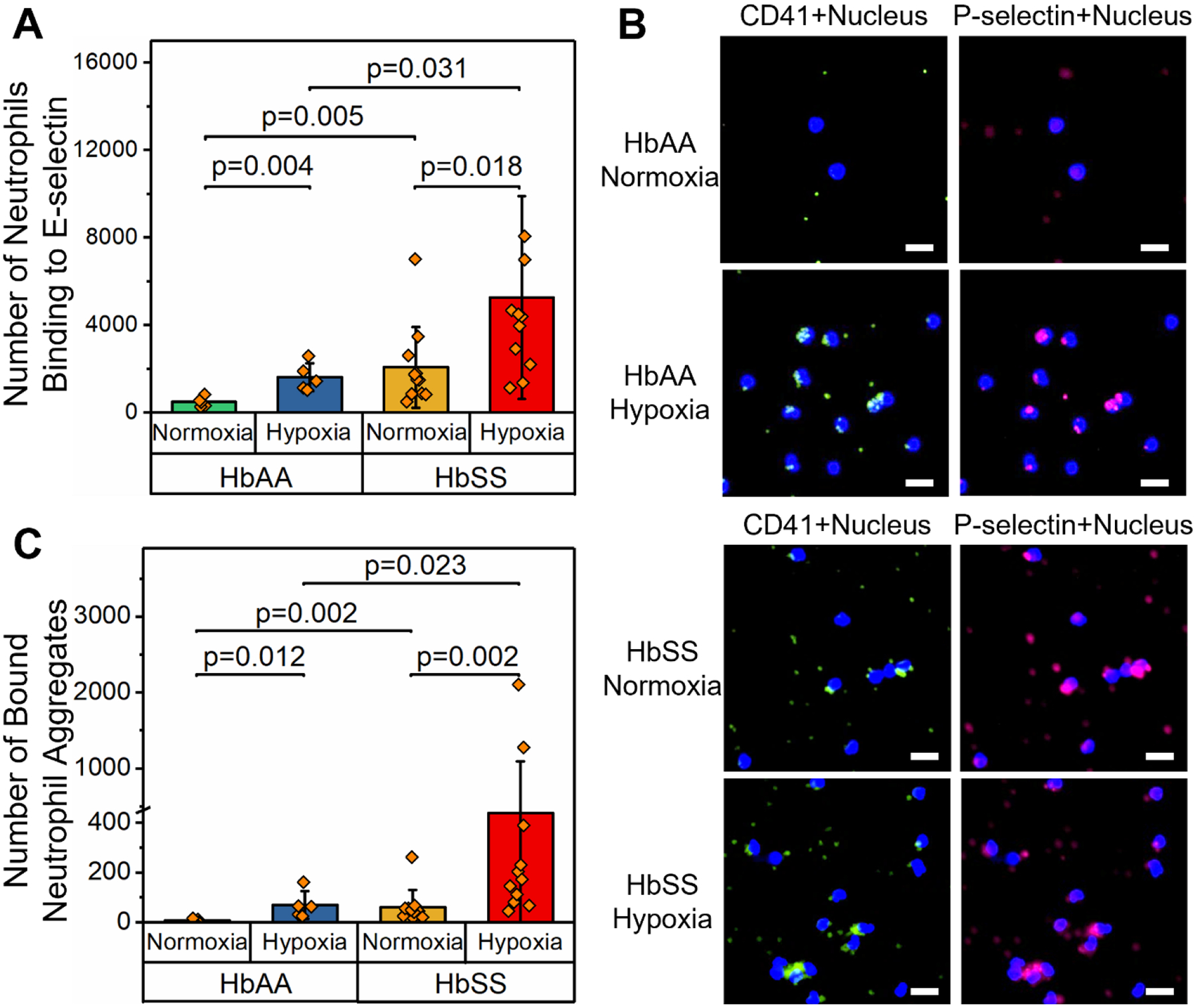
(A) The number of neutrophils binding to E-selectin was significantly greater under hypoxic conditions compared to normoxia in both HbAA (N = 5, P = 0.004, paired t-test) and HbSS (N = 11, P = 0.018, Mann-Whitney) clinical samples. Further, the number of neutrophils binding to E-selectin was significantly greater in samples from subjects with HbSS than HbAA under both normoxia (P = 0.005, Mann-Whitney) and hypoxia (P = 0.031, Mann-Whitney). (B) Representative fluorescent microscopic images showing adherent neutrophil aggregates bridged by platelets in samples from subjects with HbAA or HbSS, under hypoxic or normoxic conditions. Neutrophils were stained with DAPI, platelets were stained with FITC-conjugated anti-CD41 and Cy5-conjugated anti-P-selectin antibodies. Scale: 20 μm. (C) The number of bound neutrophil aggregates was significantly greater under hypoxia than normoxia in samples from subjects with HbAA (N = 5, P = 0.012, Mann-Whitney) and HbSS (N = 11, P = 0.002, Mann-Whitney). Further, the number of bound neutrophil aggregates was significantly greater in samples from subjects with HbSS than HbAA under both normoxia (P = 0.002, Mann-Whitney) and hypoxia (P = 0.023, Mann-Whitney). HbAA: healthy, and HbSS: homozygous SCD.
3.3. Hypoxia enhances neutrophil-platelet interactions and the formation of neutrophil aggregates in SCD
A recent study showed that acute inflammatory stimuli, including lipopolysaccharide (LPS) and heme, are sufficient to cause neutrophil-platelet microthrombi in the lungs of SCD mice but not in control mice (Bennewitz et al. 2017). In this framework, we examined if hypoxia selectively affects neutrophil aggregation in SCD subjects. We fluorescently labeled adherent cells in the microchannels to observe homotypic neutrophil-neutrophil and heterotypic neutrophil-platelet cell aggregates (Fig. 2B). Platelets were found to attach on the surface of neutrophils and bridged one neutrophil to another, propagating the size and cell number within each aggregate. In samples from subjects with HbAA, the number of bound neutrophil aggregates was significantly greater under hypoxia than normoxia (Fig. 2C, mean ± SD = 70 ± 55 vs. 6 ± 7, P = 0.012). Similarly, in HbSS samples, the number of adherent neutrophil aggregates was significantly greater in hypoxic conditions compared to normoxia (Fig. 2C, mean ± SD = 439 ± 653 vs. 60 ± 69, P = 0.002). Notably, the number of neutrophil aggregates was significantly greater in samples from subjects with HbSS than HbAA under both normoxia and hypoxia (Fig. 2C, P = 0.002 & P = 0.023, respectively). These data indicate that hypoxia enhances heterotypic neutrophil-platelet interactions and promotes the formation of neutrophil aggregates which are considerably more pronounced in subjects with SCD compared to healthy controls.
3.4. Use of the endothelialized microfluidic device to assess the therapeutic effect of E-selectin inhibition
Given that neutrophil binding to E-selectin is differentially augmented by normoxia and hypoxia in SCD samples, we next determined the therapeutic effect of E-selectin inhibition on leukocyte recruitment (including neutrophils) during vascular inflammation using endothelialized microchannels. As we wanted to capture leukocyte recruitment through all ligand-receptor interactions and elucidate the contribution of E-selectin in this process, we chose to line microchannels with HPMECs (Fig. 3A) to mimic pulmonary vaso-occlusive events in SCD. A confluent monolayer of HPMECs was achieved in the microfluidic device within 48–72 hours of culture under flow, as shown in Fig. 3A insets. Our fluorescence studies showed that following a 6-hour incubation with TNF-α, HPMECs were activated, as seen by significantly upregulated E-selectin and ICAM-1 expression compared to control unstimulated HPMECs (Fig. S2). We then performed flow adhesion experiments using heparinized clinical whole blood. Heparin was used as the anticoagulant of choice because EDTA is known to disrupt endothelial cell-cell junctions. No comparisons were performed on data generated by different anticoagulants. We observed significantly greater leukocyte adhesion to TNF-α activated HPMECs than control in samples from subjects with HbAA or HbSS (Fig. 3B, C&E, TNF-α activation vs. control in HbAA samples: 4965 ± 2072 vs. 229 ± 207, N = 3, P = 0.048; TNF-α activation vs. control in HbSS samples: 10426 ± 3671 vs. 246 ± 169, N = 6, P = 0.001). Moreover, significantly greater leukocyte adhesion to TNF-α activated HPMECs was observed in HbSS compared to HbAA (Fig. 3B, P = 0.028). Furthermore, an anti-human E-selectin antibody significantly inhibited leukocyte recruitment to TNF-α activated HPMECs in samples from 5 subjects with HbSS (Fig. 3C, D&F, TNF-α activation vs. TNF-α activation plus E-selectin antibody: 11354 ± 7184 vs. 4946 ± 3113, P = 0.029). These data suggest that inhibition of E-selectin has a beneficial effect on abnormal leukocyte recruitment to the inflamed endothelium in SCD.
Figure 3. Inhibition of E-selectin significantly reduces leukocyte recruitment to the inflamed endothelium in SCD.
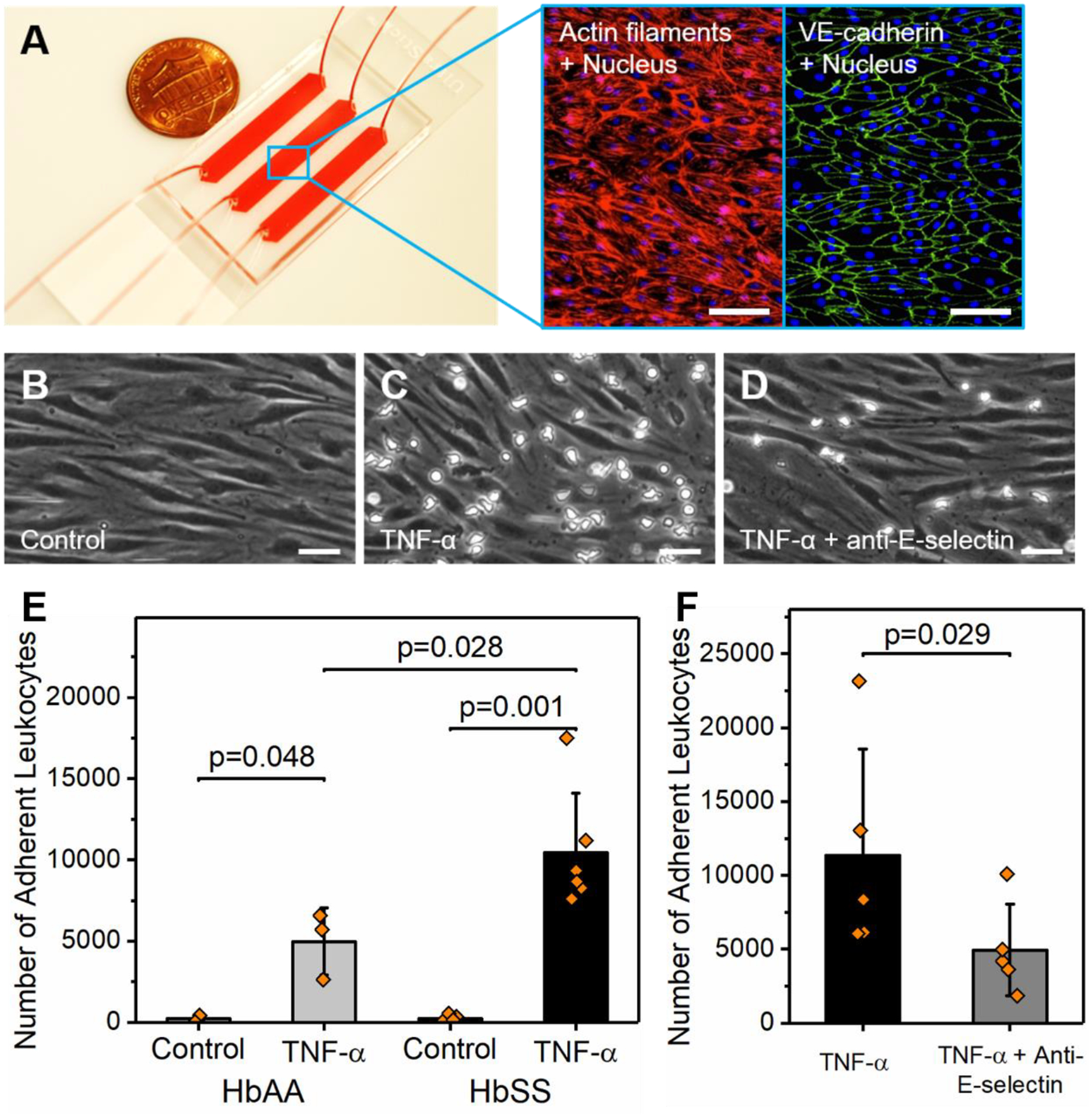
(A) The microfluidic device was lined with human pulmonary microvascular endothelial cells (HPMECs). Insets: Fluorescent labeling of HPMECs showing the uniform cellular organization, cytoskeleton, and cell-cell junctions. (B-D) Representative microscopic images of adherent leukocytes to control unstimulated HPMECs, TNF-α activated HPMECs, and TNF-α activated HPMECs pre-treated with an adhesion-blocking anti-human E-selectin antibody. (E) Overall, leukocyte adhesion to control HPMECs was negligible but significantly increased with TNF-α activation in samples from subjects with HbAA (N = 3, P = 0.048, paired-t test) and HbSS (N = 6, P = 0.001, Mann-Whitney). Further, significantly greater leukocyte adhesion to TNF-α activated HPMECs was found in samples from subjects with HbSS compared to HbAA (P = 0.028, Mann-Whitney). (F) Treatment of TNF-α activated HPMECs with an adhesion-blocking anti-human E-selectin antibody significantly decreased leukocyte adhesion in samples from subjects with HbSS (N = 5, P = 0.029, paired-t test). Scale bars indicate lengths of 100 and 50 μm, respectively. HbAA: healthy. HbSS: homozygous sickle cell disease.
3.5. Neutrophil binding to E-selectin inversely correlates with vascular hemolysis and transfusion dependency in SCD
Since SCD is a clinically heterogeneous disease, we asked if constitutive neutrophil binding to E-selectin associates with clinical variables of the disease. To address this question, blood samples obtained from 35 subjects with HbSS SCD were studied under normoxia in E-selectin functionalized microchannels. Associations with clinical variables were determined by univariate models using the K-means clustering method on two important in vivo hemolytic biomarkers in SCD, lactate dehydrogenase (LDH) levels and absolute reticulocyte counts (ARCs). The analysis identified two distinct SCD sub-populations (Fig. 4A). Subjects in HbSS Group 1 (N = 16) had significantly lower LDH levels (290 ± 18 vs. 477 ± 66, P = 0.002) and ARCs (262 ± 29 vs. 510 ± 38, P < 0.001) compared to those in HbSS Group 2 (N = 19; Table S1). When we compared the neutrophil binding profiles between these two groups, we found that subjects in the HbSS Group 1 had a significantly greater number of neutrophils binding to E-selectin than those in HbSS Group 2 (Fig. 4B, 1842 ± 532 vs. 691 ± 223, P = 0.041). We further noted that subjects in the HbSS Group 1 had significantly greater fetal hemoglobin levels (HbF%) than those in HbSS Group 2 (Fig. 4C, 10.9 ± 9.7% vs. 3.6 ± 3.6%, P = 0.043). Five subjects from each of these two groups were used to examine the neutrophil aggregate formation, where we found that subjects in HbSS Group 1 had a significantly greater number of bound neutrophil aggregates compared to subjects in the HbSS Group 2 (Fig. S3, 49 ± 38 vs. 6 ± 3, P = 0.011). Moreover, we observed significantly higher transfusion dependency in HbSS Group 2 subjects compared to HbSS Group 1 subjects (Table S1, P = 0.005). These results revealed that neutrophil binding to E-selectin inversely correlates with the degree of hemolysis and the need for blood transfusions.
Figure 4. Neutrophil binding to E-selectin inversely correlates with vascular hemolysis in SCD and is mediated by surface L-selectin.
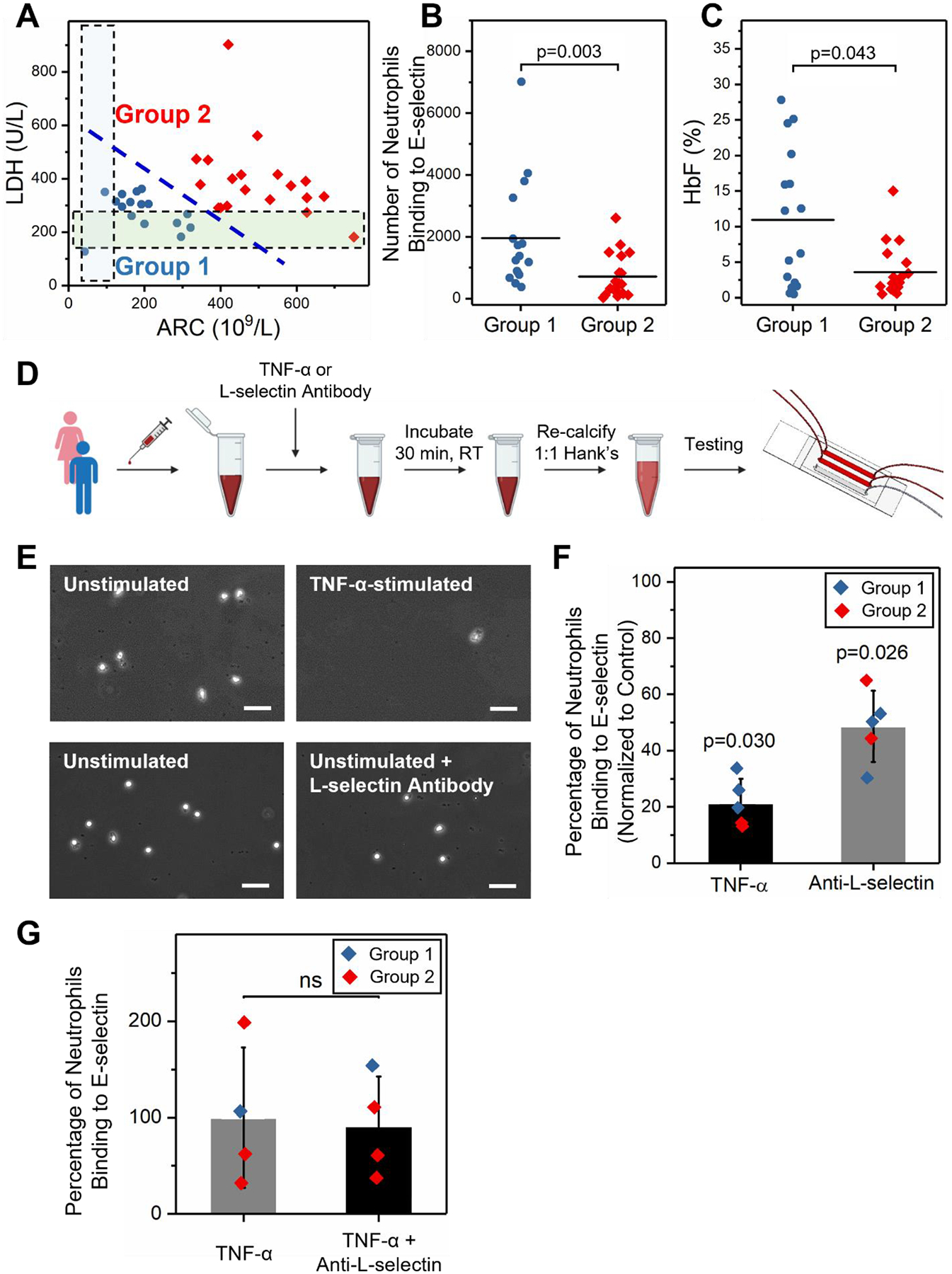
(A) Subjects were stratified into 2 sub-groups based on their serum LDH levels and absolute reticulocyte counts (ARCs) using the K-means clustering method. HbSS Group 2 (N = 19) samples had significantly higher serum LDH levels and ARCs but lower HbF% compared to HbSS Group 1 (N = 16). The shaded regions indicate reference ranges (normal) for LDH and ARC levels, respectively. (B) HbSS Group 2 subjects had significantly lower neutrophil binding to E-selectin compared to HbSS Group 1 subjects (P = 0.003, Mann-Whitney). (C) HbSS Group 2 subjects had significantly lower HbF% compared to HbSS Group 1 subjects (P = 0.043, Mann-Whitney). (D) Schematic representation of experimental conditions. Whole blood samples drawn from subjects with HbSS were incubated with 25 ng/mL TNF-α or 10 μg/mL anti-L-selectin antibody at room temperature (RT) for 30 min. Thereafter, blood samples were re-calcified and perfused through E-selectin microchannels. (E) Representative microscopic images of neutrophils adhered to E-selectin under the various conditions listed. Scale: 50 μm. (F) Neutrophil activation with TNF-α significantly reduced neutrophil binding to E-selectin (N = 5, P = 0.030, paired t-test). Similarly, L-selectin blocking antibody significantly reduced neutrophil binding to E-selectin (N = 5, P = 0.026, paired t-test). (G) Anti-L-selectin treatment did not reduce neutrophil binding of TNF-α stimulated neutrophils (N = 4, ns: P > 0.05).
3.6. Constitutive neutrophil binding to E-selectin is mediated by L-selectin and inversely correlates with vascular hemolysis in SCD
We next sought to determine the mechanism influencing neutrophil interactions with E-selectin. For these studies, we compared neutrophil binding to E-selectin in samples from subjects with SCD that were either unstimulated (control) or stimulated for 30 min with 25 ng/mL TNF-α (Fig. 4D). We determined that stimulating neutrophils with TNF-α, a known potent neutrophil agonist, led to a significant reduction in neutrophil binding to E-selectin compared to unstimulated cells (Fig. 4E&F, 21.3 ± 8.6% normalized to control unstimulated cells, P = 0.030). Importantly, when unstimulated SCD samples (N=5; Group 1–2 allocation is shown in Fig. S4) were treated with an anti-L-selectin antibody (10 μg/mL), neutrophil binding to E-selectin was reduced but not to the same extent seen with TNF-α stimulated cells (Fig. 4E&F, 48.6 ± 12.7% normalized to control, P = 0.026). In contrast, when TNF-α stimulated HbSS blood samples were treated with an anti-L-selectin antibody, neutrophil retention to E-selectin did not change (Fig. 4G, P > 0.05). These data show that neutrophil binding to E-selectin is mediated by L-selectin and the extent of neutrophil activation is inversely proportional to neutrophil interactions with E-selectin. Prior research has shown that neutrophil activation with pro-inflammatory stimuli such as formyl peptides, TNF-α or Toll-like receptor agonists leads to robust and rapid shedding of L-selectin within minutes (Hazeldine et al. 2015; Killock and Ivetic 2010). Reduced L-selectin surface expression is inversely correlated with increased αMβ2 integrin expression (Kishimoto et al. 1989). This is viewed as a coordinated mechanism to reduce adhesion receptor density and allow integrin-mediated neutrophil firm attachment to the endothelium. Combined with the results above, our working hypothesis is that lower neutrophil binding to E-selectin in HbSS Group 2 subjects is likely occurring due to constitutively higher cell activation and increased L-selectin shedding, which correlates with a more adverse disease phenotype characterized by exuberant hemolysis (greater LDH levels and ARCs) and low HbF percentage in vivo.
4. Discussion
In this study, we present a microfluidic device for the assessment of neutrophil binding to E-selectin under physiologic flow in normoxic and clinically relevant hypoxic conditions in SCD. There are three main advantages to the device: first, its cost-effectiveness (<$11 per device for 3 tests including the cost of E-selectin), which uses laser-machined PMMA and DSA and commercially available APTES-coated microscope glass slides, and therefore does not involve expensive cleanroom facilities; second, the microfluidic device has a large surface area for adhesion event quantification which is difficult to become saturated (e.g., neutrophil recruitment under hypoxia in SCD) and allows capturing of rare adhesive events (e.g., neutrophil recruitment under normoxia in healthy condition); third, the microfluidic device is clinically feasible and requires no pre-processing of whole blood. Although images were processed by manual cell counting in this work, an automated image analysis tool would significantly speed up the process, which is currently under development (unpublished work).
We employed a simple micro-gas exchanger developed in our previous work (Kim et al. 2017) to induce clinically relevant hypoxic conditions (SpO2 of 83%). We found that hypoxia enhances neutrophil-E-selectin interactions in subjects with HbAA or HbSS. While earlier studies have linked hypoxia to increased neutrophil-endothelial cell interactions through an ICAM-1-mediated mechanism (Arnould et al. 1993; Winning et al. 2010), our results indicate that in hypoxic conditions, E-selectin also contributes to inflammatory cell recruitment at the vessel wall. Further, we found that hypoxia leads to the formation of adherent homotypic neutrophil aggregates on E-selectin and to heterotypic neutrophil-platelet interactions, previously shown to be mediated by P-selectin-PSGL-1 (Li and Smyth 2019) and L-selectin-PSGL-1 (Morikis et al. 2021; Tu et al. 1996) interactions. Based on those results, we postulate that hypoxia contributes to vaso-occlusive events (VOE) by promoting neutrophil recruitment and subsequent formation of cell aggregates on the inflamed vascular endothelium in SCD, which is in line with previous studies (Dominical et al. 2014; Inwald et al. 2000; Zhang et al. 2016). Moreover, our results suggest that subjects with SCD who are likely to suffer from chronic inflammation and hypoxemia, have a greater extent of neutrophil recruitment to the vascular endothelium compared to those from healthy subjects.
Neutrophils utilize both L-selectin and ESL-1 to bind to endothelial E-selectin and PSGL-1 to bind to endothelial (and platelet) P-selectin. A seminal study by Hidalgo A et al., assessed the contribution of ESL-1 in leukocyte rolling in vivo in a model of TNF-α cremasteric venule thromboinflammation (Hidalgo et al. 2007). Briefly, the rolling frequency of transduced leukocytes was reduced by 75% in the absence of PSGL-1 but was not affected by the absence of ESL-1 alone. When ESL-1 deficiency was combined with PSGL-1 knockdown, leukocyte rolling was further reduced from 75% to ~93%. These and further studies revealed that ESL-1 primarily cooperates with PSGL-1 (and CD44) in leukocyte tethering and rolling. Both L-selectin and PSGL-1 clusters colocalize on rolling leukocytes. Still, studies evaluating if these processes depend on E-selectin showed that only L-selectin polarization was markedly reduced in E-selectin knockout mice (Hidalgo et al. 2007). These results indicate that E-selectin specifically induces L-selectin redistribution on rolling leukocytes which subsequently plays a major role in stabilizing leukocytes into steady rolling. Finally, prior research has shown that neutrophil activation with pro-inflammatory stimuli leads to robust and rapid shedding of L-selectin within minutes (Hazeldine et al. 2015; Killock and Ivetic 2010). Reduced L-selectin surface expression is inversely correlated with increased αMβ2 integrin expression (Kishimoto et al. 1989) and similarly, soluble L-selectin levels rise in individuals with chronic autoimmune diseases (Font et al. 2000; Garcia-Carrasco et al. 2000). Here, we show that i) TNF-α stimulation of SCD samples is associated with reduced neutrophil adhesion to E-selectin compared to control, unstimulated cells; ii) treating unstimulated SCD samples with anti-L-selectin antibody, significantly reduced neutrophil adhesion to E-selectin compared to unstimulated cells alone; iii) in contrast, treating TNF-α stimulated SCD samples with anti-L-selectin antibody, did not influence neutrophil retention on E-selectin (i.e., once neutrophils were already fully activated). The combination of these studies supports that neutrophil binding on endothelial E-selectin is mediated by L-selectin and neutrophil activation is accompanied by reduced neutrophil interactions with E-selectin.
Over the past few decades, considerable progress has been made in the development of novel anti-adhesive therapies for the management of SCD including P-selectin inhibitors, Crizanlizumab and Sevuparin (Manwani and Frenette 2013; Telen et al. 2019) and more recently Rivipansel (GMI-1070). Our findings in this study suggest that only a subset of subjects with SCD who have a moderate clinical phenotype, with constitutively greater levels of neutrophil binding to E-selectin are likely to benefit from a therapeutic drug targeting at E-selectin such as Rivipansel (Fig. 5). Further, those with a more severe clinical phenotype who have a significantly higher percentage of fully activated neutrophils and lower levels of neutrophil binding to E-selectin, may remain unresponsive to anti-selectin therapy (Fig. 5). Therefore, it is not entirely surprising that the Phase 3 study of Rivipansel (NCT02187003) did not meet its primary or key secondary efficacy endpoints for all accrued participants (Dampier et al. 2022. doi: 10.1182/blood.2022015797). With the growing understanding of the mechanisms of VOE development and the multifaceted and interconnected pathophysiology in SCD, it is now generally accepted that a therapeutic strategy targeted at a single adhesion molecule may not necessarily benefit the entire SCD population. A standardized microfluidic biomarker assay, as we describe herein, would be extremely useful in the identification and assessment of personalized treatment options and could improve clinical outcomes for patients with SCD.
Figure 5. Microfluidic assessment of neutrophil adhesion profile can guide therapeutic decisions in sickle cell disease.
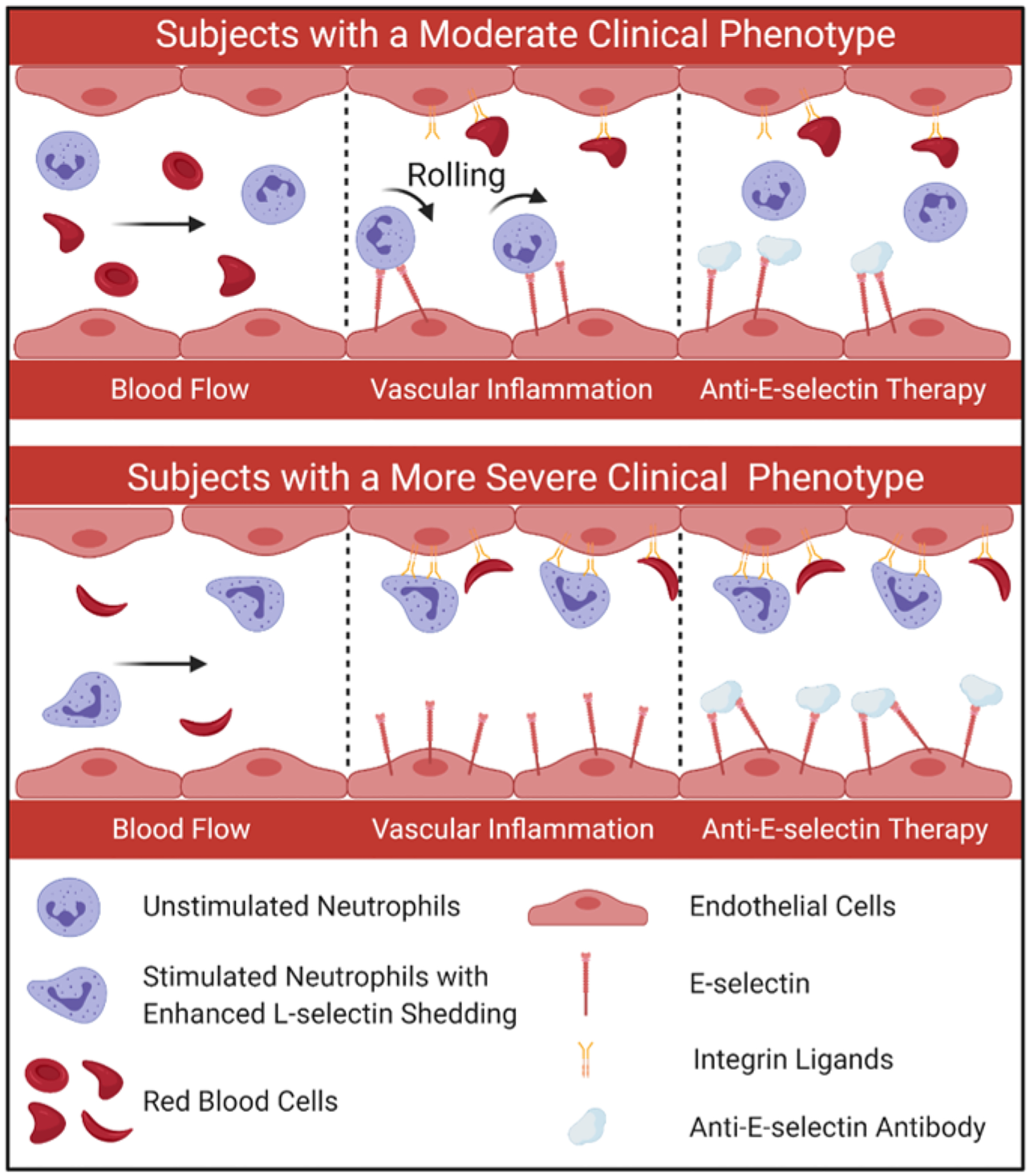
Subjects with SCD who have a moderate clinical phenotype where a smaller proportion of neutrophils are fully activated, present with relatively greater neutrophil binding to E-selectin and are more likely to benefit from therapies against E-selectin. In contrast, SCD individuals who exhibit a more severe clinical phenotype and presumably more neutrophils activated, present with lower neutrophil binding to E-selectin, likely mediated by enhanced L-selectin shedding, and may be unresponsive to therapies against E-selectin.
There are limitations to our study. First, the study population with HbSS SCD was relatively modest. A larger cohort will allow us to better assess the associations between neutrophil recruitment and patient clinical phenotypes, as well as treatments such as transfusion and the use of hydroxyurea. Second, increased L-selectin shedding on the surface of neutrophils is concordant with upregulated αMβ2 integrin expression. To link patient clinical phenotypes to neutrophil activation and reduced binding to E-selectin, flow cytometry characterization of L-selectin and αMβ2 integrin expression, as well as neutrophil adhesion to ICAM-1 among test subjects will need to be examined. Third, the mechanistic studies were performed using blood samples from patients with SCD who have an inherently larger variation in neutrophil activation than healthy samples. Fourth, the mechanism of neutrophil aggregation mediated by P-selectin or L-selectin-PSGL-1 interactions under hypoxia and its association with patient clinical phenotypes in SCD was not analyzed and warrants future investigations.
5. Conclusions
Here, we present an E-selectin functionalized microfluidic device partially mimicking the inflamed endothelium for assessment of neutrophil binding to E-selectin in physiologic flow under normoxia or clinically relevant hypoxia in SCD using diluted clinical whole blood samples. We show that hypoxia enhances neutrophil binding to E-selectin and promotes the formation of heterotypic cell aggregates in healthy subjects and significantly more in subjects with SCD. These findings highlight the impact of hypoxia on augmented neutrophil-endothelium interactions in SCD, besides its generally accepted role in inducing biophysical changes of sickle RBCs upon hemoglobin polymerization. Importantly, we identified two distinct SCD subpopulations with different profiles of neutrophil binding to E-selectin. Subjects in HbSS Group 2, with significantly greater LDH levels and ARCs but lower HbF%, had constitutively lower levels of neutrophil binding to E-selectin, compared to subjects in HbSS Group 1. And we further show that E-selectin is an important target for the inhibition of leukocyte recruitment (including neutrophils) to inflamed endothelium in SCD. Altogether, our findings strongly suggest that a well-controlled, standardized flow adhesion assay enabled by our E-selectin microfluidic device would provide valuable information for the development of a personalized therapeutic strategy (e.g., an inhibitor targeting at E-selectin) to improve the clinical outcomes in patients with SCD.
Supplementary Material
Highlights.
An E-selectin functionalized microfluidic device partially mimicking the inflamed endothelium is established to assess neutrophil binding to E-selectin in physiologic flow under normoxia or clinically relevant hypoxia, using clinical whole blood samples of individuals with sickle cell disease.
In sickle cell disease, constitutive neutrophil binding to E-selectin is mediated by L-selectin and inversely correlates with vascular hemolysis and the need for blood transfusions.
Assessment of neutrophil binding to E-selectin is informative for patient clinical phenotypes and prediction of therapeutic response to targeted therapy against E-selectin in sickle cell disease.
Acknowledgments
The authors acknowledge with gratitude the generous participation of clinical staff and patients with SCD who are followed at the University Hospitals Cleveland Medical Center Adult Sickle Cell Disease clinic. This work was supported by National Science Foundation (NSF) CAREER Award 1552782 (UAG), National Heart, Lung, and Blood Institute (NHLBI) grants R01HL137695 (EXS), R01HL133574 (UAG), R42HL160384 (UAG), OT2HL152643 (UAG), U01HL117659 (JAL), and T32HL134622 (RA), Merit Review Award BX003851 (EXS) from the Department of Veterans Affairs, and the Oscar D. Ratnoff Endowed Professorship (EXS). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, the U.S. Department of Veterans Affairs, or the United States Government.
Conflict-of-interest disclosure
U. A. G. and Case Western Reserve University have financial interests in Hemex Health Inc. U. A. G. and Case Western Reserve University have financial interests in BioChip Labs Inc. U. A. G. and Case Western Reserve University have financial interests in Xatek Inc. U. A. G. has financial interests in DxNow Inc. Financial interests include licensed intellectual property, stock ownership, research funding, employment, and consulting. Hemex Health Inc. offers point-of-care diagnostics for hemoglobin disorders, anemia, and malaria. BioChip Labs Inc. offers commercial clinical microfluidic biomarker assays for inherited or acquired blood disorders. Xatek Inc. offers point-of-care global assays to evaluate the hemostatic process. DxNow Inc. offers microfluidic and bio-imaging technologies for in vitro fertilization, forensics, and diagnostics. The competing interests of Case Western Reserve University employees are overseen and managed by the Conflict of Interests Committee according to a Conflict-of-Interest Management Plan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alapan Y, Kim C, Adhikari A, Gray KE, Gurkan-Cavusoglu E, Little JA, Gurkan UA, 2016. Sickle cell disease biochip: a functional red blood cell adhesion assay for monitoring sickle cell disease. Transl. Res 173, 74–91 e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alapan Y, Little JA, Gurkan UA, 2014. Heterogeneous red blood cell adhesion and deformability in sickle cell disease. Sci. Rep 4, 7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnould T, Michiels C, Remacle J, 1993. Increased PMN adherence on endothelial cells after hypoxia: involvement of PAF, CD18/CD11b, and ICAM-1. Am. J. Physiol 264(5 Pt 1), C1102–1110. [DOI] [PubMed] [Google Scholar]
- Bennewitz MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, Kato GJ, Gladwin MT, Sundd P, 2017. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2(1), e89761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboot JB, Allen JL, 2014. Hypoxemia in sickle cell disease: significance and management. Paediatr. Respir. Rev 15(1), 17–23. [DOI] [PubMed] [Google Scholar]
- Connes P, Alexy T, Detterich J, Romana M, Hardy-Dessources MD, Ballas SK, 2016. The role of blood rheology in sickle cell disease. Blood Rev. 30(2), 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, 2003. HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112(5), 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampier CD, Telen MJ, Wun T, Brown C, Desai PC, El Rassi F, Fuh B, Kanter J, Pastore YD, Rothman J, Taylor JG, Readett D, Sivamurthy KM, Tammara B, Tseng LJ, Lozier JN, Thackray HM, Magnani JL, Hassell K, 2022. doi: 10.1182/blood.2022015797. A Randomized Clinical Trial of the Efficacy and Safety of Rivipansel for Sickle Cell Vaso-occlusive Crisis (VOC). Blood. [DOI] [PubMed] [Google Scholar]
- Dominical VM, Samsel L, Nichols JS, Costa FF, McCoy JP Jr., Conran N, Kato GJ, 2014. Prominent role of platelets in the formation of circulating neutrophil-red cell heterocellular aggregates in sickle cell anemia. Haematologica 99(11), e214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egners A, Erdem M, Cramer T, 2016. The response of macrophages and neutrophils to hypoxia in the context of cancer and other inflammatory diseases. Mediators Inflamm. 2016, 2053646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font J, Pizcueta P, Ramos-Casals M, Cervera R, Garcia-Carrasco M, Navarro M, Ingelmo M, Engel P, 2000. Increased serum levels of soluble L-selectin (CD62L) in patients with active systemic lupus erythematosus (SLE). Clin Exp Immunol 119(1), 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Carrasco M, Pizcueta P, Cervera R, Ramos-Casals M, Siso A, de La Red G, Ingelmo M, Font J, Engel P, 2000. Circulating concentrations of soluble L-selectin (CD62L) in patients with primary Sjogren’s syndrome. Ann Rheum Dis 59(4), 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Ley K, 1999. Relevance of L-selectin shedding for leukocyte rolling in vivo. J. Exp. Med 189(6), 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JMJI, 2015. N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways 46(6), 975–984. [DOI] [PubMed] [Google Scholar]
- Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS, 2009. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nature medicine 15(4), 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo A, Peired AJ, Wild M, Vestweber D, Frenette PS, 2007. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 26(4), 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanc MT, Demirkan I, Ceylan C, Ozkan A, Gundogdu O, Goreke U, Gurkan UA, Unlu MB, 2021. Quantifying the influences of radiation therapy on deformability of human red blood cells by dual-beam optical tweezers. RSC Adv. 11(26), 15519–15527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ, 2000. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br. J. Haematol 111(2), 474–481. [DOI] [PubMed] [Google Scholar]
- Ivetic A, 2018. A head-to-tail view of L-selectin and its impact on neutrophil behaviour. Cell Tissue Res. 371(3), 437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivetic A, Green HLH, Hart SJ, 2019. L-selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol 10, 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT, 2009. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am. J. Hematol 84(9), 618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killock DJ, Ivetic A, 2010. The cytoplasmic domains of TNFalpha-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem J 428(2), 293–304. [DOI] [PubMed] [Google Scholar]
- Kim M, Alapan Y, Adhikari A, Little JA, Gurkan UA, 2017. Hypoxia-enhanced adhesion of red blood cells in microscale flow. Microcirculation 24, e12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto TK, Jutila MA, Berg EL, Butcher EC, 1989. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science 245(4923), 1238–1241. [DOI] [PubMed] [Google Scholar]
- Kong TQ, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS, 2004. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction Of beta 2 integrin gene expression. Proc. Natl. Acad. Sci. U. S. A 101(28), 10440–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukal E, Ilich A, Key NS, Little JA, Gurkan UA, 2018. Red Blood Cell Adhesion to Heme-Activated Endothelial Cells Reflects Clinical Phenotype in Sickle Cell Disease. Am. J. Hematol 93, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukal E, Man Y, Gurkan UA, Schmidt B, 2021. Blood flow velocimetry in a microchannel during coagulation using particle image velocimetry and wavelet-based optical flow velocimetry. J. Biomech. Eng 143(9), 091004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukal E, Man Y, Hill A, Liu S, Bode A, An R, Kadambi J, Little JA, Gurkan UA, 2020a. Whole blood viscosity and red blood cell adhesion: Potential biomarkers for targeted and curative therapies in sickle cell disease. Am. J. Hematol 95(11), 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukal E, Man Y, Quinn E, Tewari N, An R, Ilich A, Key NS, Little JA, Gurkan UA, 2020b. Red blood cell adhesion to ICAM-1 is mediated by fibrinogen and is associated with right-to-left shunts in sickle cell disease. Blood Adv. 4(15), 3688–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Smyth SS, 2019. Interactions between platelets, leukocytes, and the endothelium. Platelets, 295–310. [Google Scholar]
- Machado RF, Gladwin MT, 2005. Chronic sickle cell lung disease: new insights into the diagnosis, pathogenesis and treatment of pulmonary hypertension. Br. J. Haematol 129(4), 449–464. [DOI] [PubMed] [Google Scholar]
- Man Y, An R, Monchamp K, Sekyonda Z, Kucukal E, Federici C, Wulftange WJ, Goreke U, Bode A, Sheehan V, Gurkan UA, 2022. OcclusionChip: a functional microcapillary occlusion assay complementary to ektacytometry for detection of small-fraction red blood cells with abnormal deformability. Front. Physiol 13, 954106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Y, Goreke U, Kucukal E, Hill A, An R, Liu S, Bode A, Solis-Fuentes A, Nayak LV, Little JA, Gurkan UA, 2020a. Leukocyte adhesion to P-selectin and the inhibitory role of Crizanlizumab in sickle cell disease: A standardized microfluidic assessment. Blood Cells Mol. Dis 83, 102424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Y, Kucukal E, An R, Bode A, Little JA, Gurkan UA, 2020b. Standardized microfluidic assessment of red blood cell mediated microcapillary occlusion: association with clinical phenotype and hydroxyurea responsiveness in sickle cell disease. Microcirculation 28(2), e12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Y, Kucukal E, An R, Watson Q, Bosch J, Zimmerman PA, Little JA, Gurkan UA, 2020c. Microfluidic assessment of red blood cell mediated microvascular occlusion. Lab Chip 20(12), 2086–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Y, Maji D, An R, Ahuja SP, Little JA, Suster MA, Mohseni P, Gurkan UA, 2021. Microfluidic electrical impedance assessment of red blood cell-mediated microvascular occlusion. Lab Chip 21(6), 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwani D, Frenette PS, 2013. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood 122(24), 3892–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEver RP, 2015. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res 107(3), 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikis VA, Hernandez AA, Magnani JL, Sperandio M, Simon SI, 2021. Targeting Neutrophil Adhesive Events to Address Vaso-Occlusive Crisis in Sickle Cell Patients. Front Immunol 12, 663886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasimuzzaman M, Malik P, 2019. Role of the coagulation system in the pathogenesis of sickle cell disease. Blood Adv 3(20), 3170–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noomuna P, Risinger M, Zhou S, Seu K, Man Y, An R, Sheik DA, Wan J, Little JA, Gurkan UA, Turrini FM, Kalfa T, Low PS, 2020. Inhibition of Band 3 tyrosine phosphorylation: a new mechanism for treatment of sickle cell disease. Br. J. Haematol 190(4), 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou DP, Abidi SZ, Chang HY, Li X, Kato GJ, Karniadakis GE, Suresh S, Dao M, 2018. Simultaneous polymerization and adhesion under hypoxia in sickle cell disease. Proc. Natl. Acad. Sci. U. S. A 115(38), 9473–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telen MJ, Malik P, Vercellotti GM, 2019. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat. Rev. Drug Discov 18(2), 139–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L, Chen A, Delahunty MD, Moore KL, Watson SR, McEver RP, Tedder TF, 1996. L-selectin binds to P-selectin glycoprotein ligand-1 on leukocytes: interactions between the lectin, epidermal growth factor, and consensus repeat domains of the selectins determine ligand binding specificity. J Immunol 157(9), 3995–4004. [PubMed] [Google Scholar]
- Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS, 2002. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc. Natl. Acad. Sci. U. S. A 99(5), 3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER, 2005. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med 201(1), 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW, 2011. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 32(10), 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winning S, Splettstoesser F, Fandrey J, Frede S, 2010. Acute hypoxia induces HIF-independent monocyte adhesion to endothelial cells through increased intercellular adhesion molecule-1 expression: the role of hypoxic inhibition of prolyl hydroxylase activity for the induction of NF-kappa B. J. Immunol 185(3), 1786–1793. [DOI] [PubMed] [Google Scholar]
- Zarbock A, Ley K, 2008. Mechanisms and consequences of neutrophil interaction with the endothelium. Am. J. Pathol 172(1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Xu C, Manwani D, Frenette PS, 2016. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 127(7), 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöllner O, Lenter MC, Blanks JE, Borges E, Steegmaier M, Zerwes H-G, Vestweber D, 1997. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J. Cell Biol 136(3), 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zund G, Nelson DP, Neufeld EJ, Dzus AL, Bischoff J, Mayer JE, Colgan SP, 1996. Hypoxia enhances stimulus-dependent induction of E-selectin on aortic endothelial cells. Proc. Natl. Acad. Sci. U. S. A 93(14), 7075–7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.